

Abstract
Introduction
Checkpoint kinase inhibitors offer the promise of enhancing the effectiveness of widely prescribed cancer chemotherapies and radiotherapy by inhibiting the DNA damage response, as well as the potential for single agent efficacy.
Areas covered
This article surveys structural insights into the checkpoint kinases CHK1 and CHK2 that have been exploited to enhance the selectivity and potency of small molecule inhibitors. The use of mechanistic cellular assays to guide the optimisation of inhibitors is reviewed. The status of the current clinical candidates and emerging new clinical contexts for CHK1 and CHK2 inhibitors are discussed, including the prospects for single agent efficacy.
Expert opinion
Protein bound water molecules play key roles in structural features that can be targeted to gain high selectivity for either enzyme. The results of early phase clinical trials of checkpoint inhibitors have been mixed, but significant progress has been made in testing the combination of CHK1 inhibitors with genotoxic chemotherapy. Second generation CHK1 inhibitors are likely to benefit from increased selectivity and oral bioavailability. While the optimum therapeutic context for CHK2 inhibition remains unclear, the emergence of single agent preclinical efficacy for CHK1 inhibitors in specific tumour types exhibiting constitutive replication stress represents exciting progress in exploring the therapeutic potential of these agents.
Keywords: Cancer, DNA damage, cell-cycle checkpoint, kinase inhibitor, structure-based drug design
1. Introduction
The research field of checkpoint kinase (CHK) inhibitors has seen several recent developments, including publications of early phase clinical trial data for inhibitors used in combination with classical cancer chemotherapies, and also the first preclinical demonstrations of single agent efficacy for checkpoint kinase 1 (CHK1) inhibitors in specific genetic backgrounds. Previous reviews have covered the clinical development of CHK inhibitors [1-4] and the new structural classes of small molecules that have emerged from preclinical discovery [5-8]. In this article we concentrate on recently published structure-based drug design (SBDD) strategies that have enabled hit compounds against CHK1 and CHK2 to be developed to potent, selective late stage lead compounds and clinical candidates, and on advances made in understanding the determinants of inhibitor selectivity. We also focus on the cellular pharmacodynamic assays that have been used to drive optimisation of inhibitors to give mechanistically well-defined effects in relevant cancer models. We survey the development status of current clinical candidates and new potential contexts for CHK1 or CHK2 inhibition.
CHK1 and CHK2 are intracellular serine/threonine kinases that play pivotal roles in maintaining the integrity of cellular DNA. In response to intrinsic or genotoxic agent-induced DNA damage, e.g. single or double strand breaks or stalled replication forks, a network of sensors serves to activate multiple checkpoints to suspend progression through the cell cycle and simultaneously activate DNA repair mechanisms [9]. If repair is successful, checkpoints are lifted and cell duplication continues, otherwise the cell is directed to apoptosis. The DNA damage response and repair in cancer cells thus serves as a resistance mechanism to therapy with DNA-damaging cytotoxic agents or radiotherapy.
The CHK enzymes convey the signals received from upstream DNA-damage sensing proteins, principally ataxia telangiectasia mutated (ATM) and ataxia telangiectasia and Rad3 related (ATR), to downstream effectors of cell cycle arrest and DNA repair [10]. There is substantial overlap in the activation and substrates of CHK1 and CHK2. Although CHK2 activation can contribute to S- and G2/M-phase checkpoints, CHK2 is particularly important in the response to double strand DNA breaks signalled through activation of ATM and controls the p53-dependent early phase G1/S checkpoint [2, 11]. CHK2 stimulates repair of double strand DNA breaks through BRCA1 mediated processes. In contrast, CHK1 signalling is more important in response to single strand DNA breaks and stalled DNA replication signalled by activation through ATR, and resulting in later phase S and G2/M checkpoint arrest [12].
Many cancer cells harbour defects in the early phase, p53-dependent G1/S checkpoint, particularly resulting from mutation or inactivation of p53 [13-15], and as a result are more dependent on the later checkpoints, including those in S and G2/M-phases controlled by CHK1. This leads to the opportunity to selectively target cancer cells with defects in the p53-dependent checkpoint through the combination of inhibition of CHK1 with classical DNA-damaging cytotoxic drugs. Preclinical proof-of-concept of this strategy has been achieved with several CHK1 inhibitors [1-3, 16-21]. The role of CHK1 in maintaining replication fork stability has more recently been recognised as a key vulnerability for cancer cells enduring high intrinsic replication stress, providing potential contexts for single agent inhibition of CHK1 as an anticancer strategy [22-25]. In comparison to the potentiation of DNA-damaging therapies by CHK1 inhibition, where agents have reached clinical trials, the therapeutic context for selective CHK2 inhibition has not been as well defined and remains controversial. For example, inhibition of CHK2 has shown distinct and sometimes opposing effects to CHK1 inhibition when combined with DNA-damaging agents [2, 26].
2. Structure-based design applied to CHK inhibitors
2.1 CHK1 inhibitors
The majority of CHK1 inhibitors are ATP competitive and bind directly to the hinge peptide region found between the N- and C-terminal lobes of the kinase domain. The exceptions are recently described allosteric CHK1 inhibitors [27, 28]. Within the large selection of Type I inhibitors for which crystal structures have been published, similar interactions are regularly observed. With the inhibitors anchored by hydrogen bonds to one or more of Glu85, Tyr86 or Cys87 in the hinge region, they typically project polar substituents towards the ribose pocket and lipophilic groups to the selectivity surface. Beyond this surface the cleft opens up to solvent and hydrophilic groups may often be added here to balance the compound physicochemical properties. CHK1 has proven highly amenable to protein crystallography since the determination of the first apo-structure [29], thus a number of inhibitor series have been progressed using SBDD.
2.1.1 Thiophene carboxamide ureas
AZD7762 (1) is a dual CHK1/CHK2 inhibitor from AstraZeneca, for which details of the preclinical discovery using SBDD have been recently disclosed [30]. AZD7762 evolved from a thiophene carboxamide urea high-throughput screening (HTS) hit 2 [31] (Scheme 1A). Initial structure-activity studies showed the basic amine to be important for CHK1 in vitro potency, but that the series lacked activity in cellular assays quantifying abrogation of a camptothecin-induced G2/M checkpoint. Similar urea cores had been previously described as inhibiting a range of kinases [31] and prospects for gaining selectivity were based on the observation of a markedly different binding mode in CHK1.
Scheme 1.

Examples of CHK1 inhibitors generated using SBDD from initial hit to late stage leads or clinical candidates. a The structure of 25 has been drawn as it appears in the graphical abstract of the reference [51] which differs from the representation in the body of the text.
An X-ray structure of 1 (Figure 1A) represents the binding mode found for this series in CHK1, with the urea carbonyl and terminal amino functionality contacting Cys87 and Glu85 at the hinge and the amide pointing towards the ribose pocket. An alternative binding mode for this scaffold was exemplified by a crystal structure in JNK1 which showed a molecule similar to 2 binding to the hinge region in a tridentate manner through the primary amide NH and carbonyl groups as well as the urea terminal amine [30]. A set of analogues containing substituted amides to discourage the tridentate binding mode increased selectivity for CHK1 and validated the design hypothesis [30]. Cyclic amine substituents conferred increased potency due to new polar interactions between the amine and Asp148, along with dipole-dipole interactions with the backbone carbonyl of Glu134 and the amide side chain of Asn135. Removal of the original ether-linked ethylamine of 2 gave the lead compound 3 with much improved cellular activity while retaining in vitro potency.
Figure 1.

Crystal structures of CHK1 in complex with inhibitors. A) 1 (PDB 2ydj); B) Overlay of 4 (blue, PDB 2x8d), 7 (pink, PDB 2yer); C) 16 (PDB 2ym8); D) 20 (PDB 3ot3); E) 21 (PDB 3u9n); F) 23 (PDB 3tkh); Hydrogen bonds are indicated as dashed lines.
The regioisomeric thiophene seen in 1 could replace the thiophene ring of 3, and optimisation of the terminal phenyl ring was focussed on increasing selectivity for CHK1, increasing oral bioavailability and improving efficacy. A hollow fibre in vivo pharmacodynamic model was used to differentiate compounds [16], wherein polyvinylidene difluoride fibres filled with topotecan-treated HCT116 colon cancer cells were implanted into mice prior to drug treatment. After 30 h the fibres were recovered and the HCT116 cells were analysed by flow cytometry to determine the G1 and G2 cell cycle populations and assess checkpoint abrogation. 3-Fluorophenyl analogue 1 (AZD7762) was found to give the best balance of properties and was selected as a clinical candidate.
Merck have also developed CHK1 inhibitors starting from thiophene carboxamide ureas [32]. Ring formation to replace the pseudo-cycle formed by intramolecular hydrogen bonding between the amide and one of the urea amino groups gave scaffolds based around thienopyridines, thiazolopyridines and thienopyridazine cores, leading to potent CHK1 inhibitors in vitro.
2.1.2 Triazolones
In addition to the thiophene carboxamide urea 2, the HTS screen by AstraZeneca also identified triazolone 4 [31] (Scheme 1B). An X-ray structure revealed that two nitrogens of the triazolone formed a donor-acceptor interaction with the backbone Cys87 and Glu85, and the carbonyl interacted through a bridging water to Ser147 (Figure 1B) [33]. Attempts to establish polar interactions in the ribose pocket with 7-substituted phenyl triazolones led to 5 which possessed good in vitro potency but failed to abrogate a G2/M checkpoint in cells (Scheme 1B). Only with heterocycles at the 7-position, e.g. 6, designed to interact with Lys38 or the P-loop, was cellular activity observed. Crystal structures, e.g. 7 (Figure 1B), showed these compounds bound differently to 4, with the carbonyl and neighbouring NH interacting with Cys87 and Glu85, respectively [34]. This projected the pendent heterocycle towards the hinge region, resulting in an additional H-bond between Cys87 and the pyrrole NH. Superposition of the X-ray structures of these triazolones and the thiophene carboxamide urea 3 suggested appending a basic piperidine or similar group to the methyl substituent should be beneficial [34]. However the structure-activity relationships for substituents in the ribose pocket did not translate between these series and ultimately the hydroxymethyl derivative 7 gave an acceptable balance of in vitro, cellular and PK properties. Modest chemo- and radiopotentiation by 7 was observed [34]. Challenges remained with optimising the physicochemical properties and cellular potency of the triazolones.
2.1.3 Indazoles
AZD7762 (1) is a potent inhibitor of both CHK1 and CHK2. Other series of inhibitors have exploited an additional unique structural feature of the CHK1 kinase to generate selectivity for CHK1 over CHK2 and other kinases. The interior pocket of CHK1, beyond the Leu84 gatekeeper residue, contains an unusual polar residue at Asn59 instead of a hydrophobic amino acid as is more commonly found at this position. For example, the equivalent residue in CHK2 is a non-polar leucine [35]. Combined with contributions from Glu55 and the Lys38-Asp148 salt bridge, Asn59 defines a buried hydrophilic pocket [36-38]. Furthermore, crystal structures of CHK1 typically show between 1 – 3 protein bound water molecules within this pocket. Specific polar interactions from ligands to this feature can therefore provide potency and selectivity gains.
In contrast to 1 (Figure 1A), interactions to the hydrophilic features of CHK1 are exemplified in crystal structures of other inhibitors [36-38] (Figure 2). An indazole inhibitor 8 from Merck contained a pendant hydroxymethyl triazole group, where the hydroxyl replaced one of the water molecules in the hydrogen-bonding network (Figure 2A). This was associated with increased selectivity over CDK7. Indazoles such as 9 were reported by Vernalis and incorporated an ortho-methoxyphenol substituent [36]. This group displaced all three water molecules, and directly contacted Asn59 and Glu55 along with C-H…O and N-H…π-electron interactions to Val68 and Ser147, respectively (Figure 2B). This gave an increase in potency though the selectivity over other key kinases remained low. A preclinical candidate, VER-158411(structure not disclosed), has been nominated [39, 40].
Figure 2.

Crystal structures showing small molecule inhibitors interacting with the hydrophilic buried pocket in the CHK1 ATP site. A) 8 [37], (PDB 2hog); B) 9 [36], (PDB 2c3k); C) 10 [38], (PDB 2e9v); D) Chemical structures of 8, 9 and 10. Hydrogen bonds are indicated as dashed lines.
2.1.4 Pyrazines
The urea inhibitor 10 from Abbot contained a cyanopyrazine group where one of the ring nitrogens interacted with the water network and the nitrile group hydrogen bonded to the nearby Lys38 (Figure 2C) [38]. The macrocyclic scaffold of 10 was a product of structure-based design starting from the crystal structure of an acyclic bisaryl urea bound to CHK1 [41]. Macrocyclisation reinforced the intramolecular hydrogen bond observed in 10 (and the CHK1-bound acyclic progenitor) between the urea NH and pyrazine nitrogen, which contributes to locking the acyclic urea in the bioactive cis-trans conformation. Three macrocyclic analogues similar to 10 displayed high selectivity against a panel of 70 kinases [38]. The majority of kinases gave Ki > 10μM while there was still 170-fold selectivity for CHK1 over the most potent kinase inhibited, PLK1. Notably, substituted pyrazines have been used in other series of CHK1 inhibitors to increase potency and gain selectivity over other kinases [5-7], and a pyrazine urea also appears in the currently most advanced clinical candidate CHK1 inhibitor, LY2603618 (Table 1, 11) [6].
Table 1. Selected clinical trial data for CHK1 clinical candidates.
| Inhibitor | Structure | Inhibitory Activity | Status of Clinical Developmenta | Ref |
|---|---|---|---|---|
| UCN-01 (45) |
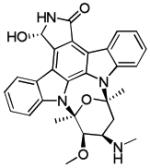
|
CHK1 IC50 11 nM | Phase II completed as single agent in relapsed T-cell lymphomas Phase II completed in combination with fluorouracil in pancreatic cancer Phase II completed in combination with topotecan for ovarian, fallopian tube and peritoneal cancers Phase II completed as single agent in metastatic melanoma Phase II completed in combination with topotecan in small cell lung cancer Multiple Phase I trials completed |
[100-103] |
| XL-844 (47) (Previously EXEL-9844) | Not disclosed | CHK1 Ki 2.2 nM CHK2 Ki 0.07 nM |
Phase I in combination with gemcitabine in advanced tumours and single agent in CLL terminated | - |
| LY2603618 (11) |
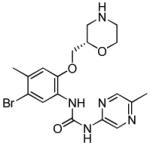
|
CHK1 IC50 7 nM | Phase I completed in combination with pemetrexed. Phase I radiolabelled drug metabolism and CYP2D6 interaction studies completed Phase II active in combination with pemetrexed or pemetrexed + cisplatin in non-small cell lung cancer Phase II active in combination with gemcitabine in pancreatic and other solid tumours active |
[74] |
| LY2606368 (48) |

|
CHK1 IC50 <1 nM CHK2 IC50 4.7 nM |
Phase I recruiting for single agent in advanced cancers, squamous cell and head and neck cancers | - |
| PF-00477736 (46) |
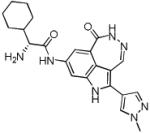
|
CHK1 Ki 0.5 nM CHK2 Ki 47 nM |
Phase I in solid tumours in combination with gemcitabine terminated. | [104] |
| AZD7762 (1) |
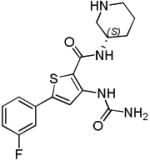
|
CHK1 IC50 5 nM CHK2 IC50 9.6 nM |
Phase I completed in solid tumours alone and in combination with gemcitabine Two additional Phase I trials terminated |
[97, 98] |
| SCH900776 (20) |
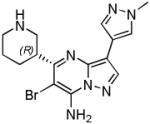
|
CHK1 IC50 3 nM CDK2 IC50 160 nM CHK2 IC50 1500 nM |
Phase I completed in combination with gemcitabine in solid tumours and lymphoma Phase 1 in combination with cytarabine in acute leukaemias terminated |
[75] |
| GDC-0575 (previously ARRY-575) | Not disclosedb | Not disclosedb | Phase I recruiting in combination with gemcitabine and as single agent in lymphoma and solid tumours | - |
| GDC-0425 | Not disclosed | Not disclosed | Phase I recruiting in combination with gemcitabine and as a single agent in lymphoma and solid tumours | - |
www.clinicatrials.gov [last accessed on 23 January 2013]
A collaboration between the Institute of Cancer Research, London and Sareum Ltd generated highly selective CHK1 inhibitors starting with virtual and high concentration biochemical screening to identify fragment hits [42]. Several fragments were pursued further using SBDD [43]. The morpholino-purine 12 (Scheme 1C) was advanced by fragment growing and scaffold morphing to the pyrazolopyridine 13 with increased potency resulting from interactions with the specificity surface of CHK1 and Glu91 in the ribose pocket [42]. Compounds were assessed for abrogation of an etoposide-induced G2/M checkpoint arrest in p53-deficient HT29 colon cells, and for single agent cytotoxicity in the same cell line. Analogues such as 13 showed encouraging 2-3 fold selectivity for the CHK1-mediated cellular effect over non-specific cytotoxicity and enhancing this differential was important in the subsequent optimisation of the compounds to ensure a selective mechanism of action.
The fusion of an additional pyridine ring to generate pyrimido[2,3-b]azaindoles, e.g. 14, was used to contact the protein bound waters in the CHK1 interior pocket [44]. A further scaffold modification to give pyridoaminopyrazines, such as 15, increased the opportunities to optimise the basic substituent and selectivity surface contacts. The pendant cyanopyrazine group of 15 interacted with Lys38 and protein bound water molecules in CHK1 with associated benefits in selectivity, exemplified by the 330-fold difference between CHK1 and CHK2 activity (Scheme 1C). The pyridine ester of 15 was exchanged for an isoquinoline in another scaffold modification, while the basic amine side chain was translocated to the pyrazine as had been demonstrated for urea-based CHK1 inhibitors [38], leading to SAR-020106 (16). The crystal structure of SAR-020106 bound to CHK1 showed extensive contacts of the cyanopyrazine substituent with Lys38 and the protein-bound water network (Figure 1C). As a result, SAR-020106 was a potent and highly selective CHK1 inhibitor. The systemic inhibitor 16 potentiated the efficacies of irinotecan and gemcitabine in SW620 human colon cancer cells in vitro and when grown as xenografts in nude mice [18]. SAR-020106 was also a potent radiosensitiser in tumour cell lines defective in p53 function [45].
To produce oral inhibitors, the metabolically stable pyridine core of 15 was hybridised with the substituted cyanopyrazine 16 to generate a new core scaffold [46]. This led to CCT244747 (17) which showed substantial oral bioavailability (F = 61%) and is the first oral CHK1 inhibitor to be fully described in the literature. CCT244747 maintained the high selectivity of SAR-020106 (at a concentration of 1 μM only 13 out of 140 representative kinases were inhibited by >50%) and significantly enhanced gemcitibine and irinotecan efficacy in human tumour xenografts in nude mice.
2.1.5 Pyrazolo[1,5-a]pyrimidines
Discovery of the Merck (previously Schering Plough) CHK1 clinical candidate SCH900776 20 started with the identification of the CDK2 inhibitor 18 from a compound library screen [47] (Scheme 1D). Medicinal chemistry exploration along two substituent vectors led to compound 19 with much improved CHK1 activity and selectivity against CDK2. A crystal structure confirmed that the lead compound bound to the hinge region of CHK1 through N1 and the C7-NH of the pyrazolo[1,5-a]pyrimidine core. The 1-methyl pyrazole at C3 contacted the protein bound waters in the interior pocket, while the piperidine interacted in the ribose pocket with Glu91 and the amide carbonyl of Glu134. These features were retained in the clinical candidate (Figure 1D). Attempts to access the specificity surface by substitution on the C7 amine proved challenging but halogen substituents in the C6 position gave a 20-fold improvement in CHK1 activity relative to the parent compound, leading ultimately to the clinical candidate SCH900776 20 [48]. A cell based assay for γ-H2AX induction, a marker for the formation of double-strand DNA breaks, was used to define the optimal cellular phenotype of compounds exhibiting varying degrees of CHK1, CHK2 and CDK selectivity during lead optimization [17].
2.1.6 Thiazole-4-carboxamides and 2-aminothiazoles
The SBDD of two new classes of CHK1 inhibitors with high in vitro potency have been disclosed by Merck [49-51]. The thiazole-4-carboxamide 21 was identified using AS-MS ALIS (Affinity Selection-Mass Spectrometry-based Automated Ligand Identification System) (Scheme 1E) [49]. A CHK1 crystal structure showed 21 bound to the ATP site through untypical CH…O interactions, whereby the thiazole C5-H and 2,3-dihydrobenzofuran C6-H interacted with the carbonyls of Glu85 and Cys87, respectively (Figure 1E). Intramolecular hydrogen bonding conferred a “U-shaped” topology to 21 which may significantly reduce the entropic penalty to binding. The amide interacted with the water network in the interior pocket. Replacement of the 2,3-dihydrobenzofuran with indole 22 conferred enhanced CHK1 potency and selectivity over CDK2.
Another series from Merck originated from the observation that the VEGFR2 (KDR) inhibitor 23 [50] was also a CHK1 inhibitor (Scheme 1F). However, compound 23 showed no activity in a cellular checkpoint escape assay measuring the release of H1299 tumour cells from DNA-damage induced cell-cycle arrest and progression into mitosis following CHK1 inhibition [37, 51]. This was attributed to the concomitant inhibition of CDK7 which, as with inhibition of other CDKs, may result in DNA-damage independent cell cycle arrest, nullifying the checkpoint abrogation activity. A crystal structure of 23 in CHK1 showed the scaffold bound to Cys87 in the hinge region via the aminothiazole (Figure 1F). This positioned the piperazine subsitutents towards solvent while the pyridine at C5 of the thiazole interacted with the water network in the interior pocket. Substituting from the meta position of this pyridine gave gains in CHK1 potency and selectivity through targeting the Glu55 and Asp148 residues. Analogues such as 24 were inhibitors of exceptional potency with very slow dissociation kinetics from the enzyme [50] (Scheme 1F). Reduction of the polar surface area of the compounds was sought to improve cell permeability and activity, leading to the difluoropiperidine 25 [51].
2.1.7 Allosteric inhibitors
Merck also developed a screening strategy to find non-ATP competitive CHK1 inhibitors [27], identifying a thioquinazolinone lead with an IC50 of 17 and 24 μM at 0.1 and 2.0 mM ATP concentrations, respectively. Medicinal chemistry optimisation gave 26 (Figure 3), with enhanced stability and potency against CHK1. The crystal structure of 26 in CHK1 showed it bound to the surface of the enzyme distant from the ATP site (Figure 3). The carbonyl from the quinazolinone ring formed a water mediated hydrogen bond to Glu134, while the piperidine amine and amide carbonyl interacted directly with Glu205 and the backbone of Leu206, respectively. The 3-chlorophenyl group fitted securely into a narrow hydrophobic cleft. Independently, Pfizer reported the discovery of potent allosteric inhibitors 27 and 28 (Figure 3) which utilised the same hydrophobic cleft but extended out in the opposite direction into a shallow groove [28].
Figure 3.

Structures of the allosteric CHK1 inhibitors 26-28 and the crystal structure of 26 bound to CHK1 (PDB 3f9n).
2.2 CHK2 inhibitors
The five main CHK2 inhibitor classes published to date are all ATP-competitive and bind to the hinge region of the kinase through interactions with one or more of Glu301, Glu302, Leu303 and Met304. Unlike CHK1 inhibitors, the interactions of CHK2 ligands fall into two distinct classes; direct hydrogen bonding [52-54] or atypical water-mediated contacts [55-59]. Several recently reported inhibitors also interact with Asp368 in the DFG motif [52, 57].
2.2.1 2-Arylbenzimidazoles
The first selective ATP-competitive CHK2 inhibitors were based on 2-arylbenzimidazoles identified from HTS [60]. The carboxylic acid hit 29 was evolved to the more potent primary amide 30 (Scheme 2A). Docking to a homology model of the ATP binding site of CHK2 was used to predict the binding of 30. One binding pose suggested the 5-amide to hydrogen bond to the hinge region. Loss of activity upon methylation of the amide nitrogen in 30 confirmed that hydrogen bond donating functionality was essential. The biaryl linker required a heteroatom, and the model suggested this was necessary to achieve a 90° twist allowing the terminal phenyl ring to maintain hydrophobic contacts in the ATP cleft. The 4-chlorophenyl analogue 31 had increased potency and it was suggested that the chlorine was partially solvent exposed. Replacement of the terminal aryl ring with alkyl-linked alcohols and amines improved solubility in selected examples [61], and the benzimidazole core was exchanged for other [6,5]-heterocycles that retained the relative spatial arrangement of the amide and terminal phenyl substituents to further refine the proposed binding mode [62].
Scheme 2.

Examples of CHK2 inhibitors generated using SBDD.
More recently, crystal structures of these benzimidazole CHK2 inhibitors have been reported and show a dramatically different binding pose than the original model [55] (Figure 4A). For 31 in CHK2, a water-mediated hydrogen bond was observed between the benzimidazole N1 and the carbonyl of Glu302 and amide NH of Met304 in the hinge. Instead of binding to the hinge region, the carboxamide substituent interacted with a network of amino acids deeper in the pocket, including Asp368. The 4-chlorophenyl substituent of 31 interacted loosely with hydrophobic residues at the entrance to the CHK2 ATP-pocket defined by Leu226, Leu236, Lys245, Leu303, and Glu305. The reported structure-activity relationships for the benzimidazoles [60-62] were better rationalised when this new binding pose involving water-mediated hydrogen bonding to the hinge was considered [55].
Figure 4.

Crystal structures of inhibitors in complex with CHK2 showing the different modes of interaction with the hinge region. A) 31 (PDB 4a9r), water-mediated hinge interaction; B) 33 (PDB 2w7x), water-mediated hinge interaction; C) 39 (PDB 2wtj), direct hinge interaction; D) 42 (PDB 2xbj), direct hinge interaction. Hydrogen bonds are indicated as dashed lines.
2.2.2 Guanidylhydrazones
Water-mediated hydrogen-bonding to the CHK2 hinge region is also a feature of a series of highly CHK2 selective guanidylhydrazone inhibitors derived from the symmetric inhibitor NSC109555 (32), the sole hit from HTS of 100,000 compounds [56, 58] (Scheme 2B). Although highly selective, 32 was not cell penetrant [58]. A crystal structure of 32 with the catalytic domain of CHK2 showed the central urea carbonyl to interact with Glu302 and Met304 in the hinge region through a mediating water molecule [56]. Again, molecular modelling had not predicted this novel binding pose. The ligand interacted with Glu273 on the C-α helix and other residues through one of the guanidylhydrazone termini. Replacement of the second guanidylhydrazone group, which had minimal contacts with the enzyme, and replacement of one side of the aryl urea with the less polar 7-nitroindole motif linked through an amide gave a cell penetrant compound, PV1019 (33) [59]. The crystal structure of 33 in CHK2 (Figure 4B) showed the nitro group facilitating the water mediated interaction with the hinge, as well as introducing a new direct interaction from the nitro group. Extensive interactions of the buried guanidylhydrazone were maintained and PV1019 showed excellent selectivity for CHK2 over other kinases.
Cyclisation of the guanidylhydrazone in 33 to give PV1115 (34) was pursued to maintain the interaction with Glu273 while removing two hydrogen bond donors to increase cell membrane permeability [57] (Scheme 2B). This also significantly enhanced CHK2 potency while maintaining high selectivity over CHK1. The crystal structure of 34 in CHK2 was similar to that of PV1019, with the difference that Lys249 was observed to move approx. 3.9 Å away from Glu273, breaking the salt bridge between these residues, to accommodate the larger cyclic guanidine group.
A superimposition of the coordinates of apo-CHK1 onto CHK2-PV1115 highlighted the presence of the bulky Tyr86 residue in the CHK1 hinge region compared to the smaller Leu303 corresponding residue in CHK2, which may render the 7-nitroindole group sterically disfavoured in CHK1. The region where the guanidine bound to Glu273 in CHK2 (Glu55 in CHK1), had an adjacent bulky residue (Tyr20) in the P-loop of CHK1 as opposed to the smaller Cys231 in CHK2. A hydrophobic pocket contacted by the methyl substituents of PV1115 was bounded by the small Leu301 gatekeeper and Leu277 in CHK2, while in CHK1 the polar Asn59 residue replaces Leu277.
Modification of the guanidylhydrazone to the N-hydroxy guanylhydrazone PV1533 (35) was carried out to lower the pKa of the ligand and improve cell permeability while maintaining the interaction to Glu273. The crystal structure of 35 in CHK2 showed the addition of the N-oxime allowed the oxygen atom to participate in water-mediated hydrogen bonding with the carboxylate side chain of Asp368 in the DFG motif. This is the first time an interaction with the DFG motif was observed in this series.
2.2.3 2-Aminopyridines
3,5-Diaryl-2-aminopyridines such as 36 (Scheme 2C) were identified by the Institute of Cancer Research, London as CHK2 inhibitors following HTS of a 7,000 member kinase-focussed library [52]. Medicinal chemistry optimization of the pyridine 5-substituent gave the 1,3-benzodioxole 37 with improved CHK2 potency and 40-fold selectivity over CHK1. Incorporation of a fused dioxole moiety onto a thienyl group gave compound 38 for which the crystal structure in CHK2 showed the heteroaromatic scaffold sandwiched between Leu309 and Leu354 in the ATP cleft, with two direct hydrogen bonds to the hinge region from the 2-aminopyridine functionality (represented in Figure 4C with 39). The terminal carboxamide of 38 accepted a hydrogen bond from Lys249, while also interacting with Glu273 in the αC-helix and Asp368 in the DFG-motif. The dioxacycle entered the solvent exposed but hydrophobic surface defined by Leu303 and Met304.
Deletion of the pyridine 3-aryl substituent and replacement with a 3-carboxamide containing a flexible alkylamine resulted in an improvement in CHK2 potency and 89-fold selectivity over CHK1 (39). A crystal structure of 39 in CHK2 revealed a water-mediated hydrogen bond between the amide and Asp368 as well as an intra-molecular hydrogen bond in the ligand between the 2-aminopyridine and the 3-carboxamide. The terminal unsubstituted amine was shown to make an additional interaction with Asp368 and through a water-mediated hydrogen bond the carboxamide interacted with Glu308. It was speculated that these new polar interactions compensated for the loss in rigidity of 39 compared to 38, leading to improved potency. Wider kinome profiling showed the 3-carboxamides represented by 39 to be more CHK2 selective than compounds such as 38.
2.2.4 2-(Quinazolin-2-yl)phenols
A distinct series of selective 2-(quinazolin-2-yl)phenol CHK2 inhibitors was discovered at the Institute of Cancer Research, London following kinome profiling of compounds from an unrelated drug discovery project (Scheme 2D) [54]. A crystal structure of the early lead 40 bound to CHK2 showed an interesting binding mode, with an intramolecular hydrogen bond between the phenol and the quinazoline N1 forming a planar pseudotetracyclic system. Furthermore, a hydrogen bond was formed between the phenolic oxygen and the amide NH of Met304 in the hinge region. The 6- and 7-positions of the quinazoline were directed out towards the solvent exposed region and the 3-aminopyrrolidine group occupied the ribose pocket, with the protonated pyrrolidine forming a charge-assisted hydrogen bond with the side chain of Asn352. These features were retained in the structure of the more optimised compound 42 bound to CHK2 (Figure 4D).
Fluorination of the phenol ring at the 5′-position increased CHK2 potency in this series. Based on the crystal structure of 40, the pyrrolidine 4- and 5-positions were substituted to add interactions to the P-loop of the kinase. The 4-(1,1-dimethyl)methyl alcohol 41 was beneficial for reducing off-target hERG inhibition and also conferred increased CHK2 selectivity over CHK1. Methoxy substitution at the solvent exposed 6- and 7-positions of the quinazoline core increased CHK2 potency and maintained low hERG activity, leading to CCT241533 (42). Although less selective for CHK2 over CHK1 compared to 41, CCT241533 showed good selectivity for CHK2 in a panel of 85 kinases. Additionally, CCT241533 had high passive permeability and inhibited CHK2 signalling in cancer cells [54]. Comparison of the binding mode of the 2-(quinazolin-2-yl)phenols with PV1019 and the benzimidazole CHK2 inhibitors shows that the hinge-binding phenolic OH of 42 occupies the same space as the mediating water molecule in the other two series.
2.2.5 Debromohymenialdisine analogues
Structure-based design was used to direct improvements to the CHK2 inhibitor debromohymeniadisine [53, 63]. Replacement of the pyrrole of the hymenialdisine core with an indole led to an indoloazepine (43, Table 2) [64]. Compound 43 showed improved CHK2 potency and selectivity for CHK2 over debromohymeniadisine. Substitution of the 2-pyrrole group in the hymenialdisine series with phenyl rings was found to reduce potency but increase selectivity over CHK1 in most examples [65].
Table 2. Structures, selectivities and cellular activities of selected CHK2 inhibitors.
| Compound | Structure | Inhibitory activity | Cellular Studies | Ref |
|---|---|---|---|---|
| 31 |

|
CHK2 IC50 15 nM CHK1 IC50 >10 μM |
Radioprotective in CD4+ and CD8+ T-cells exposed to IR. Isolated cells showed decreased apoptosis. |
[60] |
| PV1019 (33) |
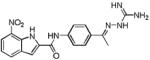
|
CHK2 IC50 138 nM CHK1 IC50 50 μM |
Abrogation of IR-induced apoptosis in mouse thymocytes. Radiosensitised U251 cells to IR. Potentiation of a topoisomerase I inhibitor in OVCAR-4 and OVCAR-5 cells. Antiproliferative effect on its own |
[59] |
| CCT241533 (42) |
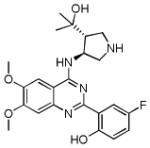
|
CHK2 IC50 3 nM CHK1 IC50 190 nM |
Radioprotective to isolated mouse thymocytes. No potentiation of the cytotoxicity of SN38, gemcitabine, etoposide, mitomycin C or bleomycin in HT29 or HeLa cancer cells Potentiation of the cytotoxicity of PARP inhibitors in BRCA-proficient HeLa and HT29 cells. |
[54] |
| (43) |

|
CHK2 IC50 8 nM CHK1 IC50 237 nM |
ATM-dependent, CHK2-mediated radioprotective effect in 184B5 p53 wild-type cells but not p53 mutant MDA-MB-231 cells | [93] |
| VRX0466617 (44) |

|
CHK2 IC50140 nM CHK1 IC50 >10 μM |
Radioprotective in LCL-N cells, BJ-hTERT fibroblasts, HCT116 cells and mouse thymocytes No potentiation of doxorubicin and cisplatin cytoxicity in MCF7 or taxol in BJ-hTERT cells |
[66] |
2.2.6 Isothiazole-4-carboxamidines
In addition to the CHK2 inhibitors developed using SBDD, Valeant Pharmaceuticals International have reported VRX0466617 (44, Table 2), as a potent and selective inhibitor of CHK2 [66]. VRX0466617 was discovered starting from an isothiazole carboxamide identified from screening. Molecular docking in CHK2 was used to guide the optimisation to give 44 [67].
3. Therapeutic contexts and clinical experience with CHK inhibitors
3.1 CHK1 inhibitor clinical development
A summary of the clinical development of selected CHK1 inhibitors is shown in Table 1. The majority of early clinical trials have investigated CHK1 inhibition in combination with DNA-damaging chemotherapies. The first generation of inhibitors to reach clinical trial were intravenous agents, and often showed low or only moderate selectivity for inhibition of CHK1 over CHK2. Despite reaching multiple Phase II trials, the development of UCN-01 (7-hydroxystaurosporine, 45) has been hindered by the low free drug levels resulting from the compound’s high avidity for human α-acid glycoprotein [68, 69]. In a recently reported Phase II trial of UCN-01 in combination with irinotecan in triple negative breast cancer patients, inconsistent inhibition of CHK1 signalling at the trial dose was demonstrated using immunochemical assessment of downstream biomarkers [99].
The development of AZD7762 (1) [30], PF-00477736 (46) [19] and XL-844 (47) [70] has been reported as discontinued by the originating organisations. While AZD7762 (1) and SCH900776 (20) were discovered using SBDD as described above, details of the medicinal chemistry strategies used to identify PF-00477736 (46), LY2603618 (11) and LY2606368 (47) have not yet been reported. Published preclinical data on PF-0047736 (46) showed it to be a potent inhibitor of CHK1 in vitro with some selectivity against CHK2 (100-fold) and CDK1 (20,000-fold) [19].
Other than UCN-01, LY2603618 (11) is apparently the most advanced CHK1 inhibitor from the first generation of compounds remaining in clinical trial (Table 1). Preclinical data on LY2603618 (11) and LY2606368 (48) have been reported in conference presentations and posters [71-73]. A phase I dose escalation study in patients with advanced solid tumours concluded that LY2603618 administered approximately 24 hours after pemetrexed showed acceptable safety and pharmacokinetic profiles [74]. Interestingly, the less progressed compound LY2606368 (48) shows no selectivity between CHK1 and CHK2 despite the presence of a pyrazine motif in the structure that is often associated with high CHK1 selectivity.
A published Phase I trial demonstrated that a combination of SCH900776 (20) with cytarabine was feasible and tolerated in adults with relapsed and/or refractory acute leukemias [75]. In addition there was preliminary evidence of some clinical activity. A dose-limiting asymptomatic prolongation of the QTcF interval was observed at doses of 80mg/m2.
GDC-0575 (previously ARRY-575) and GDC-0425 are the first orally bioavailable CHK1 inhibitors to enter Phase I clinical trials.
3.2 New potential contexts for CHK1 inhibition
While the majority of published preclinical data on the efficacy of CHK1 inhibitors has concentrated on combinations with DNA-damaging chemotherapy, it has long been recognised that abrogation of DNA damage response checkpoints and DNA repair by CHK1 inhibition also synergises with ionizing radiation (IR) [2, 76, 77]. In a recent example, the dual CHK1/CHK2 inhibitor AZD7762 (1) sensitised p53-deficient cell lines and xenografts to IR [78]. AZD7762 also sensitised MiaPaCa-2 pancreatic cancer cells to radiation in vitro, with a triple therapy of IR, gemcitabine and AZD7762 showing antitumour activity in MiaPaCa-2 and patient-derived pancreatic cancer xenografts [79]. AZD7762 has also been used recently to show that CHK1 inhibition radiosensitises xenografts of lung cancer brain metastases [80], and expression of activated (phosphorylated) CHK1 protein has been found to be elevated in radioresistant lung cancer patient-derived cell lines compared to radiosensitive cells [81]. These data suggest CHK1 inhibition may have potential in combination with radiotherapy in pancreatic and metastatic lung cancers. The more selective CHK1 inhibitor SAR-020106 (16) was shown to be a potent radiosensitiser in p53-deficient head-and-neck cancer cell lines and xenografts [45].
Combinations of CHK1 inhibition with other molecular targeted anticancer agents have been proposed as potential therapeutic contexts. Thus, inhibition of CHK1 with AZD7762 (1) or UCN-01 (45) combined with various MEK1/2 inhibitors was cytotoxic to several primary human glioma cell isolates and the CHK1 + MEK1/2 inhibitor treatment enhanced the sensitivity of glioma cells to IR [82]. The combination of PARP inhibitors and CHK1 inhibition was shown to act synergystically to suppress the growth of mammary carcinoma cells with several different genetic backgrounds, both in vitro and in xenograft models [83]. The combination of various CHK1 inhibitors and an inhibitor of the DNA damage response kinase WEE1 (MK-1775) synergistically inhibited growth and enhanced apoptosis in a variety of cancer cell lines and human tumour xenografts [84, 85, 86, 87]. Double strand breaks in DNA were observed in response to the combination of inhibitors without the application of an external genotoxic agent, which may suggest that the dual inhibition removes a survival mechanism for coping with high endogenous replication stress in cancer cells.
Perhaps the most exciting aspect of CHK1 inhibitor biology to emerge from recent work has been the discovery of defined contexts where single agent inhibition of CHK1 shows promising antitumour activity [2]. CHK1 signalling is critical in response to DNA damage resulting from defects in replication fork initiation and elongation during S-phase [24], and this key role provides a rationale for the observed high synergy seen when CHK1 inhibitors are combined with antimetabolites that generate DNA damage primarily during S-phase [88]. It has also led to the understanding that cancer cells with high intrinsic replication stress, i.e. a high level of endogenous DNA damage resulting from the highly replicative state, may come to depend on the DNA damage response and CHK1 function as a survival pathway. Cells with a complex karyotype isolated from patients with acute myeloid leukaemia (AML) were found to have elevated levels of constitutive DNA damage and CHK1 activation and to be more sensitive to CHK1 depletion by RNAi or CHK1 inhibition by UCN-01 45 than AML cells with a normal cytogenetic profile or normal granulomonocytic progenitor cells [23]. The sensitivity of melanoma cell lines to single agent CHK1 inhibition also positively correlated to the level of endogenous DNA damage [22]. Single agent activity was also observed for an oral CHK1 inhibitor (structure undisclosed) in certain cancer cell lines, with in vivo activity demonstrated in HEL92.1.7 erythroleukamia cells grown as xenografts [89], although no marker for sensitivity was described.
Importantly, there is a growing body of evidence that overexpression of the MYC transcription factors leads to intrinsic replication stress and confers sensitivity to specific inhibition of CHK1 [25, 90]. CHK1 was identified from an RNAi screen as a potential therapeutic target in MYC-N amplified neuroblastoma, with constitutive activation of signalling through CHK1 observed in cells sensitive to RNAi or CHK1 inhibition [91]. The sensitivity of MYC-N driven neuroblastoma to single agent CHK1 inhibition was also demonstrated with the oral, selective CHK1 inhibitor CCT244747 (17) which showed efficacy in a transgenic mouse model of the disease [20]. Dual inhibition of CHK1 and WEE1 kinase is effective in models of MYCN-driven neuroblastoma [87]. Deregulated expression of the oncogene c-MYC in Eμ-myc lymphoma cells was also associated with sensitivity to CHK1 inhibition [92].
3.3 The therapeutic potential of CHK2 inhibitors
No selective CHK2 inhibitor has been progressed to clinical trials to date. In part this has reflected changing views over the most appropriate therapeutic context for CHK2 inhibition [2, 11]. CHK2 inhibition was originally perceived as a means to potentiate DNA-damaging anticancer chemotherapies, in a similar fashion to CHK1 inhibitors. However, some pharmacological and siRNA studies in cancer cells have reported that dual inhibition of CHK2 and CHK1 offered no benefit over selective CHK1 inhibition, and that CHK2 inhibition may be antagonistic to CHK1-mediated potentiation of genotoxic efficacy [93-96]. Other studies have shown that the potentiation of the cytotoxicity of DNA-damaging agents by CHK2 inhibition is possible in certain cell lines [59]. Nevertheless, the selective CHK2 inhibitors from multiple chemotypes described above have proved useful chemical tools for investigating the therapeutic potential of CHK2 inhibition in cellular studies, and some patterns have emerged (Table 2).
Several selective CHK2 inhibitors have been shown to have protective effects against ionising radiation (IR) in p53 wild type cells (Table 2). Thus, benzimidazole 31 protected isolated peripheral human CD4+ and CD8+ T-cells from γ-irradiation in a concentration-dependent fashion [60]. The 2-(2-quinazolinyl)phenol CCT241533 (42) was shown to confer a radioprotective effect in isolated mouse thymocytes, with ablation of apoptosis being observed [54]. The inhibitor VRX0466617 (44) also suppressed IR-induced apoptosis in BJ-hTERT cells [66]. The negative regulator of p53, HDMX, is phosphorylated at Ser342 and Ser367 by CHK2 on exposure to IR, which results in its degradation. The extent of IR-induced HDMX degradation was diminished in a concentration-dependent fashion by 44, while responses upstream to CHK2 induced by IR were not affected by 44. PV1019 (33) was shown to ablate IR-mediated apoptosis in mouse thymocytes [59]. This reduction in apoptosis copied the behaviour of chk2(−/−) cells when exposed to IR, providing evidence of the mechanism of action of 33 through inhibition of CHK2. Cellular inhibition of CHK2 by PV1019 at relevant concentrations was demonstrated against three known substrates of the CHK2 kinase function; CHK2 autophosphorylation (IC50 = 5 μM), HDMX, and Cdc25c. Non-malignant cells were also protected from radiation-induced apoptosis by the debromohymenialdisine derivative 43 [93].
While there is clear agreement on the radioprotective effect of selective CHK2 inhibition in cells with functional p53, there are still conflicting reports on the effects of combining CHK2 inhibitors with DNA-damaging cytotoxic drugs in cancer cells. Two studies have found no potentiation despite positive evidence of inhibition of CHK2 signalling in cells through assessment of pharmacodynamic biomarkers. Thus VRX0466617 (44) did not potentiate the cytotoxicity of doxorubicin or cisplatin in MCF7 cells, nor taxol in BJ-hTERT cells [66]. More comprehensively, CCT241533 (42) was found to give no potentiation of the cytotoxicity of the DNA-damaging agents SN38, gemcitabine, etoposide, mitomycin C or bleomycin in either HT29 colon cancer or HeLa cervical cancer cell lines, both of which are deficient in p53 function [26]. A similar lack of effect of CHK2 siRNA has been reported in cancer cells [94-96].
However, it has been shown that the selective CHK2 inhibitor PV1019 (33) potentiates the activity of the topoisomerase I inhibitors topotecan and camptothecin, as well as ionising radiation, in certain human tumour cells [59]. Treatment of the OVCAR-5 cell line with PV1019 (33) and topotecan increased the growth inhibitory effect of the cytotoxic agent. Treatment of the human brain tumour cell line U251 with PV1019 and IR resulted in a dose enhancement factor of 1.4, demonstrating the only reported radiosensitisation of a tumour cell line by a CHK2 inhibitor. Depletion of CHK2 by siRNA in two ovarian tumour cell lines, OVCAR-4 and OVCAR-8, that expressed high levels of CHK2, led to increased growth inhibition compared to the control and provided evidence that CHK2 inhibition may lead to antiproliferative effects in tumour cell lines that over express CHK2. Thus, while the potentiation of DNA-damaging agents is a firmly established therapeutic context for CHK1 inhibitors, the case for CHK2 inhibition remains unclear and may depend more critically on the cell genetic background and cytotoxic agent employed.
In contrast to the lack of synergy with DNA-damaging chemotherapies exhibited by CCT241533 (42), this compound has been shown to potentiate the efficacy of two structurally distinct PARP inhibitors [26]. Both HeLa and HT29 cells displayed enhanced sensitivity to the PARP inhibitors AG14447 and olaparib in the presence of CCT241533 (42), with a sharp decrease in growth of the HeLa cells compared to the control in short and longer term colony-forming experiments. The combination of CCT241533 (42) and olaparib was shown to enhance apoptosis in HeLa cells and affect PARP inhibitor cytotoxicity through a CHK2-dependent mechanism. It is proposed that the cytotoxicity of the combination of CCT241533 and olaparib arises from inhibition of CHK2 leading to inhibition of BRCA1 phosphorylation and impairment of the homologous recombination DNA repair pathway. As PARP inhibitors prevent DNA repair through the alternative base excision repair pathway, the dual treatment produces unrepairable and lethal DNA double strand breaks.
4. Expert Opinion
There have clearly been substantial advances in the basic science and clinical progress of checkpoint kinase inhibitors, particularly in the past five years. To date, the majority of opened clinical trials have been designed to examine the preclinically well established concept of combination of CHK1 inhibition with DNA-damaging chemotherapies (Table 1). However, proof-of-concept clinical data with these agents has still to be achieved. Of the several first generation inhibitors that have entered Phase I clinical trials, the development of UCN-01 has been hindered by poor pharmacokinetic properties, while a number of other compounds have been reported not to be under further development by their originating organisations. These observations notwithstanding, at least one other first generation intravenous CHK1 inhibitor (LY2603618) has progressed to Phase II trials in the combination setting. The dose-limiting toxicities in Phase I clinical data for the different compounds reported to date vary considerably, suggesting that off-target effects differing between the chemical scaffolds tested may be major contributing factors [74, 75, 97, 98]. As yet no CHK2-specific inhibitor has been reported to enter clinical trials.
The potential importance of high selectivity for CHK1 over CHK2 and other kinases in determining the efficacy of checkpoint inhibition in combination with genotoxic agents has been highlighted through basic research with inhibitors and siRNA [2, 17, 95, 99]. Specific CHK1 inhibition in combinations with multiple DNA-damaging agents is effective in a range of tumour cell in vitro and in vivo. While certain tumour cells do appear susceptible to the potentiation of DNA-damaging agents by CHK2 inhibition, the phenomenon is less widely observed than for CHK1 inhibitors, and may be restricted to specific tumour genetic backgrounds.
Many of the first generation checkpoint inhibitors were characterised by no or modest selectivity for CHK1 over CHK2. Second generation CHK1 inhibitors are likely to be substantially more selective for CHK1 than previous agents. The need for selectivity over CDK enzymes, particularly CDK1, CDK2 and CDK7, to avoid confounding mechanistic effects is also important, and it is notable that recent preclinical research has emphasised the use of mechanistic cellular assays to drive optimization of a selective mechanism-of-action, in addition to biochemical kinome profiling [17, 46].
Structure based design has played an important role in the development of selective CHK1 and CHK2 inhibitors. Most interestingly, studies on both of these structurally distinct enzymes have called attention to the potential benefit to selectivity of incorporating protein-bound water molecules into ligand design. For CHK1, targeting the network of water molecules in the interior pocket of the kinase leads to very high selectivity across several chemical scaffolds. A set of functional groups enabling interaction with these waters have been identified, based around hydrogen-bond accepting nitrogen heterocycles such as pyridine, pyrazine, pyrazole and triazole [5, 6, 37, 38, 41, 44, 46, 47, 49, 50]. The structural element promoting the assembly of the water network in CHK1 can be traced to a single amino acid (Asn59). It is interesting to speculate that the accumulation of bound water molecules associated with this residue effectively magnifies the scale of the structural difference in the binding site between CHK1 and other enzymes without this polar substituent, which may account for the high selectivities that can be achieved through targeting this feature.
In CHK2 the discovery of water-mediated binding of inhibitors to the hinge peptide of the kinase has not been attributed to the presence of a particular amino acid. It is, however, reproduced across several inhibitor scaffolds, and the intervening water molecule can be mimicked to good effect with a phenolic group (a substitution also achieved with respect to the protein-bound water molecules in CHK1 [36]). Very high selectivity for CHK2 over CHK1 and other kinases is associated with the water-mediated hinge-binding mode. The selectivity data on CHK2 inhibitors adopting the water-mediated binding mode supports the idea that a lower reliance on strong interactions to the hinge region of kinases could be beneficial for the selectivity of type I ATP-competitive kinase inhibitors in general.
The first generation CHK1 inhibitors are intravenous agents. This would not necessarily be limiting in the clinic for combination with classical DNA-damaging chemotherapies, since the cytotoxics are typically administered intravenously over short periods in any given treatment cycle. Recent preclinical data have shown that prolonged inhibition of CHK1 after DNA-damage may be beneficial to maximise the potentiation of the anti-tumour effect of the chemotherapy [6, 20]. In this regard, the development of oral compounds [20, 46, 89] may offer advantages in the flexibility of treatment scheduling to the second generation of CHK1 inhibitors. The recent in vivo demonstrations of the expected potentiation of radiotherapy by CHK1 inhibitors [78-80] also argues for oral agents if the combination of CHK1 inhibitors with long-term fractionated radiotherapy schedules is to be optimally translated to the clinic. It is not yet clear what the most effective treatment schedules for the emerging single agent therapeutic contexts for CHK1 inhibition will be, but there is a possibility that sustained exposure would be required, for which oral inhibitors could again offer greater flexibility in the clinic.
An exciting and important development in the checkpoint kinase field has been the preclinical demonstration of single agent efficacy for inhibitors of CHK1 in specific cancer types, which could significantly enhance the clinical benefit of future drugs in this class. Intrinsic DNA damage and the activation of CHK1 signalling resulting from high endogenous replication stress appear to underlie the effectiveness of the inhibitors in many cases, mirroring the potentiation of extrinsic DNA-damaging agents observed with CHK1 inhibitors. The association of overexpression of MYC transcription factors with constitutive activation of the DNA damage response and sensitivity to CHK1 inhibition is one of the potential strategies for patient stratification [25, 90]. Preclinical data reported to date suggests that this is most strongly evidenced for paediatric MYC-N driven neuroblastoma and some B-cell lymphomas [20, 91, 92]. However, it is also notable that sensitivity to CHK1 inhibition has been observed in other cancer cell types where overexpression of MYC may not be a dominant effect [22, 23, 89]. It may still be possible to stratify cancers that are likely to be sensitive to CHK1 inhibition alone based on quantifying high intrinsic activation of the DNA damage response through CHK1, and a range of pathway biomarkers have been investigated to enable this. While progress on defining contexts where selective CHK2 inhibition may have a single agent effect is less advanced, appropriate small molecule tool compounds to address this are now available.
Article Highlights.
Structure-based design has been successfully applied to generate potent and selective inhibitors of the DNA-damage response effector kinases CHK1 and CHK2.
Highly selective CHK1 inhibition is most commonly associated with binding to a unique network of buried, protein bound water molecules in the enzyme
An unusual water-mediated hinge-binding mode for CHK2 inhibitors leads to very high selectivity for ATP-competitive kinase inhibitors.
The therapeutic potential for CHK1 inhibitors in combination with DNA-damaging chemo- and radiotherapy is well established preclinically. There have been mixed outcomes in the clinical development of CHK1 inhibitors to date, but progress has been made and second generation inhibitors are entering clinical trials.
Exciting therapeutic contexts for single agent selective CHK1 inhibition have emerged based on targeting cancer genotypes that lead to replication stress and constitutive activation of the DNA-damage response.
Acknowledgements
This work was supported by Cancer Research UK [CUK] grant number C309/A11566, and by The Institute of Cancer Research, London.
Footnotes
Declaration of interests
The authors are employees of The Institute of Cancer Research which has a commercial interest in CHK1 and CHK2 inhibitors. Authors who are, or have been, employed by The Institute of Cancer Research are subject to a ‘Rewards for Inventors Scheme’ which may reward contributors to a program that is subsequently licensed. The authors have been involved in research collaborations on CHK1 inhibitors with Sareum Ltd. and Cancer Research Technology Ltd, and on CHK2 inhibitors with Cancer Research Technology Ltd. The authors have, or have had, direct or indirect commercial interactions with Sareum Ltd., Astex Therapeutics, AstraZeneca UK Ltd., Vernalis R&D Ltd. and Novartis.
References
* Articles of interest
** Articles of high interest
- 1.Chen T, Stephens PA, Middleton FK, Curtin NJ. Targeting the S and G2 checkpoint to treat cancer. Drug Discov Today. 2012;17:194–202. doi: 10.1016/j.drudis.2011.12.009. [DOI] [PubMed] [Google Scholar]
- 2.Garrett MD, Collins I. Anticancer therapy with checkpoint inhibitors: what, where and when? Trends Pharmacol Sci. 2011;32:308–16. doi: 10.1016/j.tips.2011.02.014. [DOI] [PubMed] [Google Scholar]
- 3.Ma CX, Janetka JW, Piwnica-Worms H. Death by releasing the breaks: CHK1 inhibitors as cancer therapeutics. Trends Mol Med. 2011;17:88–96. doi: 10.1016/j.molmed.2010.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Maugeri-Saccà M, Bartucci M, De Maria R. Checkpoint kinase 1 inhibitors for potentiating systemic anticancer therapy. Cancer Treat Rev. 2012 doi: 10.1016/j.ctrv.2012.10.007. Published online; http://dx.doi.org/10.1016/j.ctrv.2012.10.007. [DOI] [PubMed] [Google Scholar]
- 5.Janetka JW, Ashwell S, Zabludoff S, Lyne P. Inhibitors of checkpoint kinases: from discovery to the clinic. Curr Opin Drug Discovery Dev. 2007;10:473–86. [PubMed] [Google Scholar]
- 6.Lainchbury M, Collins I. Checkpoint kinase inhibitors: a patent review (2009 - 2010) Expert Opin Ther Pat. 2011;21:1191–210. doi: 10.1517/13543776.2011.586632. [DOI] [PubMed] [Google Scholar]
- 7.Prudhomme M. Novel checkpoint 1 inhibitors. Recent Pat Anti-Cancer Drug Discovery. 2006;1:55–68. doi: 10.2174/157489206775246520. [DOI] [PubMed] [Google Scholar]
- 8.Nguyen TN, Tepe JJ. Current inhibitors of checkpoint kinase 2. Curr Med Chem. 2011;18:4368–74. doi: 10.2174/092986711797200390. [DOI] [PubMed] [Google Scholar]
- 9.Reinhardt HC, Yaffe MB. Kinases that control the cell cycle in response to DNA damage: Chk1, Chk2, and MK2. Curr Opin Cell Biol. 2009;21:245–55. doi: 10.1016/j.ceb.2009.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Smith J, Tho LM, Xu N, Gillespie DA. The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and cancer. Adv Cancer Res. 2010;108:73–112. doi: 10.1016/B978-0-12-380888-2.00003-0. [DOI] [PubMed] [Google Scholar]
- 11.Antoni L, Sodha N, Collins I, Garrett MD. CHK2 kinase: cancer susceptibility and cancer therapy - two sides of the same coin? Nat Rev Cancer. 2007;7:925–36. doi: 10.1038/nrc2251. [DOI] [PubMed] [Google Scholar]
- 12.Dai Y, Grant S. New insights into checkpoint kinase 1 in the DNA damage response signaling network. Clin Cancer Res. 2010;16:376–83. doi: 10.1158/1078-0432.CCR-09-1029. [** A comprehensive review of role of checkpoint kinases in the biology of the DNA damage response] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 14.Hollstein M, Sidransky D, Vogelstein B, Harris CC. p53 mutations in human cancers. Science. 1991;253:49–53. doi: 10.1126/science.1905840. [DOI] [PubMed] [Google Scholar]
- 15.Oren M. Regulation of the p53 tumor suppressor protein. J Biol Chem. 1999;274:36031–4. doi: 10.1074/jbc.274.51.36031. [DOI] [PubMed] [Google Scholar]
- 16.Zabludoff SD, Deng C, Grondine MR, et al. AZD7762, a novel checkpoint kinase inhibitor, drives checkpoint abrogation and potentiates DNA-targeted therapies. Mol Cancer Ther. 2008;7:2955–66. doi: 10.1158/1535-7163.MCT-08-0492. [DOI] [PubMed] [Google Scholar]
- 17.Guzi TJ, Paruch K, Dwyer MP, et al. Targeting the replication checkpoint using SCH 900776, a potent and functionally selective CHK1 inhibitor identified via high content screening. Mol Cancer Ther. 2011;10:591–602. doi: 10.1158/1535-7163.MCT-10-0928. [* Describes the use of high-content cellular assays to define the selectivity profile for CHK1 inhibitors during lead optimization] [DOI] [PubMed] [Google Scholar]
- 18.Walton MI, Eve PD, Hayes A, et al. The preclinical pharmacology and therapeutic activity of the novel CHK1 inhibitor SAR-020106. Mol Cancer Ther. 2010;9:89–100. doi: 10.1158/1535-7163.MCT-09-0938. [DOI] [PubMed] [Google Scholar]
- 19.Blasina A, Hallin J, Chen E, et al. Breaching the DNA damage checkpoint via PF-00477736, a novel small-molecule inhibitor of checkpoint kinase 1. Mol Cancer Ther. 2008;7:2394–404. doi: 10.1158/1535-7163.MCT-07-2391. [DOI] [PubMed] [Google Scholar]
- 20.Walton MI, Eve PD, Hayes A, et al. CCT244747 is a novel potent and selective CHK1 inhibitor with oral efficacy alone and in combination with genotoxic anticancer drugs. Clin Cancer Res. 2012;18:5650–61. doi: 10.1158/1078-0432.CCR-12-1322. [* First paper to disclose both the structure and pharmacological properties of an orally bioavailable CHK1 inhibitor] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tse AN, Rendahl KG, Sheikh T, et al. CHIR-124, a novel potent inhibitor of Chk1, potentiates the cytotoxicity of topoisomerase I poisons in vitro and in vivo. Clin Cancer Res. 2007;13:591–602. doi: 10.1158/1078-0432.CCR-06-1424. [DOI] [PubMed] [Google Scholar]
- 22.Brooks K, Oakes V, Edwards B, et al. A potent Chk1 inhibitor is selectively cytotoxic in melanomas with high levels of replicative stress. Oncogene. 2012 doi: 10.1038/onc.2012.72. published online 5 March 2012; doi:10.1038/onc.2012.72. [DOI] [PubMed] [Google Scholar]
- 23.Cavelier C, Didier C, Prade N, et al. Constitutive activation of the DNA damage signaling pathway in acute myeloid leukemia with complex karyotype: potential importance for checkpoint targeting therapy. Cancer Res. 2009;69:8652–61. doi: 10.1158/0008-5472.CAN-09-0939. [DOI] [PubMed] [Google Scholar]
- 24.Conti C, Seiler JA, Pommier Y. The mammalian DNA replication elongation checkpoint: implication of Chk1 and relationship with origin firing as determined by single DNA molecule and single cell analyses. Cell Cycle. 2007;6:2760–7. doi: 10.4161/cc.6.22.4932. [DOI] [PubMed] [Google Scholar]
- 25.Hoglund A, Nilsson LM, Muralidharan SV, et al. Therapeutic implications for the induced levels of Chk1 in Myc-expressing cancer cells. Clin Cancer Res. 2011;17:7067–79. doi: 10.1158/1078-0432.CCR-11-1198. [** Studies exploring the potential therapeutic context for selective CHK1 inhibition in MYC-overexpressing cancer cells.] [DOI] [PubMed] [Google Scholar]
- 26.Anderson VE, Walton MI, Eve PD, et al. CCT241533 is a potent and selective inhibitor of CHK2 that potentiates the cytotoxicity of PARP inhibitors. Cancer Res. 2011;71:463–72. doi: 10.1158/0008-5472.CAN-10-1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Converso A, Hartingh T, Garbaccio RM, et al. Development of thioquinazolinones, allosteric Chk1 kinase inhibitors. Bioorg Med Chem Lett. 2009;19:1240–4. doi: 10.1016/j.bmcl.2008.12.076. [* First identification and characterization of novel allosteric inhibitors of CHK1] [DOI] [PubMed] [Google Scholar]
- 28.Vanderpool D, Johnson TO, Ping C, et al. Characterization of the CHK1 allosteric inhibitor binding site. Biochemistry. 2009;48:9823–30. doi: 10.1021/bi900258v. [DOI] [PubMed] [Google Scholar]
- 29.Chen P, Luo C, Deng Y, et al. The 1.7 A crystal structure of human cell cycle checkpoint kinase Chk1: implications for Chk1 regulation. Cell. 2000;100:681–92. doi: 10.1016/s0092-8674(00)80704-7. [DOI] [PubMed] [Google Scholar]
- 30.Oza V, Ashwell S, Almeida L, et al. Discovery of checkpoint kinase inhibitor (S)-5-(3-Fluorophenyl)-N-(piperidin-3-yl)-3-ureidothiophene-2-carboxamide (AZD7762) by structure-based design and optimization of thiophenecarboxamide ureas. J Med Chem. 2012;55:5130–42. doi: 10.1021/jm300025r. [DOI] [PubMed] [Google Scholar]
- 31.Janetka JW, Almeida L, Ashwell S, et al. Discovery of a novel class of 2-ureido thiophene carboxamide checkpoint kinase inhibitors. Bioorg Med Chem Lett. 2008;18:4242–8. doi: 10.1016/j.bmcl.2008.05.016. [DOI] [PubMed] [Google Scholar]
- 32.Zhao L, Zhang Y, Dai C, et al. Design, synthesis and SAR of thienopyridines as potent CHK1 inhibitors. Bioorg Med Chem Lett. 2010;20:7216–21. doi: 10.1016/j.bmcl.2010.10.105. [DOI] [PubMed] [Google Scholar]
- 33.Oza V, Ashwell S, Brassil P, et al. Discovery of a novel class of triazolones as checkpoint kinase inhibitors--hit to lead exploration. Bioorg Med Chem Lett. 2010;20:5133–8. doi: 10.1016/j.bmcl.2010.07.015. [DOI] [PubMed] [Google Scholar]
- 34.Oza V, Ashwell S, Brassil P, et al. Synthesis and evaluation of triazolones as checkpoint kinase 1 inhibitors. Bioorg Med Chem Lett. 2012;22:2330–7. doi: 10.1016/j.bmcl.2012.01.043. [DOI] [PubMed] [Google Scholar]
- 35.Zuccotto F, Ardini E, Casale E, Angiolini M. Through the “gatekeeper door”: exploiting the active kinase conformation. J Med Chem. 2010;53:2681–94. doi: 10.1021/jm901443h. [DOI] [PubMed] [Google Scholar]
- 36.Foloppe N, Fisher LM, Francis G, et al. Identification of a buried pocket for potent and selective inhibition of Chk1: prediction and verification. Bioorg Med Chem. 2006;14:1792–804. doi: 10.1016/j.bmc.2005.10.022. [* Early paper on the identification of structural features in CHK1 that may be targeted for selectivity] [DOI] [PubMed] [Google Scholar]
- 37.Fraley ME, Steen JT, Brnardic EJ, et al. 3-(Indol-2-yl)indazoles as Chek1 kinase inhibitors: Optimization of potency and selectivity via substitution at C6. Bioorg Med Chem Lett. 2006;16:6049–53. doi: 10.1016/j.bmcl.2006.08.118. [* Early paper showing displacement of protein bound water molecules in CHK1 by the hydroxyl side chain of a potent inhibitor.] [DOI] [PubMed] [Google Scholar]
- 38.Tao Z-F,, Wang L,, Stewart KD,, et al. Structure-based design, synthesis, and biological evaluation of potent and selective macrocyclic checkpoint kinase 1 inhibitors. J Med Chem. 2007;50:1514–27. doi: 10.1021/jm061247v. [DOI] [PubMed] [Google Scholar]
- 39.Vernalis Oncology pipeline [Last accessed 28th January 2013]; V158411. Available at: http://www.vernalis.com/development/nce-pipeline.
- 40.Massey AJ, Stokes S, Browne H, et al. Abstract C207: Checkpoint abrogation and potentiation of cytotoxic chemotherapeutics with a novel checkpoint kinase 1 inhibitor. AACR - NCI - EORTC International Conference: Molecular Targets and Cancer Therapeutics - Nov 15–19, 2009; Boston, MA. Mol Canc Ther. 2009;8:C207. [Google Scholar]
- 41.Li G, Hasvold La, Tao Z-F, et al. Synthesis and biological evaluation of 1-(2,4,5-trisubstituted phenyl)-3-(5-cyanopyrazin-2-yl)ureas as potent Chk1 kinase inhibitors. Bioorg Med Chem Lett. 2006;16:2293–8. doi: 10.1016/j.bmcl.2006.01.028. [* First reported structure of a cyanopyrazine interacting with the protein bound water molecules in CHK1.] [DOI] [PubMed] [Google Scholar]
- 42.Matthews TP, Klair S, Burns S, et al. Identification of inhibitors of checkpoint kinase 1 through template screening. J Med Chem. 2009;52:4810–9. doi: 10.1021/jm900314j. [DOI] [PubMed] [Google Scholar]
- 43.Matthews TP, McHardy T, Klair S, et al. Design and evaluation of 3,6-di(hetero)aryl imidazo[1,2-a]pyrazines as inhibitors of checkpoint and other kinases. Bioorg Med Chem Lett. 2010;20:4045–9. doi: 10.1016/j.bmcl.2010.05.096. [DOI] [PubMed] [Google Scholar]
- 44.Reader JC, Matthews TP, Klair S, et al. Structure-guided evolution of potent and selective CHK1 inhibitors through scaffold morphing. J Med Chem. 2011;54:8328–42. doi: 10.1021/jm2007326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Borst GR, McLaughlin M, Kyula JN, et al. Targeted radiosensitization by the Chk1 Inhibitor SAR-020106. Int J Radiat Oncol, Biol, Phys. 2012 doi: 10.1016/j.ijrobp.2012.08.006. published online 4 September 2012, doi:10.1016/j.ijrobp.2012.08.006. [DOI] [PubMed] [Google Scholar]
- 46.Lainchbury M, Matthews TP, McHardy T, et al. Discovery of 3-alkoxyamino-5-(pyridin-2-ylamino)pyrazine-2-carbonitriles as selective, orally bioavailable CHK1 inhibitors. J Med Chem. 2012;55:10229–40. doi: 10.1021/jm3012933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dwyer MP, Paruch K, Labroli M, et al. Discovery of pyrazolo[1,5-a]pyrimidine-based CHK1 inhibitors: a template-based approach-part 1. Bioorg Med Chem Lett. 2011;21:467–70. doi: 10.1016/j.bmcl.2010.10.113. [DOI] [PubMed] [Google Scholar]
- 48.Montano R, Chung I, Garner K, et al. Preclinical development of the novel Chk1 Inhibitor SCH900776 in combination with DNA damaging agents and antimetabolites. Mol Cancer Ther. 2011:427–38. doi: 10.1158/1535-7163.MCT-11-0406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Huang X, Cheng CC, Fischmann TO, et al. Discovery of a novel series of CHK1 kinase inhibitors with a distinctive hinge binding mode. ACS Med Chem Lett. 2012;3:123–28. doi: 10.1021/ml200249h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dudkin VY, Rickert K, Kreatsoulas C, et al. Pyridyl aminothiazoles as potent inhibitors of Chk1 with slow dissociation rates. Bioorg Med Chem Lett. 2012;22:2609–12. doi: 10.1016/j.bmcl.2012.01.110. [DOI] [PubMed] [Google Scholar]
- 51.Dudkin VY, Wang C, Arrington KL, et al. Pyridyl aminothiazoles as potent Chk1 inhibitors: optimization of cellular activity. Bioorg Med Chem Lett. 2012;22:2613–9. doi: 10.1016/j.bmcl.2012.01.120. [DOI] [PubMed] [Google Scholar]
- 52.Hilton S, Naud S, Caldwell JJ, et al. Identification and characterisation of 2-aminopyridine inhibitors of checkpoint kinase 2. Bioorg Med Chem. 2010;18:707–18. doi: 10.1016/j.bmc.2009.11.058. [DOI] [PubMed] [Google Scholar]
- 53.Curman D, Cinel B, Williams DE, et al. Inhibition of the G2 DNA damage checkpoint and of protein kinases Chk1 and Chk2 by the marine sponge alkaloid debromohymenialdisine. J Biol Chem. 2001;276:17914–9. doi: 10.1074/jbc.M100728200. [DOI] [PubMed] [Google Scholar]
- 54.Caldwell JJ, Welsh EJ, Matijssen C, et al. Structure-based design of potent and selective 2-(quinazolin-2-yl)phenol inhibitors of checkpoint kinase 2. J Med Chem. 2011;54:580–90. doi: 10.1021/jm101150b. [DOI] [PubMed] [Google Scholar]
- 55.Matijssen C, Silva-Santisteban MC, Westwood IM, et al. Benzimidazole inhibitors of the protein kinase CHK2: clarification of the binding mode by flexible side chain docking and protein-ligand crystallography. Bioorg Med Chem. 2012;20:6630–9. doi: 10.1016/j.bmc.2012.09.024. [* Demonstration of an unusual water-mediated binding mode to the kinase hinge by potent and selective 2-arylbenzimidazole CHK2 inhibitors.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lountos GT, Tropea JE, Zhang D, et al. Crystal structure of checkpoint kinase 2 in complex with NSC 109555, a potent and selective inhibitor. Protein Sci. 2009;18:92–100. doi: 10.1002/pro.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lountos GT, Jobson AG, Tropea JE, et al. Structural characterization of inhibitor complexes with checkpoint kinase 2 (Chk2), a drug target for cancer therapy. J Struct Biol. 2011;176:292–301. doi: 10.1016/j.jsb.2011.09.008. [* Describes the structure-based design of a very potent and cell permeable CHK2 inhibitor using a water mediated interaction to the hinge region.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jobson AG, Cardellina JH, Scudiero D, et al. Identification of a Bis-guanylhydrazone [4,4′-Diacetyldiphenylurea-bis(guanylhydrazone); NSC 109555] as a Novel Chemotype for Inhibition of Chk2 Kinase. Mol Pharmacol. 2007;72:876–84. doi: 10.1124/mol.107.035832. [DOI] [PubMed] [Google Scholar]
- 59.Jobson AG, Lountos GT, Lorenzi PL, et al. Cellular inhibition of checkpoint kinase 2 (Chk2) and potentiation of camptothecins and radiation by the novel Chk2 inhibitor PV1019 [7-nitro-1H-indole-2-carboxylic acid {4-[1-(guanidinohydrazone)-ethyl]-phenyl}-amide] J Pharmacol Exp Ther. 2009;331:816–26. doi: 10.1124/jpet.109.154997. [* Describes the application of structure-based drug design to generate a highly selective CHK2 inhibitor which is the only reported example to show radiosensitization.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Arienti KL, Brunmark A, Axe FU, et al. Checkpoint kinase inhibitors: SAR and radioprotective properties of a series of 2-arylbenzimidazoles. J Med Chem. 2005;48:1873–85. doi: 10.1021/jm0495935. [DOI] [PubMed] [Google Scholar]
- 61.Neff DK, Lee-Dutra A, Blevitt JM, et al. 2-Aryl benzimidazoles featuring alkyl-linked pendant alcohols and amines as inhibitors of checkpoint kinase Chk2. Bioorg Med Chem Lett. 2007;17:6467–71. doi: 10.1016/j.bmcl.2007.09.098. [DOI] [PubMed] [Google Scholar]
- 62.McClure KJ, Huang L, Arienti KL, et al. Novel non-benzimidazole Chk2 kinase inhibitors. Bioorg Med Chem Lett. 2006;16:1924–8. doi: 10.1016/j.bmcl.2005.12.096. [DOI] [PubMed] [Google Scholar]
- 63.Oliver AW, Paul A, Boxall KJ, et al. Trans-activation of the DNA-damage signalling protein kinase Chk2 by T-loop exchange. The EMBO journal. 2006;25:3179–90. doi: 10.1038/sj.emboj.7601209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sharma V, Tepe JJ. Potent inhibition of checkpoint kinase activity by a hymenialdisine-derived indoloazepine. Bioorg Med Chem Lett. 2004;14:4319–21. doi: 10.1016/j.bmcl.2004.05.079. [DOI] [PubMed] [Google Scholar]
- 65.Saleem RSZ, Lansdell Ta, Tepe JJ. Synthesis and evaluation of debromohymenialdisine-derived Chk2 inhibitors. Bioorg Med Chem. 2012;20:1475–81. doi: 10.1016/j.bmc.2011.12.054. [DOI] [PubMed] [Google Scholar]
- 66.Carlessi L, Buscemi G, Larson G, et al. Biochemical and cellular characterization of VRX0466617, a novel and selective inhibitor for the checkpoint kinase Chk2. Mol Cancer Ther. 2007;6:935–44. doi: 10.1158/1535-7163.MCT-06-0567. [DOI] [PubMed] [Google Scholar]
- 67.Larson G, Yan S, Chen H, et al. Identification of novel, selective and potent Chk2 inhibitors. Bioorg Med Chem Lett. 2007;17:172–5. doi: 10.1016/j.bmcl.2006.09.067. [DOI] [PubMed] [Google Scholar]
- 68.Fuse E, Kuwabara T, Sparreboom A, et al. Review of UCN-01 development: a lesson in the importance of clinical pharmacology. J Clin Pharmacol. 2005;45:394–403. doi: 10.1177/0091270005274549. [DOI] [PubMed] [Google Scholar]
- 69.Senderowicz AM. Cyclin-dependent kinase modulators: a novel class of cell cycle regulators for cancer therapy. Cancer Chemother Biol Response Modif. 2001;19:165–88. [PubMed] [Google Scholar]
- 70.Ashwell S, Janetka JW, Zabludoff S. Keeping checkpoint kinases in line: new selective inhibitors in clinical trials. Expert Opin Investig Drugs. 2008;17:1331–40. doi: 10.1517/13543784.17.9.1331. [DOI] [PubMed] [Google Scholar]
- 71.Wu W, Bi C, Bence AK, et al. Abstract 1776: Antitumor activity of Chk1 inhibitor LY2606368 as a single agent in SW1990 human pancreas orthotopic tumor model. AACR 103rd Annual Meeting 2012, Mar 31-Apr 4, 2012; Chicago, IL. Cancer Res. 2012;72(Supplement 1) [Google Scholar]
- 72.McNeely SC, Burke TF, DurlandBusbice S, et al. Abstract A108: LY2606368, a second generation Chk1 inhibitor, inhibits growth of ovarian carcinoma xenografts either as monotherapy or in combination with standard-of-care agents. AACR-NCI-EORTC International Conference: Molecular Targets and Cancer Therapeutics, Nov 12-16, 2011; San Francisco, CA. Mol Cancer Ther. 2011;10(Supplement 1) [Google Scholar]
- 73.Calvo E, Richards D, Braiteh F, et al. Abstract A94: Dose determination of LY2603618, a Chk1 inhibitor, administered in combination with gemcitabine in patients with advanced cancer. AACR-NCI-EORTC International Conference: Molecular Targets and Cancer Therapeutics, Nov 12-16, 2011; San Francisco, CA. Mol Cancer Ther. 2011;10(Supplement 1) [Google Scholar]
- 74.Weiss GJ, Donehower RC, Iyengar T, et al. Phase I dose-escalation study to examine the safety and tolerability of LY2603618, a checkpoint 1 kinase inhibitor, administered 1 day after pemetrexed 500 mg/m(2) every 21 days in patients with cancer. Invest New Drugs. 2013;31:136–44. doi: 10.1007/s10637-012-9815-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Karp JE, Thomas BM, Greer JM, et al. Phase I and pharmacologic trial of cytosine arabinoside with the selective checkpoint 1 Inhibitor SCH900776 in refractory acute leukemias. Clin Cancer Res. 2012 doi: 10.1158/1078-0432.CCR-12-2442. published online 23 October 2012, doi:10.1158/078-0432.CCR-12-2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Luo Y, Leverson JD. New opportunities in chemosensitization and radiosensitization: modulating the DNA-damage response. Expert Rev Anticancer Ther. 2005;5:333–42. doi: 10.1586/14737140.5.2.333. [DOI] [PubMed] [Google Scholar]
- 77.Maity A, McKenna WG, Muschel RJ. The molecular basis for cell cycle delays following ionizing radiation: a review. Radiother Oncol. 1994;31:1–13. doi: 10.1016/0167-8140(94)90408-1. [DOI] [PubMed] [Google Scholar]
- 78.Mitchell JB, Choudhuri R, Fabre K, et al. In vitro and in vivo radiation sensitization of human tumor cells by a novel checkpoint kinase inhibitor, AZD7762. Clin Cancer Res. 2010;16:2076–84. doi: 10.1158/1078-0432.CCR-09-3277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Morgan MA, Parsels LA, Zhao L, et al. Mechanism of radiosensitization by the Chk1/2 inhibitor AZD7762 involves abrogation of the G2 checkpoint and inhibition of homologous recombinational DNA repair. Cancer Res. 2010;70:4972–81. doi: 10.1158/0008-5472.CAN-09-3573. [* Studies demonstrating the potential for CHK1 inhibition combined with chemo-radiation in pancreatic cancer] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yang H, Yoon SJ, Jin J, et al. Inhibition of checkpoint kinase 1 sensitises lung cancer brain metastases to radiotherapy. Biochem Biophys Res Commun. 2011;406:53–8. doi: 10.1016/j.bbrc.2011.01.106. [DOI] [PubMed] [Google Scholar]
- 81.Seol HJ, Yoo HY, Jin J, et al. The expression of DNA damage checkpoint proteins and prognostic implication in metastatic brain tumors. Oncol Res. 2011;19:381–90. doi: 10.3727/096504011x13123323849654. [DOI] [PubMed] [Google Scholar]
- 82.Tang Y, Dai Y, Grant S, Dent P. Enhancing CHK1 inhibitor lethality in glioblastoma. Cancer Biol Ther. 2012;13:379–88. doi: 10.4161/cbt.19240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Tang Y, Hamed HA, Poklepovic A, et al. Poly(ADP-ribose) polymerase 1 modulates the lethality of CHK1 inhibitors in mammary tumors. Mol Pharmacol. 2012;82:322–32. doi: 10.1124/mol.112.078907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Davies KD, Cable PL, Garrus JE, et al. Chk1 inhibition and Wee1 inhibition combine synergistically to impede cellular proliferation. Cancer Biol Ther. 2011;12:788–96. doi: 10.4161/cbt.12.9.17673. [DOI] [PubMed] [Google Scholar]
- 85.Carrassa L, Chilà R, Lupi M, et al. Combined inhibition of Chk1 and Wee1 in vitro synergistic effect translates to tumor growth inhibition in vivo. Cell Cycle. 2012;11:2507–17. doi: 10.4161/cc.20899. [DOI] [PubMed] [Google Scholar]
- 86.Guertin AD, Martin MM, Roberts B, et al. Unique functions of CHK1 and WEE1 underlie synergistic anti-tumor activity upon pharmacologic inhibition. Cancer Cell Intl. 2012;12:45. doi: 10.1186/1475-2867-12-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Russell MR, Levin K, Rader J, et al. Combination therapy targeting the chk1 and wee1 kinases shows therapeutic efficacy in neuroblastoma. Cancer Res. 2013;73:779–84. doi: 10.1158/0008-5472.CAN-12-2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.McNeely S, Conti C, Sheikh T, et al. Chk1 inhibition after replicative stress activates a double strand break response mediated by ATM and DNA-dependent protein kinase. Cell Cycle. 2010;9:995–1004. doi: 10.4161/cc.9.5.10935. [DOI] [PubMed] [Google Scholar]
- 89.Davies KD, Humphries MJ, Sullivan FX, et al. Single-agent inhibition of Chk1 is antiproliferative in human cancer cell lines in vitro and inhibits tumor xenograft growth in vivo. Oncol Res. 2011;19:349–63. doi: 10.3727/096504011x13079697132961. [DOI] [PubMed] [Google Scholar]
- 90.Murga M, Campaner S, Lopez-Contreras AJ, et al. Exploiting oncogene-induced replicative stress for the selective killing of Myc-driven tumors. Nat Struct Mol Biol. 2011;18:1331–5. doi: 10.1038/nsmb.2189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Cole KA, Huggins J, Laquaglia M, et al. RNAi screen of the protein kinome identifies checkpoint kinase 1 (CHK1) as a therapeutic target in neuroblastoma. Proc Natl Acad Sci U S A. 2011;108:3336–41. doi: 10.1073/pnas.1012351108. [** Studies that identified and validated selective inhibition of CHK1 as a target in MYCN-driven paediatric neuroblastoma] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ferrao PT, Bukczynska EP, Johnstone RW, McArthur GA. Efficacy of CHK inhibitors as single agents in MYC-driven lymphoma cells. Oncogene. 2012;31:1661–72. doi: 10.1038/onc.2011.358. [** Studies showing the sensitivity of MYC-driven lymphoma cells to CHK1 inhibition] [DOI] [PubMed] [Google Scholar]
- 93.Nguyen TNT, Saleem RSZ, Luderer MJ, et al. Radioprotection by hymenialdisine-derived checkpoint kinase 2 inhibitors. ACS Chem Biol. 2012;7:172–84. doi: 10.1021/cb200320c. [DOI] [PubMed] [Google Scholar]
- 94.Morgan MA, Parsels LA, Parsels JD, et al. The relationship of premature mitosis to cytotoxicity in response to checkpoint abrogation and antimetabolite treatment. Cell Cycle. 2006;5:1983–8. doi: 10.4161/cc.5.17.3184. [DOI] [PubMed] [Google Scholar]
- 95.Xiao Z, Xue J, Sowin TJ, Zhang H. Differential roles of checkpoint kinase 1, checkpoint kinase 2, and mitogen-activated protein kinase-activated protein kinase 2 in mediating DNA damage-induced cell cycle arrest: implications for cancer therapy. Mol Cancer Ther. 2006;5:1935–43. doi: 10.1158/1535-7163.MCT-06-0077. [DOI] [PubMed] [Google Scholar]
- 96.Azorsa DO, Gonzales IM, Basu GD, et al. Synthetic lethal RNAi screening identifies sensitizing targets for gemcitabine therapy in pancreatic cancer. J Transl Med. 2009;7:43. doi: 10.1186/1479-5876-7-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ho AL, Bendell JC, Cleary JM, et al. Phase I, open-label, dose-escalation study of AZD7762 in combination with irinotecan (irino) in patients (pts) with advanced solid tumors. J Clin Oncol. 2011;29:3033. [Google Scholar]
- 98.Sausville EA, LoRusso P, Carducci MA, et al. Phase I dose-escalation study of AZD7762 in combination with gemcitabine (gem) in patients (pts) with advanced solid tumors. J Clin Oncol. 2011;29:3058. [Google Scholar]
- 99.Stolz A, Ertych N, Bastians H. Tumor suppressor CHK2: regulator of DNA damage response and mediator of chromosomal stability. Clin Cancer Res. 2011;17:401–5. doi: 10.1158/1078-0432.CCR-10-1215. [DOI] [PubMed] [Google Scholar]
- 100.Fracasso PM, Williams KJ, Chen RC, et al. A Phase 1 study of UCN-01 in combination with irinotecan in patients with resistant solid tumor malignancies. Cancer Chemother Pharmacol. 2011;67:1225–37. doi: 10.1007/s00280-010-1410-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Li T, Christensen SD, Frankel PH, et al. A phase II study of cell cycle inhibitor UCN-01 in patients with metastatic melanoma: a California Cancer Consortium trial. Invest New Drugs. 2012;30:741–8. doi: 10.1007/s10637-010-9562-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kummar S, Gutierrez ME, Gardner ER, et al. A phase I trial of UCN-01 and prednisone in patients with refractory solid tumors and lymphomas. Cancer Chemother Pharmacol. 2010;65:383–9. doi: 10.1007/s00280-009-1154-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ma CX, Ellis MJ, Petroni GR, et al. A phase II study of UCN-01 in combination with irinotecan in patients with metastatic triple negative breast cancer. Breast Cancer Res Treat. 2013;137:483–92. doi: 10.1007/s10549-012-2378-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. [Last accessed 24 January 2013];PF-00477736 is being studied in advanced solid tumors in combination with chemotherapy with gemcitabine. Available at: http://clinicaltrials.gov/ct2/show/results/NCT00437203.
- 105.Array BioPharma, Inc. WO2010118390 Checkpoint kinase 1 inhibitors for potentiating the effect of DNA damaging agents in treatment of cancer. 2010
- 106. [Last accessed 28 January 2013];Targeting Checkpoint Kinase 1: a study in the application of preclinical data to inform clinical strategy. Presentation for the 2nd Annual Cancer Targets & Therapeutics Conference 10/21/2010. Available at: http://www.arraybiopharma.com/_documents/Publication/PubAttachment410.pdf.