

Abstract
The presence of histones acts as a barrier to protein access; thus chromatin remodeling must occur for essential processes such as transcription and replication. In conjunction with histone modifications, DNA methylation plays critical roles in gene silencing through chromatin remodeling. Chromatin remodeling is also interconnected with the DNA damage response, maintenance of stem cell properties, and cell differentiation programs. Chromatin modifications have increasingly been shown to produce long-lasting alterations in chromatin structure and transcription. Recent studies have shown environmental exposures in utero have the potential to alter normal developmental signaling networks, physiologic responses, and disease susceptibility later in life during a process known as developmental reprogramming. In this review we discuss the long-term impact of exposure to environmental compounds, the chromatin modifications that they induce, and the differentiation and developmental programs of multiple stem and progenitor cell types altered by exposure. The main focus is to highlight agents present in the human lifestyle that have the potential to promote epigenetic changes that impact developmental programs of specific cell types, may promote tumorigenesis through altering epigenetic marks, and may be transgenerational, for example, those able to be transmitted through multiple cell divisions.
Keywords: environmental toxicology, epigenetics, chromatin remodeling, in utero exposure, bioflavonoids
I. INTRODUCTION
A. Chromatin Remodeling and Epigenetics
Epigenetics is the study of heritable changes in gene expression without a change in the DNA sequence. Nucleosomes are composed of 147 base pairs of DNA wrapped around core histone proteins H2A, H2B, H3, and H4.1–3 Histone H1 acts to link histones together and to the nuclear scaffold. H3 and H4 termini extend from the nucleosome and can be modified chemically by acetylation, methylation, ubiquitination, phosphorylation, sumoylation, citrullination, and ADP-ribosylation.3 Modifications promote either open or closed chromatin,3 which in turn influences multiple cellular processes such as transcription and replication.4–6 Chromatin remodeling is also interconnected with the DNA damage response, maintenance of stem cell properties, and cell differentiation programs. Patterns of histone modification are maintained after replication and thus inherited through multiple cellular generations. DNA methylation predominantly involves the covalent addition of a methyl group (CH3) to cytosine in the context of CpG in DNA, creating a significant epigenetic marker of transcriptional inactivity. Patterns of DNA methylation are generated during development involving de novo methylation and demethylation mediated by DNA methyltransferases (DNMTs). DNMT3 regulates de novo methylation during development, and DNMT1 maintains DNA methylation patterns following replication. Global genome methylation patterns are highly specific depending on developmental stage and type of tissue.
This review focuses on commonly used environmental and dietary compounds previously known to have or suspected of having carcinogenic or mutagenic properties but recently identified as having the potential to disrupt chromatin remodeling and epigenetic regulation in stem cells, induce long-term changes in developmental programs, and promote tumorigenesis. Furthermore, we highlight recent reports that have shown that in utero exposure to these compounds can also promote epigenetic modifications that in turn induce gene expression changes that persist throughout life.7,8
B. The Connection Between Epigenetic Alterations and DNA Double-Strand Breaks
The DNA double-strand break response (DDR) is facilitated by hierarchical signaling networks that orchestrate chromatin structural changes, cell cycle checkpoints, and multiple enzymatic activities to repair broken ends of DNA. DNA double-strand breaks (DSBs) have the highest potential to promote illegitimate repair mechanisms and the accumulation of mutations and are considered the critical primary lesions in the formation of chromosomal rearrangements associated with disease and tumorigenesis. Recent advances in the understanding of the interplay between chromatin remodeling, epigenetics, and the DDR have been reviewed.9–11 New emerging evidence extends earlier findings with the potentially pathological repercussions of restoring chromatin structure, resulting in a DSB-induced epigenetic memory of damage.
Chromatin dynamics and changes in chromatin architecture that occur for repair of DSBs include nucleosome eviction from DSBs, relaxation of heterochromatin structure, and localized chromatin destabilization at DSBs.12 After DNA damage, chromatin structure is altered by adenosine triphosphate–dependent chromatin remodeling, the incorporation of histone variants into nucleosomes, and modifications to covalent histones. These histone modifications include phosphorylation of H1; acetylation of H2A and phosphorylation and ubiquitination of H2AX; acetylation and methylation of H3; and phosphorylation and acetylation of H4.3 Among the different histone modifications, phosphorylation of all 4 histones as well as the variant H2AX plays a primary role in DNA damage response by facilitating the access of repair proteins to DNA breaks.13,14 Phosphorylation of H2AX (γH2AX) spreads over large chromatin domains away from a DSB. This chromatin marking and large-scale chromatin reorganization recruits repair factors, recombination proteins, and chromatin remodeling complexes involved in DNA repair pathways.15
Heterochromatin is the tightly compacted DNA structure that acts as a barrier to DNA repair processes. As a result, heterochromatic (HC) DSBs are generally repaired more slowly than euchromatic DSBs,16 and heterochromatin and euchromatin use distinct remodeling complexes and pathways for DSB repair.17–21 DSB repair may be stalled within HC regions if a series of dynamic and localized changes fail to occur.18,22,23 The ataxia telangiectasia mutated protein and DDR mediator proteins overcome constraints posed by heterochromatin superstructure to promote repair through the modulation of 2 HC factors: KAP-1 corepressor and HP1 chromodomain protein.19 Activation of the ataxia telangiectasia mutation signaling pathway and the subsequent phosphorylation of KAP-1 trigger HC modifications required for DSB repair.24 In addition, studies have shown that histone acetyltransferase complexes act with the adenosine triphosphate–dependent switch/sucrose nonfermentable and remodel structure of chromatin (RSC)-containing chromatin remodeling complexes to facilitate DNA repair.25
Polycomb group (PcG) proteins, which have well-established roles in gene regulation, were recently found to accumulate on chromatin surrounding DNA damage.26 PcG proteins form complexes involved in the epigenetic regulation of gene expression. PcG repressive complexes catalyze posttranslational modifications critical to their gene silencing function, including histone H3K27 di- and trimethylation (H3K27me2, H3K27me3) and histone H2A ubiquitination.27–29 PcG and polycomb recessive complex (PRC) components found to respond to DNA damage include BMI-1, MEL-18, EZH2 methyltransferase, EZH1, EED, and SUZ12,30 suggesting that DNA methylation modifications occur as part of the DDR. Recruitment of PcG protein BMI1 promotes mono-ubiquitination of H2A and DNA DSB repair.31
Signal transduction pathways in DDR communicate with chromatin remodeling factors through protein-protein interactions. The chromatin remodeling protein scaffold matrix attachment region 1 (SMAR1) binds other SMAR1 elements along with histone deacetylase (HDAC) 1 and p53 to form a repressor complex to downregulate transcription.32,33 The chromatin remodeling factor Tip49 recruits Rad51, the homolog of bacterial RecA and major homologous recombination repair protein, to DNA damage sites.34 In cells depleted of Tip49, Rad51 recruitment can be restored by addition of an HDAC inhibitor, underscoring the interplay of epigenetic markers and DDR. In addition to DNA repair, the p53 signaling pathway is associated with chromatin changes that mainly involve the histone acetyltransferase Tip60 to modulate the fate of a cell between cell cycle arrest and apoptosis. 35 Numerous chromatin remodeling factors involved in DNA methylation and demethylation also play a role in DDR. In the thymus, genotoxic stress decreases global DNA methylation by a reduction in DNMT1, DNMT3a, DNMT3b, and methyl-binding proteins MeCP2 and MBD2.36 That these alterations are carried into the offspring of exposed individuals led the authors to suggest profound epigenetic dysregulation, which in turn could lead to genome destabilization and possibly serve as a precursor for transgenerational carcinogenesis.36
C. Stem Cells and Induced Pluripotent Stem Cells
Stem cells are the focus of study in many laboratories because of their unique properties of self-renewal and pluripotency that allow for both maintenance of the stem cell pool as well as generation of various cell types through differentiation. Induced pluripotent stem cells (iPSCs) have allowed for the study of the developmental processes of multiple tissue and organ systems.37–40 Stem cell models have been used in drug discovery and toxicology screens and the development of biomarker panels. 41–43
Because stem cells give rise to all of the mature cells in an organism, they have become a promising source for potential replacement therapies in the treatment of diseases,44 although both genetic integrity and a “correct” epigenome will be important factors in both their experimental and therapeutic usefulness. Extensive literature exists analyzing transcriptional and proteomic profiles as well as the chromatin modification profiles of stem cells, embryonic stem (ES) cells, iPSCs, and differentiated cell types to create signatures of “stemness.” 45–61
The genomic instability of human ES cells and iPSCs in vitro has been reported. Common abnormalities of ES cells are gains of chromosomes 12, 17, and 20.62–67 Chromosome 12 contains genes implicated in cell survival, such as STELLAR, GDF3, and NANOG, thus likely giving cells a proliferative advantage. It is interesting to note that trisomy of chromosome 12 is one of the most commonly described mosaicisms in amniocentesis68 and is characteristic of germ cell tumors in almost all cases.69 A gain of chromosome 17 is similarly associated with tumorigenesis, specifically breast cancer,70 and amplifications of chromosome 20 are observed in a wide variety of tumors.71,72 The gain of chromosome X in stem cells cultured in vitro70 is associated with trisomy 17 as well, although it is unclear whether this provides a proliferative or survival advantage; a loss of chromosome X also has been reported in vitro.64 The fact that these chromosomal abnormalities are common in both cultured stem cells and in multiple in vivo settings suggests that stem cells may be particularly susceptible to specific alterations. iPSCs have shown copy number variations, an abnormal karyotype, and point mutations.42 In addition, they have DNA methylation and histone modification defects and altered X chromosome inactivation.42 Some studies have indicated that iPSCs may “remember” their previous somatic cell fate as stored within the epigenome,49,72,74 reducing their experimental and therapeutic potential.
Transcriptional regulation and gene expression patterns occur in the context of chromatin. In general, stem cells have epigenetic signatures that are characteristic of an active chromatin state; for example, chromatin is generally decondensed. Chromatin is reorganized during differentiation programs, and chromatin immunoprecipitation (ChIP) sequencing approaches have demonstrated that chromatin structure at promoters and regulatory regions correlates with active, repressive, or poised “bivalent” transcriptional states, which in turn correlate with the state of cell differentiation.75–78
Stem cell–specific structures are regulated not only by epigenetic markers but also by higher-order chromatin structures, which are not discussed here (reviewed in Ref. 78). Global levels of DNA methylation in stem cells are similar to levels in somatic cells; however, 25% of methylation sites in stem cells are in non-CpG sites, indicating a unique methylation program.79 Multiple studies demonstrate that histone acetylation is central to the maintenance of pluripotency. H3K9 acetylation is higher overall in human stem cells and necessary to maintain an undifferentiated state, whereas H3K9 deacetylation occurs with differentiation.80 HDAC inhibitors such as butyrate and valproic acid increase overall levels of H3K9ac80 as well as the expression of pluripotency genes such as Nanog, Sox, and Oct481 and promote stem cell and iPSC survival.82,83 Other histone modifications associated with transcriptionally active chromatin also have been shown to be increased in stem cells, including H3K14 acetylation, H3K4 trimethylation (H3K4me3), and H3K36 di- and trimethylation (H3K36me2, H3K36me3).84,85 Levels of trimethylated H3K27 (H3K27me3) remain unchanged during differentiation but preferentially localize at the nuclear periphery in stem cells, and this localization decreases during differentiation.86 It is interesting to note that both H3K27me2 and H3K27me3 markers are mediated by the PRC2 and present at polycomb target genes known to be suppressed in stem cells.87,88 Taken together, these observations suggest that polycomb target genes are located at the nuclear periphery in stem cells and that this localization may serve as an additional epigenetically regulated repressive mechanism.79 Whole-genome ChIP sequencing approaches have shown that both H3K4me3 (a marker of active promoters) and H3K27me3 (a marker of silenced promoters) colocalize in ES cells for 16–22% of genes, most of which are required for developmental regulation, creating a “bivalent” marker hypothesized to poise such genes for future expression while maintaining repression in the stem cell state.75,77,78,89,90
Thus, stem cells have specific epigenetic markers important for maintaining stem cell pluripotency and self-renewal that, when disrupted, could lead to dysregulated differentiation or tumorigenesis. One well-defined example of this is in the initiation of acute myeloid leukemia with rearrangements of the mixed lineage leukemia (MLL) gene. MLL is a homologue of Drosophila melanogaster trithorax and is a positive regulator of Hox gene expression by H3K4 methylation. Hox gene expression is also negatively regulated by H3K27 methylation by polycomb group proteins, thus conferring a delicate balance of epigenetic markers. Disruption of these opposing epigenetic regulatory factors through MLL chromosomal translocation leads to hyperactivation of Hox genes and, ultimately, to leukemogenesis.91 The mechanisms by which stem cells might transform into cancer stem cells remain widely unknown; however, repeated exposure to agents that damage DNA or disrupt epigenetic gene regulation may cause stem cells to become more similar to cancer stem cells and eventually initiate disease. In support of this, repeated exposure of cultured stem cells to toxic stress and metals has been shown to promote differentiation at the expense of an accumulating stem cell pool, induce abnormal cell signaling and global proteomic alterations analogous to those observed in transformed cells, acquire multiple tumor cell characteristics, and lead to an enrichment of cancer stem cells.51,92–94
II. ENVIRONMENTAL TOXINS
A. Aldehydes and Alcohols
Carbonyl compounds are stable intermediates of photochemical oxidation of most hydrocarbons and are the precursors to free radicals and ozone; thus environmental exposure can be pervasive. Higher levels of reactive aldehydes such as acetylaldehyde and formaldehyde have been measured in ambient air samples of urban communities and are linked to toxicity, mutagenicity, and carcinogenicity95–99 (Fig. 1). Exposure to ozone during exercise results in ozonation of lipids to produce aldehydes in fluid in the epithelial lining of the airway in humans.100 Reactive aldehydes and acetaldehyde are also by-products of endogenous cellular metabolism and have been found to have genotoxic effects. Bone marrow failure in Fanconi anemia may result in part from aldehyde-mediated genotoxicity in the hematopoietic stem and progenitor cell pool. In support of this, mouse hematopoietic stem and progenitor cells are more susceptible to acetaldehyde toxicity compared with mature blood precursors.101 Hematopoietic stem cells from Aldh2−/− Fancd2−/− mice that are deficient in the Fanconi anemia pathway–mediated DNA repair and in endogenous acetaldehyde detoxification undergo a more than 600-fold reduction in numbers, display a predisposition to leukemia, and require Aldh2 for protection against acetaldehyde toxicity. 101 Another endogenous source of acetaldehyde is as the first product from the breakdown of alcohol in cells. It has been previously proposed that acetaldehyde generated from alcohol metabolism reacts in cells to generate DNA lesions that form interstrand crosslinks (ICLs).102 Since the Fanconi anemia– and breast cancer–associated DNA damage response network plays a crucial role in protecting cells against ICLs, Marietta et al.103 tested the proposed role of acetaldehyde in generating ICLs. They exposed human lymphoblastoid cells from normal individuals, a patient with xeroderma pigmentosum complementation group A, a patient with Fanconi anemia G, and a patient with Fanconi anemia A to acetaldehyde and studied the activation of the Fanconi anemia– and breast cancer–associated network. Their study reported that acetylaldehyde in a dose range of 0.1–1 mM stimulates FANCD2 monoubiquitination, BRCA1 phosphorylation at Ser1524, and γH2AX at Ser139 in a dose-dependent manner. These results demonstrate interplay between multiple DDR networks and may also support differential tissue specificity of alcohol-related carcinogenesis.103 The data also support findings of association between alcohol intake and increased breast cancer risk. Chronic exposure to ethanol induces DNA damage and an induction in the levels of the Fanconi anemia D2 (FANCD2) protein in both human neural precursor SH-SY5Y cells in culture and in the midbrain of C57BL/6J mice in vivo.104 FANCD2 response induced by alcohol thus plays a role in DDR in post-mitotic neurons and neural precursor cells.
FIG. 1.
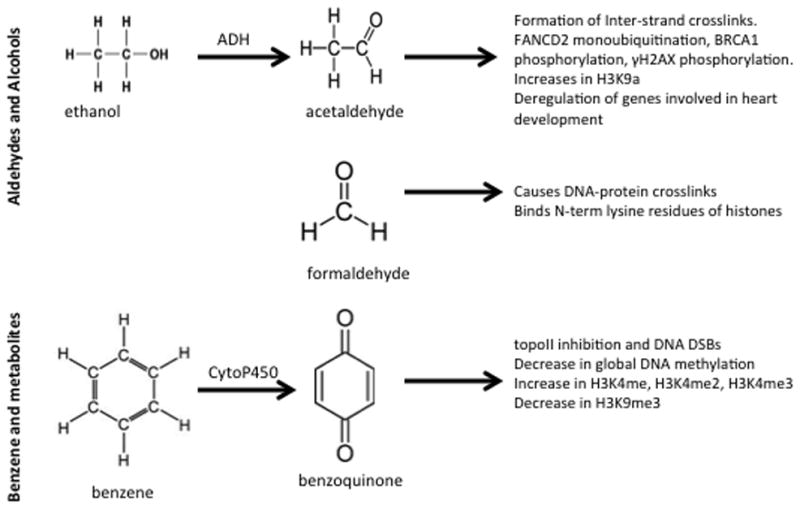
Environmental toxins. The chemical structure and biologic consequences of aldehydes and alcohols as well as benzene and its metabolites are shown.
Alcohols and aldehydes are linked with altered histone H3K9 acetylation (H3K9ac) and altered cellular differentiation in bone marrow stem cells, cardiac progenitor cells, and hepatocytes.105–110 A genome-wide reduction in H3K9ac typically occurs during human ES cell differentiation, and HDAC activity is required for ES cell differentiation. 80,111 In cardiac progenitor cells, low levels of ethanol, acetaldehyde, and acetate promoted a more than 2-fold increase in histone H3K9ac without affecting the proliferation of cells consistent with the maintenance of a progenitor state.112 High concentrations sufficient to produce a 30% reduction in cell viability also increased H3K9ac by more than 5-fold.112 In addition, high concentrations significantly elevated the expression of GATA4 and Mef2c genes related to heart development, resulting in their impaired differentiation.112 Consistent with these findings, the deregulation of genes that play a role in heart development has been proposed to be one of the mechanisms for the occurrence of congenital heart disease due to alcohol exposure during pregnancy.113
Occupational and environmental exposures to formaldehyde are prevalent. Formaldehyde is produced on a large scale in the manufacture of resins, particleboard, plywood, leather goods, paper, and pharmaceuticals. Formaldehyde is known to have genotoxic and mutagenic potential. It has been demonstrated that formaldehyde induces genotoxicity by causing DNA-protein crosslinks.114 In addition, lysine residues in the N-terminal tail and the globular fold domain of histone H4 have been identified in in vitro studies as binding sites for formaldehyde at concentrations from 5 to 100 mM, suggesting another mechanism of formaldehyde affecting epigenetic regulation.115
B. Benzene and Its Metabolites
Benzene is a ubiquitous pollutant and one of the top production chemicals in the United States (Fig. 1). It is used in the manufacturing industry and is a combustion product of cigarette smoke. Benzene is carcinogenic and causes primarily hematopoietic cancers in humans.116,117 It has been reported that it acts through its metabolites, especially 1,4-benzoquinone (1,4-BQ), as a strong topoisomerase II (topoII) poison causing DNA DSBs.118 1,4-BQ (25 μM) in vitro stimulates 8-fold DNA cleavage by topoII at sites close to defined chromosome breakpoints in leukemia. Benzene metabolites 1,4-BQ (1–10 μM) or 1,4-hydroquinone (10–100 μM) cause DNA damage and fragmentation in cultured HL60 cells though the generation of hydrogen peroxide oxidative stress, leading to apoptosis.119 Benzene and its metabolites, including benzoquinone, also influence the downstream DNA repair of DSBs. As little as 1 μM benzoquinone was sufficient to increase homologous recombination repair by 2.7-fold in a Chinese hamster ovary cell line containing a neomycin gene direct repeat recombination substrate.120 Global genomic hypomethylation is a common event in cancer and frequently observed in hematopoietic malignancies, including leukemia. Gasoline station attendants are exposed to benzene fumes, and this exposure has been thought to lead to higher rates of lymphatic and hematopoietic cancers.121 In support of this, one study showed a 1.6% decrease in global DNA methylation in these workers, suggesting an epigenetic mechanism of benzene action and cancer.122
Studies have extended earlier cell culture studies to in vivo mouse models, showing alterations in epigenetic markers and developmental reprogramming. Neonatal exposure to 1,4-bis[2-(3,5-dichloropyridyloxy)] benzene resulted in the activation of constitutive androstane receptor; a permanent increase of H3K4 mono-, di-, and trimethylation (H3K4me, H3K4me2, H3K4me3); and a decrease of H3K9 trimethylation (H3K9me3) within the Cyp2B10 locus.123 These epigenetic changes were maintained in mice throughout life and resulted in a permanent change in drug metabolism in the liver.123 Taken together, the in vitro and in vivo studies provide further support for the interplay between the DDR, DNA repair, and long-term chromatin remodeling.
C. Trace Metals
Trace amounts of metallic compounds are pervasive in the environment (Fig. 2). They are present in air, water, and food, and occupational exposure to them may occur through industrial production and waste disposal. Several studies have determined that trace metals cross the placenta,124–129 and the presence of cadmium, copper, chromium, nickel, lead, and zinc in placentas correlates with the response of the biomarker metallothionein and delta-amniolevulinic acid dehydratase, and lipid peroxidation.130 Consistent with environmental exposure to these elements, both levels of metals and biomarker responses were statistically significantly related to maternal dietary habits, consumption of canned food and bottled mineral water, as well as smoking.131 Animal models similarly have shown that sodium arsenite in drinking water administered to pregnant C57BL6/J mice resulted in dose-dependent accumulation in newborn pups.131 Trace metals elicit pleiotropic biochemical and physiological effects such as mimicry of binding in protein active sites; oxidative changes in lipid, proteins, or DNA; impaired transfer of nutrients to the fetus; low birth weight; and developmental delay. The variation of chemical properties and the reactive toxicities of each indicate that a uniform mechanism of action for all toxic metals is unlikely.
FIG. 2.
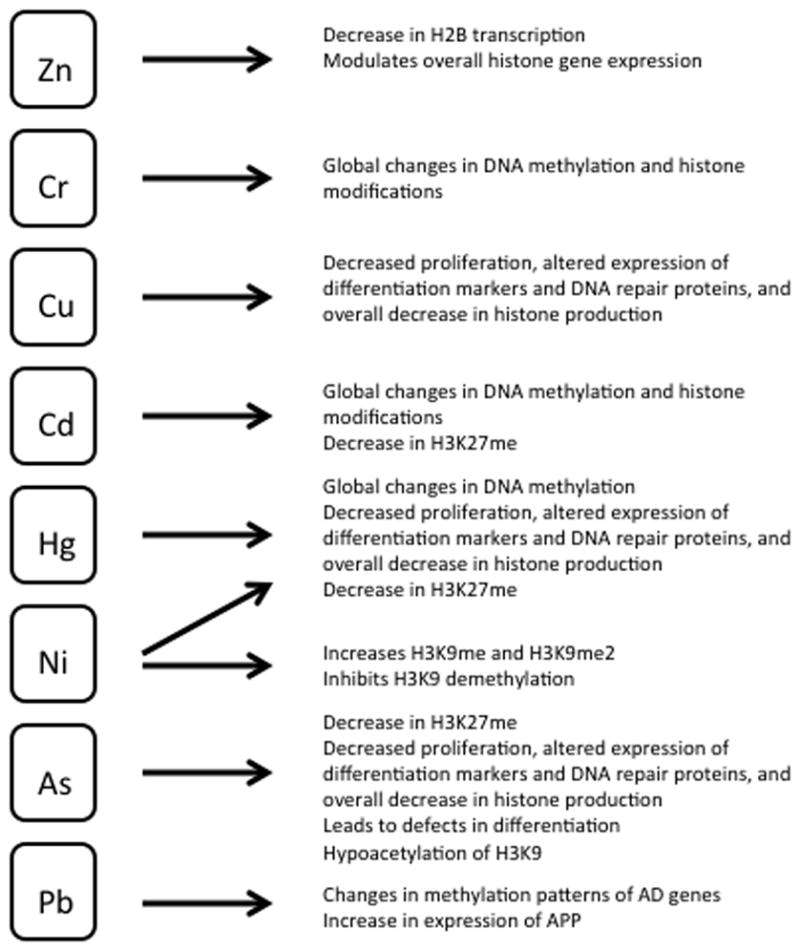
Environmental toxins: trace metals. The biologic consequences of exposure to trace metals are shown.
Recent reports have demonstrated that trace quantities of metals directly promote overall histone production, specific epigenetic modifications, and heritable changes in gene expression.132 Most important, these changes have been shown to occur in stem cells, potentially being transgenerational. Dimethylation of H3 leads to gene silencing,133 and exposure to multiple metals has been linked to this phenomenon. Zinc can modulate overall histone gene expression and possibly mediate effects on chromatin regulation.134 Treatment of human mononuclear THP-1 cells with 50 μmol/L zinc sulfate for 40 hours decreased H2B transcription by 1.58-fold. Zinc deprivation by treatment with 2.5 μmol/L of the membrane permeable zinc chelator TPEN conversely led to a 4.38-fold increase in H2B transcription. Exposure to cadmium, chromium, mercury, and nickel leads to global changes in DNA methylation and histone modifications.132 Acute in vitro exposure of mouse ES cells to arsenic, cadmium, mercury, and nickel led to a more than 50% decrease in H3K27 monomethylation (H3K27me), suggesting global induction of transcriptional repression.135 Nickel ion exposure at 250 μM or higher increased global H3K9me and H3K9me2 by 2- to 3-fold in a time-dependent manner in cell lines of different lineages, including murine ES cells and embryonic fibroblast cells and human lung carcinoma, osteosarcoma, and embryonic kidney cells.136 Furthermore, nickel ions induced gpt transgene silencing and exhibited inhibition of H3K9 demethylation, which led to, or permitted, the observed increase in H3K9me2.136 Analysis of peripheral blood mononuclear cells of humans with occupational exposure to nickel suggests that chronic exposure leads to further epigenetic changes in vivo: these samples showed an increase in H3K9me3.137 It is interesting to note that epigenetic alterations by metal exposure may be sex-specific: peripheral blood mononuclear cell analysis of women exposed to arsenic in drinking water demonstrated an increase in H3K9me2 and a decrease in H3K9ac, both markers of repression, but led to opposite changes in similarly exposed men.137
Low concentrations of trace metals are also sufficient to induce multiple cellular effects. Prolonged in vitro exposure of mouse ES cells to low concentrations (<IC50) of arsenic, cadmium, copper, lead, lithium, mercury, and nickel led to decreased cell proliferation; altered expression of cell differentiation markers Oct-4 and egfr; altered expression of DNA repair genes Rad-18, Top-3a, and Ogg-1; and overall decreased total production of histone protein.135
As a downstream result of transcriptional silencing by alterations in epigenetic markers, exposure leads to defects in cellular differentiation pathways. The arsenic derivative arsenite suppresses the expression of cellular differentiation markers to inhibit signaling pathways, maintain proliferative ability, and suppress the differentiation of keratinocyte progenitor cells, as well as transform human prostate epithelial progenitor cells into a cancer stem-cell phenotype.138–141 In one study, SCC9 human squamous carcinoma cells that exhibited a keratinocyte progenitor cell phenotype were stably transfected with constructs containing the proximal human involucrin promoter, either wild-type or mutated at both AP1 sites, were examined for their transcriptional activity using luciferase reporter activities with and without treatment with arsenate. It is notable that effects were detectable with a nontoxic concentration within the range of environmental exposures (2 μM sodium arsenate or sodium arsenite). As one marker of inhibition of differentiation, arsenite resulted in a significant reduction of c-Fos transcription factor and of acetylated H3 at the proximal and distal AP1 response elements of the involucrin gene promoter and of coactivator p300 at the proximal element of the involucrin gene promoter, as shown by chIP studies. Treatment with arsenite led to the dramatic suppression of the transcriptional activity of the involucrin gene to 2% of the level observed in the absence of any treatment.
Some research has extended cell culture studies to examine the long-term effects in vivo. Exposure of C57Bl6/J mice to 100 μg/L arsenic in drinking water from 1 week before conception until birth resulted in offspring with global H3K9 hypoacetylation, changes in functional annotation with highly significant representation of Krüppel-associated box transcription factors in brain samples, and long-term memory impairment compared to unexposed controls.142 Timed-pregnant Long–Evans hooded rats exposed to 200 ppm lead acetate in deionized drinking water during pregnancy delivered offspring with age-related neuropathological characteristics analogous to those seen in Alzheimer’s disease. These characteristics were accompanied by changes in the methylation patterns of key Alzheimer’s disease genes.143 Continued exposure to lead during the postnatal period resulted in a transient increase in β-amyloid precursor protein (APP). Messenger RNA expression during the first month after birth followed by a return to basal levels by 1 year, but a surprising subsequent delay of overexpression at 20 months after exposure to lead had ceased. These data suggest that environmental influences occurring during brain development predetermined methylation patterns, gene expression, and regulation of APP later in life, potentially altering the course of amyloidogenesis. These studies support the fetal basis of adult disease hypothesis, which states that many adult diseases have a fetal origin.144–148 Injury or environmental influences occurring at critical periods of organ development in the fetus at early stages of cell differentiation could lead to alterations in gene expression or gene imprinting, which can result in “programmatic” changes in gene expression and functional deficits evident later in life.
Epidemiological studies have well documented metals as human carcinogens associated with skin, lung, liver, and bladder cancers; however, the underlying mechanisms have not been clear. Cancer incidence increases with chronic exposure to metals such as arsenic, cadmium, chromium, and nickel.149–152 Studies associate arsenic exposure to multiple cancer types in human subjects and gene-specific DNA hypermethylation.153–157 This direct link between arsenic, tumorigenesis, and hypermethylation was further documented by low-dose (0.5 μM) exposure to arsenic trioxide that led to a transformation of BALB/c 3T3 cells and a dramatic increase in the tumor growth of these cells in a xenograft mouse model.158 Furthermore, these cells exhibited activated polycomb group proteins BMI1 and SUZ12, increased H3K27me3, and suppression of p16 and p19 that could be rescued by small hairpin RNA to either BMI1 or SUZ12.158
III. DIETARY EXPOSURES AND SUPPLEMENTS
A. Bisphenol A and Other Estrogens
Bisphenol A (BPA) is a hormonally active environmental xenoestrogen widely used in the production of polycarbonate plastics and resins, including some dental composites (Fig. 3). Human exposure to BPA comes mainly from daily diet; it leaches from food and drink packaging, water pipes, and dental sealants.159 BPA can cross the placenta and has been detected in urine, amniotic fluid, maternal and fetal plasma, placental tissue, and the breast milk of lactating mothers.159–162 Estrogens are both natural hormones produced in the body and widely used in hormone supplement therapy. 17β-Estradiol is an endogenous estrogen. Genistein is a soy phytoestrogen present in foods, particularly soybeans, and infant soy formulas163,164 (Fig. 3). Genistein and other estrogen derivatives are also available at health food stores as dietary and menopausal supplements.165 The validated and widely used ES cell test for toxicity166,167 demonstrated that exposure to BPA or genistein significantly altered genome-wide methylation patterns and decreased ES cell-to-cardiomyocyte differentiation. 168,169 Furthermore, the combination of BPA and genistein had a synergistic effect at lower concentrations, similar to those observed in human blood or sera.168
FIG. 3.
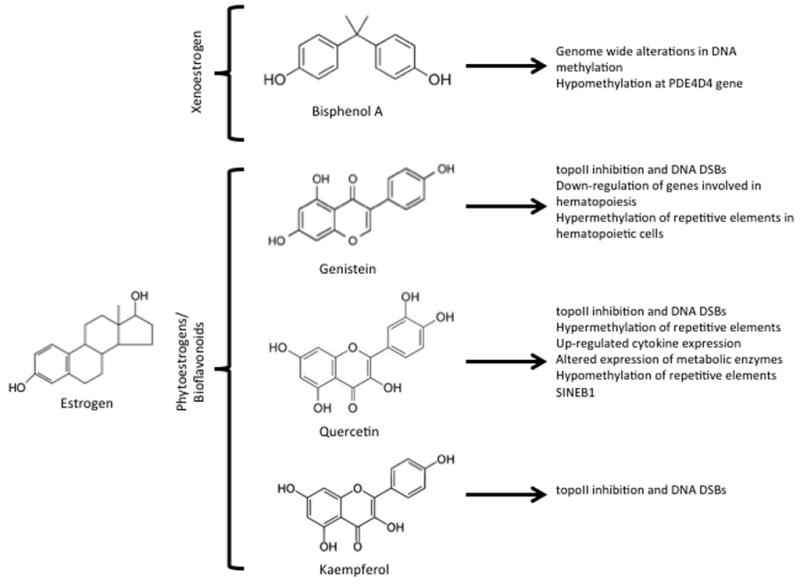
Estrogenic compounds: dietary exposures and supplements. The chemical structure and biologic consequences of xenoestrogens as well as phytoestrogens and bioflavonoids are shown. Of note is the similar phenolic ring structure of phytoestrogen and bioflavonoid compounds to etoposide, a potent inhibitor of topoII.
The epigenetically toxic effects of environmental chemicals like BPA and phthalates include DNA methylation, histone modification, and changes in the level of microRNA expression.7 Some of these effects have been found to be transgenerational. Numerous studies show that exposure to xenoestrogens can developmentally reprogram multiple organ systems. Differences in the ability of xenoestrogens to induce developmental reprogramming are likely driven by intrinsic differences in their binding to specific estrogen receptor subtypes. In the female reproductive tract, exposure is associated with alterations in morphology, hormonal response, and gene expression and promotes diverse outcomes such as obesity and cancer later in life.170–172 BPA is an endocrine disruptor, causing an adverse effect on mammalian reproduction because of the impaired development of germ cells. BPA has been reported to play a role in modulating germ cell differentiation, retinoic acid signaling, and the expression of germ cell marker genes in mouse ES cells.173 After administration of 50 μM BPA, upregulation of meiotic entry gene Stra8 (20-fold), upregulation of ovarian markers Foxl2 and Wnt4 (15- to 20-fold), and suppression of testicular markers Sox9 and Fgf9 were detected, showing that, in addition to germ cell differentiation, BPA also affects testicular and ovarian development. BPA dosing in pregnant C57BL/6J mice from embryonic day 8.5 to day 13.5 accelerated neurogenesis in the developing neurocortex and decreased the number of detectable neural stem/progenitor cells.174 Animal studies also have reported that postnatal exposure to BPA accelerates neurogenesis and causes neuronal migration defects that impair neurocortex development in embryos.175 BPA modulates adipogenic differentiation of cultured human primary adult stem cells176 and suppresses adipogenic differentiation of mouse mesenchymal stem cells.177
Physiologically relevant doses of BPA or estradiol have been reported to increase susceptibility to adult-onset prostate precancerous lesions and hormonal carcinogenesis. This imprinting involves epigenetic changes such as permanent alterations in the DNA methylation patterns of multiple cell signaling genes.178 Developmental exposure to estradiol and BPA leads to an increase in the susceptibility to prostate carcinogenesis with aging through epigenetic regulation.178 In normal prostates, gradual methylation occurs within the specific genomic cluster containing the gene for phosphodiesterase type 4 variant 4 (PDE4D4), which is an enzyme responsible for the breakdown of cyclic adenosine monophosphate. This methylation is associated with decreased expression. In contrast, exposure of neonatal Sprague-Dawley rats to BPA (10 μg/kg) or 17β-estradiol 3-benzoate (2500 μg/kg or 0.1 μg/kg) resulted in early and prolonged hypomethylation at this site and continued, elevating PDE4D4 gene expression throughout life, consistent with observed hypomethylation of this gene in prostate cancer cells. Several genes showed changes in methylation in response to neonatal estrogen treatments, many of which are permanent.
Estrogens also have been linked to the generation of DNA DSBs or inhibition of their repair. Exposure of primary gingival fibroblasts to dental adhesives containing BPA derivatives produced increased numbers of DNA breaks, marked damaged chromosomes with gH2AX, altered cell cycle profiles, and produced slower kinetics of repair. 179–181 Sensitivity to BPA derivatives may be global because exposure of keratinocytes, skin fibroblasts, intestinal cells (line LS174T), and hepatoma cells (line HepG2) all mark damaged DNA by gH2AX.182,183
B. Bioflavonoids as Topoisomerase II Inhibitors
Bioflavonoids comprise a diverse group of polyphenolic compounds divided into 3 main groups: flavones, flavonols, and isoflavones (Fig. 3). The most common sources of these bioflavonoids are fruits, vegetables, soy, tea, coffee, and wine.184 Genistein is both an estrogen derivative (discussed earlier), available at health food stores as a dietary and menopausal supplement,165 and a soy phytoestrogen present in foods, particularly soybeans, and infant soy formulas.163,164 Its effects in cells are thus likely pleiotropic, acting through both mechanisms. Because of their antioxidant capacity, flavonoids are used for their presumed health benefits, such as protection against cardiovascular diseases, cancer, and inflammation. Flavonoid supplements are available worldwide over the counter in pharmacies and drugstores.
However, accumulating evidence indicates that dietary flavonoids are potent inhibitors of topoII (topoII α and topoII β in mammalian cells185,186). DNA topoisomerases are essential cellular enzymes that cause topological changes in the DNA for processes such as replication and transcription. Inhibitors of topoII block the relegation of the transient DSBs made by topoII, leading to cell death at high doses and potentially leading to illegitimate repair, genome instability, and chromosomal abnormalities in surviving cells. The chemotherapeutic agents etoposide, doxorubicin, daunorubicin, and mitoxantrone185,187 are well-characterized topoII inhibitors that are typically used as comparison controls during the testing of other potential inhibitors. Etoposide is composed of a polycyclic ring system (rings A–D), a glycosidic moiety at the C4 position, and a pendant ring (E ring) at the C1 position.188–190 The binding of etoposide to topoII is driven by interactions with the A ring and the B ring,189 while the E ring is important for etoposide function.190 Several studies have reported that etoposide promotes MLL rearrangements in mouse ES cells,191 primitive hematopoietic stem cells, and in human fetal hematopoietic stem cells.192–195 Other anticancer agents, including teniposide, anthracyclines, and dactinomycin, also are associated with MLL rearrangements due to topoII inhibition and enhanced DNA cleavage, leading to defective DNA repair and chromosomal translocations.196
Multiple bioflavonoid compounds are also polyphenolic ring structures and thus may biochemically and mechanistically act similarly to etoposide.186 Etoposide, genistein, and quercetin contain pendant rings that feature a 4′-OH group that is essential for drug action.186,197,198 The 5′-OH group of genistein plays an important role in mediating binding to topoII, and the 4′-OH plays a significant role in function.186 TopoII inhibition by bioflavonoids was investigated in an in vitro plasmid DNA cleavage assay using purified recombinant wild-type human topoIIα and IIβ; it showed that these compounds were active against topoIIβ.186 Genistein (50 μM) was shown to be the most effective of the bioflavonoids tested and stimulated enzyme-mediated DNA cleavage ~10-fold,186 whereas 100 μM genistein efficiently induced topoII-DNA cleavage complexes in both cultured mouse myeloid progenitor cells (32Dc13) and Top2β knockout mouse embryonic fibroblasts, and it was suggested that these complexes are processed by proteasome, which led to chromosome rearrangements.199 Cultured human lymphocytes treated with 50 μM genistein display chromosome abnormalities in metaphase karyotypic analyses.200 DSBs with the MLL gene breakpoint cluster region were induced by bioflavonoid exposure both in primary human progenitor hematopoietic cells from healthy newborns and adults as well as in hematopoietic progenitor cell lines (BV173 and K562).201 Quercetin, genistein, and kaempferol induced DSBs in primary human hematopoietic CD34+ stem cell–enriched cells (at doses of 25 and 50 μM).202 Besides chromosomal translocations, monosomy or trisomy of MLL also was reported in cells exposed to quercetin.202
It is important to note that synthetic flavonoids are able to cross the placenta in the rat and are found in all fetal tissues (17% of the initial dose), including the fetal brain.203 Maternal and fetal distributions of the synthetic radioactively labeled bioflavonoid EMD-49209 were detectable 1–24 hours after intravenous injection into pregnant Wistar rats. Transplacental exposure to high but biological amounts of the flavonoids genistein and quercetin in Atm-ΔSRI mutant mice with an impaired capacity for DSB repair led to the detection (using inverse polymerase chain reaction) of a 2-fold higher number of MLL rearrangements compared with their wild-type siblings.184 Parallel in vitro studies with bone marrow cells exposed to genistein (50 μM) or quercetin (50 μM) showed 2.1–5.0 rearrangements/80 ng genomic DNA (1 per 13,000 cells) for quercetin or genistein compared with 0.2 translocations/80 ng genomic DNA for wild-type cells. Thus, the risk of these rearrangements due to in utero exposure to these bioflavonoids increases in the presence of compromised DNA repair, although in that study MLL rearrangements were detectable in all samples regardless of diet or mutational status.184
The epigenetic and transgenerational effects of these dietary compounds were addressed in a study that showed that exposure of progeny to genistein through maternal diet during pregnancy can have long-lasting effects on the progeny.204 Mice (129/SvJ:C57BL/6J background) ~8 weeks old were given genistein (270 mg/kg of feed) from conception until birth. Genistein induced epigenetic changes and altered the color of the coat of agouti mice. The progeny of these mice had a significant downregulation of genes involved in hematopoiesis of bone marrow cells, increased erythropoiesis, and a permanent signature hypermethylation of repetitive elements in hematopoietic lineages. Thus prenatal exposure to genistein affected the epigenetic signature of hematopoietic cells and caused long-lasting alterations in gene expression.
Exposure to the flavonoid quercetin during pregnancy can result in long-term changes in iron homeostasis during adulthood.205 Quercetin is a strong iron chelator and has the ability to cross the placenta and accumulate in the fetus. In this study, female mice (129/SvJ:C57BL/6J background) were given quercetin (302 mg/kg feed) from 3 days before conception until the end of gestation. Mice prenatally exposed to quercetin had upregulated iron-associated cytokine expression and significantly increased iron storage in the liver (~94 ng/mg for quercetin exposure vs. ~62 ng/mg for control). Quercetin exposure was associated with hypermethylation of repetitive elements, and these epigenetic modifications could cause these long-term changes in cytokine gene expression. All of these changes led to a shift toward a higher expression of cytokines associated with inflammation in the liver of adult mice that were prenatally exposed to quercetin.
Quercetin also has been shown to affect the xenobiotic metabolism of chemical carcinogens in mice that were prenatally exposed to this compound. 206 Mice (129/SvJ:C57BL/6J background) were given quercetin (1 mmol or 302 mg/kg of feed) from 3 days before conception until the end of gestation. Mice exposed to quercetin showed altered biotransformation of the environmental pollutant benzo[a]pyrene. This occurred because of the altered gene expression of metabolic enzymes such as Cyp1a1, Cyb1b1, Nqo1, and Ugt1a6, which persisted into adulthood in a tissue- and sex-dependent manner. These long-lasting changes were associated with epigenetic alterations since prenatal quercetin exposure led to hypomethylation of repetitive elements SINEB1. These persistent alterations in the metabolic enzymes of adult mice may affect cancer risk because of environmental chemical carcinogens.
IV. CONCLUDING REMARKS
The exposure to environmental and dietary agents discussed above is widespread today. Because of the beneficial effects assumed to be associated with the use of bioflavonoids, their use as dietary supplements is increasingly popular and widespread. However, a growing body of evidence is emerging regarding the long-term implications and adverse effects of using these compounds in an unrestricted manner. Multiple environmental toxins and dietary agents have the potential to cause long-term epigenetic changes, leading to dysregulation of multiple cellular functions and developmental pathways. Epigenetic modulation, cell differentiation, gene expression, signal transduction, and illegitimate DNA repair all are associated with human diseases and cancer.207 It is important to note that epigenetic alterations following exposure will continue to affect cellular development long after exposure has ceased. In view of the variety of adverse effects these agents have in exposed individuals, it is important to raise public awareness, set guidelines, and regulate the use and market availability of such compounds to reduce the risk of disease. Since the role of in utero exposures in causing long-term transgenerational effects has been demonstrated to be critical, it is important to address the susceptibility of different stages of cell differentiation to the deleterious molecular changes induced by these agents. However, a system to directly and rapidly examine the role of a large number of different compounds, both individually and in combination, in inducing the genetic/epigenetic changes discussed above is lacking, and thus testing has been limited to small isolated studies. Even the inherent genomic instability of existing stem cell lines or the memory of previous epigenetic codes of iPSCs suggest that they may not always mimic in vivo consequences. The development of model systems for cells at different stages of differentiation with different susceptibilities to epigenetic alterations will provide useful insights into the comparative risk to human populations and how the timing or stage of development may affect in vivo consequences to individuals.
References
- 1.Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature. 1997;389:251–60. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- 2.Luger K, Rechsteiner TJ, Flaus AJ, Waye MM, Richmond TJ. Characterization of nucleosome core particles containing histone proteins made in bacteria. J Mol Biol. 1997;272:301–11. doi: 10.1006/jmbi.1997.1235. [DOI] [PubMed] [Google Scholar]
- 3.Campos EI, Reinberg D. Histones: annotating chromatin. Annu Rev Genet. 2009;43:559–99. doi: 10.1146/annurev.genet.032608.103928. [DOI] [PubMed] [Google Scholar]
- 4.Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403:41–5. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- 5.Turner BM. Histone acetylation and an epigenetic code. Bioessays. 2000;22:836–45. doi: 10.1002/1521-1878(200009)22:9<836::AID-BIES9>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 6.Workman JL. Nucleosome displacement in transcription. Genes Dev. 2006;20:2009–17. doi: 10.1101/gad.1435706. [DOI] [PubMed] [Google Scholar]
- 7.Singh S, Li SS. Epigenetic effects of environmental chemicals bisphenol a and phthalates. Int J Mol Sci. 2012;13:10143–53. doi: 10.3390/ijms130810143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Baccarelli A, Bollati V. Epigenetics and environmental chemicals. Curr Opin Pediatr. 2009;21:243–51. doi: 10.1097/mop.0b013e32832925cc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lai W, Li H, Liu S, Tao Y. Connecting chromatin modifying factors to DNA damage response. Int J Mol Sci. 2013;14:2355–69. doi: 10.3390/ijms14022355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pandita TK, Richardson C. Chromatin remodeling finds its place in the DNA double-strand break response. Nucleic Acids Res. 2009;37:1363–77. doi: 10.1093/nar/gkn1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Orlowski C, Mah LJ, Vasireddy RS, El-Osta A, Karagiannis TC. Double-strand breaks and the concept of short- and long-term epigenetic memory. Chromosoma. 2011;120:129–49. doi: 10.1007/s00412-010-0305-6. [DOI] [PubMed] [Google Scholar]
- 12.Xu Y, Price BD. Chromatin dynamics and the repair of DNA double strand breaks. Cell Cycle. 2011;10:261–7. doi: 10.4161/cc.10.2.14543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bao Y. Chromatin response to DNA double-strand break damage. Epigenomics. 2011;3:307–21. doi: 10.2217/epi.11.14. [DOI] [PubMed] [Google Scholar]
- 14.Celeste A, Difilippantonio S, Difilippantonio MJ, Fernandez-Capetillo O, Pilch DR, Sedelnikova OA, Eckhaus M, Ried T, Bonner WM, Nussenzweig A. H2AX haploinsufficiency modifies genomic stability and tumor susceptibility. Cell. 2003;114:371–83. doi: 10.1016/s0092-8674(03)00567-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Scully R, Xie A. Double strand break repair functions of histone H2AX. Mutat Res. 2013 Jul 31; doi: 10.1016/j.mrfmmm.2013.07.007. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goodarzi AA, Noon AT, Deckbar D, Ziv Y, Shiloh Y, Löbrich M, Jeggo PA. ATM signaling facilitates repair of DNA double-strand breaks associated with heterochromatin. Mol Cell. 2008;31:167–77. doi: 10.1016/j.molcel.2008.05.017. [DOI] [PubMed] [Google Scholar]
- 17.Cann KL, Dellaire G. Heterochromatin and the DNA damage response: the need to relax. Biochem Cell Biol. 2011;89:45–60. doi: 10.1139/O10-113. [DOI] [PubMed] [Google Scholar]
- 18.Chiolo I, Tang J, Georgescu W, Costes SV. Nuclear dynamics of radiation-induced foci in euchromatin and heterochromatin. Mutat Res. 2013 Aug 16; doi: 10.1016/j.mrfmmm.2013.08.001. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Goodarzi AA, Jeggo P, Lobrich M. The influence of heterochromatin on DNA double strand break repair: getting the strong, silent type to relax. DNA Repair (Amst) 2010;9:1273–82. doi: 10.1016/j.dnarep.2010.09.013. [DOI] [PubMed] [Google Scholar]
- 20.Jakob B, Splinter J, Conrad S, Voss KO, Zink D, Durante M, Löbrich M, Taucher-Scholz G. DNA double-strand breaks in heterochromatin elicit fast repair protein recruitment, histone H2AX phosphorylation and relocation to euchromatin. Nucleic Acids Res. 2011;39:6489–99. doi: 10.1093/nar/gkr230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lorat Y, Schanz S, Schuler N, Wennemuth G, Rube C, Rube CE. Beyond repair foci: DNA double-strand break repair in euchromatic and heterochromatic compartments analyzed by transmission electron microscopy. PLoS One. 2012;7:e38165. doi: 10.1371/journal.pone.0038165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chiolo I, Minoda A, Colmenares SU, Polyzos A, Costes SV, Karpen GH. Double-strand breaks in heterochromatin move outside of a dynamic HP1a domain to complete recombinational repair. Cell. 2011;144:732–44. doi: 10.1016/j.cell.2011.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Goodarzi AA, Jeggo PA. The heterochromatic barrier to DNA double strand break repair: how to get the entry visa. Int J Mol Sci. 2012;13:11844–60. doi: 10.3390/ijms130911844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Noon AT, Shibata A, Rief N, Löbrich M, Stewart GS, Jeggo PA, Goodarzi AA. 53BP1-dependent robust localized KAP-1 phosphorylation is essential for heterochromatic DNA double-strand break repair. Nat Cell Biol. 2010;12:177–84. doi: 10.1038/ncb2017. [DOI] [PubMed] [Google Scholar]
- 25.Kimura A, Horikoshi M. Tip60 acetylates six lysines of a specific class in core histones in vitro. Genes Cells. 1998;3:789–800. doi: 10.1046/j.1365-2443.1998.00229.x. [DOI] [PubMed] [Google Scholar]
- 26.Gieni RS, Ismail IH, Campbell S, Hendzel MJ. Polycomb group proteins in the DNA damage response: a link between radiation resistance and “stemness”. Cell Cycle. 2011;10:883–94. doi: 10.4161/cc.10.6.14907. [DOI] [PubMed] [Google Scholar]
- 27.Cao R, Wang L, Wang H, Xia L, Erdjument-Bromage H, Tempst P, Jones RS, Zhang Y. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science. 2002;298:1039–43. doi: 10.1126/science.1076997. [DOI] [PubMed] [Google Scholar]
- 28.Endoh M, Endo TA, Endoh T, Isono K, Sharif J, Ohara O, Toyoda T, Ito T, Eskeland R, Bickmore WA, Vidal M, Bernstein BE, Koseki H. Histone H2A mono-ubiquitination is a crucial step to mediate PRC1-dependent repression of developmental genes to maintain ES cell identity. PLoS Genet. 2012;8(7):e1002774. doi: 10.1371/journal.pgen.1002774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kuzmichev A, Nishioka K, Erdjument-Bromage H, Tempst P, Reinberg D. Histone methyltransferase activity associated with a human multiprotein complex containing the Enhancer of Zeste protein. Genes Dev. 2002;16:2893–905. doi: 10.1101/gad.1035902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vissers JH, van Lohuizen M, Citterio E. The emerging role of Polycomb repressors in the response to DNA damage. J Cell Sci. 2012;125(Pt 17):3939–48. doi: 10.1242/jcs.107375. [DOI] [PubMed] [Google Scholar]
- 31.Ginjala V, Nacerddine K, Kulkarni A, Oza J, Hill SJ, Yao M, Citterio E, van Lohuizen M, Ganesan S. BMI1 is recruited to DNA breaks and contributes to DNA damage-induced H2A ubiquitination and repair. Mol Cell Biol. 2011;31:1972–82. doi: 10.1128/MCB.00981-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pavithra L, Mukherjee S, Sreenath K, Kar S, Sakaguchi K, Roy S, Chattopahdyay S. SMAR1 forms a ternary complex with p53-MDM2 and negatively regulates p53-mediated transcription. J Mol Biol. 2009;388:691–702. doi: 10.1016/j.jmb.2009.03.033. [DOI] [PubMed] [Google Scholar]
- 33.Sinha S, Malonia SK, Mittal SP, Mathai J, Pal JK, Chattopadhyay S. Chromatin remodelling protein SMAR1 inhibits p53 dependent transactivation by regulating acetyl transferase p300. Int J Biochem Cell Biol. 2012;44:46–52. doi: 10.1016/j.biocel.2011.10.020. [DOI] [PubMed] [Google Scholar]
- 34.Gospodinov A, Tsaneva I, Anachkova B. RAD51 foci formation in response to DNA damage is modulated by TIP49. Int J Biochem Cell Biol. 2009;41:925–33. doi: 10.1016/j.biocel.2008.09.004. [DOI] [PubMed] [Google Scholar]
- 35.Tang Y, Luo J, Zhang W, Gu W. Tip60-dependent acetylation of p53 modulates the decision between cell-cycle arrest and apoptosis. Mol Cell. 2006;24:827–39. doi: 10.1016/j.molcel.2006.11.021. [DOI] [PubMed] [Google Scholar]
- 36.Koturbash I, Baker M, Loree J, Kutanzi K, Hudson D, Pogribny I, Sedelnikova O, Bonner W, Kovalchuk O. Epigenetic dysregulation underlies radiation-induced transgenerational genome instability in vivo. Int J Radiat Oncol Biol Phys. 2006;66:327–30. doi: 10.1016/j.ijrobp.2006.06.012. [DOI] [PubMed] [Google Scholar]
- 37.Nakagawa M, Koyanagi M, Tanabe K, Takahashi K, Ichisaka T, Aoi T, Okita K, Mochiduki Y, Takizawa N, Yamanaka S. Generation of induced pluripotent stem cells without Myc from mouse and human fibroblasts. Nat Biotechnol. 2008;26:101–6. doi: 10.1038/nbt1374. [DOI] [PubMed] [Google Scholar]
- 38.Takahashi K, Okita K, Nakagawa M, Yamanaka S. Induction of pluripotent stem cells from fibroblast cultures. Nat Protoc. 2007;2:3081–9. doi: 10.1038/nprot.2007.418. [DOI] [PubMed] [Google Scholar]
- 39.Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–72. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 40.Yu J, Vodyanik MA, Smuga-Otto K, Antosiewicz-Bourget J, Frane JL, Tian S, Nie J, Jonsdottir GA, Ruotti V, Stewart R, Slukvin II, Thomson JA. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007;318:1917–20. doi: 10.1126/science.1151526. [DOI] [PubMed] [Google Scholar]
- 41.Klaric M, Winkler J, Vojnits K, Meganathan K, Jagtap S, Ensenat-Waser R, Hescheler J, Sachinidis A, Bremer-Hoffmann S. Current status of human pluripotent stem cell based in vitro toxicity tests. Front Biosci (Schol Ed) 2013;5:118–33. doi: 10.2741/s361. [DOI] [PubMed] [Google Scholar]
- 42.Liu W, Deng Y, Liu Y, Gong W, Deng W. Stem cell models for drug discovery and toxicology studies. J Biochem Mol Toxicol. 2013;27:17–27. doi: 10.1002/jbt.21470. [DOI] [PubMed] [Google Scholar]
- 43.Scott CW, Peters MF, Dragan YP. Human induced pluripotent stem cells and their use in drug discovery for toxicity testing. Toxicol Lett. 2013;219:49–58. doi: 10.1016/j.toxlet.2013.02.020. [DOI] [PubMed] [Google Scholar]
- 44.Prikrylova T, Pachernik J, Kozubek S, Bartova E. Epigenetics and chromatin plasticity in embryonic stem cells. World J Stem Cells. 2013;5:73–85. doi: 10.4252/wjsc.v5.i3.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bibikova M, Chudin E, Wu B, Zhou L, Garcia EW, Liu Y, Shin S, Plaia TW, Auerbach JM, Arking DE, Gonzalez R, Crook J, Davidson B, Schulz TC, Robins A, Khanna A, Sartipy P, Hyllner J, Vanquri P, Savant-Bhonsale S, Smith AK, Chakravarti A, Maitra A, Rao M, Barker DL, Loring JF, Fan JB. Human embryonic stem cells have a unique epigenetic signature. Genome Res. 2006;16:1075–83. doi: 10.1101/gr.5319906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Calhoun JD, Rao RR, Warrenfeltz S, Rekaya R, Dalton S, McDonald J, Stice SL. Transcriptional profiling of initial differentiation events in human embryonic stem cells. Biochem Biophys Res Commun. 2004;323:453–64. doi: 10.1016/j.bbrc.2004.08.117. [DOI] [PubMed] [Google Scholar]
- 47.Gesslbauer B, Krenn E, Zenzmaier C, Preisegger KH, Kungl AJ. Lessons from the stem cell proteome. Curr Stem Cell Res Ther. 2006;1:395–409. doi: 10.2174/157488806778226867. [DOI] [PubMed] [Google Scholar]
- 48.Hanna J, Cheng AW, Saha K, Kim J, Lengner CJ, Soldner F, Cassady JP, Muffat J, Carey BW, Jaenisch R. Human embryonic stem cells with biological and epigenetic characteristics similar to those of mouse ESCs. Proc Natl Acad Sci U S A. 2010;107:9222–7. doi: 10.1073/pnas.1004584107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hanna JH, Saha K, Jaenisch R. Pluripotency and cellular reprogramming: facts, hypotheses, unresolved issues. Cell. 2010;143:508–25. doi: 10.1016/j.cell.2010.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lagarkova MA, Volchkov PY, Lyakisheva AV, Philonenko ES, Kiselev SL. Diverse epigenetic profile of novel human embryonic stem cell lines. Cell Cycle. 2006;5:416–20. doi: 10.4161/cc.5.4.2440. [DOI] [PubMed] [Google Scholar]
- 51.Mouzannar R, McCafferty J, Benedetto G, Richardson C. Transcriptional and phospho-proteomic screens reveal stem cell activation of insulin-resistance and transformation pathways following a single minimally toxic episode of Ros. Int J Genomics Proteomics. 2011;2(1):34–49. [PMC free article] [PubMed] [Google Scholar]
- 52.Munoz J, Heck AJ. Quantitative proteome and phosphoproteome analysis of human pluripotent stem cells. Methods Mol Biol. 2011;767:297–312. doi: 10.1007/978-1-61779-201-4_22. [DOI] [PubMed] [Google Scholar]
- 53.Nishino K, Ohgane J, Suzuki M, Hattori N, Shiota K. Methylation in embryonic stem cells in vitro. Methods Mol Biol. 2006;329:421–45. doi: 10.1385/1-59745-037-5:421. [DOI] [PubMed] [Google Scholar]
- 54.Novak A, Amit M, Ziv T, Segev H, Fishman B, Admon A, Itskovitz-Eldor J. Proteomics profiling of human embryonic stem cells in the early differentiation stage. Stem Cell Rev. 2012;8:137–49. doi: 10.1007/s12015-011-9286-y. [DOI] [PubMed] [Google Scholar]
- 55.Rao RR, Stice SL. Gene expression profiling of embryonic stem cells leads to greater understanding of pluripotency and early developmental events. Biol Reprod. 2004;71:1772–8. doi: 10.1095/biolreprod.104.030395. [DOI] [PubMed] [Google Scholar]
- 56.Richards M, Tan SP, Tan JH, Chan WK, Bongso A. The transcriptome profile of human embryonic stem cells as defined by SAGE. Stem Cells. 2004;22:51–64. doi: 10.1634/stemcells.22-1-51. [DOI] [PubMed] [Google Scholar]
- 57.Sato N, Sanjuan IM, Heke M, Uchida M, Naef F, Brivanlou AH. Molecular signature of human embryonic stem cells and its comparison with the mouse. Dev Biol. 2003;260:404–13. doi: 10.1016/s0012-1606(03)00256-2. [DOI] [PubMed] [Google Scholar]
- 58.Sato S, Yagi S, Arai Y, Hirabayashi K, Hattori N, Iwatani M, Okita K, Ohgane J, Tanaka S, Wakayama T, Yamanaka S, Shiota K. Genome-wide DNA methylation profile of tissue-dependent and differentially methylated regions (T-DMRs) residing in mouse pluripotent stem cells. Genes Cells. 2010;15:607–18. doi: 10.1111/j.1365-2443.2010.01404.x. [DOI] [PubMed] [Google Scholar]
- 59.Schulze M, Ungefroren H, Bader M, Fandrich F. Derivation, maintenance, and characterization of rat embryonic stem cells in vitro. Methods Mol Biol. 2006;329:45–58. doi: 10.1385/1-59745-037-5:45. [DOI] [PubMed] [Google Scholar]
- 60.Uchida S, Gellert P, Braun T. Deeply dissecting stemness: making sense to non-coding RNAs in stem cells. Stem Cell Rev. 2012;8:78–86. doi: 10.1007/s12015-011-9294-y. [DOI] [PubMed] [Google Scholar]
- 61.Ulloa-Montoya F, Kidder BL, Pauwelyn KA, Chase LG, Luttun A, Crabbe A, Geraerts M, Sharov AA, Piao Y, Ko MS, Hu WS, Verfaillie CM. Comparative transcriptome analysis of embryonic and adult stem cells with extended and limited differentiation capacity. Genome Biol. 2007;8:R163. doi: 10.1186/gb-2007-8-8-r163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Draper JS, Moore HD, Ruban LN, Gokhale PJ, Andrews PW. Culture and characterization of human embryonic stem cells. Stem Cells Dev. 2004;13:325–36. doi: 10.1089/scd.2004.13.325. [DOI] [PubMed] [Google Scholar]
- 63.Draper JS, Smith K, Gokhale P, Moore HD, Maltby E, Johnson J, Meisner L, Zwaka TP, Thomson JA, Andrews PW. Recurrent gain of chromosomes 17q and 12 in cultured human embryonic stem cells. Nat Biotechnol. 2004;22:53–4. doi: 10.1038/nbt922. [DOI] [PubMed] [Google Scholar]
- 64.Lefort N, Perrier AL, Laabi Y, Varela C, Peschanski M. Human embryonic stem cells and genomic instability. Regen Med. 2009;4:899–909. doi: 10.2217/rme.09.63. [DOI] [PubMed] [Google Scholar]
- 65.Maitra A, Arking DE, Shivapurkar N, Ikeda M, Stastny V, Kassauei K, Sui G, Cutler DJ, Liu Y, Brimble SN, Noaksson K, Hyllner J, Schulz TC, Zeng X, Freed WJ, Crook J, Abraham S, Colman A, Sartipy P, Matsui S, Carpenter M, Gazdar AF, Rao M, Chakravarti A. Genomic alterations in cultured human embryonic stem cells. Nat Genet. 2005;37:1099–103. doi: 10.1038/ng1631. [DOI] [PubMed] [Google Scholar]
- 66.Rosler ES, Fisk GJ, Ares X, Irving J, Miura T, Rao MS, Carpenter MK. Long-term culture of human embryonic stem cells in feeder-free conditions. Dev Dyn. 2004;229:259–74. doi: 10.1002/dvdy.10430. [DOI] [PubMed] [Google Scholar]
- 67.Spits C, Mateizel I, Geens M, Mertzanidou A, Staessen C, Vandeskelde Y, Van der Elst J, Liebaers I, Sermon K. Recurrent chromosomal abnormalities in human embryonic stem cells. Nat Biotechnol. 2008;26:1361–3. doi: 10.1038/nbt.1510. [DOI] [PubMed] [Google Scholar]
- 68.Chen CP, Su YN, Su JW, Chern SR, Chen YT, Chen LF, Wang W. Mosaic trisomy 12 at amniocentesis: prenatal diagnosis and molecular genetic analysis. Taiwan J Obstet Gynecol. 2013;52:97–105. doi: 10.1016/j.tjog.2013.01.012. [DOI] [PubMed] [Google Scholar]
- 69.Korkola JE, Houldsworth J, Bosl GJ, Chaganti RS. Molecular events in germ cell tumours: linking chromosome- 12 gain, acquisition of pluripotency and response to cisplatin. BJU Int. 2009;104(9 Pt B):1334–8. doi: 10.1111/j.1464-410X.2009.08855.x. [DOI] [PubMed] [Google Scholar]
- 70.Reinholz MM, Bruzek AK, Visscher DW, Lingle WL, Schroeder MJ, Perez EA, Jenkins RB. Breast cancer and aneusomy 17: implications for carcinogenesis and therapeutic response. Lancet Oncol. 2009;10:267–77. doi: 10.1016/S1470-2045(09)70063-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hodgson JG, Chin K, Collins C, Gray JW. Genome amplification of chromosome 20 in breast cancer. Breast Cancer Res Treat. 2003;78:337–45. doi: 10.1023/a:1023085825042. [DOI] [PubMed] [Google Scholar]
- 72.Knuutila S, Bjorkqvist AM, Autio K, Tarkkanen M, Wolf M, Monni O, Szymanska J, Larramendy ML, Tapper J, Pere H, El-Rifai W, Hemmer S, Wasenius VM, Vidgren V, Zhu Y. DNA copy number amplifications in human neoplasms: review of comparative genomic hybridization studies. Am J Pathol. 1998;152:1107–23. [PMC free article] [PubMed] [Google Scholar]
- 73.Kim K, Doi A, Wen B, Ng K, Zhao R, Cahan P, Kim J, Aryee MJ, Ji H, Ehrlich LI, Yabuuchi A, Takeuchi A, Cunniff KC, Hongguang H, McKinney-Freeman S, Naveiras O, Yoon TJ, Irizarry RA, Jung N, Seita J, Hanna J, Murakami P, Jaenisch R, Weissleder R, Orkin SH, Weissman IL, Feinberg AP, Daley GQ. Epigenetic memory in induced pluripotent stem cells. Nature. 2010;467:285–90. doi: 10.1038/nature09342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tobin SC, Kim K. Generating pluripotent stem cells: differential epigenetic changes during cellular reprogramming. FEBS Lett. 2012;586:2874–81. doi: 10.1016/j.febslet.2012.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, Fry B, Meissner A, Wernig M, Plath K, Jaenisch R, Wagschal A, Feil R, Schreiber SL, Lander ES. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006;125:315–26. doi: 10.1016/j.cell.2006.02.041. [DOI] [PubMed] [Google Scholar]
- 76.Meshorer E, Misteli T. Chromatin in pluripotent embryonic stem cells and differentiation. Nat Rev Mol Cell Biol. 2006;7:540–6. doi: 10.1038/nrm1938. [DOI] [PubMed] [Google Scholar]
- 77.Mikkelsen TS, Ku M, Jaffe DB, Issac B, Lieberman E, Giannoukos G, Alvarez P, Brockman W, Kim TK, Koche RP, Lee W, Mendenhall E, O’Donovan A, Presser A, Russ C, Xie X, Meissner A, Wernig M, Jaenisch R, Nusbaum C, Lander ES, Bernstein BE. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature. 2007;448:553–60. doi: 10.1038/nature06008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Serrano L, Vazquez BN, Tischfield J. Chromatin structure, pluripotency and differentiation. Exp Biol Med (Maywood) 2013;238:259–70. doi: 10.1177/1535370213480718. [DOI] [PubMed] [Google Scholar]
- 79.Mattout A, Meshorer E. Chromatin plasticity and genome organization in pluripotent embryonic stem cells. Curr Opin Cell Biol. 2010;22:334–41. doi: 10.1016/j.ceb.2010.02.001. [DOI] [PubMed] [Google Scholar]
- 80.Krejci J, Uhlirova R, Galiova G, Kozubek S, Smigova J, Bartova E. Genome-wide reduction in H3K9 acetylation during human embryonic stem cell differentiation. J Cell Physiol. 2009;219:677–87. doi: 10.1002/jcp.21714. [DOI] [PubMed] [Google Scholar]
- 81.Kidder BL, Palmer S. HDAC1 regulates pluripotency and lineage specific transcriptional networks in embryonic and trophoblast stem cells. Nucleic Acids Res. 2012;40:2925–39. doi: 10.1093/nar/gkr1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Liang G, Taranova O, Xia K, Zhang Y. Butyrate promotes induced pluripotent stem cell generation. J Biol Chem. 2010;285:25516–21. doi: 10.1074/jbc.M110.142059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Mali P, Chou BK, Yen J, Ye Z, Zou J, Dowey S, Brodsky RA, Ohm JE, Yu W, Baylin SB, Yusa K, Bradley A, Meyers DJ, Mukherjee C, Cole PA, Cheng L. Butyrate greatly enhances derivation of human induced pluripotent stem cells by promoting epigenetic remodeling and the expression of pluripotency-associated genes. Stem Cells. 2010;28:713–20. doi: 10.1002/stem.402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Efroni S, Carmel L, Schaefer CG, Buetow KH. Superposition of transcriptional behaviors determines gene state. PLoS One. 2008;3:e2901. doi: 10.1371/journal.pone.0002901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Efroni S, Duttagupta R, Cheng J, Dehghani H, Hoeppner DJ, Dash C, Bazett-Jones DP, Le Grice S, McKay RD, Buetow KH, Gingeras TR, Misteli T, Meshorer E. Global transcription in pluripotent embryonic stem cells. Cell Stem Cell. 2008;2:437–47. doi: 10.1016/j.stem.2008.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Luo L, Gassman KL, Petell LM, Wilson CL, Bewersdorf J, Shopland LS. The nuclear periphery of embryonic stem cells is a transcriptionally permissive and repressive compartment. J Cell Sci. 2009;122(Pt 20):3729–37. doi: 10.1242/jcs.052555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Margueron R, Reinberg D. The Polycomb complex PRC2 and its mark in life. Nature. 2011;469:343–9. doi: 10.1038/nature09784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Boyer LA, Plath K, Zeitlinger J, Brambrink T, Medeiros LA, Lee TI, Levine SS, Wernig M, Tajonar A, Ray MK, Bell GW, Otte AP, Vidal M, Gifford DK, Young RA, Jaenisch R. Polycomb complexes repress developmental regulators in murine embryonic stem cells. Nature. 2006;441:349–53. doi: 10.1038/nature04733. [DOI] [PubMed] [Google Scholar]
- 89.Pan G, Tian S, Nie J, Yang C, Ruotti V, Wei H, Jonsdottir GA, Stewart R, Thomson JA. Whole-genome analysis of histone H3 lysine 4 and lysine 27 methylation in human embryonic stem cells. Cell Stem Cell. 2007;1:299–312. doi: 10.1016/j.stem.2007.08.003. [DOI] [PubMed] [Google Scholar]
- 90.Vastenhouw NL, Schier AF. Bivalent histone modifications in early embryogenesis. Curr Opin Cell Biol. 2012;24:374–86. doi: 10.1016/j.ceb.2012.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Abramovich C, Humphries RK. Hox regulation of normal and leukemic hematopoietic stem cells. Curr Opin Hematol. 2005;12:210–6. doi: 10.1097/01.moh.0000160737.52349.aa. [DOI] [PubMed] [Google Scholar]
- 92.Rappolee DA, Xie Y, Slater JA, Zhou S, Puscheck EE. Toxic stress prioritizes and imbalances stem cell differentiation: implications for new biomarkers and in vitro toxicology tests. Syst Biol Reprod Med. 2012;58:33–40. doi: 10.3109/19396368.2011.647381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Qu W, Tokar EJ, Kim AJ, Bell MW, Waalkes MP. Chronic cadmium exposure in vitro causes acquisition of multiple tumor cell characteristics in human pancreatic epithelial cells. Environ Health Perspect. 2012;120:1265–71. doi: 10.1289/ehp.1205082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wang L, Chen F, Zhang Z, Chen G, Luo J, Shi X. Cancer stem cells in the mechanism of metal carcinogenesis. J Environ Pathol Toxicol Oncol. 2012;31:245–63. doi: 10.1615/jenvironpatholtoxicoloncol.v31.i3.60. [DOI] [PubMed] [Google Scholar]
- 95.Andreini B, Baroni R, Galimberi E, Sesana G. Aldehydes in the atmospheric environment: evaluation of human exposure in the north-west area of Mila. Microchem J. 2000;67:11–9. [Google Scholar]
- 96.Bunkoed O, Thavarungkul P, Thammakhet C, Kanatharana P. A simple and high collection efficiency sampling method for monitoring of carbonyl compounds in a workplace environment. J Environ Sci Health A Tox Hazard Subst Environ Eng. 2012;47:167–75. doi: 10.1080/10934529.2012.640244. [DOI] [PubMed] [Google Scholar]
- 97.Lu H, Cai QY, Wen S, Chi Y, Guo S, Sheng G, Fu J. Seasonal and diurnal variations of carbonyl compounds in the urban atmosphere of Guangzhou, China. Sci Total Environ. 2010;408:3523–9. doi: 10.1016/j.scitotenv.2010.05.013. [DOI] [PubMed] [Google Scholar]
- 98.Seo YK, Baek SO. Characterization of carbonyl compounds in the ambient air of an industrial city in Korea. Sensors (Basel) 2011;11:949–63. doi: 10.3390/s110100949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wang HK, Huang CH, Chen KS, Peng YP, Lai CH. Measurement and source characteristics of carbonyl compounds in the atmosphere in Kaohsiung city, Taiwan. J Hazard Mater. 2010;179(1–3):1115–21. doi: 10.1016/j.jhazmat.2010.03.122. [DOI] [PubMed] [Google Scholar]
- 100.Frampton MW, Pryor WA, Cueto R, Cox C, Morrow PE, Utell MJ. Ozone exposure increases aldehydes in epithelial lining fluid in human lung. Am J Respir Crit Care Med. 1999;159(4 Pt 1):1134–7. doi: 10.1164/ajrccm.159.4.9807057. [DOI] [PubMed] [Google Scholar]
- 101.Garaycoechea JI, Crossan GP, Langevin F, Daly M, Arends MJ, Patel KJ. Genotoxic consequences of endogenous aldehydes on mouse haematopoietic stem cell function. Nature. 2012;489:571–5. doi: 10.1038/nature11368. [DOI] [PubMed] [Google Scholar]
- 102.Theruvathu JA, Jaruga P, Nath RG, Dizdaroglu M, Brooks PJ. Polyamines stimulate the formation of mutagenic 1,N2-propanodeoxyguanosine adducts from acetaldehyde. Nucleic Acids Res. 2005;33:3513–20. doi: 10.1093/nar/gki661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Marietta C, Thompson LH, Lamerdin JE, Brooks PJ. Acetaldehyde stimulates FANCD2 monoubiquitination, H2AX phosphorylation, and BRCA1 phosphorylation in human cells in vitro: implications for alcohol-related carcinogenesis. Mutat Res. 2009;664(1–2):77–83. doi: 10.1016/j.mrfmmm.2009.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Rulten SL, Hodder E, Ripley TL, Stephens DN, Mayne LV. Alcohol induces DNA damage and the Fanconi anemia D2 protein implicating FANCD2 in the DNA damage response pathways in brain. Alcohol Clin Exp Res. 2008;32:1186–96. doi: 10.1111/j.1530-0277.2008.00673.x. [DOI] [PubMed] [Google Scholar]
- 105.Backs J, Olson EN. Control of cardiac growth by histone acetylation/deacetylation. Circ Res. 2006;98:15–24. doi: 10.1161/01.RES.0000197782.21444.8f. [DOI] [PubMed] [Google Scholar]
- 106.Gong Z, Wezeman FH. Inhibitory effect of alcohol on osteogenic differentiation in human bone marrow-derived mesenchymal stem cells. Alcohol Clin Exp Res. 2004;28:468–79. doi: 10.1097/01.alc.0000118315.58404.c1. [DOI] [PubMed] [Google Scholar]
- 107.Karamboulas C, Swedani A, Ward C, Al-Madhoun AS, Wilton S, Boisvenue S, Ridgeway AG, Skerjanc IS. HDAC activity regulates entry of mesoderm cells into the cardiac muscle lineage. J Cell Sci. 2006;119(Pt 20):4305–14. doi: 10.1242/jcs.03185. [DOI] [PubMed] [Google Scholar]
- 108.Kim JS, Shukla SD. Histone H3 modifications in rat hepatic stellate cells by ethanol. Alcohol Alcohol. 2005;40:367–72. doi: 10.1093/alcalc/agh170. [DOI] [PubMed] [Google Scholar]
- 109.Park PH, Lim RW, Shukla SD. Involvement of histone acetyltransferase (HAT) in ethanol-induced acetylation of histone H3 in hepatocytes: potential mechanism for gene expression. Am J Physiol Gastrointest Liver Physiol. 2005;289:G1124–36. doi: 10.1152/ajpgi.00091.2005. [DOI] [PubMed] [Google Scholar]
- 110.1Strasak L, Bartova E, Harnicarova A, Galiova G, Krejci J, Kozubek S. H3K9 acetylation and radial chromatin positioning. J Cell Physiol. 2009;220:91–101. doi: 10.1002/jcp.21734. [DOI] [PubMed] [Google Scholar]
- 111.Lee JH, Hart SR, Skalnik DG. Histone deacetylase activity is required for embryonic stem cell differentiation. Genesis. 2004;38:32–8. doi: 10.1002/gene.10250. [DOI] [PubMed] [Google Scholar]
- 112.Zhong L, Zhu J, Lv T, Chen G, Sun H, Yang X, Huang X, Tian J. Ethanol and its metabolites induce histone lysine 9 acetylation and an alteration of the expression of heart development-related genes in cardiac progenitor cells. Cardiovasc Toxicol. 2010;10:268–74. doi: 10.1007/s12012-010-9081-z. [DOI] [PubMed] [Google Scholar]
- 113.Haycock PC. Fetal alcohol spectrum disorders: the epigenetic perspective. Biol Reprod. 2009;81:607–17. doi: 10.1095/biolreprod.108.074690. [DOI] [PubMed] [Google Scholar]
- 114.Merk O, Speit G. Significance of formaldehyde-induced DNA-protein crosslinks for mutagenesis. Environ Mol Mutagen. 1998;32:260–8. doi: 10.1002/(sici)1098-2280(1998)32:3<260::aid-em9>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 115.Lu K, Boysen G, Gao L, Collins LB, Swenberg JA. Formaldehyde-induced histone modifications in vitro. Chem Res Toxicol. 2008;21:1586–93. doi: 10.1021/tx8000576. [DOI] [PubMed] [Google Scholar]
- 116.Snyder R. Leukemia and benzene. Int J Environ Res Public Health. 2012;9:2875–93. doi: 10.3390/ijerph9082875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Wang L, He X, Bi Y, Ma Q. Stem cell and benzene-induced malignancy and hematotoxicity. Chem Res Toxicol. 2012;25:1303–15. doi: 10.1021/tx3001169. [DOI] [PubMed] [Google Scholar]
- 118.Lindsey RH, Jr, Bromberg KD, Felix CA, Osheroff N. 1,4-Benzoquinone is a topoisomerase II poison. Biochemistry. 2004;43:7563–74. doi: 10.1021/bi049756r. [DOI] [PubMed] [Google Scholar]
- 119.Hiraku Y, Kawanishi S. Oxidative DNA damage and apoptosis induced by benzene metabolites. Cancer Res. 1996;56:5172–8. [PubMed] [Google Scholar]
- 120.Winn LM. Homologous recombination initiated by benzene metabolites: a potential role of oxidative stress. Toxicol Sci. 2003;72:143–9. doi: 10.1093/toxsci/kfg008. [DOI] [PubMed] [Google Scholar]
- 121.Keenan JJ, Gaffney S, Gross SA, Ronk CJ, Paustenbach DJ, Galbraith D, Kerger BD. An evidence-based analysis of epidemiologic associations between lymphatic and hematopoietic cancers and occupational exposure to gasoline. Hum Exp Toxicol. 2013;32:1007–27. doi: 10.1177/0960327113476909. [DOI] [PubMed] [Google Scholar]
- 122.Fustinoni S, Rossella F, Polledri E, Bollati V, Campo L, Byun HM, Agnello L, Consonni D, Pesatori AC, Baccarelli A, Bertazzi PA. Global DNA methylation and low-level exposure to benzene. Med Lav. 2012;103:84–95. [PubMed] [Google Scholar]
- 123.Chen WD, Fu X, Dong B, Wang YD, Shiah S, Moore DD, Huang W. Neonatal activation of the nuclear receptor CAR results in epigenetic memory and permanent change of drug metabolism in mouse liver. Hepatology. 2012;56:1499–509. doi: 10.1002/hep.25766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Chen CY, Lin TH. Nickel toxicity to human term placenta: in vitro study on lipid peroxidation. J Toxicol Environ Health A. 1998;54:37–47. doi: 10.1080/009841098159015. [DOI] [PubMed] [Google Scholar]
- 125.Iyengar GV, Rapp A. Human placenta as a “dual” biomarker for monitoring fetal and maternal environment with special reference to potentially toxic trace elements. Part 3: toxic trace elements in placenta and placenta as a biomarker for these elements. Sci Total Environ. 2001;280(1–3):221–38. doi: 10.1016/s0048-9697(01)00827-0. [DOI] [PubMed] [Google Scholar]
- 126.Iyengar GV, Rapp A. Human placenta as a “dual” biomarker for monitoring fetal and maternal environment with special reference to potentially toxic trace elements. Part 2: essential minor, trace and other (non-essential) elements in human placenta. Sci Total Environ. 2001;280(1–3):207–19. doi: 10.1016/s0048-9697(01)00826-9. [DOI] [PubMed] [Google Scholar]
- 127.Iyengar GV, Rapp A. Human placenta as a “dual” biomarker for monitoring fetal and maternal environment with special reference to potentially toxic trace elements. Part 1: physiology, function and sampling of placenta for elemental characterisation. Sci Total Environ. 2001;280(1–3):195–206. doi: 10.1016/s0048-9697(01)00825-7. [DOI] [PubMed] [Google Scholar]
- 128.Osman K, Akesson A, Berglund M, Bremme K, Schutz A, Ask K, Vahter M. Toxic and essential elements in placentas of Swedish women. Clin Biochem. 2000;33:131–8. doi: 10.1016/s0009-9120(00)00052-7. [DOI] [PubMed] [Google Scholar]
- 129.Vargas Zapata CL, Trugo NM, Donangelo CM. Zinc uptake by human placental microvillous membrane vesicles: effects of gestational age and maternal serum zinc levels. Biol Trace Elem Res. 2000;73:127–37. doi: 10.1385/bter:73:2:127. [DOI] [PubMed] [Google Scholar]
- 130.Serafim A, Company R, Lopes B, Rosa J, Cavaco A, Castela G, Gastela E, Olea N, Bebianno MJ. Assessment of essential and nonessential metals and different metal exposure biomarkers in the human placenta in a population from the south of Portugal. J Toxicol Environ Health A. 2012;75(13–15):867–77. doi: 10.1080/15287394.2012.690704. [DOI] [PubMed] [Google Scholar]
- 131.Markowski VP, Currie D, Reeve EA, Thompson D, Wise JP., Sr Tissue-specific and dose-related accumulation of arsenic in mouse offspring following maternal consumption of arsenic-contaminated water. Basic Clin Pharmacol Toxicol. 2011;108:326–32. doi: 10.1111/j.1742-7843.2010.00660.x. [DOI] [PubMed] [Google Scholar]
- 132.Martinez-Zamudio R, Ha HC. Environmental epigenetics in metal exposure. Epigenetics. 2011;6:820–7. doi: 10.4161/epi.6.7.16250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Jackson JP, Johnson L, Jasencakova Z, Zhang X, Perez- Burgos L, Singh PB, Cheng X, Schubert I, Jenuwein T, Jacobsen SE. Dimethylation of histone H3 lysine 9 is a critical mark for DNA methylation and gene silencing in Arabidopsis thaliana. Chromosoma. 2004;112:308–15. doi: 10.1007/s00412-004-0275-7. [DOI] [PubMed] [Google Scholar]
- 134.Mazzatti DJ, Uciechowski P, Hebel S, Engelhardt G, White AJ, Powell JR, Rink L, Haase H. Effects of long-term zinc supplementation and deprivation on gene expression in human THP-1 mononuclear cells. J Trace Elem Med Biol. 2008;22:325–36. doi: 10.1016/j.jtemb.2008.06.002. [DOI] [PubMed] [Google Scholar]
- 135.Gadhia SR, Calabro AR, Barile FA. Trace metals alter DNA repair and histone modification pathways concurrently in mouse embryonic stem cells. Toxicol Lett. 2012;212:169–79. doi: 10.1016/j.toxlet.2012.05.013. [DOI] [PubMed] [Google Scholar]
- 136.Chen H, Ke Q, Kluz T, Yan Y, Costa M. Nickel ions increase histone H3 lysine 9 dimethylation and induce transgene silencing. Mol Cell Biol. 2006;26:3728–37. doi: 10.1128/MCB.26.10.3728-3737.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Arita A, Shamy MY, Chervona Y, Clancy HA, Sun H, Hall MN, Qu Q, Gamble MV, Costa M. The effect of exposure to carcinogenic metals on histone tail modifications and gene expression in human subjects. J Trace Elem Med Biol. 2012;26(2–3):174–8. doi: 10.1016/j.jtemb.2012.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Kachinskas DJ, Qin Q, Phillips MA, Rice RH. Arsenate suppression of human keratinocyte programming. Mutat Res. 1997;386:253–61. doi: 10.1016/s1383-5742(97)00015-x. [DOI] [PubMed] [Google Scholar]
- 139.Sinitsyna NN, Reznikova TV, Qin Q, Song H, Phillips MA, Rice RH. Arsenite suppression of involucrin transcription through AP1 promoter sites in cultured human keratinocytes. Toxicol Appl Pharmacol. 2010;243:275–82. doi: 10.1016/j.taap.2009.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Tokar EJ, Diwan BA, Waalkes MP. Arsenic exposure transforms human epithelial stem/progenitor cells into a cancer stem-like phenotype. Environ Health Perspect. 2010;118:108–15. doi: 10.1289/ehp.0901059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Tokar EJ, Qu W, Waalkes MP. Arsenic, stem cells, and the developmental basis of adult cancer. Toxicol Sci. 2011;120(Suppl 1):S192–203. doi: 10.1093/toxsci/kfq342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Cronican AA, Fitz NF, Carter A, Saleem M, Shiva S, Barchowsky A, Koldamova R, Schug J, Lefterov I. Genome- wide alteration of histone H3K9 acetylation pattern in mouse offspring prenatally exposed to arsenic. PLoS One. 2013;8(2):e53478. doi: 10.1371/journal.pone.0053478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Basha MR, Wei W, Bakheet SA, Benitez N, Siddiqi HK, Ge YW, Lahiri DK, Zawia NH. The fetal basis of amyloidogenesis: exposure to lead and latent overexpression of amyloid precursor protein and beta-amyloid in the aging brain. J Neurosci. 2005;25:823–9. doi: 10.1523/JNEUROSCI.4335-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Barker DJ. Fetal programming of coronary heart disease. Trends Endocrinol Metab. 2002;13:364–8. doi: 10.1016/s1043-2760(02)00689-6. [DOI] [PubMed] [Google Scholar]
- 145.Barker DJ, Winter PD, Osmond C, Margetts B, Simmonds SJ. Weight in infancy and death from ischaemic heart disease. Lancet. 1989;2:577–80. doi: 10.1016/s0140-6736(89)90710-1. [DOI] [PubMed] [Google Scholar]
- 146.Ozanne SE. Metabolic programming in animals. Br Med Bull. 2001;60:143–52. doi: 10.1093/bmb/60.1.143. [DOI] [PubMed] [Google Scholar]
- 147.Valdez R, Athens MA, Thompson GH, Bradshaw BS, Stern MP. Birthweight and adult health outcomes in a biethnic population in the USA. Diabetologia. 1994;37:624–31. doi: 10.1007/BF00403383. [DOI] [PubMed] [Google Scholar]
- 148.Yarbrough DE, Barrett-Connor E, Kritz-Silverstein D, Wingard DL. Birth weight, adult weight, and girth as predictors of the metabolic syndrome in postmenopausal women: the Rancho Bernardo Study. Diabetes Care. 1998;21:1652–8. doi: 10.2337/diacare.21.10.1652. [DOI] [PubMed] [Google Scholar]
- 149.Doll R, Morgan LG, Speizer FE. Cancers of the lung and nasal sinuses in nickel workers. Br J Cancer. 1970;24:623–32. doi: 10.1038/bjc.1970.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Gibb HJ, Lees PS, Pinsky PF, Rooney BC. Lung cancer among workers in chromium chemical production. Am J Ind Med. 2000;38:115–26. doi: 10.1002/1097-0274(200008)38:2<115::aid-ajim1>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 151.Smith AH, Hopenhayn-Rich C, Bates MN, Goeden HM, Hertz-Picciotto I, Duggan HM, Wood R, Kosnett MJ, Smith MT. Cancer risks from arsenic in drinking water. Environ Health Perspect. 1992;97:259–67. doi: 10.1289/ehp.9297259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Yuan Y, Marshall G, Ferreccio C, Steinmaus C, Selvin S, Liaw J, Bates MN, Smith AH. Acute myocardial infarction mortality in comparison with lung and bladder cancer mortality in arsenic-exposed region II of Chile from 1950 to 2000. Am J Epidemiol. 2007;166:1381–91. doi: 10.1093/aje/kwm238. [DOI] [PubMed] [Google Scholar]
- 153.Chanda S, Dasgupta UB, Guhamazumder D, Gupta M, Chaudhuri U, Lahiri S, Das S, Ghosh N, Chatterjee D. DNA hypermethylation of promoter of gene p53 and p16 in arsenic-exposed people with and without malignancy. Toxicol Sci. 2006;89:431–7. doi: 10.1093/toxsci/kfj030. [DOI] [PubMed] [Google Scholar]
- 154.Chen WT, Hung WC, Kang WY, Huang YC, Chai CY. Urothelial carcinomas arising in arsenic-contaminated areas are associated with hypermethylation of the gene promoter of the death-associated protein kinase. Histopathology. 2007;51:785–92. doi: 10.1111/j.1365-2559.2007.02871.x. [DOI] [PubMed] [Google Scholar]
- 155.Liu J, Benbrahim-Tallaa L, Qian X, Yu L, Xie Y, Boos J, Qu W, Waalkes MP. Further studies on aberrant gene expression associated with arsenic-induced malignant transformation in rat liver TRL1215 cells. Toxicol Appl Pharmacol. 2006;216:407–15. doi: 10.1016/j.taap.2006.06.006. [DOI] [PubMed] [Google Scholar]
- 156.Marsit CJ, Karagas MR, Danaee H, Liu M, Andrew A, Schned A, Nelson HH, Kelsey KT. Carcinogen exposure and gene promoter hypermethylation in bladder cancer. Carcinogenesis. 2006;27:112–6. doi: 10.1093/carcin/bgi172. [DOI] [PubMed] [Google Scholar]
- 157.Marsit CJ, Karagas MR, Schned A, Kelsey KT. Carcinogen exposure and epigenetic silencing in bladder cancer. Ann N Y Acad Sci. 2006;1076:810–21. doi: 10.1196/annals.1371.031. [DOI] [PubMed] [Google Scholar]
- 158.Kim HG, Kim DJ, Li S, Lee KY, Li X, Bode AM, Dong Z. Polycomb (PcG) proteins, BMI1 and SUZ12, regulate arsenic-induced cell transformation. J Biol Chem. 2012;287:31920–8. doi: 10.1074/jbc.M112.360362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Vandenberg LN, Hauser R, Marcus M, Olea N, Welshons WV. Human exposure to bisphenol A (BPA) Reprod Toxicol. 2007;24:139–77. doi: 10.1016/j.reprotox.2007.07.010. [DOI] [PubMed] [Google Scholar]
- 160.Balakrishnan B, Henare K, Thorstensen EB, Ponnampalam AP, Mitchell MD. Transfer of bisphenol A across the human placenta. Am J Obstet Gynecol. 2010;202:393, e1–7. doi: 10.1016/j.ajog.2010.01.025. [DOI] [PubMed] [Google Scholar]
- 161.He Y, Miao M, Herrinton LJ, Wu C, Yuan W, Zhou Z, Li DK. Bisphenol A levels in blood and urine in a Chinese population and the personal factors affecting the levels. Environ Res. 2009;109:629–33. doi: 10.1016/j.envres.2009.04.003. [DOI] [PubMed] [Google Scholar]
- 162.Mørck TJ, Sorda G, Bechi N, Rasmussen BS, Nielsen JB, Ietta F, Rytting E, Mathiesen L, Paulesu L, Knudsen LE. Placental transport and in vitro effects of Bisphenol A. Reprod Toxicol. 2010;30:131–7. doi: 10.1016/j.reprotox.2010.02.007. [DOI] [PubMed] [Google Scholar]
- 163.Setchell KD, Zimmer-Nechemias L, Cai J, Heubi JE. Exposure of infants to phyto-oestrogens from soy-based infant formula. Lancet. 1997;350:23–7. doi: 10.1016/S0140-6736(96)09480-9. [DOI] [PubMed] [Google Scholar]
- 164.Whitten PL, Patisaul HB. Cross-species and interassay comparisons of phytoestrogen action. Environ Health Perspect. 2001;109(Suppl 1):5–20. doi: 10.1289/ehp.01109s15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Williamson-Hughes PS, Flickinger BD, Messina MJ, Empie MW. Isoflavone supplements containing predominantly genistein reduce hot flash symptoms: a critical review of published studies. Menopause. 2006;13:831–9. doi: 10.1097/01.gme.0000227330.49081.9e. [DOI] [PubMed] [Google Scholar]
- 166.Balls M, Hellsten E. Statement of the scientific validity of the embryonic stem cell test (EST)–an in vitro test for embryotoxicity. Altern Lab Anim. 2002;30:265–8. [PubMed] [Google Scholar]
- 167.Pennings JL, van Dartel DA, Robinson JF, Pronk TE, Piersma AH. Gene set assembly for quantitative prediction of developmental toxicity in the embryonic stem cell test. Toxicology. 2011;284(1–3):63–71. doi: 10.1016/j.tox.2011.03.017. [DOI] [PubMed] [Google Scholar]
- 168.Kong D, Xing L, Liu R, Jiang J, Wang W, Shang L, Wei X, Hao W. Individual and combined developmental toxicity assessment of bisphenol A and genistein using the embryonic stem cell test in vitro. Food Chem Toxicol. 2013;60:497–505. doi: 10.1016/j.fct.2013.08.006. [DOI] [PubMed] [Google Scholar]
- 169.Sato N, Yamakawa N, Masuda M, Sudo K, Hatada I, Muramatsu M. Genome-wide DNA methylation analysis reveals phytoestrogen modification of promoter methylation patterns during embryonic stem cell differentiation. PLoS One. 2011;6(4):e19278. doi: 10.1371/journal.pone.0019278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Delclos KB, Bucci TJ, Lomax LG, Latendresse JR, Warbritton A, Weis CC, Newbold RR. Effects of dietary genistein exposure during development on male and female CD (Sprague-Dawley) rats. Reprod Toxicol. 2001;15:647–63. doi: 10.1016/s0890-6238(01)00177-0. [DOI] [PubMed] [Google Scholar]
- 171.Newbold RR, Banks EP, Bullock B, Jefferson WN. Uterine adenocarcinoma in mice treated neonatally with genistein. Cancer Res. 2001;61:4325–8. [PubMed] [Google Scholar]
- 172.Palmer JR, Wise LA, Hatch EE, Troisi R, Titus-Ernstoff L, Strohsnitter W, Kaufman R, Herbst AL, Noller KL, Hyer M, Hoover RN. Prenatal diethylstilbestrol exposure and risk of breast cancer. Cancer Epidemiol Biomarkers Prev. 2006;15:1509–14. doi: 10.1158/1055-9965.EPI-06-0109. [DOI] [PubMed] [Google Scholar]
- 173.Aoki T, Takada T. Bisphenol A modulates germ cell differentiation and retinoic acid signaling in mouse ES cells. Reprod Toxicol. 2012;34:463–70. doi: 10.1016/j.reprotox.2012.06.001. [DOI] [PubMed] [Google Scholar]
- 174.Komada M, Asai Y, Morii M, Matsuki M, Sato M, Nagao T. Maternal bisphenol A oral dosing relates to the acceleration of neurogenesis in the developing neocortex of mouse fetuses. Toxicology. 2012;295(1–3):31–8. doi: 10.1016/j.tox.2012.02.013. [DOI] [PubMed] [Google Scholar]
- 175.Nakamura K, Itoh K, Yaoi T, Fujiwara Y, Sugimoto T, Fushiki S. Murine neocortical histogenesis is perturbed by prenatal exposure to low doses of Bisphenol A. J Neurosci Res. 2006;84:1197–205. doi: 10.1002/jnr.21020. [DOI] [PubMed] [Google Scholar]
- 176.Linehan C, Gupta S, Samali A, O’Connor L. Bisphenol A-mediated suppression of LPL gene expression inhibits triglyceride accumulation during adipogenic differentiation of human adult stem cells. PLoS One. 2012;7(5):e36109. doi: 10.1371/journal.pone.0036109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Biemann R, Navarrete Santos A, Navarrete Santos A, Riemann D, Knelangen J, Blüher M, Koch H, Fischer B. Endocrine disrupting chemicals affect the adipogenic differentiation of mesenchymal stem cells in distinct ontogenetic windows. Biochem Biophys Res Commun. 2012;417:747–52. doi: 10.1016/j.bbrc.2011.12.028. [DOI] [PubMed] [Google Scholar]
- 178.Ho SM, Tang WY, Belmonte de Frausto J, Prins GS. Developmental exposure to estradiol and bisphenol A increases susceptibility to prostate carcinogenesis and epigenetically regulates phosphodiesterase type 4 variant 4. Cancer Res. 2006;66:5624–32. doi: 10.1158/0008-5472.CAN-06-0516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Urcan E, Scherthan H, Styllou M, Haertel U, Hickel R, Reichl FX. Induction of DNA double-strand breaks in primary gingival fibroblasts by exposure to dental resin composites. Biomaterials. 2010;31:2010–4. doi: 10.1016/j.biomaterials.2009.11.065. [DOI] [PubMed] [Google Scholar]
- 180.Blasiak J, Synowiec E, Tarnawska J, Czarny P, Poplawski T, Reiter RJ. Dental methacrylates may exert genotoxic effects via the oxidative induction of DNA double strand breaks and the inhibition of their repair. Mol Biol Rep. 2012;39:7487–96. doi: 10.1007/s11033-012-1582-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Szczepanska J, Pawlowska E, Synowiec E, Czarny P, Rekas M, Blasiak J, Szaflik JP. Protective effect of chitosan oligosaccharide lactate against DNA double-strand breaks induced by a model methacrylate dental adhesive. Med Sci Monit. 2011;17:BR201–8. doi: 10.12659/MSM.881898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Audebert M, Dolo L, Perdu E, Cravedi JP, Zalko D. Use of the gammaH2AX assay for assessing the genotoxicity of bisphenol A and bisphenol F in human cell lines. Arch Toxicol. 2011;85:1463–73. doi: 10.1007/s00204-011-0721-2. [DOI] [PubMed] [Google Scholar]
- 183.Ibuki Y, Tani Y, Toyooka T. UVB-exposed chlorinated bisphenol A generates phosphorylated histone H2AX in human skin cells. Chem Res Toxicol. 2008;21:1770–6. doi: 10.1021/tx800129n. [DOI] [PubMed] [Google Scholar]
- 184.Vanhees K, de Bock L, Godschalk RW, van Schooten FJ, van Waalwijk van Doorn-Khosrovani SB. Prenatal exposure to flavonoids: implication for cancer risk. Toxicol Sci. 2011;120:59–67. doi: 10.1093/toxsci/kfq388. [DOI] [PubMed] [Google Scholar]
- 185.Wang JC. Cellular roles of DNA topoisomerases: a molecular perspective. Nat Rev Mol Cell Biol. 2002;3:430–40. doi: 10.1038/nrm831. [DOI] [PubMed] [Google Scholar]
- 186.Bandele OJ, Osheroff N. Bioflavonoids as poisons of human topoisomerase II alpha and II beta. Biochemistry. 2007;46:6097–108. doi: 10.1021/bi7000664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Pommier Y, Leo E, Zhang H, Marchand C. DNA topoisomerases and their poisoning by anticancer and antibacterial drugs. Chem Biol. 2010;17:421–33. doi: 10.1016/j.chembiol.2010.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Baldwin EL, Osheroff N. Etoposide, topoisomerase II and cancer. Curr Med Chem Anticancer Agents. 2005;5:363–72. doi: 10.2174/1568011054222364. [DOI] [PubMed] [Google Scholar]
- 189.Bender RP, Jablonksy MJ, Shadid M, Romaine I, Dunlap N, Anklin C, Graves DE, Osheroff N. Substituents on etoposide that interact with human topoisomerase IIalpha in the binary enzyme-drug complex: contributions to etoposide binding and activity. Biochemistry. 2008;47:4501–9. doi: 10.1021/bi702019z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Wilstermann AM, Bender RP, Godfrey M, Choi S, Anklin C, Berkowitz DB, Osheroff N, Graves DE. Topoisomerase II - drug interaction domains: identification of substituents on etoposide that interact with the enzyme. Biochemistry. 2007;46:8217–25. doi: 10.1021/bi700272u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Blanco JG, Edick MJ, Relling MV. Etoposide induces chimeric Mll gene fusions. FASEB J. 2004;18:173–5. doi: 10.1096/fj.03-0638fje. [DOI] [PubMed] [Google Scholar]
- 192.Libura J, Ward M, Solecka J, Richardson C. Etoposide-initiated MLL rearrangements detected at high frequency in human primitive hematopoietic stem cells with in vitro and in vivo long-term repopulating potential. Eur J Haematol. 2008;81:185–95. doi: 10.1111/j.1600-0609.2008.01103.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Sung PA, Libura J, Richardson C. Etoposide and illegitimate DNA double-strand break repair in the generation of MLL translocations: new insights and new questions. DNA Repair (Amst) 2006;5(9–10):1109–18. doi: 10.1016/j.dnarep.2006.05.018. [DOI] [PubMed] [Google Scholar]
- 194.Moneypenny CG, Shao J, Song Y, Gallagher EP. MLL rearrangements are induced by low doses of etoposide in human fetal hematopoietic stem cells. Carcinogenesis. 2006;27:874–81. doi: 10.1093/carcin/bgi322. [DOI] [PubMed] [Google Scholar]
- 195.Libura J, Slater DJ, Felix CA, Richardson C. Therapy-related acute myeloid leukemia-like MLL rearrangements are induced by etoposide in primary human CD34+ cells and remain stable after clonal expansion. Blood. 2005;105:2124–31. doi: 10.1182/blood-2004-07-2683. [DOI] [PubMed] [Google Scholar]
- 196.Felix CA. Leukemias related to treatment with DNA topoisomerase II inhibitors. Med Pediatr Oncol. 2001;36:525–35. doi: 10.1002/mpo.1125. [DOI] [PubMed] [Google Scholar]
- 197.Elsea SH, McGuirk PR, Gootz TD, Moynihan M, Osheroff N. Drug features that contribute to the activity of quinolones against mammalian topoisomerase II and cultured cells: correlation between enhancement of enzyme-mediated DNA cleavage in vitro and cytotoxic potential. Antimicrob Agents Chemother. 1993;37:2179–86. doi: 10.1128/aac.37.10.2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Long BH, Musial ST, Brattain MG. Comparison of cytotoxicity and DNA breakage activity of congeners of podophyllotoxin including VP16-213 and VM26: a quantitative structure-activity relationship. Biochemistry. 1984;23:1183–8. doi: 10.1021/bi00301a024. [DOI] [PubMed] [Google Scholar]
- 199.Azarova AM, Lin RK, Tsai YC, Liu LF, Lin CP, Lyu YL. Genistein induces topoisomerase IIbeta- and proteasome- mediated DNA sequence rearrangements: Implications in infant leukemia. Biochem Biophys Res Commun. 2010;399:66–71. doi: 10.1016/j.bbrc.2010.07.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Abe T. Infantile leukemia and soybeans–a hypothesis. Leukemia. 1999;13:317–20. doi: 10.1038/sj.leu.2401344. [DOI] [PubMed] [Google Scholar]
- 201.Strick R, Strissel PL, Borgers S, Smith SL, Rowley JD. Dietary bioflavonoids induce cleavage in the MLL gene and may contribute to infant leukemia. Proc Natl Acad Sci U S A. 2000;97:4790–5. doi: 10.1073/pnas.070061297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Barjesteh van Waalwijk van Doorn-Khosrovani S, Janssen J, Maas LM, Godschalk RW, Nijhuis JG, van Schooten FJ. Dietary flavonoids induce MLL translocations in primary human CD34+ cells. Carcinogenesis. 2007;28:1703–9. doi: 10.1093/carcin/bgm102. [DOI] [PubMed] [Google Scholar]
- 203.Schroder-van der Elst JP, van der Heide D, Rokos H, Morreale de Escobar G, Kohrle J. Synthetic flavonoids cross the placenta in the rat and are found in fetal brain. Am J Physiol. 1998;274(2 Pt 1):E253–6. doi: 10.1152/ajpendo.1998.274.2.E253. [DOI] [PubMed] [Google Scholar]
- 204.Vanhees K, Coort S, Ruijters EJ, Godschalk RW, van Schooten FJ, Barjesteh van Waalwijk van Doorn-Khosrovani S. Epigenetics: prenatal exposure to genistein leaves a permanent signature on the hematopoietic lineage. FASEB J. 2011;25:797–807. doi: 10.1096/fj.10-172155. [DOI] [PubMed] [Google Scholar]
- 205.Vanhees K, Godschalk RW, Sanders A, van Waalwijk van Doorn-Khosrovani SB, van Schooten FJ. Maternal quercetin intake during pregnancy results in an adapted iron homeostasis at adulthood. Toxicology. 2011;290(2–3):350–8. doi: 10.1016/j.tox.2011.10.017. [DOI] [PubMed] [Google Scholar]
- 206.Vanhees K, van Schooten FJ, Moonen EJ, Maas LM, van Waalwijk van Doorn-Khosrovani SB, Godschalk RW. Maternal intake of quercetin during gestation alters ex vivo benzo[a]pyrene metabolism and DNA adduct formation in adult offspring. Mutagenesis. 2012;27:445–51. doi: 10.1093/mutage/ges002. [DOI] [PubMed] [Google Scholar]
- 207.Dawson MA, Kouzarides T. Cancer epigenetics: from mechanism to therapy. Cell. 2012;150:12–27. doi: 10.1016/j.cell.2012.06.013. [DOI] [PubMed] [Google Scholar]