

Abstract
In the title compound, C17H18Cl3NO5·H2O, intramolecular N—H⋯O and C—H⋯Cl hydrogen bonds form S(6) and S(5) ring motifs, respectively. The chiral organic molecule is connected to the solvent water molecule by a short O—H⋯O hydrogen bond. In the crystal, a weak C—H⋯Cl interaction connects the organic molecules along [100] while the water molecules act as bridges between the organic molecules in both the [100] and [010] directions, generating layers parallel to the ab plane.
Related literature
For the synthesis of the title compound and a similar crystal structure, see: Flores et al. (2008 ▶). For information about levulinic acid and the biological properties of its derivatives, see: Flores et al. (2013 ▶); Hachuła et al. (2013 ▶); Lo & Ng (2008 ▶). For short intermolecular hydrogen-bond interactions, see: Pojarová et al. (2010 ▶). For intramolecular hydrogen-bonding systems, see: da Costa et al. (2013 ▶).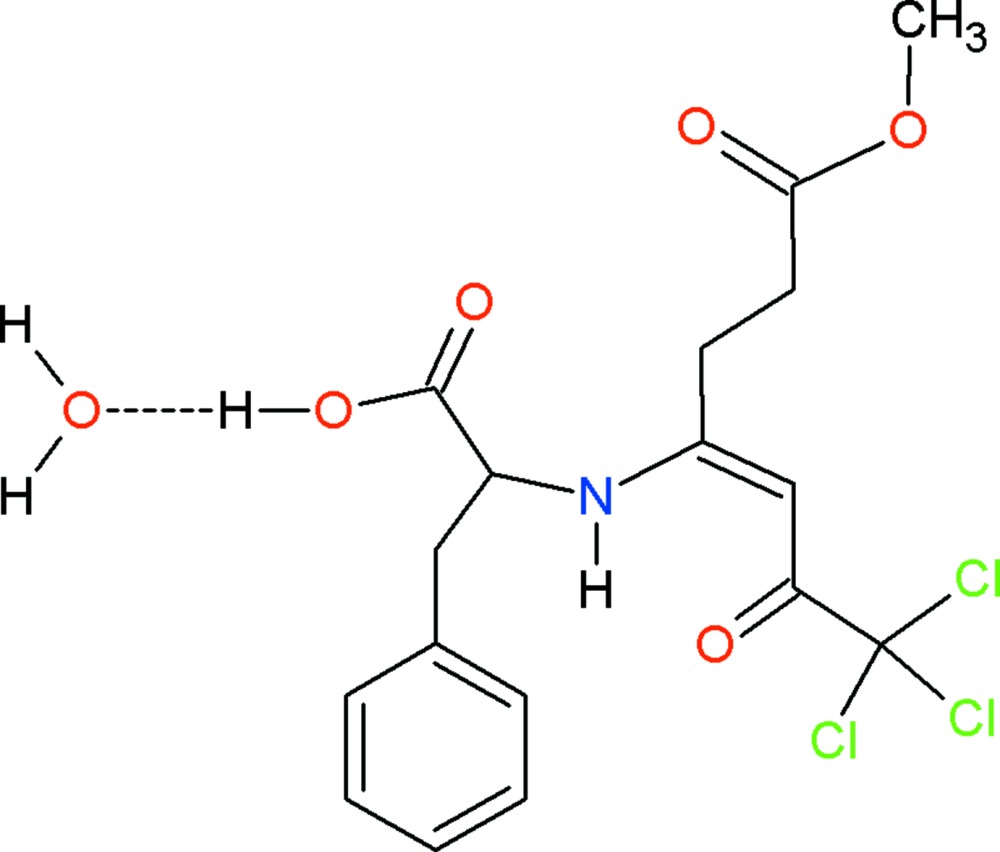
Experimental
Crystal data
C17H18Cl3NO5·H2O
M r = 440.69
Triclinic,
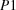
a = 5.6684 (16) Å
b = 8.601 (3) Å
c = 10.336 (3) Å
α = 87.720 (19)°
β = 85.696 (17)°
γ = 85.649 (17)°
V = 500.8 (2) Å3
Z = 1
Mo Kα radiation
μ = 0.49 mm−1
T = 296 K
0.98 × 0.30 × 0.12 mm
Data collection
Bruker APEXII CCD diffractometer
Absorption correction: Gaussian (XPREP; Bruker, 2006 ▶) T min = 0.881, T max = 1
13424 measured reflections
6020 independent reflections
4784 reflections with I > 2σ(I)
R int = 0.024
Refinement
R[F 2 > 2σ(F 2)] = 0.040
wR(F 2) = 0.105
S = 1.04
6020 reflections
256 parameters
3 restraints
H atoms treated by a mixture of independent and constrained refinement
Δρmax = 0.41 e Å−3
Δρmin = −0.32 e Å−3
Absolute structure: Flack parameter determined using 1984 quotients [(I +)−(I −)]/[(I +)+(I −)] (Parsons et al., 2013 ▶)
Absolute structure parameter: 0.04 (2)
Data collection: APEX2 (Bruker, 2009 ▶); cell refinement: SAINT (Bruker, 2009 ▶); data reduction: SAINT; program(s) used to solve structure: SHELXS97 (Sheldrick, 2008 ▶); program(s) used to refine structure: SHELXL2013 (Sheldrick, 2008 ▶); molecular graphics: DIAMOND (Brandenburg, 2006 ▶); software used to prepare material for publication: publCIF (Westrip, 2010 ▶).
Supplementary Material
Crystal structure: contains datablock(s) I. DOI: 10.1107/S1600536814000154/pk2509sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S1600536814000154/pk2509Isup2.hkl
Supporting information file. DOI: 10.1107/S1600536814000154/pk2509Isup3.cml
CCDC reference: http://scripts.iucr.org/cgi-bin/cr.cgi?rm=csd&csdid=979612
Additional supporting information: crystallographic information; 3D view; checkCIF report
Table 1. Hydrogen-bond geometry (Å, °).
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| C6—H6A⋯Cl1i | 0.97 | 2.94 | 3.774 (3) | 145 |
| O33—H33B⋯O91ii | 0.87 (6) | 1.89 (6) | 2.766 (4) | 177 (5) |
| N41—H41⋯O21 | 0.83 (5) | 2.05 (6) | 2.672 (3) | 131 (5) |
| O33—H33A⋯O21iii | 0.76 (6) | 2.06 (6) | 2.815 (3) | 171 (6) |
| O92—H92⋯O33 | 0.89 (5) | 1.66 (5) | 2.542 (4) | 175 (5) |
| C3—H3⋯Cl1 | 0.93 | 2.55 | 3.031 (3) | 112 |
Symmetry codes: (i) 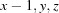 ; (ii)
; (ii) 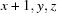 ; (iii)
; (iii) 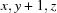 .
.
Acknowledgments
The authors are grateful for financial support from the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq, Universal grant 6577818477962764–01), the Fundação de Amparo à Pesquisa do Estado do Rio Grande do Sul (FAPERGS, PqG grant 1016236) and the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES-PROEX).
supplementary crystallographic information
1. Comment
Dielectrophiles derived from levulinic acid (Hachuła et al., 2013; Lo & Ng, 2008) belong to an important class of organic synthetic intermediates for the synthesis of a variety of heterocyclic compounds. Such precursors are used to produce pyrrolidinones, pyrrolones, pyrazoles and pyrimidines with very interesting biological activities (Flores et al., 2008; Flores et al., 2013). As a part of our studies, we report in this paper the crystal structure of (S,Z)-3-phenyl-2-(1,1,1-trichloro-7–2,7-dioxo-3-hepten- 4-ylamine)propanoic acid, obtained from the reaction between methyl 7,7,7-trichloro-4-methoxy-6-oxo-3-heptenoate and L-phenylalanine.
In the crystal structure of the title compound, the asymmetric unit is composed of the whole chiral organic molecule, C17H18Cl3NO5, connected to a water molecule (Fig.1). This connection consists of a short intermolecular hydrogen bond interaction involving the hydrogen atom of the carboxylic acid fragment [O92—H92···O33, 2.542 (4) Å; Pojarová et al., 2010]. Additionally, S(6) and S(5) ring motifs are formed by two distinct intramolecular hydrogen bonding systems, N41—H41···O21 [2.672 (3) Å] and C3—H3···Cl1 [3.031 (3) Å], respectively, thereby stabilizing the structure (da Costa et al., 2013).
There is also a weak C6—H6A···Cl1i intermolecular interaction [3.774 (3) Å] connecting organic molecules along the [100] crystallographic direction. The water molecules act as a bridging element in the crystal structure by expanding its dimensionality in both [100] and [010] crystallographic directions. The intermolecular hydrogen bond interactions generate bidimensional layers parallel to the ab plane. Each atom of the water molecule is connected to different groups on adjacent organic molecules: carboxylic acid [O92—H92···O33, 2.542 (4) Å and O33—H33B···O91ii, 2.766 (4) Å] and ketone [O33—H33A···O21iii, 2.815 (3) Å]. Symmetry codes: (i) x–1, y, z; (ii) x + 1, y, z; (iii) x, y + 1, z. A super cell central projection of the crystal structure can be viewed in Fig. 2, which depicts a crystal packing diagram as viewed along the crystallographic a axis.
2. Experimental
To a stirred solution of methyl 7,7,7-trichloro-4-methoxy-6-oxo-3-heptenoate (5 mmol, 1.52 g) and L-phenylalanine (5.5 mmol, 0.91 g), at 25 °C, was added a solution of 1 mol·L-1 NaOH. There was an immediate formation of a yellow precipitate and the mixture was further stirred for 30 minutes. A solution of 50% HCl was added until the pH ≈ 1, when there was complete precipitation of the yellow solid. The solid was extracted with ethyl acetate, and this solution was dried over anhydrous MgSO4. The ethyl acetate was removed on a rotary evaporator to give the product as a yellow solid. Yield: 79%. m. p. 120 – 123 °C. 1H NMR (400 MHz, DMSO-D6, TMS): δ 2.17 (m, 2H, CH2), 2.44 (m, 2H, CH2), 3.06 (dd, 1H, 3J=9.1 Hz, 2J=14 Hz, CH2Ph), 3.37 (dd, 1H, 3J=9.1 Hz, 2J=14 Hz, CH2Ph), 3.66 (s, 3H, OMe), 4.53 (m, 1H, CHchiral), 5.60 (s, 1H, =CH), 7.22–7.33 (m, 5H, Ph), 10.9 (d, 1H, 3J = 10 Hz, NH) p.p.m.; 13C NMR (100 MHz, DMSO-D6): δ 26.8, 31.5, 39.9, 52.2, 58.1, 86.0, 96.9, 127.5, 128.9, 129.5, 135.4, 169.9, 172.0, 173.2, 181.2 p.p.m.. Crystals were grown from a methanol solution, which was slowly evaporated at room temperature.
3. Refinement
All H atoms attached to carbon were positioned with idealized geometry and were refined isotropically. For H atoms of CH3 group, Uiso(H) was set to 1.5Ueq(C) using a riding model with C—H = 0.96 Å. For all remaining H atoms attached to C atoms, Uiso(H) was set to 1.2Ueq(C) using a riding model with the following C—H distances: C—H (CH) = 0.93 Å, C—H (CHchiral) = 0.98 Å and C—H (CH2) = 0.97 Å. H atoms attached to nitrogen, H atoms of the water molecule and the H atom of the carboxylic acid fragment were located in difference Fourier maps, and were refined with Uiso values set to 1.5Ueq of the parent atom. Reflections (001) and (001) were omitted due to the large difference observed between Fo2 and Fc2.
Figures
Fig. 1.

An ellipsoid plot (50% probability) showing the asymmetric unit. Hydrogen bonds are represented as dashed lines. Symmetry codes: (i) x–1, y, z; (ii) x + 1, y, z; (iii) x, y + 1, z.
Fig. 2.

Packing of molecules along the [100] direction through intermolecular hydrogen bonds, represented with dashed lines. Some hydrogen atoms were omitted for clarity.
Crystal data
| C17H18Cl3NO5·H2O | F(000) = 228 |
| Mr = 440.69 | Dx = 1.461 Mg m−3 |
| Triclinic, P1 | Melting point: 393 K |
| a = 5.6684 (16) Å | Mo Kα radiation, λ = 0.71073 Å |
| b = 8.601 (3) Å | Cell parameters from 3866 reflections |
| c = 10.336 (3) Å | θ = 3.0–25.5° |
| α = 87.720 (19)° | µ = 0.49 mm−1 |
| β = 85.696 (17)° | T = 296 K |
| γ = 85.649 (17)° | Blade, colorless |
| V = 500.8 (2) Å3 | 0.98 × 0.30 × 0.12 mm |
| Z = 1 |
Data collection
| Bruker APEXII CCD diffractometer | 6020 independent reflections |
| Radiation source: fine-focus sealed tube | 4784 reflections with I > 2σ(I) |
| Graphite monochromator | Rint = 0.024 |
| φ and ω scans | θmax = 30.7°, θmin = 2.4° |
| Absorption correction: gaussian (XPREP; Bruker, 2006) | h = −8→8 |
| Tmin = 0.881, Tmax = 1 | k = −12→12 |
| 13424 measured reflections | l = −14→14 |
Refinement
| Refinement on F2 | Hydrogen site location: mixed |
| Least-squares matrix: full | H atoms treated by a mixture of independent and constrained refinement |
| R[F2 > 2σ(F2)] = 0.040 | w = 1/[σ2(Fo2) + (0.0502P)2 + 0.0376P] where P = (Fo2 + 2Fc2)/3 |
| wR(F2) = 0.105 | (Δ/σ)max < 0.001 |
| S = 1.04 | Δρmax = 0.41 e Å−3 |
| 6020 reflections | Δρmin = −0.32 e Å−3 |
| 256 parameters | Absolute structure: Flack parameter determined using 1984 quotients [(I+)-(I-)]/[(I+)+(I-)] (Parsons et al., 2013) |
| 3 restraints | Absolute structure parameter: 0.04 (2) |
Special details
| Experimental. Absorption correction: XPREP (Bruker, 2006) was used to perform the Gaussian absorption correction based on the face-indexed crystal size. |
| Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| O33 | 0.4813 (5) | 1.0147 (3) | 0.9230 (3) | 0.0554 (6) | |
| H33B | 0.629 (11) | 0.977 (6) | 0.919 (5) | 0.083* | |
| H41 | 0.147 (9) | 0.465 (6) | 0.794 (5) | 0.083* | |
| H33A | 0.458 (10) | 1.092 (7) | 0.887 (5) | 0.083* | |
| H92 | 0.352 (9) | 0.869 (6) | 0.864 (5) | 0.083* | |
| Cl1 | 0.75049 (16) | 0.24572 (10) | 0.42096 (9) | 0.0652 (3) | |
| Cl2 | 0.50084 (18) | 0.01516 (11) | 0.57338 (11) | 0.0759 (3) | |
| Cl3 | 0.88358 (15) | 0.16349 (14) | 0.67617 (11) | 0.0742 (3) | |
| N41 | 0.1053 (4) | 0.5403 (3) | 0.7462 (2) | 0.0403 (5) | |
| C1 | 0.6395 (5) | 0.1924 (3) | 0.5785 (3) | 0.0432 (6) | |
| C7 | −0.1436 (5) | 0.7761 (4) | 0.3642 (3) | 0.0424 (6) | |
| C9 | 0.0625 (5) | 0.7901 (3) | 0.8625 (3) | 0.0439 (6) | |
| C3 | 0.3699 (5) | 0.4434 (3) | 0.5739 (3) | 0.0412 (6) | |
| H3 | 0.4282 | 0.4569 | 0.4880 | 0.049* | |
| C10 | −0.0576 (5) | 0.6530 (3) | 0.8145 (3) | 0.0402 (6) | |
| H10 | −0.1704 | 0.6956 | 0.7526 | 0.048* | |
| C5 | 0.1234 (5) | 0.6912 (3) | 0.5398 (3) | 0.0414 (6) | |
| H5A | 0.2526 | 0.7135 | 0.4764 | 0.050* | |
| H5B | 0.0854 | 0.7828 | 0.5913 | 0.050* | |
| C111 | −0.0514 (5) | 0.4758 (4) | 1.0193 (3) | 0.0455 (6) | |
| C2 | 0.4540 (5) | 0.3128 (3) | 0.6460 (3) | 0.0386 (5) | |
| C6 | −0.0917 (6) | 0.6567 (4) | 0.4700 (3) | 0.0472 (7) | |
| H6A | −0.0645 | 0.5545 | 0.4328 | 0.057* | |
| H6B | −0.2285 | 0.6543 | 0.5321 | 0.057* | |
| C11 | −0.2023 (5) | 0.5712 (4) | 0.9264 (3) | 0.0475 (7) | |
| H11A | −0.3005 | 0.6496 | 0.9745 | 0.057* | |
| H11B | −0.3071 | 0.5033 | 0.8897 | 0.057* | |
| C112 | 0.0981 (7) | 0.5453 (4) | 1.0959 (3) | 0.0584 (8) | |
| H112 | 0.1040 | 0.6532 | 1.0916 | 0.070* | |
| C8 | −0.4279 (7) | 0.8699 (5) | 0.2188 (4) | 0.0649 (10) | |
| H8A | −0.5833 | 0.8501 | 0.1953 | 0.097* | |
| H8B | −0.3165 | 0.8573 | 0.1446 | 0.097* | |
| H8C | −0.4299 | 0.9745 | 0.2481 | 0.097* | |
| C116 | −0.0616 (8) | 0.3156 (4) | 1.0306 (4) | 0.0638 (9) | |
| H116 | −0.1627 | 0.2664 | 0.9813 | 0.077* | |
| C115 | 0.0793 (10) | 0.2279 (5) | 1.1156 (4) | 0.0792 (13) | |
| H115 | 0.0718 | 0.1202 | 1.1222 | 0.095* | |
| C113 | 0.2391 (9) | 0.4563 (6) | 1.1789 (4) | 0.0734 (11) | |
| H113 | 0.3422 | 0.5039 | 1.2280 | 0.088* | |
| C114 | 0.2259 (9) | 0.2964 (6) | 1.1884 (4) | 0.0774 (13) | |
| H114 | 0.3183 | 0.2363 | 1.2450 | 0.093* | |
| O72 | −0.3587 (4) | 0.7608 (3) | 0.3218 (2) | 0.0535 (5) | |
| O92 | 0.2915 (4) | 0.7852 (3) | 0.8366 (3) | 0.0554 (5) | |
| O91 | −0.0517 (4) | 0.8944 (3) | 0.9190 (3) | 0.0591 (6) | |
| O71 | −0.0124 (5) | 0.8698 (3) | 0.3220 (3) | 0.0637 (7) | |
| C4 | 0.2004 (5) | 0.5547 (3) | 0.6269 (3) | 0.0370 (5) | |
| O21 | 0.3934 (4) | 0.2806 (2) | 0.7613 (2) | 0.0473 (5) |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| O33 | 0.0494 (13) | 0.0492 (13) | 0.0673 (15) | −0.0017 (11) | −0.0076 (11) | 0.0051 (11) |
| Cl1 | 0.0759 (6) | 0.0560 (5) | 0.0586 (5) | 0.0031 (4) | 0.0241 (4) | −0.0061 (4) |
| Cl2 | 0.0741 (6) | 0.0476 (4) | 0.1050 (8) | −0.0204 (4) | 0.0308 (5) | −0.0288 (5) |
| Cl3 | 0.0413 (4) | 0.0946 (7) | 0.0838 (6) | 0.0154 (4) | −0.0058 (4) | −0.0032 (5) |
| N41 | 0.0439 (13) | 0.0350 (11) | 0.0398 (12) | 0.0079 (10) | −0.0005 (9) | 0.0012 (9) |
| C1 | 0.0375 (14) | 0.0379 (14) | 0.0531 (16) | −0.0024 (11) | 0.0056 (11) | −0.0056 (11) |
| C7 | 0.0410 (14) | 0.0435 (15) | 0.0412 (14) | 0.0029 (12) | −0.0024 (11) | 0.0028 (11) |
| C9 | 0.0441 (14) | 0.0397 (14) | 0.0459 (15) | 0.0077 (11) | −0.0048 (11) | 0.0056 (12) |
| C3 | 0.0417 (14) | 0.0403 (14) | 0.0398 (13) | 0.0013 (11) | 0.0024 (10) | 0.0017 (11) |
| C10 | 0.0375 (13) | 0.0376 (13) | 0.0444 (14) | 0.0073 (11) | −0.0052 (11) | −0.0035 (11) |
| C5 | 0.0419 (14) | 0.0338 (13) | 0.0481 (15) | −0.0020 (10) | −0.0039 (11) | 0.0043 (11) |
| C111 | 0.0480 (15) | 0.0477 (16) | 0.0385 (13) | 0.0000 (13) | 0.0078 (12) | −0.0018 (12) |
| C2 | 0.0349 (12) | 0.0363 (13) | 0.0440 (14) | 0.0022 (10) | −0.0012 (10) | −0.0049 (10) |
| C6 | 0.0478 (16) | 0.0414 (15) | 0.0532 (16) | −0.0051 (12) | −0.0114 (13) | 0.0081 (13) |
| C11 | 0.0387 (14) | 0.0508 (17) | 0.0516 (16) | 0.0032 (12) | 0.0001 (12) | −0.0028 (13) |
| C112 | 0.075 (2) | 0.0510 (19) | 0.0489 (17) | −0.0038 (17) | −0.0081 (16) | 0.0032 (14) |
| C8 | 0.063 (2) | 0.081 (3) | 0.0495 (18) | 0.0133 (19) | −0.0149 (15) | 0.0064 (17) |
| C116 | 0.082 (3) | 0.0495 (19) | 0.059 (2) | −0.0072 (17) | 0.0021 (18) | −0.0027 (15) |
| C115 | 0.115 (4) | 0.049 (2) | 0.069 (3) | 0.007 (2) | 0.006 (3) | 0.0063 (18) |
| C113 | 0.084 (3) | 0.085 (3) | 0.052 (2) | −0.004 (2) | −0.0171 (19) | 0.0069 (19) |
| C114 | 0.093 (3) | 0.078 (3) | 0.055 (2) | 0.026 (2) | −0.002 (2) | 0.013 (2) |
| O72 | 0.0482 (12) | 0.0614 (14) | 0.0511 (12) | −0.0012 (10) | −0.0121 (9) | 0.0063 (10) |
| O92 | 0.0449 (12) | 0.0501 (13) | 0.0709 (14) | −0.0013 (10) | −0.0017 (10) | −0.0073 (11) |
| O91 | 0.0540 (13) | 0.0445 (12) | 0.0778 (16) | 0.0107 (10) | −0.0050 (11) | −0.0166 (11) |
| O71 | 0.0538 (14) | 0.0628 (15) | 0.0738 (16) | −0.0076 (11) | −0.0098 (12) | 0.0244 (12) |
| C4 | 0.0379 (12) | 0.0331 (13) | 0.0400 (13) | −0.0025 (10) | −0.0037 (10) | 0.0014 (10) |
| O21 | 0.0524 (12) | 0.0423 (11) | 0.0432 (11) | 0.0133 (9) | 0.0038 (8) | 0.0029 (9) |
Geometric parameters (Å, º)
| O33—H33B | 0.87 (6) | C5—H5B | 0.9700 |
| O33—H33A | 0.76 (6) | C111—C116 | 1.384 (5) |
| Cl1—C1 | 1.757 (3) | C111—C112 | 1.385 (5) |
| Cl2—C1 | 1.772 (3) | C111—C11 | 1.510 (4) |
| Cl3—C1 | 1.770 (3) | C2—O21 | 1.241 (4) |
| N41—C4 | 1.314 (4) | C6—H6A | 0.9700 |
| N41—C10 | 1.453 (3) | C6—H6B | 0.9700 |
| N41—H41 | 0.83 (5) | C11—H11A | 0.9700 |
| C1—C2 | 1.564 (4) | C11—H11B | 0.9700 |
| C7—O71 | 1.186 (4) | C112—C113 | 1.385 (5) |
| C7—O72 | 1.343 (4) | C112—H112 | 0.9300 |
| C7—C6 | 1.500 (4) | C8—O72 | 1.448 (4) |
| C9—O91 | 1.209 (4) | C8—H8A | 0.9600 |
| C9—O92 | 1.303 (4) | C8—H8B | 0.9600 |
| C9—C10 | 1.523 (4) | C8—H8C | 0.9600 |
| C3—C2 | 1.397 (4) | C116—C115 | 1.393 (6) |
| C3—C4 | 1.401 (4) | C116—H116 | 0.9300 |
| C3—H3 | 0.9300 | C115—C114 | 1.344 (7) |
| C10—C11 | 1.545 (4) | C115—H115 | 0.9300 |
| C10—H10 | 0.9800 | C113—C114 | 1.382 (7) |
| C5—C4 | 1.509 (4) | C113—H113 | 0.9300 |
| C5—C6 | 1.517 (4) | C114—H114 | 0.9300 |
| C5—H5A | 0.9700 | O92—H92 | 0.89 (5) |
| H33B—O33—H33A | 115 (6) | C7—C6—H6A | 109.2 |
| C4—N41—C10 | 126.9 (2) | C5—C6—H6A | 109.2 |
| C4—N41—H41 | 121 (4) | C7—C6—H6B | 109.2 |
| C10—N41—H41 | 112 (4) | C5—C6—H6B | 109.2 |
| C2—C1—Cl1 | 116.0 (2) | H6A—C6—H6B | 107.9 |
| C2—C1—Cl3 | 107.9 (2) | C111—C11—C10 | 113.8 (2) |
| Cl1—C1—Cl3 | 107.55 (16) | C111—C11—H11A | 108.8 |
| C2—C1—Cl2 | 107.0 (2) | C10—C11—H11A | 108.8 |
| Cl1—C1—Cl2 | 109.06 (17) | C111—C11—H11B | 108.8 |
| Cl3—C1—Cl2 | 109.22 (17) | C10—C11—H11B | 108.8 |
| O71—C7—O72 | 124.4 (3) | H11A—C11—H11B | 107.7 |
| O71—C7—C6 | 125.1 (3) | C111—C112—C113 | 120.9 (4) |
| O72—C7—C6 | 110.5 (2) | C111—C112—H112 | 119.6 |
| O91—C9—O92 | 124.1 (3) | C113—C112—H112 | 119.6 |
| O91—C9—C10 | 121.0 (3) | O72—C8—H8A | 109.5 |
| O92—C9—C10 | 114.9 (3) | O72—C8—H8B | 109.5 |
| C2—C3—C4 | 122.1 (3) | H8A—C8—H8B | 109.5 |
| C2—C3—H3 | 119.0 | O72—C8—H8C | 109.5 |
| C4—C3—H3 | 119.0 | H8A—C8—H8C | 109.5 |
| N41—C10—C9 | 113.6 (2) | H8B—C8—H8C | 109.5 |
| N41—C10—C11 | 110.4 (2) | C111—C116—C115 | 120.1 (4) |
| C9—C10—C11 | 111.3 (2) | C111—C116—H116 | 119.9 |
| N41—C10—H10 | 107.1 | C115—C116—H116 | 119.9 |
| C9—C10—H10 | 107.1 | C114—C115—C116 | 121.0 (4) |
| C11—C10—H10 | 107.1 | C114—C115—H115 | 119.5 |
| C4—C5—C6 | 110.9 (2) | C116—C115—H115 | 119.5 |
| C4—C5—H5A | 109.5 | C114—C113—C112 | 119.7 (4) |
| C6—C5—H5A | 109.5 | C114—C113—H113 | 120.1 |
| C4—C5—H5B | 109.5 | C112—C113—H113 | 120.1 |
| C6—C5—H5B | 109.5 | C115—C114—C113 | 119.9 (4) |
| H5A—C5—H5B | 108.0 | C115—C114—H114 | 120.1 |
| C116—C111—C112 | 118.3 (3) | C113—C114—H114 | 120.1 |
| C116—C111—C11 | 120.3 (3) | C7—O72—C8 | 115.5 (3) |
| C112—C111—C11 | 121.4 (3) | C9—O92—H92 | 111 (3) |
| O21—C2—C3 | 125.9 (3) | N41—C4—C3 | 122.1 (2) |
| O21—C2—C1 | 115.3 (2) | N41—C4—C5 | 120.6 (2) |
| C3—C2—C1 | 118.7 (3) | C3—C4—C5 | 117.4 (2) |
| C7—C6—C5 | 112.0 (2) | ||
| C4—N41—C10—C9 | −76.6 (4) | N41—C10—C11—C111 | 54.1 (3) |
| C4—N41—C10—C11 | 157.5 (3) | C9—C10—C11—C111 | −73.0 (3) |
| O91—C9—C10—N41 | 178.7 (3) | C116—C111—C112—C113 | 1.9 (6) |
| O92—C9—C10—N41 | −0.6 (4) | C11—C111—C112—C113 | −178.7 (4) |
| O91—C9—C10—C11 | −56.0 (3) | C112—C111—C116—C115 | −1.1 (5) |
| O92—C9—C10—C11 | 124.7 (3) | C11—C111—C116—C115 | 179.5 (4) |
| C4—C3—C2—O21 | −0.5 (5) | C111—C116—C115—C114 | 0.3 (7) |
| C4—C3—C2—C1 | 178.8 (3) | C111—C112—C113—C114 | −1.9 (7) |
| Cl1—C1—C2—O21 | −173.6 (2) | C116—C115—C114—C113 | −0.3 (7) |
| Cl3—C1—C2—O21 | −53.0 (3) | C112—C113—C114—C115 | 1.0 (7) |
| Cl2—C1—C2—O21 | 64.5 (3) | O71—C7—O72—C8 | −0.8 (5) |
| Cl1—C1—C2—C3 | 7.0 (4) | C6—C7—O72—C8 | −179.7 (3) |
| Cl3—C1—C2—C3 | 127.7 (3) | C10—N41—C4—C3 | 175.1 (3) |
| Cl2—C1—C2—C3 | −114.9 (3) | C10—N41—C4—C5 | −7.0 (4) |
| O71—C7—C6—C5 | 13.3 (5) | C2—C3—C4—N41 | −2.2 (5) |
| O72—C7—C6—C5 | −167.8 (3) | C2—C3—C4—C5 | 179.8 (3) |
| C4—C5—C6—C7 | −168.9 (3) | C6—C5—C4—N41 | −86.6 (3) |
| C116—C111—C11—C10 | −115.5 (3) | C6—C5—C4—C3 | 91.4 (3) |
| C112—C111—C11—C10 | 65.1 (4) |
Hydrogen-bond geometry (Å, º)
| D—H···A | D—H | H···A | D···A | D—H···A |
| C6—H6A···Cl1i | 0.97 | 2.94 | 3.774 (3) | 145 |
| O33—H33B···O91ii | 0.87 (6) | 1.89 (6) | 2.766 (4) | 177 (5) |
| N41—H41···O21 | 0.83 (5) | 2.05 (6) | 2.672 (3) | 131 (5) |
| O33—H33A···O21iii | 0.76 (6) | 2.06 (6) | 2.815 (3) | 171 (6) |
| O92—H92···O33 | 0.89 (5) | 1.66 (5) | 2.542 (4) | 175 (5) |
| C3—H3···Cl1 | 0.93 | 2.55 | 3.031 (3) | 112 |
Symmetry codes: (i) x−1, y, z; (ii) x+1, y, z; (iii) x, y+1, z.
Footnotes
Supporting information for this paper is available from the IUCr electronic archives (Reference: PK2509).
References
- Brandenburg, K. (2006). DIAMOND Crystal Impact GbR, Bonn, Germany.
- Bruker (2006). XPREP Bruker AXS Inc., Madison, Wisconsin, USA.
- Bruker (2009). APEX2 and SAINT Bruker AXS Inc., Madison, Wisconsin, USA.
- Costa, D. P. da, Nobre, S. M., Lisboa, B. G., Vicenti, J. R. de M. & Back, D. F. (2013). Acta Cryst. E69, o201. [DOI] [PMC free article] [PubMed]
- Flores, A. F. C., Flores, D. C., Oliveira, G., Pizzuti, L., Silva, R. M. S., Martins, M. A. P. & Bonacorso, H. G. (2008). J. Braz. Chem. Soc. 19, 184–193.
- Flores, A. F. C., Malavolta, J. L., Souto, A. A., Goularte, R. B., Flores, D. C. & Piovesan, L. A. (2013). J. Braz. Chem. Soc. 24, 580–584.
- Hachuła, B., Polasz, A., Dzida, M., Nowak, M. & Kusz, J. (2013). Acta Cryst. E69, o1406. [DOI] [PMC free article] [PubMed]
- Lo, K. M. & Ng, S. W. (2008). Acta Cryst. E64, m722–m723. [DOI] [PMC free article] [PubMed]
- Parsons, S., Flack, H. D. & Wagner, T. (2013). Acta Cryst. B69, 249–259. [DOI] [PMC free article] [PubMed]
- Pojarová, M., Fejfarová, K. & Makrlík, E. (2010). Acta Cryst. E66, o3341–o3342. [DOI] [PMC free article] [PubMed]
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Westrip, S. P. (2010). J. Appl. Cryst. 43, 920–925.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablock(s) I. DOI: 10.1107/S1600536814000154/pk2509sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S1600536814000154/pk2509Isup2.hkl
Supporting information file. DOI: 10.1107/S1600536814000154/pk2509Isup3.cml
CCDC reference: http://scripts.iucr.org/cgi-bin/cr.cgi?rm=csd&csdid=979612
Additional supporting information: crystallographic information; 3D view; checkCIF report