

Abstract
A highly efficient palladium-catalyzed decarbonylative dehydration reaction of carboxylic acids is reported. This method transforms abundant and renewable even-numbered natural fatty acids into valuable and expensive odd-numbered alpha olefins. Additionally, the chemistry displays a high functional group tolerance. The process employs low loading of palladium catalyst and proceeds under solvent-free and relatively mild conditions.
Keywords: fatty acids, decarbonylative dehydration, linear alpha olefins, renewable feedstocks, palladium, large scale process
Linear alpha olefins represent an important class of industrial chemicals with a wide range of applications. They are used as co-monomers for ethylene polymerization[1] as well as precursors to plasticizers, lubricants, and surfactants.[2] Currently, these olefins are mainly produced by oligomerization of ethylene,[3] which, in turn, is derived from petroleum. As the world's oil reserves continue to diminish, development of renewable feedstocks for the production of alpha olefins becomes increasingly important. One obvious choice is ethylene from biomass-derived ethanol.[4] A potentially more direct method is the decarbonylative dehydration of long chain fatty acids. The latter route is particularly attractive because fatty acids are inexpensive and readily available starting materials derived from many natural sources. Since natural fatty acids contain an even number of carbon atoms, their corresponding alpha olefins will be odd-numbered after decarbonylative dehydration. Moreover, conventional ethylene oligomerization processes deliver only even-numbered alpha olefins,[3] and odd-numbered olefins are largely inaccessible and prohibitively expensive.[5] These odd-numbered olefins are valuable building blocks in the synthesis of various fine chemicals such as lepidopteran insect pheromones, but are currently far too costly to be practical.[6] Therefore, the development of an efficient and economic process for fatty acid decarbonylative dehydration is highly desirable.
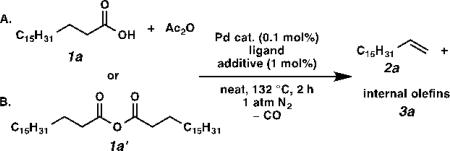
Many strategies to convert fatty acids to alpha olefins have been pursued. Lead tetraacetate-mediated oxidative decarboxylation is a classical method.[7] Alternative protocols that avoid stoichiometric toxic reagents have also been developed, such as Kolbe electrolysis[8] and silver-catalyzed oxidative decarboxylation.[9] However, these reactions proceed through highly reactive radical intermediates, and thus suffer from low yields due to many side reactions. A more recent approach entails the transition metal-catalyzed decarbonylative dehydration of fatty acids. A variety of transition metals including rhodium,[10] iridium,[11] palladium,[12] and iron[13] have been shown to catalyze decarbonylative dehydration reactions. To date, palladium has demonstrated the highest activity, and catalyst loadings as low as 0.01 mol% have been reported independently by Miller[12a] and Kraus[12b] (Scheme 1A). Unfortunately, their methods require very high temperatures (230–250 °C). In addition, it is necessary to distill the olefin product from the reaction mixture as soon as it is formed in order to prevent double bond isomerization, and therefore only volatile olefins can be produced this way. Decarbonylation processes under milder conditions have been developed independently by Gooßen[12c] and Scott[12d] (Scheme 1B). Although their reactions proceed at 110 °C, much higher palladium catalyst loading (3 mol%) and an expensive, high-boiling-point solvent (DMPU) are required. We envisioned that by judicious choice of ligand set for palladium and other parameters, the most advantageous aspects of these two systems could be combined. Herein we report a palladium-catalyzed decarbonylative dehydration using low catalyst loading under relatively mild and solvent-free conditions to produce alpha olefins in good yield and high selectivity (Scheme 1C).
Scheme 1.

Palladium-catalyzed decarbonylative dehydration. (A) High temperature processes (Miller, Kraus). (B) Low temperature processes (Gooßen, Scott). (C) This research.
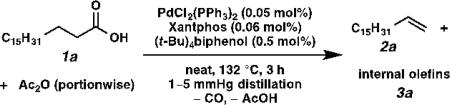
At the outset of our study, we examined the palladium-catalyzed decarbonylative dehydration of stearic acid 1a in neat acetic anhydride as the dehydrating agent (Table 1). A preliminary survey of phosphine ligands revealed Xantphos to be an optimal and unique ligand for the transformation (entries 1–4). It is believed that acetic anhydride converts the stearic acid into stearic anhydride in situ, which then undergoes oxidative addition by Pd(0) to initiate the catalytic cycle.[12] Considering that acetic anhydride itself could compete with stearic anhydride for oxidative addition at the metal center, we sought to simplify the system by using pre-formed stearic anhydride 1a’ alone. To our surprise, the decarbonylation of neat stearic anhydride with the same catalyst was exceedingly slow (only 12% yield, 120 TON, in 2 h; entry 5). Comparing the two systems, we found that the former had one equivalent of acid in it, while the latter was acid-free. Thus, we posited that acid might play a role in promoting reactivity. Consequently we added 1 mol% isophthalic acid to the system, and the yield rose to 22% (entry 6). When the ligand-to-metal ratio was reduced from 4:1 to 1.2:1, the yield dramatically increased to 92%, but the selectivity dropped to 31% (entry 7). Since it has been shown previously that triphenylphosphine can inhibit olefin isomerization,[12a] we used PdCl2(PPh3)2 instead of PdCl2(nbd) (nbd = norbornadiene), and we were delighted to observe an increase in selectivity to 54% with negligible erosion in yield (90%; entry 8). Finally, we examined a number of protic additives (entries 9–12), and found that (t-Bu)4biphenol gave the optimal overall performance to furnish the highest yield of alpha olefin (entry 12).
Table 1.
Effects of catalyst, ligand, and additive.[a]
| |||||||
|---|---|---|---|---|---|---|---|
| Entry | Rxn | Pd cat. | Ligand (mol%) | Additive | Yield (%)[b] | Alpha (%)[b] | Y × A (%)[c] |
| 1 | A | PdCl2(nbd) | PPh3 (0.8) | -- | 0 | -- | 0 |
| 2 | A | PdCl2(nbd) | dppp (04) | -- | 0 | -- | 0 |
| 3 | A | PdCl2(nbd) | DPEphos (0.4) | -- | 43 | 59 | 25 |
| 4 | A | PdCl2(nbd) | Xantphos (0.4) | -- | 60 | 55 | 33 |
| 5 | B | PdCl2(nbd) | Xantphos (0.4) | -- | 12 | 100 | 12 |
| 6 | B | PdCl2(nbd) | Xantphos (0.4) | isophthalic acid | 22 | 96 | 21 |
| 7 | B | PdCl2(nbd) | Xantphos (0.12) | isophthalic acid | 92 | 31 | 29 |
| 8 | B | PdCl2(PPh3)2 | Xantphos (0.12) | isophthalic acid | 90 | 54 | 49 |
| 9 | B | PdCl2(PPh3)2 | Xantphos (0.12) | p-TsOH·H2O | 86 | 5 | 4 |
| 10 | B | PdCl2(PPh3)2 | Xantphos (0.12) | salicylamide | 60 | 90 | 54 |
| 11 | B | PdCl2(PPh3)2 | Xantphos (0.12) | 2,2′-biphenol | 59 | 91 | 54 |
| 12 | B | PdCl2(PPh3)2 | Xantphos (0.12) | (t-Bu)4biphenol | 84 | 70 | 59 |
Conditions: A) 1 equiv 1a (5 mmol), 2 equiv Ac2O; B) 1 equiv 1a′ (5 mmol).
Determined by 1H NMR with methyl benzoate as internal standard. Alpha = 2a/(2a+3a).
Y × A = Yield × Alpha.
One major limitation of using pre-formed stearic anhydride as the substrate is that only half of the molecule can be converted to the olefin while the other half is wasted as stearic acid, so the maximum theoretical yield based on the acid is 50%. Control experiments also revealed that the buildup of acid in the reaction mixture was responsible for olefin isomerization and erosion of alpha selectivity. We envisioned that portionwise addition of acetic anhydride to the reaction mixture and distillation of acetic acid in situ could transform stearic acid back into its anhydride, thereby reducing acid concentration and increasing the yield and selectivity (Table 2). When two portions of acetic anhydride were added to the reaction mixture with immediate distillation of acetic acid, once every 1.5 hours, the olefin product was obtained in 69% yield based on stearic acid and 62% selectivity (entry 1). Furthermore, when three portions of acetic anhydride were added, once every hour, the olefin product was obtained in 67% yield and 86% alpha selectivity (entry 2). Finally, when six portions of acetic anhydride were added, once every half hour, the olefin product was obtained in 67% isolated yield and 89% alpha selectivity (entry 3). The selectivity trend clearly shows that the more frequently acetic anhydride is added to dehydrate stearic acid, the higher the alpha selectivity. This is a remarkable result in that it is possible to maintain high selectivity without having to distill the olefin product out of the reaction mixture as it forms.
Table 2.
Portionwise addition of acetic anhydride.[a]
| |||
|---|---|---|---|
| Entry | Equiv of Ac2O | Yield (%)[b] | Alpha (%)[b] |
| 1 | 1+0.5 (once every 1.5 hours) | 69 | 62 |
| 2 | 1+0.5+0.25 (once every hour) | 67 | 86 |
| 3 | 1+0.14+0.12+0.1+0.09+0.08 (once every half hour) | 68 (67) | 8 |
20 mmol 1a.
Determined by 1H NMR (isolated yield in parentheses).
With the optimized conditions in hand, we explored the decarbonylative dehydration of a variety of fatty acid substrates, as shown in Table 3. Common saturated fatty acids with carbon numbers from 12 to 18 all provided the corresponding olefin in good yield and high alpha selectivity (entries 1–4). In particular, volatile olefins were formed with exceptionally high selectivities (entries 3 and 4). Terminally functionalized fatty acids were also competent substrates, and functional groups such as esters, chlorides, imides, silyl ethers, ketones, terminal olefins, and substituted aromatics were all well tolerated (entries 5–13). Notably, allylbenzene derivative 2k was formed with 91% alpha selectivity (entry 11), which is impressive considering the significant thermodynamic driving force for isomerization into conjugation with the aromatic ring. Carboxylic acids with α- or β-substituents were considerably less reactive (entries 14–16). Nevertheless, catalyst turnovers up to 400 could be achieved for these challenging substrates. Compared to previous reports by Miller[12a] and Kraus,[12b] our reaction exhibits a much broader scope, does not require distillation of the olefin products to maintain high selectivity, and is compatible with various heteroatom-containing functional groups.
Table 3.
Substrate scope study.[b]
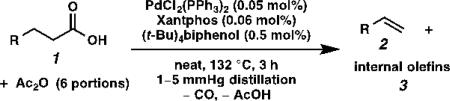
| |||||
|---|---|---|---|---|---|
| Entry | Substrate | Product | Yield (%)[b] | TON | Alpha (%)[c] |
| 1 |
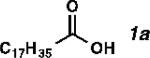
|
|
67 | 1340 | 89 |
| 2 |
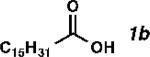
|
|
41 | 820 | 97 |
| 3 |
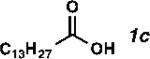
|
|
65 | 1300 | 99 |
| 4 |
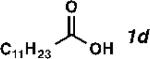
|
|
73 | 1460 | 99 |
| 5[d,e] |
|
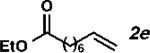
|
63 | 1260 | 98 |
| 6[d] |
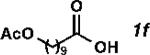
|
|
67 | 1340 | 96 |
| 7[d] |
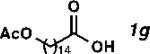
|
|
60 | 1200 | 89 |
| 8 |
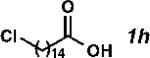
|
|
75 | 1500 | 86 |
| 9 |
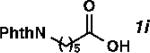
|
|
76 | 1520 | 83 |
| 10 |
|
|
64 | 1280 | 80 |
| 11 |
|
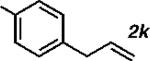
|
80 | 1600 | 91 |
| 12 |
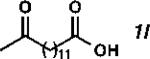
|
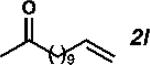
|
49 | 980 | 88 |
| 13 |
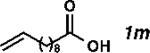
|
|
59 | 1180 | 87 |
| 14[f] |
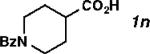
|
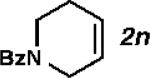
|
20 80[h] |
400 320[h] |
--[g] --[h] |
| 15 |
|
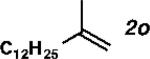
|
19 | 380 | --[g] |
| 16[i] |
|
|
71 | 71 | --[i] |
Conditions: 20 mmol 1, 6 portions of Ac2O, 1+0.14+0.12+0.10+0.09+0.08 equiv, added every 30 min.
Isolated yield (column chromatography).
Determined by 1H NMR.
Purified by distillation.
18.5 mmol 1e.
PdCl2(nbd) (0.05 mol%), PPh3 (0.05 mol%), Xantphos (0.06 mol%), 1.5 h, 3 portions of Ac2O (1+0.15+0.10 equiv).
Single isomer observed.
PdCl2(PPh3)2 (0.25 mol%), Xantphos (0.30 mol%), (t-Bu)4biphenol (1 mol%), 2n:3n = 49:51.
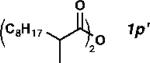
2-methyldecanoic anhydride (10 mmol), no Ac2O, PdCl2(nbd) (1 mol%), Xantphos (1.1 mol%), salicylamide (2 mol%), 160 °C, 10 mmHg distillation, 10 h, 3p:2p = 73:27.
Since this process requires no solvent and low catalyst loading, it can be readily scaled up (Scheme 2). In a laboratory setting, a 100 mmol scale (28.4 g stearic acid or 23.0 g 10-acetoxydecanoic acid) decarbonylative dehydration was easily carried out in a 100 mL round-bottom flask. Compared with small-scale reactions, the large-scale ones afforded products in similar yields and slightly higher alpha selectivity. When the olefin is sufficiently volatile, it is distilled out together with the acetic acid (Table 3, entries 3–6, 11, and 13). Although distillation of olefin is not necessary in order to maintain high selectivity (e.g. Scheme 2A), it is convenient to do so for volatile olefins (Scheme 2B).
Scheme 2.

Large-scale decarbonylative dehydration of stearic acid and 10-acetoxydecanoic acid.
The olefin products thus obtained are important building blocks in chemical synthesis. For example, 8-nonenyl acetate (2f) is a precursor to insect pheromones 4a (Oriental fruit moth pheromone) [14] and 4b (Figure-of-Eight moth pheromone).[15] Aided by Z-selective cross metathesis catalyst 5, olefin 2f reacts with 1-pentene or 1-hexene to afford pheromones 4a or 4b in 58% and 48% yield, respectively, and >98% Z-selectivity (Table 4).[6b]
Table 4.
Synthesis of pheromones 4a and 4b from olefin 2f.
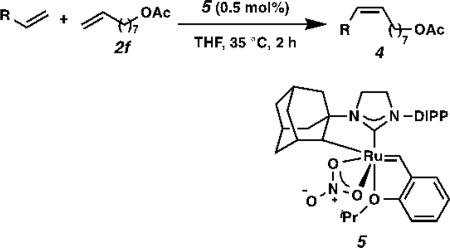
4.8 mmol 2f.
Determined by 1H NMR.
In summary, we have developed a highly efficient palladium-catalyzed decarbonylative dehydration process that converts carboxylic acids (e.g. fatty acids) to linear alpha olefins in good yield and with high selectivity. The reaction requires low palladium catalyst loading and proceeds under solvent-free and relatively mild conditions. In situ distillation of the olefin product is not necessary, and a wide range of functionalized and unfunctionalized carboxylic acids can be transformed into their corresponding olefins. Process development for large-scale production is underway and will be reported in due course.
Experimental Section
A 15 mL round-bottom flask was charged with PdCl2(PPh3)2 (0.01 mmol, 0.05 mol%), Xantphos (0.012 mmol, 0.06 mol%), (t-Bu)4biphenol (0.1 mmol, 0.5 mol%), and fatty acid substrate (20 mmol, 1 equiv). The flask was equipped with a distillation head and a 25 mL round-bottom receiving flask. The closed system was connected to a vacuum manifold, equipped with a needle valve and a digital vacuum gauge. The system was evacuated and refilled with N2 (x 3), and the first portion of acetic anhydride (20 mmol, 1 equiv) was added via syringe through the septum that seals the top of the distillation head. The flask was lowered into a 20 °C oil bath and gradually heated to 132 °C in 23 min. When oil bath temperature rose to 122 °C, the needle valve was closed, switched to vacuum, and the needle valve carefully and slowly opened to allow distillation of acetic acid into a receiving flask, which was cooled to –78 °C. When the oil bath temperature reached 130 °C, time was recorded as t = 0. After distillation ceased (about t = 3 min), the needle valve was opened fully and a vacuum of 1–5 mmHg was drawn. At t = 30 min, the system was refilled with N2, and the second portion of acetic anhydride (2.8 mmol, 0.14 equiv) was added via syringe. The system was then gradually (t = 35 min) resubjected to a vacuum of 1–5 mmHg. Acetic anhydride was added as follows (0.12, 0.10, 0.09. 0.08 equiv) in the same manner every 30 min. The reaction was stopped at t = 3 h and allowed to cool to ambient temperature. The residual reaction mixture was purified by flash chromatography. If it contained solids, it was suction-filtered first and the solids washed with hexanes, and the filtrate was concentrated and purified by chromatography. In cases where the product was distilled together with acetic acid, the distillate was added dropwise to a saturated NaHCO3 solution, stirred for 30 min, and the resulting mixture was extracted with dichloromethane (30 mL x 3). The combined extracts were dried over Na2SO4, filtered and concentrated. The crude product was then subjected to flash chromatography or distillation to afford the olefin in pure form.
Supplementary Material
Acknowledgements
The authors wish to thank the Resnick Sustainability Institute at Caltech (graduate fellowship to Y.L.), BP (postdoctoral fellowship to A.F. under the XC2 program), NIH-NIGMS (R01GM080269, B.M.S.), NIH (NIH 5R01GM031332-27, R.H.G.), the Gordon and Betty Moore Foundation, the Caltech Center for Catalysis and Chemical Synthesis, and Caltech for financial support. Materia, Inc. is thanked for the generous donation of ruthenium catalysts. Dr. David VanderVelde is acknowledged for NMR spectroscopy assistance. Dr. Mona Shahgholi and Naseem Torian are acknowledged for high-resolution mass spectrometry assistance. Dr. Scott Virgil is acknowledged for helpful discussions.
Footnotes
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/adsc.201######. ((Please delete if not appropriate))
References
- 1.Ittel SD, Johnson LK, Brookhart M. Chem. Rev. 2000;100:1169–1203. doi: 10.1021/cr9804644. [DOI] [PubMed] [Google Scholar]
- 2.a Marquis DM, Sharman SH, House R, Sweeney WA. J. Am. Oil Chem. Soc. 1966;43:607–614. [Google Scholar]; b Franke R, Selent D, Börner A. Chem. Rev. 2012;112:5675–5732. doi: 10.1021/cr3001803. [DOI] [PubMed] [Google Scholar]
- 3.a Al-Jarallah AM, Anabtawi JA, Siddiqui MAB, Aitani AM, Al-Sa'doun AW. Catal. Today. 1992;14:1–121. [Google Scholar]; b Small BL, Brookhart M. J. Am. Chem. Soc. 1998;120:7143–7144. [Google Scholar]
- 4.Ouyang J, Kong F, Su G, Hu Y, Song Q. Catal. Lett. 2009;132:64–74. [Google Scholar]
-
5.Prices of acids and olefins (Sigma-Aldrich, 10/5/2013):
Acid $/kg Olefin $/kg Decanoic (C10) 16.28 1-Nonene (C9) 12,080.00 1-Decene (C10) 55.87 Laurie (C12) 14.20 1-Undecene (C11) 7,240.00 1-Dodecene (C12) 187.60 Myristic (C14) 11.76 1-Tridecene (C13) 23,800.00 1-Tetradecene (C14) 47.74 Palmitic (C16) 7.68 1-Pentadecene (C15) 24,722.58 1-Hexadecene (C16) 2,758.62 Stearic (C18) 13.20 1-Heptadecene (C17) 69,500.00 - 6.a Herbert MB, Marx VM, Pederson RL, Grubbs RH. Angew. Chem. 2013;125:328–332. doi: 10.1002/anie.201206079. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2013;52:310–314. [Google Scholar]; b Rosebrugh LE, Herbert MB, Marx VM, Keitz BK, Grubbs RH. J. Am. Chem. Soc. 2013;135:1276–1279. doi: 10.1021/ja311916m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bacha JD, Kochi JK. Tetrahedron. 1968;24:2215–2226. [Google Scholar]
- 8.Kolbe H. Justus Liebigs Ann. Chem. 1849;69:257–294. [Google Scholar]
- 9.a Anderson JM, Kochi JK. J. Org. Chem. 1969;35:986–989. [Google Scholar]; b van der Klis F, van den Hoorn MH, Blaauw R, van Haveren J, van Es DS. Eur. J. Lipid Sci. Technol. 2011;113:562–571. [Google Scholar]
- 10.Foglia TA, Barr PA, Am J. Oil Chem. Soc. 1976;53:737–741. doi: 10.1007/BF02637388. [DOI] [PubMed] [Google Scholar]
- 11.Maetani S, Fukuyama T, Suzuki N, Ishihara D, Ryu I. Organometallics. 2011;30:1389–1394. [Google Scholar]
- 12.a Miller JA, Nelson JA, Byrne MP. J. Org. Chem. 1993;58:18–20. [Google Scholar]; b Kraus GA, Riley S. Synthesis. 2012;44:3003–3005. [Google Scholar]; c Gooßen LJ, Rodríguez N. Chem. Commun. 2004:724–725. doi: 10.1039/b316613a. [DOI] [PubMed] [Google Scholar]; d Le Nôtre J, Scott EL, Franssen MCR, Sanders JPM. Tetrahedron Lett. 2010;51:3712–3715. [Google Scholar]; e Miranda MO, Pietrangelo A, Hillmyer MA, Tolman WB. Green Chem. 2012;14:490–494. [Google Scholar]
- 13.Maetani S, Fukuyama T, Suzuki N, Ishihara D, Ryu I. Chem. Commun. 2012;48:2552–2554. doi: 10.1039/c2cc18093f. [DOI] [PubMed] [Google Scholar]
- 14.Baker TC, Cardé RT, Miller JR. J. Chem. Ecol. 1980;6:749–758. [Google Scholar]
- 15.Subchev M, Toth M, Wu D, Stanimirova L, Toshova T, Karpati Z. J. Appl. Ent. 2000;124:197–199. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.