

Abstract
In the title hydrated salt [systematic name: 1-(1,3-benzodioxol-5-ylmethyl)piperazin-1-ium 4-nitrobenzoate monohydrate], C12H17N2O2 +·C7H4NO4 −·H2O, the piperazinium ring of the cation adopts a slightly distorted chair conformation. The piperonyl and piperazine rings are rotated with respect to each other with an N—C—C—C torsion angle of 45.6 (2)°. In the anion, the nitro group is almost coplanar with the adjacent benzene ring, forming a dihedral angle of only 3.9 (4)°. In the crystal, the cations, anions and water molecules are linked through N—H⋯O and O—H⋯O hydrogen bonds into chains along the a axis. In addition, weaker intermolecular C—H⋯O interactions are also observed within the chains. The anions form centrosymmetric couples through π-stacking interactions, with an intercentroid distance of 3.681 (4) Å between the benzene rings.
Related literature
For the drug, piribedil {systematic name: 2-[4-(benzo[1,3]dioxol-5-ylmethyl)piperazin-1-yl]pyrimidine}, an antiparkinsonian agent, see: Millan et al. (2001 ▶). For piperonylpiperazine derivatives with α-adrenergic antagonist and vasodilator properties, see: Gobert et al. (2003 ▶); Gilbert et al. (1968 ▶). For the use of piperazine in the construction of various bioactive molecules, see: Choudhary et al. (2006 ▶). For the antimicrobial activity of piperazine derivatives, see: Kharb et al. (2012 ▶). For related biologically active compounds, see: Brockunier et al. (2004 ▶); Bogatcheva et al. (2006 ▶). For a review on the current pharmacological and toxicological information for piperazine derivatives, see: Elliott (2011 ▶). For a related structure, see: Capuano et al. (2000 ▶). For puckering parameters, see Cremer & Pople (1975 ▶). For standard bond lengths, see: Allen et al. (1987 ▶).
Experimental
Crystal data
C12H17N2O2 +·C7H4NO4 −·H2O
M r = 405.40
Triclinic,
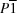
a = 6.0745 (5) Å
b = 12.0617 (11) Å
c = 13.4817 (10) Å
α = 92.561 (7)°
β = 98.753 (7)°
γ = 93.326 (7)°
V = 973.20 (14) Å3
Z = 2
Cu Kα radiation
μ = 0.90 mm−1
T = 173 K
0.42 × 0.36 × 0.24 mm
Data collection
Agilent Xcalibur (Eos, Gemini) diffractometer
Absorption correction: multi-scan (CrysAlis PRO and CrysAlis RED; Agilent, 2012 ▶) T min = 0.882, T max = 1.000
6403 measured reflections
3761 independent reflections
3196 reflections with I > 2σ(I)
R int = 0.021
Refinement
R[F 2 > 2σ(F 2)] = 0.043
wR(F 2) = 0.120
S = 1.03
3761 reflections
263 parameters
H-atom parameters constrained
Δρmax = 0.27 e Å−3
Δρmin = −0.20 e Å−3
Data collection: CrysAlis PRO (Agilent, 2012 ▶); cell refinement: CrysAlis PRO; data reduction: CrysAlis RED (Agilent, 2012 ▶); program(s) used to solve structure: SUPERFLIP (Palatinus & Chapuis, 2007 ▶; Palatinus & van der Lee, 2008 ▶; Palatinus et al., 2012 ▶); program(s) used to refine structure: SHELXL97 (Sheldrick, 2008 ▶); molecular graphics: OLEX2 (Dolomanov et al., 2009 ▶); software used to prepare material for publication: OLEX2.
Supplementary Material
Crystal structure: contains datablock(s) I. DOI: 10.1107/S160053681400261X/ld2118sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S160053681400261X/ld2118Isup2.hkl
Supporting information file. DOI: 10.1107/S160053681400261X/ld2118Isup3.cml
CCDC reference: http://scripts.iucr.org/cgi-bin/cr.cgi?rm=csd&csdid=985155
Additional supporting information: crystallographic information; 3D view; checkCIF report
Table 1. Hydrogen-bond geometry (Å, °).
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| N2A—H2AA⋯O1W i | 0.94 | 1.84 | 2.7800 (16) | 172 |
| N2A—H2AB⋯O1B ii | 0.93 | 1.80 | 2.7262 (16) | 175 |
| C9A—H9AA⋯O2A iii | 0.99 | 2.58 | 3.3260 (19) | 132 |
| C10A—H10A⋯O1W iv | 0.99 | 2.51 | 3.2833 (19) | 135 |
| O1W—H1WA⋯O2B v | 0.90 | 1.76 | 2.6526 (16) | 170 |
| O1W—H1WB⋯O1B ii | 0.92 | 1.90 | 2.7867 (16) | 163 |
Symmetry codes: (i) 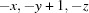 ; (ii)
; (ii) 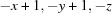 ; (iii)
; (iii) 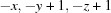 ; (iv)
; (iv) 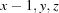 ; (v)
; (v) 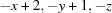 .
.
Acknowledgments
CNK thanks the University of Mysore for research facilities and is also grateful to the Principal, Maharani’s Science College for Women, Mysore, for giving permission to undertake research. JPJ acknowledges the NSF–MRI program (grant No. CHE-1039027) for funds to purchase the X-ray diffractometer.
supplementary crystallographic information
1. Comment
1-(3,4-Methylenedioxybenzyl)piperazine or 1-piperonylpiperazine is a psychoactive drug of the piperazine class and is used to synthesise the drug, piribedil, an antiparkinsonian agent (Millan et al., 2001). Piperonylpiperazine derivatives also has α-adrenergic antagonist properties (Gobert et al., 2003) and peripheral vasodilator properties (Gilbert et al., 1968). The piperazine moiety is extensively employed to construct various bioactive molecules with anti-bacterial, antimalarial activity and as antipsychotic agents (Choudhary et al., 2006). A valuable insight into recent advances on antimicrobial activity of piperazine derivatives is reported (Kharb et al., 2012). Piperazines are among the most important building blocks in today's drug discovery and are found in biologically active compounds across a number of different therapeutic areas (Brockunier et al., 2004; Bogatcheva et al., 2006). A review on the current pharmacological and toxicological information for piperazine derivatives is available (Elliott, 2011). The crystal structure of an N-piperonyl analogue of the atypical antipsychotic clozapine (Capuano et al., 2000) is reported. In continuation of our work on salts of piperonylpiperazines, this paper reports the crystal structure of the title compound, (I), C12H17N2O2+ . C7H4NO4- . H2O.
The asymmetric unit of the title compound, (I), contains one independent 1-piperonylpiperazinium monocation, one 4-nitrobenzoate monoanion and one water molecule (Fig. 1). The piperazine ring in the cation adopts a slightly disordered chair conformation (puckering parameters Q, θ, and φ = 0.590 (2)Å, 3.8 (6)° and 1.68 (4)°; (Cremer & Pople, 1975). The piperonyl and piperazine rings are twisted with respect to each other with an N1A/C1A/C2A/C8A torsion angle of 45.6 (2)°. In the anion, the nitro substituent is slightly twisted from the mean plane of the phenyl ring with a dihedral angle of 3.9 (4)°. Bond lengths are in normal ranges (Allen et al., 1987). In the crystal, the cations and anions interact through N—H···O intermolecular hydrogen bonds while weak C—H···O intermolecular interactions are observed between the cations (Fig. 2). The crystal packing is stabilized by these N—H···O and O—H···O intermolecular hydrogen bonds and weak C—H···O intermolecular interactions (Table 1) involving the water molecules which form 1D chains along [1 0 0]. In addition, weak Cg5–Cg5 π–π stacking interactions with an intercentroid distance of 3.681 (4)Å (Symmetry operation 2-x, -y, -z; Cg5 is the centroid between the phenyl rings, C1B–C6B, of the anions) contribute to the crystal packing.
2. Experimental
1-piperonylpiperazine ( 2.2g, 0.01 mol) and p-nitrobenzoic acid (1.67 g, 0.01 mol) were dissolved in hot N,N-dimethylformamide and stirred for 10 mins at 323 K. The resulting solution was allowed to cool slowly at room temperature. The crystals of the title salt appeared after a few days was used as such for x-ray studies (m. p:448-451 K).
3. Refinement
All of the H atoms were placed in their calculated positions and then refined using the riding model with Atom—H lengths of 0.95Å (CH) , 0.99Å (CH2), 0.92 or 0.94Å (NH2), 0.89 or 0.91Å (OH2). Isotropic displacement parameters for these atoms were set to 1.2 (CH, CH2, NH2) or 1.5 (OH2) times Ueq of the parent atom.
Figures
Fig. 1.

ORTEP drawing of one independent monocation-monoanion-water molecule unit in the asymmetric unit of (I) (C12H17N2O2+ . C7H4NO4- . H2O) showing the labeling scheme with 30% probability displacement ellipsoids.
Fig. 2.

Molecular packing for (I) viewed along the b axis. Dashed lines indicate N—H···O, O—H···O intermolecular hydrogen bonds and weak C—H···O intermolecular interactions. H atoms not involved in hydrogen bonding have been removed for clarity.
Crystal data
| C12H17N2O2+·C7H4NO4−·H2O | Z = 2 |
| Mr = 405.40 | F(000) = 428 |
| Triclinic, P1 | Dx = 1.383 Mg m−3 |
| a = 6.0745 (5) Å | Cu Kα radiation, λ = 1.54184 Å |
| b = 12.0617 (11) Å | Cell parameters from 2866 reflections |
| c = 13.4817 (10) Å | θ = 3.3–72.4° |
| α = 92.561 (7)° | µ = 0.90 mm−1 |
| β = 98.753 (7)° | T = 173 K |
| γ = 93.326 (7)° | Irregular, colourless |
| V = 973.20 (14) Å3 | 0.42 × 0.36 × 0.24 mm |
Data collection
| Agilent Xcalibur (Eos, Gemini) diffractometer | 3761 independent reflections |
| Radiation source: Enhance (Cu) X-ray Source | 3196 reflections with I > 2σ(I) |
| Detector resolution: 16.0416 pixels mm-1 | Rint = 0.021 |
| ω scans | θmax = 72.4°, θmin = 3.3° |
| Absorption correction: multi-scan (CrysAlis PRO and CrysAlis RED; Agilent, 2012) | h = −7→7 |
| Tmin = 0.882, Tmax = 1.000 | k = −14→13 |
| 6403 measured reflections | l = −16→15 |
Refinement
| Refinement on F2 | Hydrogen site location: mixed |
| Least-squares matrix: full | H-atom parameters constrained |
| R[F2 > 2σ(F2)] = 0.043 | w = 1/[σ2(Fo2) + (0.0563P)2 + 0.0984P] where P = (Fo2 + 2Fc2)/3 |
| wR(F2) = 0.120 | (Δ/σ)max < 0.001 |
| S = 1.03 | Δρmax = 0.27 e Å−3 |
| 3761 reflections | Δρmin = −0.20 e Å−3 |
| 263 parameters | Extinction correction: SHELXL97 (Sheldrick, 2008), Fc*=kFc[1+0.001xFc2λ3/sin(2θ)]-1/4 |
| 0 restraints | Extinction coefficient: 0.0049 (6) |
| Primary atom site location: structure-invariant direct methods |
Special details
| Geometry. All esds (except the esd in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell esds are taken into account individually in the estimation of esds in distances, angles and torsion angles; correlations between esds in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell esds is used for estimating esds involving l.s. planes. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| O1A | 0.5807 (2) | 0.15502 (11) | 0.58196 (11) | 0.0590 (4) | |
| O2A | 0.5429 (2) | 0.34467 (10) | 0.59022 (10) | 0.0501 (3) | |
| N1A | −0.1474 (2) | 0.41016 (10) | 0.31368 (9) | 0.0318 (3) | |
| N2A | −0.1698 (2) | 0.54167 (10) | 0.14110 (9) | 0.0332 (3) | |
| H2AA | −0.2433 | 0.4918 | 0.0891 | 0.040* | |
| H2AB | −0.1248 | 0.6053 | 0.1115 | 0.040* | |
| C1A | −0.1964 (3) | 0.31162 (14) | 0.36785 (13) | 0.0403 (4) | |
| H1AA | −0.2762 | 0.2532 | 0.3195 | 0.048* | |
| H1AB | −0.2965 | 0.3308 | 0.4166 | 0.048* | |
| C2A | 0.0109 (3) | 0.26602 (13) | 0.42328 (11) | 0.0369 (3) | |
| C3A | 0.0366 (3) | 0.15269 (14) | 0.41979 (13) | 0.0446 (4) | |
| H3A | −0.0772 | 0.1047 | 0.3811 | 0.054* | |
| C4A | 0.2234 (3) | 0.10638 (14) | 0.47100 (14) | 0.0500 (4) | |
| H4A | 0.2393 | 0.0285 | 0.4678 | 0.060* | |
| C5A | 0.3822 (3) | 0.17849 (14) | 0.52603 (12) | 0.0426 (4) | |
| C6A | 0.6796 (3) | 0.25825 (15) | 0.62731 (13) | 0.0466 (4) | |
| H6AA | 0.8318 | 0.2712 | 0.6106 | 0.056* | |
| H6AB | 0.6907 | 0.2575 | 0.7013 | 0.056* | |
| C7A | 0.3587 (3) | 0.29153 (13) | 0.53067 (11) | 0.0375 (3) | |
| C8A | 0.1769 (3) | 0.33840 (13) | 0.48093 (12) | 0.0378 (3) | |
| H8A | 0.1632 | 0.4165 | 0.4851 | 0.045* | |
| C9A | −0.3555 (2) | 0.45704 (13) | 0.27135 (11) | 0.0337 (3) | |
| H9AA | −0.4451 | 0.4722 | 0.3254 | 0.040* | |
| H9AB | −0.4436 | 0.4028 | 0.2212 | 0.040* | |
| C10A | −0.3064 (3) | 0.56353 (13) | 0.22182 (12) | 0.0353 (3) | |
| H10A | −0.4481 | 0.5943 | 0.1927 | 0.042* | |
| H10B | −0.2245 | 0.6190 | 0.2726 | 0.042* | |
| C11A | 0.0375 (2) | 0.48756 (13) | 0.18124 (12) | 0.0349 (3) | |
| H11A | 0.1350 | 0.5397 | 0.2300 | 0.042* | |
| H11B | 0.1200 | 0.4685 | 0.1256 | 0.042* | |
| C12A | −0.0217 (3) | 0.38327 (12) | 0.23225 (11) | 0.0336 (3) | |
| H12A | −0.1124 | 0.3295 | 0.1826 | 0.040* | |
| H12B | 0.1165 | 0.3483 | 0.2598 | 0.040* | |
| O1B | 1.02663 (19) | 0.27812 (9) | −0.04752 (9) | 0.0437 (3) | |
| O2B | 1.3475 (2) | 0.26959 (12) | 0.05605 (11) | 0.0591 (4) | |
| O3B | 0.8956 (2) | −0.16582 (12) | 0.28622 (10) | 0.0616 (4) | |
| O4B | 0.5845 (2) | −0.15818 (12) | 0.18715 (11) | 0.0595 (4) | |
| N1B | 0.7773 (2) | −0.12428 (11) | 0.21838 (10) | 0.0409 (3) | |
| C1B | 1.0487 (2) | 0.14427 (11) | 0.07692 (11) | 0.0301 (3) | |
| C2B | 1.1819 (2) | 0.09318 (13) | 0.15270 (12) | 0.0357 (3) | |
| H2B | 1.3335 | 0.1195 | 0.1723 | 0.043* | |
| C3B | 1.0959 (3) | 0.00431 (13) | 0.19982 (12) | 0.0369 (3) | |
| H3B | 1.1868 | −0.0319 | 0.2505 | 0.044* | |
| C4B | 0.8739 (2) | −0.02983 (12) | 0.17066 (11) | 0.0319 (3) | |
| C5B | 0.7362 (2) | 0.02062 (12) | 0.09760 (11) | 0.0329 (3) | |
| H5B | 0.5832 | −0.0040 | 0.0803 | 0.039* | |
| C6B | 0.8256 (2) | 0.10799 (12) | 0.04996 (11) | 0.0329 (3) | |
| H6B | 0.7341 | 0.1431 | −0.0013 | 0.039* | |
| C7B | 1.1503 (3) | 0.23837 (12) | 0.02427 (12) | 0.0355 (3) | |
| O1W | 0.34602 (17) | 0.60766 (9) | 0.01811 (8) | 0.0378 (3) | |
| H1WA | 0.4609 | 0.6478 | −0.0004 | 0.057* | |
| H1WB | 0.2443 | 0.6571 | 0.0317 | 0.057* |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| O1A | 0.0637 (8) | 0.0486 (8) | 0.0625 (8) | 0.0261 (6) | −0.0051 (7) | −0.0006 (6) |
| O2A | 0.0471 (7) | 0.0435 (7) | 0.0574 (8) | 0.0115 (5) | −0.0010 (6) | −0.0032 (6) |
| N1A | 0.0333 (6) | 0.0342 (6) | 0.0301 (6) | 0.0046 (5) | 0.0096 (5) | 0.0081 (5) |
| N2A | 0.0372 (7) | 0.0298 (6) | 0.0332 (6) | −0.0004 (5) | 0.0060 (5) | 0.0086 (5) |
| C1A | 0.0417 (8) | 0.0422 (9) | 0.0399 (8) | 0.0020 (7) | 0.0123 (7) | 0.0149 (7) |
| C2A | 0.0452 (9) | 0.0383 (8) | 0.0305 (7) | 0.0060 (7) | 0.0124 (6) | 0.0105 (6) |
| C3A | 0.0591 (10) | 0.0377 (9) | 0.0374 (8) | 0.0035 (7) | 0.0081 (7) | 0.0026 (7) |
| C4A | 0.0716 (12) | 0.0331 (8) | 0.0468 (10) | 0.0140 (8) | 0.0095 (9) | 0.0039 (7) |
| C5A | 0.0532 (10) | 0.0405 (9) | 0.0366 (8) | 0.0172 (7) | 0.0085 (7) | 0.0059 (7) |
| C6A | 0.0491 (10) | 0.0522 (10) | 0.0404 (9) | 0.0139 (8) | 0.0073 (7) | 0.0065 (8) |
| C7A | 0.0456 (9) | 0.0368 (8) | 0.0326 (8) | 0.0072 (7) | 0.0123 (6) | 0.0029 (6) |
| C8A | 0.0468 (9) | 0.0317 (8) | 0.0384 (8) | 0.0078 (6) | 0.0138 (7) | 0.0081 (6) |
| C9A | 0.0312 (7) | 0.0382 (8) | 0.0332 (7) | 0.0041 (6) | 0.0081 (6) | 0.0055 (6) |
| C10A | 0.0354 (7) | 0.0349 (8) | 0.0368 (8) | 0.0078 (6) | 0.0065 (6) | 0.0053 (6) |
| C11A | 0.0315 (7) | 0.0378 (8) | 0.0375 (8) | 0.0017 (6) | 0.0105 (6) | 0.0094 (6) |
| C12A | 0.0374 (8) | 0.0328 (7) | 0.0335 (7) | 0.0074 (6) | 0.0114 (6) | 0.0067 (6) |
| O1B | 0.0434 (6) | 0.0370 (6) | 0.0553 (7) | 0.0035 (5) | 0.0167 (5) | 0.0182 (5) |
| O2B | 0.0420 (7) | 0.0654 (9) | 0.0692 (9) | −0.0174 (6) | 0.0100 (6) | 0.0172 (7) |
| O3B | 0.0658 (9) | 0.0635 (9) | 0.0555 (8) | 0.0021 (7) | 0.0009 (7) | 0.0353 (7) |
| O4B | 0.0504 (7) | 0.0568 (8) | 0.0712 (9) | −0.0115 (6) | 0.0075 (7) | 0.0281 (7) |
| N1B | 0.0471 (8) | 0.0380 (7) | 0.0393 (7) | 0.0017 (6) | 0.0096 (6) | 0.0128 (6) |
| C1B | 0.0330 (7) | 0.0255 (7) | 0.0334 (7) | 0.0031 (5) | 0.0107 (6) | −0.0002 (6) |
| C2B | 0.0300 (7) | 0.0373 (8) | 0.0395 (8) | 0.0008 (6) | 0.0050 (6) | 0.0015 (6) |
| C3B | 0.0381 (8) | 0.0400 (8) | 0.0327 (8) | 0.0077 (6) | 0.0022 (6) | 0.0080 (6) |
| C4B | 0.0390 (8) | 0.0286 (7) | 0.0299 (7) | 0.0032 (6) | 0.0092 (6) | 0.0056 (6) |
| C5B | 0.0302 (7) | 0.0327 (7) | 0.0350 (8) | −0.0017 (6) | 0.0032 (6) | 0.0062 (6) |
| C6B | 0.0338 (7) | 0.0309 (7) | 0.0338 (7) | 0.0033 (6) | 0.0027 (6) | 0.0068 (6) |
| C7B | 0.0367 (8) | 0.0295 (7) | 0.0436 (9) | 0.0019 (6) | 0.0170 (7) | 0.0028 (6) |
| O1W | 0.0347 (5) | 0.0365 (6) | 0.0422 (6) | −0.0020 (4) | 0.0061 (5) | 0.0073 (5) |
Geometric parameters (Å, º)
| O1A—C5A | 1.373 (2) | C9A—C10A | 1.509 (2) |
| O1A—C6A | 1.421 (2) | C10A—H10A | 0.9900 |
| O2A—C6A | 1.431 (2) | C10A—H10B | 0.9900 |
| O2A—C7A | 1.3802 (19) | C11A—H11A | 0.9900 |
| N1A—C1A | 1.4619 (19) | C11A—H11B | 0.9900 |
| N1A—C9A | 1.4617 (18) | C11A—C12A | 1.511 (2) |
| N1A—C12A | 1.4648 (18) | C12A—H12A | 0.9900 |
| N2A—H2AA | 0.9422 | C12A—H12B | 0.9900 |
| N2A—H2AB | 0.9268 | O1B—C7B | 1.2622 (19) |
| N2A—C10A | 1.4888 (19) | O2B—C7B | 1.2400 (19) |
| N2A—C11A | 1.4913 (18) | O3B—N1B | 1.2192 (18) |
| C1A—H1AA | 0.9900 | O4B—N1B | 1.2231 (18) |
| C1A—H1AB | 0.9900 | N1B—C4B | 1.4693 (19) |
| C1A—C2A | 1.509 (2) | C1B—C2B | 1.393 (2) |
| C2A—C3A | 1.384 (2) | C1B—C6B | 1.388 (2) |
| C2A—C8A | 1.408 (2) | C1B—C7B | 1.516 (2) |
| C3A—H3A | 0.9500 | C2B—H2B | 0.9500 |
| C3A—C4A | 1.395 (2) | C2B—C3B | 1.387 (2) |
| C4A—H4A | 0.9500 | C3B—H3B | 0.9500 |
| C4A—C5A | 1.366 (3) | C3B—C4B | 1.379 (2) |
| C5A—C7A | 1.379 (2) | C4B—C5B | 1.379 (2) |
| C6A—H6AA | 0.9900 | C5B—H5B | 0.9500 |
| C6A—H6AB | 0.9900 | C5B—C6B | 1.385 (2) |
| C7A—C8A | 1.367 (2) | C6B—H6B | 0.9500 |
| C8A—H8A | 0.9500 | O1W—H1WA | 0.8987 |
| C9A—H9AA | 0.9900 | O1W—H1WB | 0.9158 |
| C9A—H9AB | 0.9900 | ||
| C5A—O1A—C6A | 106.07 (13) | C10A—C9A—H9AA | 109.6 |
| C7A—O2A—C6A | 105.74 (13) | C10A—C9A—H9AB | 109.6 |
| C1A—N1A—C12A | 111.33 (12) | N2A—C10A—C9A | 109.95 (12) |
| C9A—N1A—C1A | 109.81 (12) | N2A—C10A—H10A | 109.7 |
| C9A—N1A—C12A | 108.99 (11) | N2A—C10A—H10B | 109.7 |
| H2AA—N2A—H2AB | 107.3 | C9A—C10A—H10A | 109.7 |
| C10A—N2A—H2AA | 113.1 | C9A—C10A—H10B | 109.7 |
| C10A—N2A—H2AB | 113.7 | H10A—C10A—H10B | 108.2 |
| C10A—N2A—C11A | 110.93 (11) | N2A—C11A—H11A | 109.7 |
| C11A—N2A—H2AA | 104.7 | N2A—C11A—H11B | 109.7 |
| C11A—N2A—H2AB | 106.6 | N2A—C11A—C12A | 109.91 (12) |
| N1A—C1A—H1AA | 109.0 | H11A—C11A—H11B | 108.2 |
| N1A—C1A—H1AB | 109.0 | C12A—C11A—H11A | 109.7 |
| N1A—C1A—C2A | 112.76 (13) | C12A—C11A—H11B | 109.7 |
| H1AA—C1A—H1AB | 107.8 | N1A—C12A—C11A | 110.19 (12) |
| C2A—C1A—H1AA | 109.0 | N1A—C12A—H12A | 109.6 |
| C2A—C1A—H1AB | 109.0 | N1A—C12A—H12B | 109.6 |
| C3A—C2A—C1A | 120.29 (15) | C11A—C12A—H12A | 109.6 |
| C3A—C2A—C8A | 119.60 (15) | C11A—C12A—H12B | 109.6 |
| C8A—C2A—C1A | 120.09 (14) | H12A—C12A—H12B | 108.1 |
| C2A—C3A—H3A | 118.8 | O3B—N1B—O4B | 123.48 (14) |
| C2A—C3A—C4A | 122.45 (17) | O3B—N1B—C4B | 117.98 (14) |
| C4A—C3A—H3A | 118.8 | O4B—N1B—C4B | 118.52 (13) |
| C3A—C4A—H4A | 121.6 | C2B—C1B—C7B | 119.39 (13) |
| C5A—C4A—C3A | 116.74 (16) | C6B—C1B—C2B | 119.79 (14) |
| C5A—C4A—H4A | 121.6 | C6B—C1B—C7B | 120.82 (13) |
| O1A—C5A—C7A | 110.07 (15) | C1B—C2B—H2B | 119.6 |
| C4A—C5A—O1A | 128.41 (16) | C3B—C2B—C1B | 120.75 (14) |
| C4A—C5A—C7A | 121.52 (16) | C3B—C2B—H2B | 119.6 |
| O1A—C6A—O2A | 108.35 (14) | C2B—C3B—H3B | 121.1 |
| O1A—C6A—H6AA | 110.0 | C4B—C3B—C2B | 117.81 (14) |
| O1A—C6A—H6AB | 110.0 | C4B—C3B—H3B | 121.1 |
| O2A—C6A—H6AA | 110.0 | C3B—C4B—N1B | 119.33 (13) |
| O2A—C6A—H6AB | 110.0 | C3B—C4B—C5B | 122.89 (14) |
| H6AA—C6A—H6AB | 108.4 | C5B—C4B—N1B | 117.78 (13) |
| C5A—C7A—O2A | 109.55 (14) | C4B—C5B—H5B | 120.7 |
| C8A—C7A—O2A | 127.90 (14) | C4B—C5B—C6B | 118.61 (13) |
| C8A—C7A—C5A | 122.54 (15) | C6B—C5B—H5B | 120.7 |
| C2A—C8A—H8A | 121.4 | C1B—C6B—H6B | 119.9 |
| C7A—C8A—C2A | 117.14 (14) | C5B—C6B—C1B | 120.13 (13) |
| C7A—C8A—H8A | 121.4 | C5B—C6B—H6B | 119.9 |
| N1A—C9A—H9AA | 109.6 | O1B—C7B—C1B | 117.18 (13) |
| N1A—C9A—H9AB | 109.6 | O2B—C7B—O1B | 125.94 (15) |
| N1A—C9A—C10A | 110.19 (12) | O2B—C7B—C1B | 116.88 (14) |
| H9AA—C9A—H9AB | 108.1 | H1WA—O1W—H1WB | 106.6 |
| O1A—C5A—C7A—O2A | 0.01 (19) | C9A—N1A—C1A—C2A | −173.67 (12) |
| O1A—C5A—C7A—C8A | 179.33 (15) | C9A—N1A—C12A—C11A | 61.80 (15) |
| O2A—C7A—C8A—C2A | 179.03 (14) | C10A—N2A—C11A—C12A | 55.02 (16) |
| N1A—C1A—C2A—C3A | −135.98 (16) | C11A—N2A—C10A—C9A | −55.19 (16) |
| N1A—C1A—C2A—C8A | 45.6 (2) | C12A—N1A—C1A—C2A | 65.54 (16) |
| N1A—C9A—C10A—N2A | 58.74 (16) | C12A—N1A—C9A—C10A | −61.96 (15) |
| N2A—C11A—C12A—N1A | −58.34 (16) | O3B—N1B—C4B—C3B | 3.6 (2) |
| C1A—N1A—C9A—C10A | 175.85 (12) | O3B—N1B—C4B—C5B | −176.66 (15) |
| C1A—N1A—C12A—C11A | −176.93 (12) | O4B—N1B—C4B—C3B | −175.27 (15) |
| C1A—C2A—C3A—C4A | −178.95 (15) | O4B—N1B—C4B—C5B | 4.5 (2) |
| C1A—C2A—C8A—C7A | 178.88 (13) | N1B—C4B—C5B—C6B | −178.38 (13) |
| C2A—C3A—C4A—C5A | 0.3 (3) | C1B—C2B—C3B—C4B | −1.4 (2) |
| C3A—C2A—C8A—C7A | 0.5 (2) | C2B—C1B—C6B—C5B | −0.6 (2) |
| C3A—C4A—C5A—O1A | −179.27 (16) | C2B—C1B—C7B—O1B | 176.26 (13) |
| C3A—C4A—C5A—C7A | 0.0 (3) | C2B—C1B—C7B—O2B | −4.0 (2) |
| C4A—C5A—C7A—O2A | −179.42 (16) | C2B—C3B—C4B—N1B | 179.53 (13) |
| C4A—C5A—C7A—C8A | −0.1 (3) | C2B—C3B—C4B—C5B | −0.2 (2) |
| C5A—O1A—C6A—O2A | −4.64 (19) | C3B—C4B—C5B—C6B | 1.3 (2) |
| C5A—C7A—C8A—C2A | −0.2 (2) | C4B—C5B—C6B—C1B | −0.9 (2) |
| C6A—O1A—C5A—C4A | −177.74 (18) | C6B—C1B—C2B—C3B | 1.8 (2) |
| C6A—O1A—C5A—C7A | 2.89 (19) | C6B—C1B—C7B—O1B | −3.2 (2) |
| C6A—O2A—C7A—C5A | −2.88 (18) | C6B—C1B—C7B—O2B | 176.51 (14) |
| C6A—O2A—C7A—C8A | 177.85 (16) | C7B—C1B—C2B—C3B | −177.71 (13) |
| C7A—O2A—C6A—O1A | 4.63 (19) | C7B—C1B—C6B—C5B | 178.89 (13) |
| C8A—C2A—C3A—C4A | −0.5 (3) |
Hydrogen-bond geometry (Å, º)
| D—H···A | D—H | H···A | D···A | D—H···A |
| N2A—H2AA···O1Wi | 0.94 | 1.84 | 2.7800 (16) | 172 |
| N2A—H2AB···O1Bii | 0.93 | 1.80 | 2.7262 (16) | 175 |
| C9A—H9AA···O2Aiii | 0.99 | 2.58 | 3.3260 (19) | 132 |
| C10A—H10A···O1Wiv | 0.99 | 2.51 | 3.2833 (19) | 135 |
| O1W—H1WA···O2Bv | 0.90 | 1.76 | 2.6526 (16) | 170 |
| O1W—H1WB···O1Bii | 0.92 | 1.90 | 2.7867 (16) | 163 |
Symmetry codes: (i) −x, −y+1, −z; (ii) −x+1, −y+1, −z; (iii) −x, −y+1, −z+1; (iv) x−1, y, z; (v) −x+2, −y+1, −z.
Footnotes
Supporting information for this paper is available from the IUCr electronic archives (Reference: LD2118).
References
- Agilent (2012). CrysAlis PRO and CrysAlis RED Agilent Technologies, Yarnton, England.
- Allen, F. H., Kennard, O., Watson, D. G., Brammer, L., Orpen, A. G. & Taylor, R. (1987). J. Chem. Soc. Perkin Trans. 2, pp. S1–19.
- Bogatcheva, E., Hanrahan, C., Nikonenko, B., Samala, R., Chen, P., Gearhart, J., Barbosa, F., Einck, L., Nacy, C. A. & Protopopova, M. (2006). J. Med. Chem. 49, 3045–3048. [DOI] [PMC free article] [PubMed]
- Brockunier, L. L., He, J., Colwell, L. F. Jr, Habulihaz, B., He, H., Leiting, B., Lyons, K. A., Marsilio, F., Patel, R. A., Teffera, Y., Wu, J. K., Thornberry, N. A., Weber, A. E. & Parmee, E. R. (2004). Bioorg. Med. Chem. Lett. 14, 4763–4766. [DOI] [PubMed]
- Capuano, B., Crosby, I. T., Gable, R. W. & Lloyd, E. J. (2000). Acta Cryst. C56, 339–340. [DOI] [PubMed]
- Choudhary, P., Kumar, R. & Verma, K. (2006). Bioorg. Med. Chem. 14, 1819–1826. [DOI] [PubMed]
- Cremer, D. & Pople, J. A. (1975). J. Am. Chem. Soc. 97, 1354–1358.
- Dolomanov, O. V., Bourhis, L. J., Gildea, R. J., Howard, J. A. K. & Puschmann, H. (2009). J. Appl. Cryst. 42, 339–341.
- Elliott, S. (2011). Drug Test Anal. 3, 430–438. [DOI] [PubMed]
- Gilbert, R., Canevari, R. J. M. J., Laubie, M. J. & Le Douarec, J. C. (1968). J. Med. Chem. 11, 1151–1155. [DOI] [PubMed]
- Gobert, A., Di Cara, B., Cistarelli, L. & Millan, M. J. (2003). J. Pharmacol. Exp. Ther. 305, 338–46. [DOI] [PubMed]
- Kharb, R., Bansal, K. & Sharma, A. K. (2012). Pharma Chem. 4, 2470–2488.
- Millan, M. J., Cussac, D. & Milligan, G. (2001). J. Pharmacol. Exp. Ther. 297, 876–887. [PubMed]
- Palatinus, L. & Chapuis, G. (2007). J. Appl. Cryst. 40, 786–790.
- Palatinus, L., Prathapa, S. J. & van Smaalen, S. (2012). J. Appl. Cryst. 45, 575–580.
- Palatinus, L. & van der Lee, A. (2008). J. Appl. Cryst. 41, 975–984.
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablock(s) I. DOI: 10.1107/S160053681400261X/ld2118sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S160053681400261X/ld2118Isup2.hkl
Supporting information file. DOI: 10.1107/S160053681400261X/ld2118Isup3.cml
CCDC reference: http://scripts.iucr.org/cgi-bin/cr.cgi?rm=csd&csdid=985155
Additional supporting information: crystallographic information; 3D view; checkCIF report