

Abstract
MATERIALS AND METHODS: We examined pretreatment magnetic resonance imaging (MRI) examinations from 32 patients with glioblastoma multiforme (GBM) enrolled in The Cancer Genome Atlas (TCGA). Spatial variations in T1 post-gadolinium and either T2-weighted or fluid attenuated inversion recovery sequences from each tumor MRI study were used to characterize each small region of the tumor by its local contrast enhancement and edema/cellularity (“habitat”). The patient cohort was divided into group 1 (survival < 400 days, n = 16) and group 2 (survival > 400 days, n = 16). RESULTS: Histograms of relative values in each sequence demonstrated that the tumor regions were consistently divided into high and low blood contrast enhancement, each of which could be subdivided into regions of high, low, and intermediate cell density/interstitial edema. Group 1 tumors contained greater volumes of habitats with low contrast enhancement but intermediate and high cell density (not fully necrotic) than group 2. Both leave-one-out and 10-fold cross-validation schemes demonstrated that individual patients could be correctly assigned to the short or long survival group with 81.25% accuracy. CONCLUSION: We demonstrate that novel image analytic techniques can characterize regional habitat variations in GBMs using combinations of MRI sequences. A preliminary study of 32 patients from the TCGA database found that the distribution of MRI-defined habitats varied significantly among the different survival groups. Radiologically defined ecological tumor analysis may provide valuable prognostic and predictive biomarkers in GBM and other tumors.
Introduction
Intratumoral and intertumoral heterogeneities are well recognized at molecular, cellular, and tissue scales [1–6]. This is clearly evident in the imaging characteristics of glioblastoma multiforme, which typically include regions of high and low contrast enhancement as well as high and low interstitial edema and cell density. Several recent molecular studies have clearly demonstrated that there is also significant genetic variation among cells in different tumors and even in different regions of the same tumor [3–5]. In one study, for example, samples from spatially separated sites within glioblastoma multiforme (GBM) tumors found that multiple molecular subtypes were present in all of the examined tumors [3]. It is clear that this molecular heterogeneity may significantly limit efforts to personalize cancer treatment based on the use of molecular profiling to identify druggable targets [7–9]. However, there has, thus far, been little effort to relate the spatial heterogeneity observed in clinical imaging with the genetic heterogeneity found in molecular studies.
Temporal and spatial cellular heterogeneities are typically ascribed to clonal evolution generated by accumulating random mutations in cancer cell populations [3,10,11]. However, Darwinian dynamics are ultimately governed by the interactions of local environmental selection forces with cell phenotypes (not genotypes) [12,13]. That is, while mutations may occur randomly, proliferation of that clone will proceed only if its corresponding phenotype is more fit than extant populations within the context of the local adaptive landscape. Because of this evolutionary triage of each heritable (i.e., genetic or epigenetic) event, we have proposed that intratumoral evolution is fundamentally linked to the regional variations in microenvironmental selection forces that ultimately determine the fitness of any genotype/phenotype [12–14]. We hypothesize that the Darwinian dynamics that link genetic changes with environmental conditions will permit the characterization of regional variations in the molecular properties of cancer cells with environmental conditions (such as blood flow, edema, and cell density) that can be determined with clinical imaging [15].
Spatial heterogeneity in tumor characteristics is well recognized in cross-sectional clinical imaging (Figure 1). Many tumors exhibit significant regional differences in contrast enhancement along with variations in cellular density, water content, fibrosis, and necrosis. In clinical practice, this heterogeneity is typically described in non-quantitative terms. More recently, metrics [16,17] of heterogeneity, such as Shannon entropy, have been developed and can be correlated with tumor molecular features [18–21] and clinical outcomes [22–24]. However, metrics that assign a single value to heterogeneity tacitly assume that the tumor is “well mixed” and thus does not capture spatial distributions of specific tumor properties.
Figure 1.
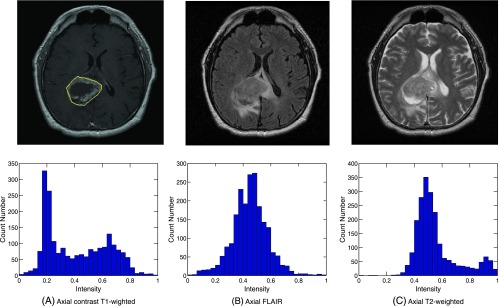
An example of an analysis from a single axial plane MRI image from a patient with GBM. For the first row: (A) Yellow boundary outlines the tumor region including enhancement and non-enhancement. (B, C) The corresponding axial plane FLAIR and T2 scans, respectively. The second row illustrates the associated pixel histogram distribution (tumor region).
Here, our general goal is to develop a spatially explicit approach that identifies and quantifies specific subregions of the tumor based on clinical imaging metrics that may provide information about the underlying evolutionary dynamics. In this approach, we hypothesize that tumors will generally possess subregions with variable Darwinian dynamics, including environmental selection forces and phenotypic adaptation to those forces [15]. Our approach here generates radio-logically defined “habitats” by spatially superimposing two different magnetic resonance imaging (MRI) sequences from the same tumor. Our goal in this initial work is to examine regional variations in perfusion/extravasation based on T1 post-gadolinium images and interstitial edema/cell density determined by fluid attenuated inversion recovery (FLAIR) and T2 images. Clearly, a full characterization of Darwinian dynamics in intratumor habitats will require more extensive imaging probably from multiple modalities [e.g., positron emission tomography (PET), MRI, and computed tomography (CT)] or other MRI sequences (particularly apparent diffusion coefficient maps). Nevertheless, in this preliminary study, we ask the following questions: 1) Can GBMs be consistently divided into some small number of specific radiologically defined habitats based on combinations of images sensitive to blood flow and edema? 2) Does the distribution of these regions vary among tumors in different survival groups?
This work builds on prior studies using regional variations in MRI appearance to correlate with genetic and histologic tumor characteristics [25–29]. Here, we explicitly apply a novel ecological/evolutionary perspective that allows clinical imaging characteristics to define regional variations in intratumoral Darwinian dynamics that govern intratumoral molecular heterogeneity. In the preliminary studies, we demonstrate that the distribution of these radiologically defined habitats can be correlated with clinical outcomes.
Materials and Methods
Patient Information
A data set of 32 patients was collected between January and December 2012 from the publicly available The Cancer Genome Atlas (TCGA; http://cancerimagingarchive.net/) database for our preliminary study. Although the database contains more than 500 cases, full imaging sets were often not available. Patients were included if they 1) had complete MRI studies that included post-contrast T1-weighted, FLAIR, and T2-weighted sequences and 2) had clinical survival time. Patients were excluded if they had multiple tumors or if the tumor was too small to analyze (<2 cm in diameter). A subset of 66 cases (43 cases with less than 400 days survival and 23 cases with more than 400 days survival) satisfied these two conditions. In our initial analysis, we selected a balanced data set (Figure 2) that included 16 cases each in group 1 (survival time below 400 days) and group 2 (survival time above 400 days), respectively. The latter 16 were arbitrarily chosen. All of the images had a 200 mm x 200 mm field of view and 5-mm slice thickness, with 256 x 256 or 512 x 512 acquisition matrices. To ensure uniform resolution of intravariation of each sequence, for each case, three channels were registered by bilinear interpolation. Since the enrolled patients were from multiple institutions, the studies were performed on a wide range of MRI units with some variations in technique.
Figure 2.

Survival time distribution demonstrating a broad scale from 16 to 1730 days.
Image Normalization
To enable consistent evaluations for all cases, the obtained MRI imaging data were processed by standardizing the intensity scales [30]. We employed the linear normalization on each volume. The voxels of each volume of the tumor region were independently normalized into the scale from 0 to 1. Thus, the normalization captured the local tumor variations of specific patients in the standard range.
Tumor Identification
For consistency, the regions of interest were segmented by manually drawn masks on the post-gadolinium T1-weighted images as shown in Figure 1. Although automated tumor segmentation methods have been described [31,32], they can be unpredictably inaccurate and appeared to offer no advantage over manual segmentation in GBMs where tumor edges are characteristically well defined in T1 post-gadolinium sequences.
Histogram Analysis
For our initial analysis, we generated two-dimensional (2D) histograms (Figures 3 and 4) of the cumulative voxel intensities for all tumors. To perform a cohort analysis, the frequencies were rescaled into a range [0,1] using the normalization described above. In the histograms, the y-axis represent the frequency with which a particular MRI sequence intensity (x-axis) was observed.
Figure 3.

Two-dimensional histogram distribution. In the responding group, the distribution of normalized values of T1 post-gadolinium, T2-weighted, and FLAIR images was plotted, respectively. Each group was shown as a normalized cumulative histogram.
Figure 4.

In the top figure, the frequency of the normalized values of all T1 post-gadolinium images was plotted. Using a Gaussian mixture model [34], the histogram was divided into two Gaussian populations with a separation point of 0.26. The normalized FLAIR signal was then plotted in the high and low groups. The result suggests that GBMs consist of five dominant habitats—two with high blood flow and high and low cell density and three with low blood flow and high, low, and intermediate cell densities.
In addition, 3D histograms (Figure 5) were used to visually observe variations in tumor heterogeneity. The x- and y-axes consisted of the available pairs of MRI modalities: post-gadolinium T1-weighted and FLAIR, post-gadolinium T1-weighted and T2-weighted, or FLAIR and T2-weighted. We used a joint histogram that considered the cross-distribution of each modality. For instance, considering the pair of post-gadolinium T1-weighted and FLAIR modalities, an aggregated histogram was formed by counting the joint intensity of each voxel (xi, xj), where xi was denoted as the T1-weighted intensity signal and xj was the associated FLAIR intensity signal. The z-axis dimension represented the joint distribution of each voxel. The remaining combinations followed a similar process to obtain the 3D histogram representation.
Figure 5.
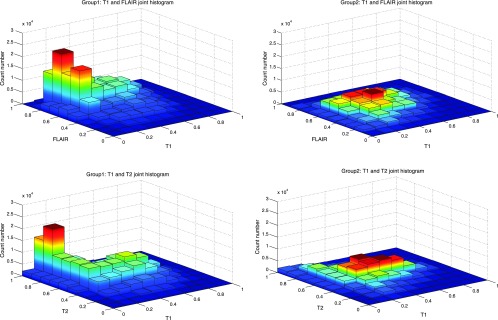
Three-dimensional histogram distribution shows the relative distribution of combinations of perfusion and cell density in the two groups of GBMs. For each group, we plotted the joint cumulative 3D histogram by summing all cases of 3D histograms of each class. The third dimension is the frequency distribution. Group 2 cases are relatively homogenous with most regions clustering in habitats of high perfusion and intermediate cell density. Group 1 cases show greater heterogeneity with more areas of decreased perfusion with mixed cell density.
Survival Time Criterion
The clinical survival time (Figure 2) was defined as the number of days between the date of the initial pathologic diagnosis and the time to death obtained from the patient demographics in the TCGA database. We used the MRI imaging data that were obtained at the initial diagnosis, thus the possible influence of the following clinical therapy was not accounted for in our study. Since there was no explicit prior study suggesting the precise survival threshold for different survival groups, we chose to approximately follow the overall statistics in the study [33], where a reported median value of survival time for malignant brain tumor was 12 to 14 months. In our study, the patient cohort was initially divided into two equal groups: group 1(survival time <400 days) and group 2 (survival time > 400 days), thus the criterion used here differs from that used in another study [34], which defined long-term survival as more than 3 years (36 months); only 2% to 5% of patients were in this group.
Results
Demographic Data
Figure 2 and Tables 1–3 summarize the clinical and molecular data. The group with survival time < 400 days were slightly older (mean age 62) and had more total mutations (n = 29) than the group with survival time > 400 days (mean age 60, total mutations = 25). Only limited information was available on molecular subtype, although we note that recent investigations have shown that multiple subtypes are characteristically observed in each tumor [4]. The mean tumor diameter and overall volume was slightly greater in group 1, but the difference was not statistically significant. All patients were treated with radiation therapy, chemotherapy, and surgery, although details were not available.
Table 1.
Data Set of Demographic Information.
| Survival < 400 Days (n = 16) | Survival > 400 Days (n = 16) | |
| Age: Range, median | 47–80, 62 | 18–78, 60 |
| Sex | 8M, 8F | 10M, 6F |
| Histology | Available in n = 9 | Available in n = 11 |
| Classical | 1/9 | 2/11 |
| Mesenchymal | 4/9 | 6/11 |
| Proneural | 3/9 | 2/11 |
| Neural | 1/9 | 1/11 |
| Mutations | ||
| CDKN2A | 8 | 11 |
| EGFR | 6 | 8 |
| PTEN | 4 | 3 |
| PDGFRA | 2 | 2 |
| TP53 | 3 | 1 |
| CDK4 | 3 | 0 |
| NF1 | 2 | 1 |
| CDK6 | 1 | 0 |
Table 3.
Distribution of Gene Mutations in Group 2.
| Tumor ID | CDKN2A | EGFR | PTEN | PDGFRA | TP53 | CDK4 | NF1 | CDK6 | Total |
| 1 | X | X | 2 | ||||||
| 2 | X | X | 2 | ||||||
| 3 | X | X | X | X | X | 5 | |||
| 4 | X | X | 2 | ||||||
| 5 | X | X | 2 | ||||||
| 6 | X | 1 | |||||||
| 7 | X | X | 2 | ||||||
| 8 | 0 | ||||||||
| 9 | 0 | ||||||||
| 10 | X | X | 2 | ||||||
| 11 | X | X | 2 | ||||||
| 12 | 0 | ||||||||
| 13 | X | X | 2 | ||||||
| 14 | X | X | 2 | ||||||
| 15 | X | 1 | |||||||
| 16 | X | 1 | |||||||
| Total | 11 | 8 | 3 | 2 | 1 | 0 | 1 | 0 | 25 |
Variations in Blood Flow and Cellular Density
Figure 3 demonstrates variation in the normalized values of T1 post-gadolinium, T2-weighted, and FLAIR images in different survival groups. In the T1 post-gadolinium images, there are two populations that are roughly Gaussian distributions around high and low means. This suggests that GBMs are generally divided into regions of high and low contrast enhancement that we view as an approximate measure of blood flow. That is, while the dynamics leading to contrast enhancement includes blood flow and vascular integrity (extravasation), we assume in our bifurcated classification that the nonenhancing regions have poorer blood flow than the enhancing regions. Group 1 demonstrates a shift in the distribution of these enhancement regions from high to low.
The T2-weighted and FLAIR distributions suggest that group 1 tumors actually contain habitats that are either not present or rare within long-term survival tumors. For both FLAIR and T2-weighted histograms, the tumor volume is dominated by a single population, probably with one other smaller population leading to some asymmetry of the Gaussian distribution. In group 1, tumor set distribution is significantly more heterogeneous with at least three distinct regions.
Initial Spatial Analysis
Since the T1 post-gadolinium images were consistently divided into two regions, we used this as a starting point for combining sequences. All of the tumors were divided spatially into high and low enhancement regions using a normalized intensity of 0.26 as the dividing point. The threshold was found by fitting a Gaussian mixture model [35,36] to a cumulative histogram of all T1 post-gadolinium images and finding where two classes intersected. After this spatial division, FLAIR values were projected onto the high and low enhancement groups. As shown in Figure 4, this resulted in clear separation of the GBM images into five distinct radiologically defined combinations of contrast enhancement and interstitial edema. In the high enhancement (i.e., high T1 post-gadolinium) regions, there is a region with low FLAIR signal indicating cell density and interstitial edema comparable to normal brain tissue. However, a second habitat with higher FLAIR signal indicates that some tumor regions with high levels of enhancement have lower cell density and higher interstitial edema than normal tissue. Similarly, in the low enhancement regions of the tumor, one subregion shows very high FLAIR signal representing necrosis. However, two additional subregions each with less water and more apparent cellularity are also present. We interpret this to indicate the presence of viable cell populations that have adapted to local environmental conditions generated by low flow (e.g., hypoxia and acidosis).
Applying Spatial Analysis to Clinical Response
The two groups were analyzed using 3D graphs that plotted the relative frequency of regions with specific combinations of T1 post-gadolinium signal and either FLAIR or T2-weighted signal. As shown in Figure 4, group 2 tumors typically consist of tumor habitats with high enhancement (i.e., >0.26) and relatively high cell density. Group 1 tumors had increased regions of low enhancement. Interestingly, while these often corresponded to high T2-weighted or FLAIR signal indicating necrosis, regions with low enhancement and relatively high cell density were frequently present.
Statistical Analysis and Clinical Survival Time Group Prediction
To test the predicative capability, a binary classification scheme (e.g., group 1 and group 2) was formulated. We used the machine learning classifier, support vector machines [38,39], to classify samples by using a Gaussian kernel function to project features into a high-dimensional space. Both leave-one-out and 10-fold cross-validation schemes were used for performance evaluation. We determined the accuracy (81.25% for leave-one-out), specificity, and sensitivity values with results summarized in Table 4. In addition, we show receiver operator curve (ROC) curves in Figure 6, and the associated area under the curve values are also given.
Table 4.
Prediction Performance.
| Cross-Validation | Accuracy | Specificity | Sensitivity | Area Under the Curve Values |
| Leave-one-out | 81.25% | 77.78% | 85.71% | 0.86 |
| 10-fold | 78.13% | 73.68% | 84.62% | 0.83 |
Figure 6.

Prediction results. Both leave-one-out and 10-fold cross-validation schemes were used to validate the prediction performance.
Spatial Mapping
To examine the spatial clustering of habitats, we divided the combined imaging data sets into four arbitrary habitats—high and low blood flow and high and low cell density. These were then projected back onto the MRI studies. In detail, the nonparametric Otsu segmentation approach [40] was used for the intratumor segmentation. Given a modality, after setting the number of groups (two groups in our study) to be segmented, the Otsu algorithm iteratively searched for an optimal decision boundary until convergence. As shown in Figure 7, habitats generally clustered into spatial groups after an intersection operation between two MRI modalities. The choice of modalities and mapping procedure can be varied according to the needs of the specific task. In short, as a tool for brain tumor heterogeneity analysis, the design of the spatial habitat concept gave rise to various opportunities for quantitative measurement (i.e., using these habitats to quantitatively observe tumor evolution progress). The detailed spatial relationships of the different groups will be the subject of future investigations.
Figure 7.

The block diagram of spatial mapping. For input brain tumor data (e.g., T1-weighted and FLAIR), two intratumor habitats were obtained by using the nonparametric Otsu algorithm [39] from each modality separately (as seen from blue and red dashed curves). The following spatial mapping procedure was conducted by an intersection operation. The visual habitats are shown in the last block with color codes.
Discussion
Multiple recent studies have demonstrated marked genetic heterogeneity between and within tumors. This is typically ascribed to clonal evolution driven by random mutations. However, genetic mutations simply represent one component (“a mechanism of inheritance”) of Darwinian dynamics, which are ultimately governed by phenotypic heterogeneity and variations in environmental selection forces [12]. While genetic heterogeneity clearly poses a challenge to molecularly based targeted therapy, we hypothesize that these variations, rather than a stochastic process governed by random mutations, may represent predictable and reproducible outcomes from identifiable Darwinian dynamics.
In our model, intratumoral evolution is fundamentally governed by variations in environmental selection forces that are largely dependent on local blood flow. That is, while changes in cancer cells may be the result of random genetic or epigenetic events, clonal expansion of each new genotype is entirely dependent on its fitness within the context of the local environment and the fitness of the competing tumor populations. Thus, the dominant cancer phenotype and genotype within each tumor region is largely determined by their ability to adapt to environmental conditions that are generally governed by blood flow including oxygen, glucose, H+, and serum growth factors. This suggests that only limited numbers of general adaptive strategies are necessary—i.e., evolving the capacity to survive and proliferate in hypoxia. However, the phenotypic expression of those strategies is a much larger set of possibilities and the genetic pathway to those phenotypesis likely very much larger [13]. Thus, the genetic variation among cancer cells could look chaotic even when the underlying evolutionary dynamics are fairly straightforward.
This connection between environmental selection forces and phenotypic adaptations/genetic heterogeneity provides a theoretical bridge between radiologic imaging and cellular evolution within tumors. Thus, we hypothesized that radiographic manifestation of blood flow and interstitial edema can identify and map distinctive variations in environmental selection forces (“habitats”) within each tumor.
To evaluate the potential role of habitat variations in survival, we arbitrarily divided our study group into two groups based on survival time. Our results demonstrate that group 1 and group 2 GBMs have distinctly different patterns of vascularity and cellular density. As shown in Figure 4, GBMs consistently divide into five MRI-defined combinations of blood flow and cellular density. At present, the underlying evolutionary dynamics cannot be determined unambiguously. Clearly, the expected patterns are high blood flow and high cell density and low blood flow with low cell density. The three additional regions of apparent mismatch between blood flow and cellular density will require further investigation. In general, it is likely that they represent two possible “ecologies”: 1) cellular evolution. This could result in adaptive strategies that permit increased proliferation in regions of poor perfusion or increased utilization of substrate (because of Warburg physiology, for example) that increases glucose uptake and toxic acid production in regions that are well perfused. 2) Temporal variations in regional perfusion. This would result in cycles of normoxia and hypoxia so that the average perfusion results in greater or lesser cellularity than expected based on a single observation.
As shown in Figure 5, we find that group 2 tumors are more homogeneous with a dominant habitat in which there is high blood flow and intermediate cell density. Group 1 tumors, however, contain relatively high volumes of low blood flow habitats that may have very low cell density indicating necrosis but often exhibit cell densities comparable to those seen in well-perfused regions.
This suggests a clear need to further investigate the reason(s) these habitats confer a poor prognosis. Multiple factors may be involved including poor perfusion and hypoxia, which may limit the effectiveness of chemotherapy or radiation therapy. Furthermore, hypoxia-adapted cells often exhibit more stem-like behavior with up-regulation of survival pathways that confers resistance to treatments.
Our study represents only a preliminary test of our underlying hypothesis and suffers from a number of limitations. Since this is an initial retrospective study using a publicly available database, clinical and imaging data were significantly limited. In particular, the absence of apparent diffusion coefficient maps did not permit estimates of regional variations in cell density. The MRI scans were performed on a wide range of imaging platforms with some variations in technique. The details of therapy were not available and so we cannot estimate the potential role of treatment variations in survival data. Furthermore, a large number of factors in addition to the tumor characteristics may influence survival. In particular, some of the short-term survivors may have died from complications of therapy. Finally, the analyzed cohort is relatively small and our results will need to be confirmed in larger retrospective and prospective studies.
Nevertheless, we show here that clinical imaging can be used to gain insight into the evolutionary dynamics within tumors. Our results suggest that combinations of sequences from standard MRI imaging can define spatially and physiologically distinct regions or habitats within the ecology of GBMs and that this may be useful as a patient-specific prognostic biomarker. Ultimately, many other combinations of imaging characteristics including other modalities such as FDG PET should be investigated and may provide greater information regarding intratumoral evolution. Finally, we note that changes in intratumoral habitats during therapy may provide useful information regarding response and the evolution of adaptive strategies.
Table 2.
Distribution of Gene Mutations in Group 1.
| Tumor ID | CDKN2A | EGFR | PTEN | PDGFRA | TP53 | CDK4 | NF1 | CDK6 | Total |
| 1 | X | X | X | 3 | |||||
| 2 | X | 1 | |||||||
| 3 | X | 1 | |||||||
| 4 | X | X | X | 3 | |||||
| 5 | X | X | X | X | 4 | ||||
| 6 | X | X | 2 | ||||||
| 7 | 0 | ||||||||
| 8 | X | X | X | 2 | |||||
| 9 | X | 1 | |||||||
| 10 | X | 1 | |||||||
| 11 | X | X | X | 3 | |||||
| 12 | 0 | ||||||||
| 13 | X | X | 2 | ||||||
| 14 | 0 | ||||||||
| 15 | X | X | 2 | ||||||
| 16 | X | X | X | 3 | |||||
| Total | 8 | 6 | 4 | 2 | 3 | 3 | 2 | 1 | 29 |
Footnotes
This work is supported by the following National Institutes of Health/National Cancer Institute (NIH/NCI) grants: U54CA143970-01, U01CA143062, R01CA077575 and R01CA170595.
References
- 1.Yancovitz M, Litterman A, Yoon J, Ng E, Shapiro RL, Berman RS, Pavlick AC, Darvishian F, Christos P, Mazumdar M, et al. Intra- and inter-tumor heterogeneity of BRAFV600E mutations in primary and metastatic melanoma. PLoS One. 2012;7:e29336. doi: 10.1371/journal.pone.0029336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Inda MM, Bonavia R, Mukasa A, Narita Y, Sah D, Vandenberg S, Brennan C, Johns TG, Bachoo R, Hadwiger P, et al. Tumor heterogeneity is an active process maintained by a mutant EGFR-induced cytokine circuit in glioblastoma. Genes Dev. 2010;24:1731–1745. doi: 10.1101/gad.1890510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, Martinez P, Matthews N, Steward A, Tarpey P, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. NEngl JMed. 2012;366:883–892. doi: 10.1056/NEJMoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sottoriva A, Spiteri I, Piccirillo SG, Touloumis A, Collins VP, Marioni JC, Curtis C, Watts C, Tavaré S. Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc Natl Acad Sci USA. 2013;110:4009–4014. doi: 10.1073/pnas.1219747110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Marusyk A, Almendro V, Polyak K. Intra-tumour heterogeneity: a looking glass for cancer? Nat Rev Cancer. 2012;12:323–334. doi: 10.1038/nrc3261. [DOI] [PubMed] [Google Scholar]
- 6.Yachida S, Jones S, Bozic I, Antal T, Leary R, Fu B, Kamiyama M, Hruban RH, Eshleman JR, Nowak MA, et al. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature. 2010;467:1114–1117. doi: 10.1038/nature09515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gerlinger M, Swanton C. How Darwinian models inform therapeutic failure initiated by clonal heterogeneity in cancer medicine. Br J Cancer. 2010;103:1139–1143. doi: 10.1038/sj.bjc.6605912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mirnezami R, Nicholson J, Darzi A. Preparing for precision medicine. N Engl J Med. 2012;266:489–490. doi: 10.1056/NEJMp1114866. [DOI] [PubMed] [Google Scholar]
- 9.Kern SE. Why your new cancer biomarker may never work: recurrent patterns and remarkable diversity in biomarker failures. Cancer Res. 2012;72:1–5. doi: 10.1158/0008-5472.CAN-12-3232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nowell PC. The clonal evolution of tumor cell populations. Science. 1976;194:23–28. doi: 10.1126/science.959840. [DOI] [PubMed] [Google Scholar]
- 11.Greaves M, Maley CC. Clonal evolution in cancer. Nature. 2012;481:306–313. doi: 10.1038/nature10762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gatenby RA, Gillies RJ. A microenvironmental model of carcinogenesis. Nat Rev Cancer. 2008;8:56–61. doi: 10.1038/nrc2255. [DOI] [PubMed] [Google Scholar]
- 13.Gatenby R. Finding cancer's first principles. Nature. 2012;49:55. doi: 10.1038/491s55a. [DOI] [PubMed] [Google Scholar]
- 14.Gillies RJ, Verduzco D, Gatenby RA. Evolutionary dynamics of carcinogenesis and why targeted therapy does not work. Nat Rev Cancer. 2012;12:487–493. doi: 10.1038/nrc3298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gatenby R, Grove O, Gillies R. Quantitative imaging in cancer evolution and ecology. Radiology. 2013;269:8–15. doi: 10.1148/radiol.13122697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Asselin MC, O'Connor JP, Boellaard R, Thacker NA, Jackson A. Quantifying heterogeneity in human tumours using MRI and PET. Eur J Cancer. 2012;48:447–455. doi: 10.1016/j.ejca.2011.12.025. [DOI] [PubMed] [Google Scholar]
- 17.Chicklore S, Goh V, Siddique M, Roy A, Marsden PK, Cook GJ. Quantifying tumour heterogeneity in 18F-FDG PET/CT imaging by texture analysis. Eur J Nucl Med Mol Imaging. 2012;40:133–140. doi: 10.1007/s00259-012-2247-0. [DOI] [PubMed] [Google Scholar]
- 18.Lambin P, Rios-Velazquez E, Leijenaar R, Carvalho S, van Stiphout RG, Granton P, Zegers CM, Gillies R, Boellard R, Dekker A, et al. Radiomics: extracting more information from medical images using advanced feature analysis. Eur J Cancer. 2012;30:1234–1248. doi: 10.1016/j.ejca.2011.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nair VS, Gevaert O, Davidzon G, Napel S, Graves EE, Hoang CD, Shrager JB, Quon A, Rubin DL, Plevritis SK. Prognostic PET 18F-FDG uptake imaging features are associated with major oncogenomic alterations in patients with resected non-small cell lung cancer. Cancer Res. 2012;72:3725–3734. doi: 10.1158/0008-5472.CAN-11-3943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Diehn M, Nardini C, Wang DS, McGovern S, Jayaraman M, Liang Y, Aldape K, Cha S, Kuo MD. Identification of noninvasive imaging surrogates for brain tumor gene-expression modules. Proc Natl Acad Sci USA. 2008;105:5213–5218. doi: 10.1073/pnas.0801279105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Segal E, Sirlin CB, Ooi C, Adler AS, Gollub J, Chen X, Chan BK, Matcuk GR, Barry CT, Chang HY, et al. Decoding global gene expression programs in liver cancer by noninvasive imaging. Nat Biotechnol. 2007;25:675–680. doi: 10.1038/nbt1306. [DOI] [PubMed] [Google Scholar]
- 22.Tixier F, Le Rest CC, Hatt M, Albarghach N, Pradier O, Metges JP, Corcos L, Visvikis D. Intratumor heterogeneity characterized by textural features on baseline 18F-FDG PET images predicts response to concomitant radiochemotherapy in esophageal cancer. J Nucl Med. 2011;52:369–378. doi: 10.2967/jnumed.110.082404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pang KK, Hughes T. MR imaging of the musculoskeletal soft tissue mass: is heterogeneity a sign of malignancy? J Chin Med Assoc. 2003;66:655–661. [PubMed] [Google Scholar]
- 24.Ganeshan B, Panayiotou E, Burnand K, Dizdarevic S, Miles K. Tumour heterogeneity in non-small cell lung carcinoma assessed by CT texture analysis: a potential marker of survival. Eur Radiol. 2012;22:796–802. doi: 10.1007/s00330-011-2319-8. [DOI] [PubMed] [Google Scholar]
- 25.Zhou M, Hall LO, Goldgof DB, Gillies RJ, Gatenby RA. Survival time prediction of patients with glioblastoma multiforme tumors using spatial distance measurement. SPIE Medical Imaging Conference on Computer-Aid Diagnosis. 2013;8670:7. id867020. [Google Scholar]
- 26.Cancer Genome Atlas Research Network, author. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–1068. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zinn PO, Majadan B, Sathyan P, Singh SK, Majumder S, Jolesz FA, Colen RR. Radiogenomic mapping of edema/cellular invasion MRI-phenotypes in glioblastoma multiforme. PLoS One. 2011;10:e2541. doi: 10.1371/journal.pone.0025451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Barajas RF, Philips JJ, Parvataneni R, Molinaro A, Essock-Burns E, Bourned G, Parsa AT, Aghi MK, McDermott MW, Berger MS, et al. Regional variation in histopathologic features of tumor specimens from treatment-naive glioblastoma correlates with anatomic and physiologic MR imaging. Neuro Oncol. 2012;14:942–954. doi: 10.1093/neuonc/nos128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Barajas RF, Hodgson JG, Chang JS, Vandenberg SR, Yeh RG, Parsa AT, McDermott MW, Berger MS, Dillon WP, Cha S. Glioblastoma multiforme regional genetic and cellular expression patterns: influence on anatomic and physiologic MR imaging. Radiology. 2010;254:564–576. doi: 10.1148/radiol.09090663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shah M, Xiao Y, Subbamma N, Francis S, Arnold DL, Collins DL, Arbel T. Evaluating intensity normalization on MRIs of human brain with multiple sclerosis. Med Image Anal. 2011;15:267–282. doi: 10.1016/j.media.2010.12.003. [DOI] [PubMed] [Google Scholar]
- 31.Clark MC, Hall LO, Goldgof DB, Velthuizen R, Murtagh R, Silbiger MS. Automatic tumor segmentation using knowledge-based techniques. IEEE Trans Med Imag. 1998;17:187–201. doi: 10.1109/42.700731. [DOI] [PubMed] [Google Scholar]
- 32.Prastawa M, Bullitt E, Ho S, Gerig G. A brain tumor segmentation framework based on outlier detection. Med Image Anal. 2004;8:275–283. doi: 10.1016/j.media.2004.06.007. [DOI] [PubMed] [Google Scholar]
- 33.Fine HA. The basis for current treatment recommendations for malignant gliomas. JNeurooncol. 1994;20:111–120. doi: 10.1007/BF01052722. [DOI] [PubMed] [Google Scholar]
- 34.Sonoda Y, Kumabe T, Watanabe M, Nakazato Y, Inoue T, Kanamori M, Tominaga T. Long-term survivors of glioblastoma: clinical features and molecular analysis. Acta Neurochir. 2009;151:1349–1358. doi: 10.1007/s00701-009-0387-1. [DOI] [PubMed] [Google Scholar]
- 35.Figueiredo M, Jain A. Unsupervised learning of finite mixture models. IEEE Trans Pattern Anal Mach Intell. 2002;24:381–396. [Google Scholar]
- 36.Banfield JD, Raferty AE. Model-based Gaussian and non-Gaussian clustering. Biometrics. 1993;49:803–821. [Google Scholar]
- 37.Leung T, Malik J. Representing and recognizing the visual appearance of materials using three-dimensional textons. JComput Vis. 2001;43:29–44. [Google Scholar]
- 38.Chang CC, Lin CJ. LIBSVM: a library for support vector machines. ACM Trans Intell Syst Technol. 2011;2:1–27. [Google Scholar]
- 39.Cortes C, Vapnik V. Support-vector network. Mach Learn. 1995;20:273–297. [Google Scholar]
- 40.Otsu N. A threshold selection method from gray-level histogram. IEEE Trans Syst Man Cybern. 1979;9:62–66. [Google Scholar]