

Abstract
Most mouse models of diabetes do not fully reproduce features of human diabetic nephropathy, limiting their utility in inferring mechanisms of human disease. Here we performed detailed phenotypic and genetic characterization of leptin-receptor (Lepr) deficient mice on the FVB/NJ background (FVBdb/db), an obese model of type II diabetes, to determine their suitability to model human diabetic nephropathy. These mice have sustained hyperglycemia, significant albuminuria and characteristic diabetic renal findings including mesangial sclerosis and nodular glomerulosclerosis after 6 months of age. In contrast, equally obese, hyperglycemic Lepr/Sur1 deficient C57BL/6J (Sur1 has defective insulin secretion) mice have minimal evidence of nephropathy. A genome-wide scan in 165 Lepr deficient backcross progeny derived from FVB/NJ and C57BL/6J identified a major locus influencing nephropathy and albuminuria on chromosome 8B1-C5 (Dbnph1 locus, peak lod score 5.0). This locus was distinct from those contrasting susceptibility to beta cell hypertrophy and HIV-nephropathy between the same parental strains, indicating specificity to diabetic kidney disease. Genome-wide expression profiling showed that high and low risk Dbnph1 genotypes were associated with significant enrichment for oxidative phosphorylation and lipid clearance, respectively; molecular pathways shared with human diabetic nephropathy. Hence, we found that the FVBdb/db mouse recapitulates many clinical, histopathological and molecular features of human diabetic nephropathy. Identifying underlying susceptibility gene(s) and downstream dysregulated pathways in these mice may provide insight into the disease pathogenesis in humans.
INTRODUCTION
Diabetic nephropathy (DN) is the most common cause of kidney failure worldwide 1. DN has characteristic histopathologic features and is associated with dysregulation of a large number of cellular and molecular pathways2. Interindividual variability in risk of DN and familial aggregation of the trait have suggested significant genetic contribution to this disease3–12. For example, only one-third of diabetics eventually develop nephropathy, and up to one quarter of DN patients have a relative with end-stage renal failure3–12. Genetic and epidemiologic studies have also clearly demonstrated that DN has complex inheritance and the development of disease is determined by the interplay of many risk alleles 3–12. Human linkage studies have mapped susceptibility loci for DN to multiple segments of the genome 3–9. Critically, these studies have indicated that DN risk alleles are distinct from those conferring susceptibility to diabetes or other forms of nephropathy 3–12 and that discovery of underlying genes would necessitate very large populations concordant for diabetes duration but discordant for nephropathy outcome. More recently, a genome-wide association study (GWAS) has identified four suggestive susceptibility loci that impart risk ratios of 1.3–1.5 on disease12.
As the result of additional GWAS studies emerge in the near future, relevant experimental models will be necessary to characterize the role of implicated genes in disease pathogenesis. Genetic studies of murine models can provide a powerful complement to human studies because they permit rigorous control of genetic and environmental parameters, and also provide the ability to readily access relevant tissues, enabling careful dissection of disease-contributing pathways. However, the best studied diabetes models, such as the streptozotocin model or obese mice on the C57BL/B6 background (e.g. Leptin/Leptin receptor deficient mice) only manifest mild proteinuria, minimal glomerulosclerosis and no progression to renal failure, limiting inference to human disease13,14. It is not clear whether the absence of classic DN in these models is attributable to genetic resistance to diabetic renal complications, insufficient duration of hyperglycemia, or a combination of both factors 13–22.
The Leptin receptor (Lepr) deficient mouse on the FVB/NJ genetic background (FVBdb/db) is a validated model of sustained type 2 diabetes 21–23. Lepr deficiency causes hyperphagia, insulin resistance, defective thermogenesis, and infertility but the development of hyperglycemia is strain dependent 16,21,22,24,25. The Lepr deficient C57BL/6J mice (B6db/db) only exhibit transient hyperglycemia 13,19, while the Lepr-deficient C57BLKS/J mice develop severe hyperglycemia, which produces islet cell toxicity, rapidly leading to severe metabolic derangements and early demise 16,25. In contrast, the FVB/NJ beta cells are resistant to toxic effects of hyperglycemia and expand sufficiently to secrete insulin and permit survival 21,22. Consequently, the FVBdb/db mice exhibit sustained diabetes for over 6 month, providing a powerful model for examining the long-term complications of hyperglycemia23. Our previous genetic studies have shown that the differential ability to expand beta cell mass between the FVB/NJ and C57BL/6J strains is attributable to a major gene on chromosome 5 22. Interestingly, these same strains exhibit contrasting susceptibility to HIV-1 associated nephropathy (HIVAN) and our mapping studies have recently shown that these HIVAN susceptibility loci encode transregulators of podocyte gene expression24.
Initial characterization of the FVBdb/db model had shown development of glomerulosclerosis with several classic DN features, as well as significantly increased renal lipid accumulation23. Here, we determined whether the divergent DN phenotype between the B6db/db and FVBdb/db strains is simply attributable to differences in duration of hyperglycemia or reflected variation in genetic susceptibility to kidney damage. Since we had previously characterized determinants of susceptibility to diabetes and HIVAN between the same strains, the present study was also able to also distinguish whether the murine DN susceptibility loci and downstream pathways are the same or distinct from genetic factors mediating susceptibility to diabetes or other forms of nephropathy.
RESULTS
Characterization of strains with contrasting genetic susceptibility to DN
Both sexes of FVBdb/db mice show obesity, persistently elevated blood glucose concentrations starting at weaning (Table 1, ref. 21,22). Consistent with previous findings23, longitudinal study revealed the development of massive albuminuria after 4 to 5 months of age (Figure 1). By six months of age, the FVBdb/db had ten-fold more albuminuria compared to non-diabetic counterparts. This was accompanied by capillary basement membrane thickening, diffuse mesangial sclerosis and nodular glomerulosclerosis (Figure 2). In addition, the kidneys displayed widespread tulubointerstitial disease, with microsystic tubular dilatation due to protein cast formation (Figure 2). The sclerotic lesions involved 15–50% of glomeruli. In contrast to other models of DN13,14, there was no significant increase in blood pressure or heart rate in obese FVBdb/db compared to lean heterozygous counterparts (Figure 1). These data demonstrate that the FVBdb/db strain is an excellent model for studying the pathogenesis of DN.
Table 1.
Characteristics of mouse cohorts mice at 6 months of age.
| FVBdb/db | B6db/db | B6db/db Sur 1−/− | (FVB×B6) F1 db/db |
Lean | |
|---|---|---|---|---|---|
| Male | |||||
| N | 10 | 8 | 9 | 14 | 3 |
| Weight (grams) | 71.3 ± 6.3** | 61.4 ± 9.3* | 59.5 ± 9.0* | 70.8 ± 6.1 | 32.4 ± 3.0 |
| Glucose (mg/dl) | 369 ± 173* | 191 ± 159 | 327 ± 141* | 561 ± 55** | 95 ± 33 |
| Insulin (ng/ml) | 47.0 ± 26* | 58.6 ± 45 | 38.6 ± 33 | 10.9 ± 12 | 2.9 ± 1.6 |
| Histology Scores | 4.3 ± 0.9 # | 0.5 ± 0.5 | 0.8 ± 0.7 | 0.8 ± 0.8 | 0.1 ± 0.4 |
| Female | |||||
| N | 16 | 9 | 9 | 21 | 5 |
| Weight (grams) | 73.0 ± 6.8*** | 60.8 ± 5.5*** | 54.7 ± 6.1 *** | 74.0 ± 6.9 *** | 32.4 ± 3.0 |
| Glucose (mg/dl) | 239 ± 107* | 133 ± 26 | 266 ± 148* | 411 ± 81*** | 115 ± 15 |
| Insulin (ng/ml) | 485 ± 154 *** | 76.3 ± 60* | 24.8 ± 33.1 | 60.9 ± 31** | 1.0 ± 0.2 |
| Histology Scores | 3.7 ± 0.6 # | 0.3 ± 0.5 | 0.5 ± 0.5 | 0.5 ± 0.5 | 0.2 ± 0.4 |
Male and female mice were sacrificed at 6 months of age. Data are presented as means and standard deviations. The group size refers to number of mice evaluated for weight, glucose and insulin. The group size for histology scores are listed in supplementary table 1. Blood glucose was determined in the ad lib state. Lean mice are db+/− genotype on the B6db+/− or FVBdb+/− mice.
p<0.05,
p<0.001, p<0.0001 vs. lean mice.
p<0.001 vs. any other genotype group (two sided t-tests). The scores for subcomponents are shown in supplemental data.
Figure 1.
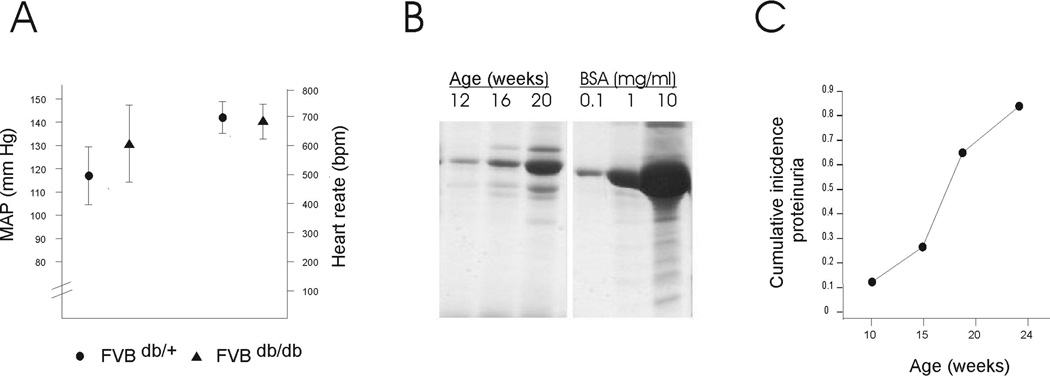
A. Tail Cuff measurements show no differences in systolic blood pressure and heart rate between FVB db/db and lean FVBdb/+ mice B. Serial SDS-PAGE of spot urine samples from one FVB db/db mouse at three different ages. BSA standards are shown on the right panel. C. Progressive increase in the proportion of mice showing albuminuria >1mg/ml, as measured by SDS-PAGE of spot urine samples compared to a dilution of BSA standard (N=12–18 male and famel mice per age group). D. Urine albumin/creatinine ratio at 28 and 52 weeks of age (*p<0.05 FVB db/db vs. FVBdb/+)
Figure 2. Histopathology of FVBdb/db Kidneys.

A. Diffuse and global mesangial sclerosis affecting all glomeruli (20X). B. Glomerulus with nodular mesangial sclerosis (60X). C. Hyaline accumulation in Bowman's capsule forming a "capsular drop" lesion (60X). D. Severe mesangial sclerosis occluding glomerular capillary lumina in continuity with arteriosclerosis of the preglomerular arteriole (60X).
In contrast to FVBdb/db, B6db/db mice only exhibit transient hyperglycemia (~1–2 months) and minimal kidney lesions (table 1). This was confirmed in independent cohort of FVBdb/db and B6db/db mice studied at Albert Einstein College of Medicine (supplementary table 1). To distinguish whether this difference in severity of nephropathy is attributable to variation in duration of hyperglycemia or genetic susceptibility to kidney injury, we generated an equally obese and hyperglycemic C57BL/6J model by combining db and Sur1 null alleles in the C57BL/6J genetic background (B6 Sur1 −/−,db/db mice). The Sur1 gene encodes one of the two subunits that compose the ATP sensitive K-channel in the beta cell, and the loss of SUR1 causes a defect in insulin secretion 25. The addition of Sur1 null alleles on the B6db/db background resulted in sustained hyperglycemia and glucose intolerance comparable to the FVBdb/db strain (Table 1). However, detailed histologic examination of both B6db/db and B6 Sur1 −/−,db/db kidneys at 6 months of age only revealed glomerulomegaly and mild mesangial matrix expansion without any evidence of glomerulosclerosis or tubulointerstitial disease (Table 1, Figure 3). These data demonstrated that the B6 genome confers protective alleles that prevent the development of nephropathy. To determine the likely mode of inheritance, we generated (FVB × B6)db/db F1 hybrids. Similar to B6 Sur1 −/−,db/db mice, F1 hybrids were hyperglycemic but did not demonstrate any significant evidence of renal disease at 6 months of age (Table 1). These data indicated dominant inheritance of diabetes susceptibility alleles, but recessive inheritance of nephropathy susceptibility, suggesting that a backcross of the F1 hybrids to FVB strain would localize DN susceptibility genes.
Figure 3. Comparison of histopathology between FVBdb/db (A–C) and B6db/db Sur1−/− (D–F).

There is diffuse and global mesangial sclerosis in FVBdb/db mice (A, 40X) but minimal mesangial sclerosis in B6db/db Sur1−/− glomeruli (C, 40X). Proteinaceous casts dilate the outer medullary tubules in FVB db/db mice (B, 60X), but not B6db/db Sur1−/− mice (D, 60X). Electron micrograph of FVBdb/db glomerulus shows thickened glomerular basement membranes and effaced foot processes (C). A representative capillary of B6db/db Sur1−/− glomerular capillary shows normally thin glomerular basement membrane with intact foot processes (F).
A major susceptibility locus for nephropathy maps to mouse chromosome 8
To map nephropathy susceptibility loci, we generated 165 obese [(FVBxB6)F1 X FVB]db/db backcross progeny. Animals were sacrificed at six months of age, and scored for multiple phenotypes related to diabetic nephropathy (histopathology, albuminuria, BUN). Virtually all of backcross progeny developed sustained hyperglycemia by 6 weeks of age (95% with glucose >250 mg/dl). Despite uniform exposure to hyperglycemia, 61/165 of mice (38%) had no evidence of renal involvement based on normal renal histology and absence of proteinuria. Similarly we observed a wide distribution of albumin creatinine ratio (ACR) in the cross, consistent with increased genetic variance (ACR range 5–4891 ug/mg). These data demonstrated segregation of nephropathy predisposing genes in this cross and further suggested that these genes are distinct from those contributing to diabetes susceptibility.
A genome-wide scan was performed with 103 informative markers across the autosomes. A nonparametric analysis was performed for the histology score, because this trait could not be transformed to normality; both parametric (EM mapping) and nonparametric analyses were used for albuminuria and BUN. There was no linkage to the chromosome 5 locus that mediates pancreatic beta cell hypertrophy and insulinemia in FVB, demonstrating that insulinemia is not a likely causal factor for nephropathy in our model 22. Consistent with uniform hyperglycemia in the backcross and the dominant effect of the chr. 5 locus on persistent hyperglycemia 22, we found no major loci for glucose levels in our cross. In contrast, examination of renal phenotypes revealed significant linkage of the global histology score and albuminuria to chr 8 with lod scores of 4.2 and 2.9 respectively. To maximize inheritance information and confirm linkage, we fine mapped the region with 7 additional markers. This fine-mapping procedure confirmed our initial findings, yielding nonparametric lods scores of 5.0 and 3.6 for the global histology score and albuminuria (peak at 40cM between D8Mit178 and D8Mit248, figure 4). This linkage was robust to each component of histology scores, with glomerulosclerosis and the tubular dilatation/casts scores yielding lod scores of 4.5 and 3.3 respectively (Figure 5). In parametric analysis, the ln transformed ACR also yielded a lod score of 3.3 on chromosome 8. These phenotypes all reach genome-wide significance by traditional criteria for a backcross (i.e. LOD ≥ 3.3). Moreover, these traits exceed empiric significance thresholds based on 10,000 permutations of phenotypes on genotype (genome-wide p-value <0.0001 for global histology score and p=0.004 for ACR respectively). Remarkably, if global renal histology score was treated as a binary trait, grouping as unaffected the 61 mice with no evidence of albuminuria and kidney injury and classifying all others as affected, there was still significant evidence of linkage to chromosome 8 with lod= 3.0 at D8Mit248 (Figure 4B, E), suggesting a quasi-Mendelian effect on phenotype. We have named this locus Dbnph1 (Diabetic nephropathy 1). At the Dbnph1 locus, the FVB allele significantly increased the risk of disease, conferring a doubling of the global histology and 60% increase in albuminuria traits (Figure 5). There were no other traits demonstrating linkage to the Dbnph1 locus. Although significantly correlated with histology and albuminuria, BUN demonstrated a suggestive signal on chromosome 6 (peak lod =2.5 at rs3688920). Thus, the linkage to chromosome 8 remained the only significant signal across the genome.
Figure 4. Linkage of diabetic nephropathy to chromosome 8 in backcross mice.

Distribution of histology score as quantitative trait (A) and binary trait (B, 1= no disease, 2 = any disease), and Ln-transformed albumin/creatinine ratio (C). Below each histogram, the genome-wide lod plots for these traits for nonparametric analysis of linkage (after fine-mapping with 7 microsatellite markers on chromosome 8) are shown (panels D–F). The X axis denotes genetic distance in centimorgans (cM), chromosomes are listed on the bottom. The Y axis shows the lod score. The dashed lines represent the genome-wide significance thresholds based on 10,000 permutations.
Figure 5. Linkage of nephropathy subphenotypes to chromosome 8.

A. Lod plot demonstrating linkage of all nephropathy subphenotypes to chromosome 8 after fine-mapping with 7 microsatellite markers. The X axis denotes genetic distance in centimorgans (cM). The Y axis shows the lod score. The location of genotyped markers is indicated above the plots. The horizontal line above the lod plots shows the lod −2 interval for the Dbnph1 locus B. Phenotypic values associated with FVB/FVB vs FVB/B6 genotype at the peak lod score (D8Mit248).
Genome-wide gene expression profiling reveals differentially activated pathways in mice carrying Dbnph1 susceptibility and resistance alleles
To gain insight into pathogenic pathways that are dysregulated in the diabetic mice, we performed gene expression profiling of whole kidney in 10 male obese backcross mice. We selected backcross mice without glomerulosclerosis, that harbored either FVB/FVB or FVB/B6 genotype across the entire length of chromosome 8. Because of the large effect of the Dbnph1 locus on nephropathy phenotypes, the mice with the high risk FVB/FVB genotype on chromosome 8 had significantly higher ACR compared to mice with the low risk FVB/B6 genotype (215 + 132 vs. 12 + 6, p=0.009). This design enabled comparison of expression profile in mice with high and low likelihood of progression to overt DN, while minimizing detection of “secondary” gene expression changes due to overt kidney injury. To detect dysregulated pathways, we utilized gene set enrichment analysis (GSEA)26, a sensitive analytic method for detecting coordinated enrichment for genes belonging to known molecular pathways (gene sets). Using GSEA, we interrogated 639 canonical pathways curated by the Molecular Signature Database26. GSEA demonstrated that mice with the FVB/FVB genotype on chr. 8 have significant enrichment for oxidative phosphorylation (OXPHOS) pathways (FWER p-value =0.009 based on 10,000 permutations, table 2), whereas mice with the protective FVB/B6 genotype on chr. 8 displayed significant enrichment for complement and coagulation pathways (FWER p<0.0001), statin/ABC transporters and peroxisome proliferator-activated receptor (PPAR) signaling (FWER p<0.0001), and several overlapping amino acid synthetic /metabolic pathways (FWER p<0.0001). Among these, the OXPHOS and PPAR signaling pathways were particularly noteworthy because they have been respectively implicated in initiation and amelioration of human DN 27–31. The leading edge subsets for both pathways (i.e. the subset of genes that accounted for most of the enrichment score) included many canonical genes in the OXPHOS (e.g. cytochrome c oxidase and NADH dehydrogenase subunits) and PPAR signaling (e.g. PPARγ and PPARα). These findings were confirmed by repeating GSEA with 5 independently curated gene sets representing OXPHOS and PPAR signaling pathways (p<0.001 for each, supplemental table 2). Because the components of these pathways are located on other chromosomes, these changes can only be attributed to trans-regulation by the chromosome 8 nephropathy susceptibility alleles. Moreover, because we profiled mice prior to development of glomerulosclerosis, these data suggest that activation of OXPHOS and PPAR signaling characterize high and low risk of progression to overt DN lesions.
Table 2.
Pathways enriched in Backcross mice with contrasting genotypes at the Dbnph1 locus
| Gene Set Name | no gene |
NES | Nominal p-value |
FDR q-value |
FWER p-value |
|---|---|---|---|---|---|
| Enriched in mice with FVB/FVB genotype | |||||
| Hsa00190 oxidative phosphorylation | 116 | 2.02 | <0.0001 | 0.006 | 0.009 |
| Oxidative phosphorylation | 55 | 1.885 | 0.0003 | 0.02 | 0.05 |
| Enriched in mice with B6/FVB genotype | |||||
| Hsa04610 complement and coagulation cascades | 62 | −2.55 | <0.0001 | <0.0001 | <0.0001 |
| Hsa00980 metabolism of xenobiotics by cytochrome p450 | 48 | −2.35 | <0.0001 | <0.0001 | <0.0001 |
| Hsa03320 PPAR signaling pathway | 67 | −2.23 | <0.0001 | <0.0001 | <0.0001 |
| Tryptophan metabolism | 49 | −2.22 | <0.0001 | <0.0001 | <0.0001 |
| Statin pathway PHARMGKB | 16 | −2.15 | <0.0001 | <0.0001 | <0.0001 |
| Hsa00120 bile acid biosynthesis | 35 | −2.13 | <0.0001 | <0.0001 | 0.0002 |
| Intrinsic pathway | 21 | −2.10 | <0.0001 | <0.0001 | 0.0004 |
| Hsa00150 androgen and estrogen metabolism | 28 | −2.1 | <0.0001 | <0.0001 | 0.0005 |
| Blood clotting cascade | 19 | −2.09 | <0.0001 | 0.0001 | 0.001 |
| Hsa00500 starch and sucrose metabolism | 65 | −2.09 | <0.0001 | 0.0004 | 0.002 |
| Gamma hexachlorocyclohexane degradation | 25 | −2.02 | <0.0001 | 0.0004 | 0.004 |
| Hsa00591 linoleic acid metabolism | 28 | −2.01 | <0.0001 | 0.0004 | 0.003 |
| Hsa02010 ABC transporters general | 41 | −1.99 | <0.0001 | 0.0004 | 0.005 |
| Hsa04640 hematopoietic cell lineage | 74 | −1.97 | <0.0001 | 0.001 | 0.02 |
The name and the number of genes in each enriched gene set are indicated. NES= normalized enrichment score, a metric for GSEA; FDR= false discovery rate, FWER= Family-wise error rate.
Annotation and prioritization of positional candidates on chromosome 8
The lod-2 interval for the chromosome 8 QTL spans >40 Mb between D8Mit65 and D8Mit211 and contains over 545 positional candidates. To resolve this region further, we performed haplotype comparisons between the FVB and B6 strains to identify segments that are non- identical by descent. We identified 270 candidates with different haplotypes between the two strains. We annotated these genes based on three approaches (supplementary table 3). We first searched for genes that are differential expressed between mice with the FVB/B6 and FVB/FVB genotypes at the chromosome 8 locus, identifying 14 genes at a nominal p<0.05 (i.e. likely to harbor cis-eQTLs). We also determined which positional candidate harbor potentially pathogenic missense variants. This identified 144 missense variants in 81 positional candidates; analysis with SIFT predicted 9/144 missense SNPs to be damaging. Finally, we examined which positional candidates demonstrate the highest correlation with leading edge subsets in the OXPHOS and PPAR signaling pathways. We further confirmed these annotation results by measuring gene expression in an expanded set of 20 backcross mice without glomerulosclerosis for three positional candidates that had a cis-eQTLs based on microarray data (Ndufa13, JunD and Ocel1). This confirmed that the expression of these genes is regulated by a cis-eQTL, validating the microarray results (figure 6). We next sequenced the promoter region, coding segments and 3’ UTR of these three positional candidates. We identified all SNPs annotated in NCBI and the Mouse Phenome Database, but also uncovered several noncoding sequence variants that were not annotated in databases (Supplemental table 4), consistent with incomplete polymorphism discovery among inbred strains32. The full annotated gene list is presented in supplementary Table 3, representing the initial characterization of positional candidates, which can be studied further to identify the underlying susceptibility gene(s)
Figure 6.

Genotypic differences in expression of selected transcripts among mice with no evidence of glomerulosclerosis. Jund, Ocel1 Ndufa13. Y axis expression level normalized to a wild type B6 mouse. FF= FVB/FVB genotype (n=7) and FB= FVB/B6 genotype (N=13) at the Dbnph1 locus on chromosome 8. * p<0.05 by T-test.
DISCUSSION
Most murine models of diabetes do not reproduce the full features of human DN 13,16,17. Inadequate exposure to hyperglycemia and genetic resistance likely contributes to the lack of advanced DN lesions13,14. In the current study, we describe the FVBdb/db mouse as a very useful tool for dissecting genetic pathways underlying DN. The distinguishing feature of this model is the resistance of beta cells to toxic effects of hyperglycemia and continued increase in insulin secretion from beta-cell mass expansion, which partially compensates for mounting insulin resistance. This results in sustained diabetes and reliable onset of DN with cardinal histological features of disease after 6 months of age. By generating hyperglycemic B6db/db Sur1−/− and F1 hybrids with comparable levels of hyperglycemia, we further demonstrated that the development of DN in FVBdb/db was not simply due to the duration of diabetes but attributable to intrinsic genetic vulnerability to this diabetic complication.
We identified a major locus on chromosome 8B1-C5 (Dbnph1) influencing the development of nephropathy in the FVBdb/db strain. Remarkably, this locus was the only significant signal across the genome and conferred a very large effect, almost doubling the global histology score. The linkage findings were robust to parametric and nonparametric analytic methods and affected all major nephropathy phenotypes. In addition, the Dbnph1 locus was also distinct from loci influencing contrasting susceptibility to beta cell mass expansion (chr 5.) and the development of HIVAN (Chr. 4 and 13) between FVB and B6 strains22,24. There was no evidence suggesting that the Dbnph1 locus promotes the development of nephropathy by influencing serum glucose levels: there were no correlations between glucose levels and any renal histopathology or albuminuria parameters; our previous study using the same parental strains had demonstrated that variation in severity of diabetes between FVB and B6 is attributable to a dominant locus on chromosome 524; consistent with the above finding and the dominance of the chr 05 locus, all mice in our backcross cohort were diabetic and linkage scans for serum glucose levels did not reveal any loci influencing glucose levels; and incorporation of glucose levels as a covariate in the linkage analysis did not change linkage of nephropathy parameters to chromosome 8. Taken together, these data demonstrates that independent genetic factors influence insulinemia, DN and HIVAN phenotypes between FVB and B6 strains, further recapitulating the situation in humans. Our previous studies had also shown that HIVAN susceptibility alleles produce differential expression of many podocyte genes, demonstrating that HIVAN loci transregulate podocyte gene expression 24. Therefore, to gain insight into dysregulated pathways, we followed up the current mapping experiments by gene expression profiling in backcross mice with nephropathy susceptibility or resistance alleles. We identified several differentially expressed pathways that had been previously implicated in human DN. In particular, the activation of PPAR signaling in mice with the DN protective alleles is consistent with recent studies showing that PPAR agonists reduce proteinuria and kidney injury in humans and Lepr mice 27–29. These data are also consistent with studies implicating lipid accumulation in the pathogenesis of nephropathy in FVBdb/db and other DN models, as well as humans23,33. In addition, the upregulation of OXPHOS pathways in mice with the susceptibility alleles recapitulates a recent study that identified upregulation of this pathway in skin fibroblasts of patients with rapid progression of DN 30. Altogether, the finding of a locus that is specific for DN and is enriched for genetic pathways previously implicated in differential susceptibility to human DN provide compelling evidence for the relevance of the FVBdb/db model and the Dbnph1 locus to human disease.
Recent studies have shown that combining gene expression profiling with QTL analysis can stratify genes by identification of cis- expression QTLs34. We therefore performed an initial annotation of cis-eQTLs within the Dbnph1 interval and found several positional candidates that have been implicated in the pathogenesis of nephropathy. For example, Jund, a component of the AP-1 complex, has been implicated in preventing the wave of proliferative lesions subsequent to nephron reduction surgery, through activation of the TGF-alpha 35. In addition, variation in Jund was recently shown to mediate glomerular macrophage infiltration and inflammation in experimental glomerulonephritis in mice36. Ndufa13 (aka Grim19) is a mitochondrial complex I protein that is important for cellular respiration and regulation of apoptotic signaling through interactions with Stat3 37, a transcription factor implicated in diabetic nephropathy 38. Finally, Ocel1, a gene of unknown function, was also notable because it displayed the strongest cis-eQTL at the chromosome 8 locus, and expression was highly correlated with the OXPHOS pathway. These data can be pursued by generation of congenic strains that capture and isolate the linkage interval, enabling refinement of the DN susceptibility gene(s) and better detection of additional cis-eQTLs.
To the date, the FVBdb/db has been independently studied by investigators at four different sites (Columbia University, Albert Einstein College of Medicine, University of Denver and at Jackson laboratories for AMDCC)14,21–23. All four sites have found significant evidence of alburminuria in this strain after 6 months of age (> 10-fold increase compared to lean counterparts after 6 months, satisfying a major AMDCC criterion for DN). In addition, all sites, except for the AMDCC study, also found histopathologic evidence of nephropathy, as described in this study. The reasons for absence of histopathologic lesions in the AMDCC study are not clear, and may be due to variation in environment, diet or microflora. However, the severity of albuminuria in the FVBdb/db colony studied by AMDCC completely recapitulates the findings in our study, suggesting that with a longer follow-up period, histopathologic lesions consistent with our observations would have been observed. These data highlight the complexity of mouse genetic studies of diabetic complications, suggesting that variation in gene, environment and in particular, length of follow-up should be considered in evaluation of models.
Genome scans among diabetic and nondiabetic human populations have identified a number of loci influencing albuminuria, glomerular filtration rate (GFR) or the risk of renal failure 3–9. These studies have strongly suggested multifactorial determination of disease 3–9. Most recently, a GWAS for DN has identified suggestive signals in the CPVL/CHN2, FRMD3, CARS genes and several intergenic loci 12. Association studies have also identified genes or loci underlying GFR (UMOD, SHROOM3 and STC139 and nondiabetic kidney failure (MYH9, ref.10,11). None of these genes map to mouse chromosome 8 nor showed differential expression based on Dbnph1 genotype. However, as we develop Dbnph1 congenic strains and more GWAS loci become available, the Dbnph1 locus can be better clarified in the context of human disease. In addition, it is likely that future GWAS projects will uncover more signals encompassing multiple genes or intergenic regions of unknown function, necessitating follow-up studies in validated experimental models. Thus the FVBdb/db model will likely prove very relevant for identification of pathways leading to human kidney failure but also for functional characterization of loci identified in human genetics studies.
RESEARCH DESIGN AND METHODS
Animal breeding and Phenotypes
The FVBdb/db strain was produced by >10 generation of backcrossing of the db allele from the C57BLKS/J to the FVB/NJ strain. A genome-scan with 103 informative markers in the 10th generation detected no C57BLKS/J derived alleles, demonstrating successful introgression of the db allele into the FVB/NJ genome. We obtained Sur1 knockout mice from Drs. Joe and Lydia Aguilar-Bryan (Baylor College of Medicine) and backcrossed the Sur1 KO allele to C57BL/6J-db mice for >10 generations. A mapping cohort was produced by backcrossing FVBxB6 db/+ or FVBxB6 db-3J/+ F1 hybrids to FVBdb/+ mice and their obese progeny phenotyped mice at 24 weeks of age. Both the db and db-3J alleles encode loss of function mutations in the LEPR, resulting in obesity and metabolic phenotypes that are indistinguishable on the B6 background40. Three primary phenotypes related to renal disease were characterized: renal histology, blood urea nitrogen (BUN) and proteinuria. Urine albumin/creatinine ratio (ACR) was quantitated by ELISA (Albuwell and creatinine companion kits from Exocell, Philadelphia, PA); ACR values were Ln transformed to achieve a normal distribution for linkage analysis and comparison between genotypes. Urine samples from the various F1 hybrids were not available because they were lost during an institutional move (SC). Renal histology was scored independently by an investigator (VDD) blinded to genetic background and other traits. We adapted a semi-quantitative scoring system that we have validated for four previous mouse mapping studies of nephropathy. We performed a standard grading for mesangial sclerosis and supplemented this with additional entries for progression to more extensive glomerulosclerosis (segmental or global) as a measure of glomerular chronicity. We also included tubulointerstitial lesions that may accompany proteinuria (such as casts) and tubular atrophy/interstitial fibrosis (which are measures of progression to chronicity). These additional parameters may be seen in mouse models of diabetic nephropathy with potential to progress (such as the Ove26 mouse) as well as human diabetic nephropathy. For each animal, we counted 100 glomeruli and scored three parameters: 1) the percent of glomeruli that exhibit any degree of glomerulosclerosis; 2) the percent of glomeruli that exhibit any degree of mesangial sclerosis; 3) the percent of tubules exhibiting dilatation or casts using a semi-quantitative scale: 0= no disease, 1= 1–25% of tissue showing abnormalities, 2= 26–50%, 3= >50% of tissue affected. Because these values were highly correlated, we also computed a global histologic score as sum of three histology injury parameters (range 0–9). Blood pressure was measured using non-invasive tail cuff method; mice were acclimated for 5 days prior to measurements. The protocol was approved by the IACUC committee at the Columbia University Medical Center and the Albert Einstein College of Medicine.
Genotyping and analysis of linkage
The mapping cohort was genotyped using 103 informative SNPs across mouse autosomes (Kbioscience, UK). The SNPs were interpolated on the Whitehead Institute genetic map by correlating their physical position with the nearest microsatellite marker on NCBI build 37. Primary phenotypes analyzed were the global histology score, BUN and urine albumin/creatinine ratio. Multipoint lod scores were calculated using the R/QTL package 41. Because all histology traits were skewed and could not be transformed to a normal distribution, nonparametric analyses were performed for these traits. However, both parametric (Expectation/Maximization (EM) mapping method) and nonparametric analyses were performed after Ln transformation for urine albumin/creatinine ratio and BUN (which resulted in a normal distribution). Traditional thresholds for significance in the backcross were used (lod score > 3.3, reference 42). In addition, we conducted 10,000 permutations of phenotypes on genotype to determine the empirical significance of linkage findings.
Microarray analysis
We performed microarray analysis with the Affymetrix ST 1.0 exon arrays (Santa Clara, CA). Total kidney RNA was extracted from 10 backcross mice with no glomerulosclerosis or proteinuria and selected to have FVB/FVB or FVB/B6 genotypes across chromosome 8 (5 of each genotype). Sample preparation, labeling and hybridization will be performed as per Affymetrix recommended protocol. Signal intensities were normalized using the GCRMA method. Gene Set Enrichment analysis (GSEA) was performed as implemented in the Gene Pattern (Broad Institute, reference 26). For this analysis, we utilized 639 canonical pathways curated by the Molecular Signature Database (http://www.broad.mit.edu/gsea/msigdb/index.jsp). To detect empiric significance of enrichment results, we performed 10,000 permutation of gene labels in the arrays. P-values were corrected for multiple testing based on the Family-Wise Error rate (FWER), with FWER p<0.05 considered significant evidence of enrichment.
Bioinformatics
Phenotype comparisons between inbred mouse mice were conducted using 2-sided t-tests assuming unequal variance. In-silico analysis of haplotype on chromosome 8 was performed by downloading all SNPs that were polymorphic between FVB and B6 and that were located within the chromosome 8 lod −2 interval (Mouse Phenome Database). These SNPs were cross-referenced with the NCBI build 37 to identify positional candidates with differing haplotypes, yielding a list of 14,633 SNPs located within 270 genes. This SNP list was further filtered to identify missense sequence variants (144 identified). All missense variants were analyzed with SIFT 43 to predict potentially pathogenic substitutions. To detect cis-eQTLs on chromosome 8, compared microarray expression values of positional candidates in backcross mice based on genotypes on chromosome 8, using a two-sided t-test.
Supplementary Material
Acknowledgments
This study was supported by a pilot award from the Columbia University Diabetes and Endocrinology Center (NIH P30-DK63608), the AECOM Diabetes Research and Training Center P60 DK20541, New York Obesity Research Center PO1DK26687 and a gift from the Columbia University Center for Glomerular Diseases.
Footnotes
This paper was presented in part at the 2009 meeting of the American Society of Nephrology.
REFERENCES
- 1.Maisonneuve P, et al. Distribution of primary renal diseases leading to end-stage renal failure in the United States, Europe, and Australia/New Zealand: results from an international comparative study. Am J Kidney Dis. 2000;35:157–165. doi: 10.1016/S0272-6386(00)70316-7. [DOI] [PubMed] [Google Scholar]
- 2.Sheetz MJ, King GL. Molecular understanding of hyperglycemia's adverse effects for diabetic complications. Jama. 2002;288:2579–2588. doi: 10.1001/jama.288.20.2579. [DOI] [PubMed] [Google Scholar]
- 3.Moczulski DK, Rogus JJ, Antonellis A, Warram JH, Krolewski AS. Major susceptibility locus for nephropathy in type 1 diabetes on chromosome 3q: results of novel discordant sib-pair analysis. Diabetes. 1998;47:1164–1169. doi: 10.2337/diabetes.47.7.1164. [DOI] [PubMed] [Google Scholar]
- 4.Vardarli I, et al. Gene for susceptibility to diabetic nephropathy in type 2 diabetes maps to 18q22.3-23. Kidney Int. 2002;62:2176–2183. doi: 10.1046/j.1523-1755.2002.00663.x. [DOI] [PubMed] [Google Scholar]
- 5.Osterholm AM, et al. Genome-wide scan for type 1 diabetic nephropathy in the Finnish population reveals suggestive linkage to a single locus on chromosome 3q. Kidney Int. 2007;71:140–145. doi: 10.1038/sj.ki.5001933. [DOI] [PubMed] [Google Scholar]
- 6.Schelling JR, et al. Genome-wide scan for estimated glomerular filtration rate in multi-ethnic diabetic populations: the Family Investigation of Nephropathy and Diabetes (FIND) Diabetes. 2008;57:235–243. doi: 10.2337/db07-0313. [DOI] [PubMed] [Google Scholar]
- 7.Freedman BI, et al. Genome-wide linkage scans for renal function and albuminuria in Type 2 diabetes mellitus: the Diabetes Heart Study. Diabet Med. 2008;25:268–276. doi: 10.1111/j.1464-5491.2007.02361.x. [DOI] [PubMed] [Google Scholar]
- 8.Iyengar SK, et al. Genome-wide scans for diabetic nephropathy and albuminuria in multiethnic populations: the family investigation of nephropathy and diabetes (FIND) Diabetes. 2007;56:1577–1585. doi: 10.2337/db06-1154. [DOI] [PubMed] [Google Scholar]
- 9.Bowden DW, et al. A genome scan for diabetic nephropathy in African Americans. Kidney Int. 2004;66:1517–1526. doi: 10.1111/j.1523-1755.2004.00915.x. [DOI] [PubMed] [Google Scholar]
- 10.Kopp JB, et al. MYH9 is a major-effect risk gene for focal segmental glomerulosclerosis. Nat Genet. 2008 doi: 10.1038/ng.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kao WH, et al. MYH9 is associated with nondiabetic end-stage renal disease in African Americans. Nat Genet. 2008 doi: 10.1038/ng.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pezzolesi MG, et al. Genome-wide association scan for diabetic nephropathy susceptibility genes in type 1 diabetes. Diabetes. 2009;58:1403–1410. doi: 10.2337/db08-1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Breyer MD, et al. Mouse models of diabetic nephropathy. J Am Soc Nephrol. 2005;16:27–45. doi: 10.1681/ASN.2004080648. [DOI] [PubMed] [Google Scholar]
- 14.Brosius FC, 3rd, et al. Mouse Models of Diabetic Nephropathy. J Am Soc Nephrol. 2009 doi: 10.1681/ASN.2004080648. [DOI] [PubMed] [Google Scholar]
- 15.Sharma K, McCue P, Dunn SR. Diabetic kidney disease in the db/db mouse. Am J Physiol Renal Physiol. 2003;284:F1138–F1144. doi: 10.1152/ajprenal.00315.2002. [DOI] [PubMed] [Google Scholar]
- 16.Qi Z, et al. Characterization of susceptibility of inbred mouse strains to diabetic nephropathy. Diabetes. 2005;54:2628–2637. doi: 10.2337/diabetes.54.9.2628. [DOI] [PubMed] [Google Scholar]
- 17.Gurley SB, et al. Impact of genetic background on nephropathy in diabetic mice. Am J Physiol Renal Physiol. 2006;290:F214–F222. doi: 10.1152/ajprenal.00204.2005. [DOI] [PubMed] [Google Scholar]
- 18.Leiter EH, Chapman HD, Coleman DL. The influence of genetic background on the expression of mutations at the diabetes locus in the mouse. V. Interaction between the db gene and hepatic sex steroid sulfotransferases correlates with gender-dependent susceptibility to hyperglycemia. Endocrinology. 1989;124:912–922. doi: 10.1210/endo-124-2-912. [DOI] [PubMed] [Google Scholar]
- 19.Haluzik M, et al. Genetic background (C57BL/6J versus FVB/N) strongly influences the severity of diabetes and insulin resistance in ob/ob mice. Endocrinology. 2004;145:3258–3264. doi: 10.1210/en.2004-0219. [DOI] [PubMed] [Google Scholar]
- 20.Qiu J, Ogus S, Mounzih K, Ewart-Toland A, Chehab FF. Leptin-deficient mice backcrossed to the BALB/cJ genetic background have reduced adiposity, enhanced fertility, normal body temperature, and severe diabetes. Endocrinology. 2001;142:3421–3425. doi: 10.1210/endo.142.8.8323. [DOI] [PubMed] [Google Scholar]
- 21.Chua S, Jr, et al. Differential beta cell responses to hyperglycaemia and insulin resistance in two novel congenic strains of diabetes (FVB- Lepr (db)) and obese (DBA- Lep (ob)) mice. Diabetologia. 2002;45:976–990. doi: 10.1007/s00125-002-0880-z. [DOI] [PubMed] [Google Scholar]
- 22.Luo N, et al. Allelic variation on chromosome 5 controls beta-cell mass expansion during hyperglycemia in leptin receptor-deficient diabetes mice. Endocrinology. 2006;147:2287–2295. doi: 10.1210/en.2005-0853. [DOI] [PubMed] [Google Scholar]
- 23.Wang Z, et al. Regulation of renal lipid metabolism, lipid accumulation, and glomerulosclerosis in FVBdb/db mice with type 2 diabetes. Diabetes. 2005;54:2328–2335. doi: 10.2337/diabetes.54.8.2328. [DOI] [PubMed] [Google Scholar]
- 24.Papeta N, et al. Susceptibility loci for murine HIV-associated nephropathy encode trans-regulators of podocyte gene expression. J Clin Invest. 2009;119:1178–1188. doi: 10.1172/JCI37131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Seghers V, Nakazaki M, DeMayo F, Aguilar-Bryan L, Bryan J. Sur1 knockout mice. A model for K(ATP) channel-independent regulation of insulin secretion. J Biol Chem. 2000;275:9270–9277. doi: 10.1074/jbc.275.13.9270. [DOI] [PubMed] [Google Scholar]
- 26.Subramanian A, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Park CW, et al. Accelerated diabetic nephropathy in mice lacking the peroxisome proliferator-activated receptor alpha. Diabetes. 2006;55:885–893. doi: 10.2337/diabetes.55.04.06.db05-1329. [DOI] [PubMed] [Google Scholar]
- 28.Cha DR, et al. Peroxisome proliferator activated receptor alpha/gamma dual agonist tesaglitazar attenuates diabetic nephropathy in db/db mice. Diabetes. 2007;56:2036–2045. doi: 10.2337/db06-1134. [DOI] [PubMed] [Google Scholar]
- 29.Miyazaki Y, Cersosimo E, Triplitt C, DeFronzo RA. Rosiglitazone decreases albuminuria in type 2 diabetic patients. Kidney Int. 2007;72:1367–1373. doi: 10.1038/sj.ki.5002516. [DOI] [PubMed] [Google Scholar]
- 30.Huang C, et al. Diabetic nephropathy is associated with gene expression levels of oxidative phosphorylation and related pathways. Diabetes. 2006;55:1826–1831. doi: 10.2337/db05-1438. [DOI] [PubMed] [Google Scholar]
- 31.Forbes JM, Coughlan MT, Cooper ME. Oxidative stress as a major culprit in kidney disease in diabetes. Diabetes. 2008;57:1446–1454. doi: 10.2337/db08-0057. [DOI] [PubMed] [Google Scholar]
- 32.Frazer KA, et al. A sequence-based variation map of 8.27 million SNPs in inbred mouse strains. Nature. 2007;448:1050–1053. doi: 10.1038/nature06067. [DOI] [PubMed] [Google Scholar]
- 33.Proctor G, et al. Regulation of renal fatty acid and cholesterol metabolism, inflammation, and fibrosis in Akita and OVE26 mice with type 1 diabetes. Diabetes. 2006;55:2502–2509. doi: 10.2337/db05-0603. [DOI] [PubMed] [Google Scholar]
- 34.Hubner N, et al. Integrated transcriptional profiling and linkage analysis for identification of genes underlying disease. Nat Genet. 2005;37:243–253. doi: 10.1038/ng1522. [DOI] [PubMed] [Google Scholar]
- 35.Pillebout E, et al. JunD protects against chronic kidney disease by regulating paracrine mitogens. J Clin Invest. 2003;112:843–852. doi: 10.1172/JCI17647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Behmoaras J, et al. Jund is a determinant of macrophage activation and is associated with glomerulonephritis susceptibility. Nat Genet. 2008;40:553–559. doi: 10.1038/ng.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang J, et al. The cell death regulator GRIM-19 is an inhibitor of signal transducer and activator of transcription 3. Proc Natl Acad Sci U S A. 2003;100:9342–9347. doi: 10.1073/pnas.1633516100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lu TC, et al. Knockdown of Stat3 activity in vivo prevents diabetic glomerulopathy. Kidney Int. 2009;76:63–71. doi: 10.1038/ki.2009.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kottgen A, et al. Multiple loci associated with indices of renal function and chronic kidney disease. Nat Genet. 2009 doi: 10.1038/ng.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li C, Ioffe E, Fidahusein N, Connolly E, Friedman JM. Absence of soluble leptin receptor in plasma from dbPas/dbPas and other db/db mice. J Biol Chem. 1998;273:10078–10082. doi: 10.1074/jbc.273.16.10078. [DOI] [PubMed] [Google Scholar]
- 41.Broman KW, Wu H, Sen S, Churchill GA. R/qtl: QTL mapping in experimental crosses. Bioinformatics. 2003;19:889–890. doi: 10.1093/bioinformatics/btg112. [DOI] [PubMed] [Google Scholar]
- 42.Lander E, Kruglyak L. Genetic dissection of complex traits: guidelines for interpreting and reporting linkage results. Nat Genet. 1995;11:241–247. doi: 10.1038/ng1195-241. [DOI] [PubMed] [Google Scholar]
- 43.Ng PC, Henikoff S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31:3812–3814. doi: 10.1093/nar/gkg509. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.