

Abstract
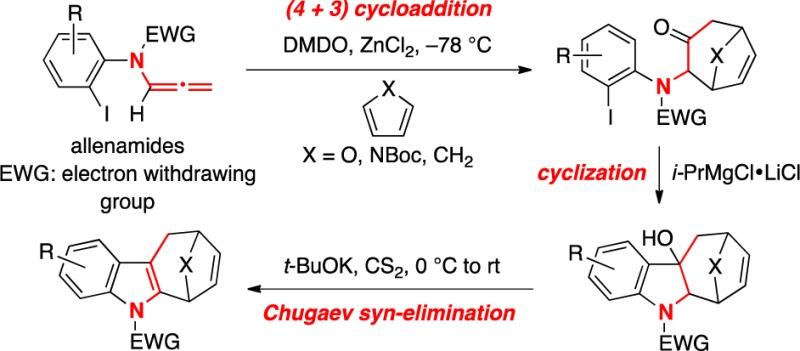
A strategy for synthesizing highly functionalized cyclohepta[b]indoles through a concise (4 + 3) cycloaddition–cyclization–elimination sequence is described. The cycloaddition features nitrogen-stabilized oxyallyl cations derived from epoxidations of N-aryl-N-sulfonyl-substituted allenamides, while the cyclization and elimination employed an intramolecular Grignard addition and a one-step Chugaev process, respectively.
Cyclohepta[b]indoles represent a prevalent structural motif among pharmaceutical entities as well as bioactive natural products such as ervitsine,1 aristolasol,2 silicine,3 ambigune E,4 and actinophyllic acid (Figure 1).5 Cyclohepta[b]indole derivatives have been shown to inhibit adipocyte fatty acid binding protein (A-FABP)6 and of deacetylase SIRT1.7 Consequently, cyclohepta[b]indoles have received much attention from the synthetic community. Previous approaches8 include Fischer indole syntheses,9 ring expansions,10 and intramolecular cyclizations.11 Recently, (4 + 3) cycloaddition12 approaches have been developed. Wu13 reported an elegant three-component (4 + 3) cycloaddition for constructing cyclohepta[b]indoles, while Tang14 and Iwasawa15 envisaged rhodium- and platinum-catalyzed (4 + 3) transformations. A formal (4 + 3) cycloaddition reaction was a key step in Martin’s total synthesis of actinophyllic acid.16
Figure 1.
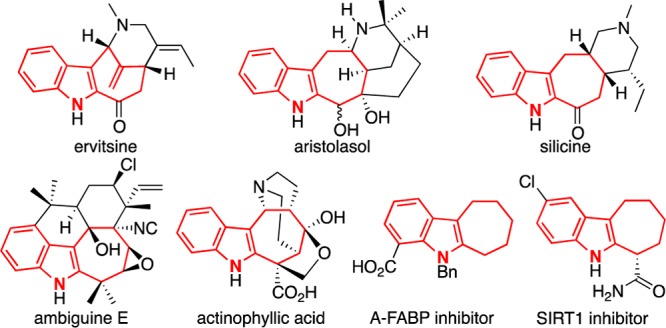
Representative cyclohepta[b]indole compounds.
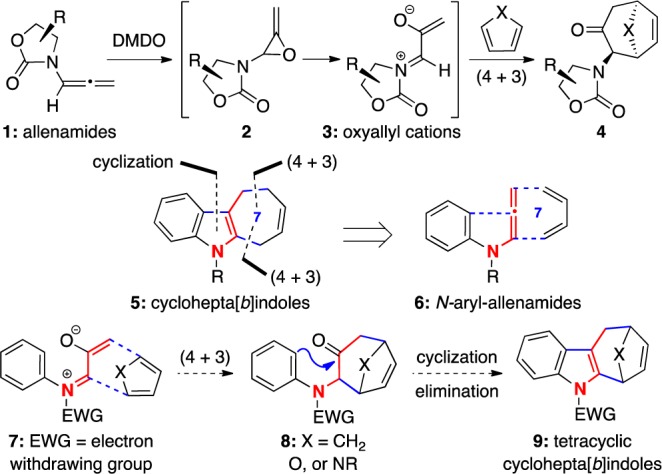
We became interested in cyclohepta[b]indoles because of our long-standing program in (4 + 3) cycloaddition reactions17 employing nitrogen-stabilized oxyallyl cations (see 3)18 derived from epoxidations of allenamides 1 (Scheme 1).19−21 Structurally, only three C–C bonds separate cyclohepta[b]indoles 5 from N-arylallenamides 6. Thus, we envisioned that while it would be difficult to include the indole formation concomitant with the (4 + 3) cycloaddition, an ensuing intramolecular cyclization from the aryl group onto the carbonyl group in (4 + 3) cycloadducts 8 should accomplish such a purpose, leading to tetracyclic cyclohepta[b]indoles 9. We wish to communicate here our success in achieving a strategy for constructing cyclohepta[b]indoles via a concise (4 + 3) cycloaddition–cyclization–elimination sequence.
Scheme 1. (4 + 3) Cycloaddition–Cyclization Strategy.
Our approach toward cyclohepta[b]indoles commenced with a (4 + 3) cycloaddition reaction of N-aryl-N-sulfonylallenamide 10a(22) as shown in Scheme 2. It is noteworthy that although we have reported intramolecular (4 + 3) cycloaddition reactions of N-sulfonylallenamides,23 the current attempt represents the first example in which an N-sulfonyl-substituted allenamide is used in an intermolecular (4 + 3) cycloaddition manifold. More significantly, allenamide 10a is an N-aryl- or aniline-substituted allenamide. While this appears to be a minor structural perturbation from previous allenamides, it is well known that the stability and reactivity of allenamides are closely regulated by the substitution pattern on the nitrogen atom, especially when a substitution can impact on its ability to delocalize or donate toward the allenic motif.19,24 This constitutes the first challenge in our strategy because an N-aryl group allows the delocalization of the nitrogen lone pair, thereby deactivating the allenamide reactivity. However, when N-aryl-N-sulfonylsulfonamide 10a was subjected to standard conditions (ZnCl2, DMDO, furan, and 4 Å MS in CH2Cl2), it reacted successfully with furan to afford the desired cycloadduct 11a in 70% yield as a single diastereomer (Scheme 2).
Scheme 2. Cycloaddition of an N-Aryl-N-sulfonylallenamide.

With this result in hand, the scope of (4 + 3) cycloaddition reactions of N-aryl-N-sulfonylallenamides could be explored (Figure 2). Allenamides with different substituents on the aryl ring worked well to give products 11b–g in moderate to good yields. In addition, Boc-protected pyrrole and cyclopentadiene can also serve as suitable dienes to furnish cycloadducts 11h and 11i, respectively. Allenamides stabilized by a carbamate group also proved to be efficient in this reaction (see 11j and 11k), although 11k was obtained as an inseparable mixture with a modest 3:1 diastereomeric ratio [stereochemistry of the major isomer unassigned]. When allenamide bearing a 4-methoxybenzenesulfonyl substituent was used, the desired cycloadduct 11l was obtained in a relatively lower yield (11l, 38%), thereby implying that these cycloadditions may favor allenamides with strong electron-withdrawing groups.
Figure 2.

Scope of the (4 + 3) cycloaddition.
Having succeeded in this first step, we proceeded to complete the cyclohepta[b]indole ring system. As indicated above, we envisioned that an intramolecular addition of aryl anion to the carbonyl carbon should fulfill this task. To accomplish this task, we elected to go with a strategy adopted by Kobayashi and co-workers for constructing quinoline and indole rings.25 By using i-PrMgCl·LiCl complex as the magnesium–halogen exchange reagent,25,26 an intramolecular Grignard addition could take place effectively to give the tetracyclic alcohol 12a in 63% yield (Scheme 3). Other tetracyclic alcohols 12b–l could also be obtained utilizing the same reaction condition as shown in Figure 3, thereby demonstrating the generality of this cyclization. The structural as well as stereochemical integrity of 12b was unambiguously assigned through its X-ray single-crystal structure (Figure 4).
Scheme 3. Intramolecular Grignard Addition.

Figure 3.

Intramolecular Grignard addition products.
Figure 4.
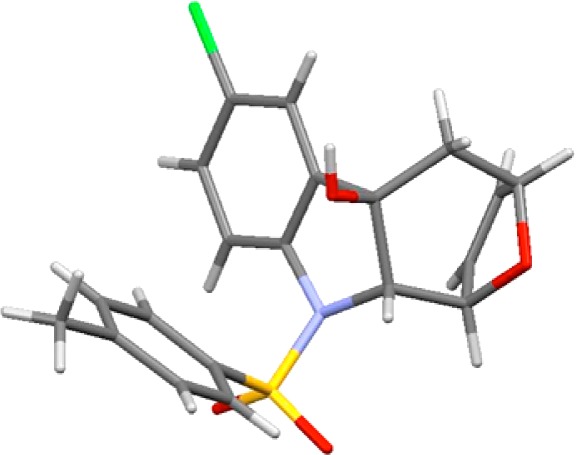
X-ray structure of tetracyclic alcohol 12b.
While tetracycle alcohols 12a–l represent highly functionalized structural manifolds with multistereogenic centers that can be useful in further transformations and evolution of complexity, to truly complete the indole synthesis, elimination of the tertiary alcohol should be accomplished here. We had hoped that a simple acid-induced dehydrative elimination–aromatization sequence should furnish the desired 14a (Scheme 4). However, despite being stable to silica gel column chromatography, when tetracyclic alcohol 12a was treated with catalytic amount of p-TsOH at 0 °C, we found complete unraveling of the tetracycle. The only product obtained was 3-furfurylindole 17, likely derived form an acid-induced fragmentation process via intermediates 15 and 16.27
Scheme 4. Acid-Induced Grob-Type Fragmentation.
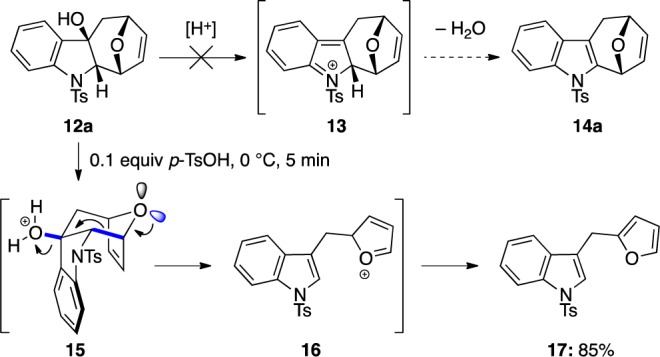
This fragmentation reaction is likely favored for two reasons: (a) The perfect anti-periplanar alignments between C–OH bond and the C–C bond and the C–C bond and the equatorial lone pair of the bridging oxygen (all in blue) or the perfect overlap and delocalization into the respective σ* and (b) the formation of two aromatic systems (indole and furan) as the thermodynamic driving force. Thus, we reasoned that if we first functionalize the cycloheptenone double bond in 12a, we should prevent the formation of furan and reduce the driving force of the fragmentation. As shown in Scheme 5, when triol 18, which could be prepared via a simple dihydroxylation from 12a, was submitted to the same acidic condition, the elimination product 19 was obtained in 30% yield. While this result strongly supported our hypothesis and gave a highly functionalized cyclohepta[b]indoles in 19, we recognized that the overall process is not as efficient.
Scheme 5. Successful Eliminations.

To be more effective in completing the construction of cyclohepta[b]indoles, we pursued a direct elimination method via syn-hydrogen abstraction mechanism and hoped we could circumvent the fragmentation facilitated by the anti-periplanar alignments. However, SeO2 did not react with tetracyclic alcohol 12a under basic condition, while Burgess reagent provided again only fragmentation product 17. We subsequently turned our attention to transforming tetracyclic alcohol 12a into a xanthate intermediate for a potential Chugaev’s syn-elimination. When 12a was treated with base and CS2, the desired cyclohepta[b]indole 14a was obtained directly without the isolation of any xanthate intermediates (Scheme 5). This result could be explained by a direct intramolecular hydrogen abstraction of the xanthate anion 20. It is noteworthy that base played an important role in this elimination reaction. While using t-BuOK led to 100% conversion, KH could only provide a conversion of 70%, and conversions were less than 10% when employing NaH and LiHMDS.
We subsequently applied this effective one-step Chugaev elimination to other tetracyclic alcohols 12b–l (Figure 5). This reaction in general gave excellent yields to afford an array of cyclohepta[b]indoles 14b–e,i,l. Cyclohepta[b]indoles 14f and 14g could be obtained only when using KH as the base because t-BuOK-induced methyl ester hydrolysis in these two examples. Cyclohepta[b]indole 14h was also prepared under the KH conditions since t-BuOK led to complete decomposition of the corresponding starting material 12h; we are not clear of the rationale at this point. In addition, cyclohepta[b]indoles 14j and 14k with indole nitrogen protected as carbamates were obtained with slightly lower yields. These last three examples suggest that the N-protecting groups could be important in this elimination process.
Figure 5.
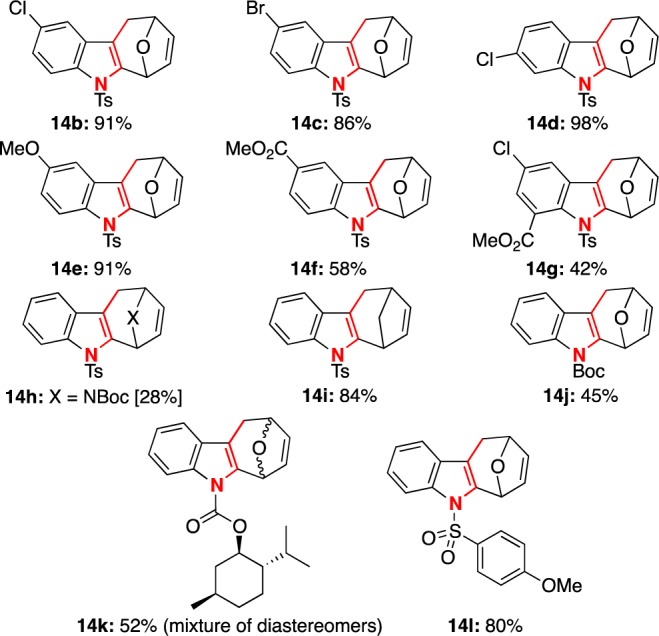
Synthetic scope of cyclohepta[b]indoles.
We have developed a strategy for synthesizing highly functionalized cyclohepta[b]indoles through an efficient sequence of a (4 + 3) cycloaddition–cyclization–elimination. The cycloaddition features nitrogen-stabilized oxyallyl cations derived from epoxidations of N-aryl-N-sulfonyl-substituted allenamides, while the cyclization and elimination employed an intramolecular Grignard addition and a useful one-step Chugaev process, respectively. Applications of this strategy in natural product synthesis are currently underway.
Acknowledgments
We thank the NIH (GM066055) for financial support and Dr. Victor Young at the University of Minnesota for X-ray structural analysis.
Supporting Information Available
Experimental procedures as well as NMR spectra, characterizations, and X-ray structural files. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Andriantsiferana M.; Besselievre R.; Riche C.; Husson H. P. Tetrahedron Lett. 1977, 30, 2587. [Google Scholar]
- Quirion J. C.; Kan-Fan C.; Bick I. R. C.; Husson H. P. Phytochemistry 1988, 27, 3337. [Google Scholar]
- Shafiee A.; Ahond A.; Bui A. M.; Langlois Y.; Riche C.; Potier P. Tetrahedron Lett. 1976, 17, 921. [Google Scholar]
- Smitka T. A.; Bonjouklian R.; Doolin L.; Jones N. D.; Deeter J. B.; Yoshida W. Y.; Prinsep M. R.; Moore R. E.; Patterson G. M. L. J. Org. Chem. 1992, 57, 857. [Google Scholar]
- Carrol A. R.; Hyde E.; Smith J.; Quinn R. J.; Guymer G.; Foster P. I. J. Org. Chem. 2005, 70, 1096. [DOI] [PubMed] [Google Scholar]
- Barf T.; Lehmann F.; Hammer K.; Haile S.; Axen E.; Medina C.; Uppenberg J.; Svensson S.; Rondahl L.; Lundbaeck T. Bioorg. Med. Chem. Lett. 2009, 19, 1745. [DOI] [PubMed] [Google Scholar]
- Napper A. D.; Hixon J.; McDonagh T.; Keavey K.; Pons J. F.; Barker J.; Yau W. T.; Amouzegh P.; Flegg A.; Hamelin E.; Thomas R. T.; Kates M.; Jones S.; Navia M. A.; Saunders J.; DiStefano P. S.; Curtis R. J. Med. Chem. 2005, 48, 8045. [DOI] [PubMed] [Google Scholar]
- For general examples, see:; a Willis M. C.; Brace G. N.; Holmes I. P. Angew. Chem., Int. Ed. 2005, 44, 403. [DOI] [PubMed] [Google Scholar]; b Barluenga J.; Jiménez-Aquino A.; Valdés C.; Aznar F. Angew. Chem., Int. Ed. 2007, 46, 1529. [DOI] [PubMed] [Google Scholar]; c Barluenga J.; García- Rodríguez J.; Suárez-Sobrino Á. L.; Tomás M. Chem.—Eur. J. 2009, 15, 8800. [DOI] [PubMed] [Google Scholar]; d Silvanus A. C.; Heffernan S. J.; Liptrot D. J.; Kociok-Köhn G.; Andrews B. I.; Carbery D. R. Org. Lett. 2009, 11, 1175. [DOI] [PubMed] [Google Scholar]
- a Mewshaw R. E.; Silverman L. S.; Mathew R. M.; Kaiser C.; Sherrill R. G.; Cheng M.; Tiffany C. W.; Karbon E. W.; Bailey M. A.; Borosky S. A.; Ferkany J. W.; Abreu M. E. J. Med. Chem. 1993, 36, 1488. [DOI] [PubMed] [Google Scholar]; b Napper A. D.; Hixon J.; McDonagh T.; Keavey K.; Pons J.; Barker J.; Yau W. T.; Amouzegh P.; Flegg A.; Hamelin E.; Thomas R. J.; Kates M.; Jones S.; Navia M. A.; Saunders J. O.; DiStefano P. S.; Curtis R. J. Med. Chem. 2005, 48, 8045. [DOI] [PubMed] [Google Scholar]
- a Horwell D. C.; McKiernan M. J.; Osborne S. Tetrahedron Lett. 1998, 39, 8729. [Google Scholar]; b Sun K.; Liu S.; Bec P. M.; Driver T. G. Angew. Chem., Int. Ed. 2011, 50, 1702. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Gritsch P. J.; Stempel E.; Gaich T. Org. Lett. 2013, 15, 5472. [DOI] [PubMed] [Google Scholar]
- a Harris M.; Grierson D. S.; Riche C.; Husson H. Tetrahedron Lett. 1980, 21, 1957. [Google Scholar]; b Joseph B.; Alagille D.; Rousseau C.; Mérour J. Tetrahedron 1999, 55, 4341. [Google Scholar]; c Ishikura M.; Kato H. Tetrahedron 2002, 58, 9827. [Google Scholar]; d Liu C.; Widenhoefer R. A. J. Am. Chem. Soc. 2004, 126, 10250. [DOI] [PubMed] [Google Scholar]; e Martin C. L.; Overman L. E.; Rohde J. M. J. Am. Chem. Soc. 2008, 130, 7568. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Amat M.; Checa B.; Llor N.; Molins E.; Bosch J. Chem. Commun. 2009, 45, 2935. [DOI] [PubMed] [Google Scholar]; g Lu Y.; Du X.; Jia X.; Liu Y. Adv. Synth. Catal. 2009, 351, 1517. [Google Scholar]; h Butin A.; Kostyukova O. N.; Tsiunchik F. A.; Uchuskin M. G.; Serdyuk O. V.; Trushkov I. V. J. Heterocycl. Chem. 2010, 48, 684. [Google Scholar]; i Oda M.; Ito K.; Takagi H.; Fujiwara Y. Heterocycles 2012, 86, 623. [Google Scholar]; j Bennasar M.-L.; Zulaica E.; Solé D.; Alonso S. Tetrahedron 2012, 68, 4641. [Google Scholar]; k Kuznetsov A.; Makarov A.; Rubtsov A. E.; Butin A. V.; Gevorgyan V. J. Org. Chem. 2013, 78, 12144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For general reviews of oxyallyl cycloadditions, see:; a Rigby J. H.; Pigge F. C. Org. React. 2004, 351. [Google Scholar]; b Davies H. M. L. In Advances in Cycloaddition; Harmata M., Ed.; JAI: Stamford, 1999; Vol. 5, p 119. [Google Scholar]; c Hartung I. V.; Hoffmann H. M. R. Angew. Chem., Int. Ed. 2004, 43, 1934. [DOI] [PubMed] [Google Scholar]; d Battiste M. A.; Pelphrey P. M.; Wright D. L. Chem.—Eur. J. 2006, 12, 3438. [DOI] [PubMed] [Google Scholar]; e Harmata M. Adv. Synth. Catal. 2006, 348, 2297. [Google Scholar]; f Harmata M. Chem. Commun. 2010, 46, 8886. [DOI] [PubMed] [Google Scholar]; g Harmata M. Chem. Commun. 2010, 46, 8904. [DOI] [PubMed] [Google Scholar]
- Han X.; Li H.; Hughes R. P.; Wu J. Angew. Chem., Int. Ed. 2012, 51, 10390. [DOI] [PubMed] [Google Scholar]
- Shu D.; Song W.; Li X.; Tang W. Angew. Chem., Int. Ed. 2013, 52, 3237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusama H.; Sogo H.; Saito K.; Suga T.; Iwasawa N. Synlett 2013, 24, 1364. [Google Scholar]
- Granger B. A.; Jewett I. T.; Butler J. D.; Hua B.; Knezevic C. E.; Parkinson E. I.; Hergenrother P. J.; Martin S. F. J. Am. Chem. Soc. 2013, 135, 12984. [DOI] [PubMed] [Google Scholar]
- For a leading review, see:Lohse A. G.; Hsung R. P. Chem.—Eur. J. 2011, 17, 3812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For some examples, see:; a Walters M. A.; Arcand H. R.; Lawrie D. J. Tetrahedron Lett. 1995, 36, 23. [Google Scholar]; b Sung M. J.; Lee H. I.; Chong Y.; Cha J. K. Org. Lett. 1999, 1, 2017. [DOI] [PubMed] [Google Scholar]; c Myers A. G.; Barbay J. K. Org. Lett. 2001, 3, 425. [DOI] [PubMed] [Google Scholar]; d Xiong H.; Hsung R. P.; Berry C. R.; Rameshkumar C. J. Am. Chem. Soc. 2001, 123, 7174. [DOI] [PubMed] [Google Scholar]; e Harmata M.; Ghosh S. K.; Hong X.; Wacharasindhu S.; Kirchhoefer P. J. Am. Chem. Soc. 2003, 125, 2058. [DOI] [PubMed] [Google Scholar]; f Xiong H.; Huang J.; Ghosh S. K.; Hsung R. P. J. Am. Chem. Soc. 2003, 125, 12694. [DOI] [PubMed] [Google Scholar]; g Huang J.; Hsung R. P. J. Am. Chem. Soc. 2005, 127, 50. [DOI] [PubMed] [Google Scholar]; h MaGee D. I.; Godineau E.; Thornton P. D.; Walters M. A.; Sponholtz D. J. Eur. J. Org. Chem. 2006, 3667. [Google Scholar]
- For reviews on allenamide chemistry, see:; a Lu T.; Lu Z.; Ma Z.-X.; Zhang Y.; Hsung R. P. Chem. Rev. 2013, 130, 4862. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Hsung R. P.; Wei L.-L.; Xiong H. Acc. Chem. Res. 2003, 36, 773. [DOI] [PubMed] [Google Scholar]; Also see:; c Standen P. E.; Kimber M. C. Curr. Opin. Drug Discovery Dev. 2010, 13, 645. [PubMed] [Google Scholar]; d Deagostino A.; Prandi C.; Tabasso S.; Venturello P. Molecules 2010, 15, 2667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For general reviews on allenes, see:Krause N.; Hashmi A. S. K.. Modern Allene Chemistry; Wiley–VCH Verlag: Weinheim, 2004; Vols. 1 and 2. [Google Scholar]
- For our recent work, see:; a Krenske E. H.; Houk K. N.; Lohse A. G.; Antoline J. E.; Hsung R. P. Chem. Sci. 2010, 1, 387. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Lohse A. G.; Krenske E. H.; Antoline J. E.; Houk K. N.; Hsung R. P. Org. Lett. 2010, 12, 5506. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Antoline J. E.; Krenske E. H.; Lohse A. G.; Houk K. N.; Hsung R. P. J. Am. Chem. Soc. 2011, 133, 14443. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Du Y.; Krenske E. H.; Antoline J. E.; Lohse A. G.; Houk K. N.; Hsung R. P. J. Org. Chem. 2013, 78, 1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- See the Supporting Information.
- Loshe A. G.; Hsung R. P.; Leider M. D.; Ghosh S. K. J. Org. Chem. 2011, 76, 3246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei L.-L.; Hsung R. P.; Xiong H.; Mulder J. A.; Nkansah N. T. Org. Lett. 1999, 1, 2145. [Google Scholar]
- Kobayashi Y.; Igarashi J.; Feng C.; Tojo T. Tetrahedron Lett. 2012, 53, 3742. [Google Scholar]
- We found that a quick addition of i-PrMgCl·LiCl complex is crucial for achieving a good conversion. The addition of i-PrMgCl·LiCl THF solution should be conducted within 1 s, or all in one shot. Low conversions were found (<20%) when i-PrMgCl·LiCl was added dropwise via syringe pump during a period of 30 min. Prolonging the reaction time, raising the reaction temperature, or adding starting materials into i-PrMgCl·LiCl solution did not help improve the conversion.
- When employing acids such as CSA, CF3CO2H, or PPTS, we obtained the same fragmentation product.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.