

Abstract
The alkyl chain length of quaternary ammonium/PEG copolyoxetanes has been varied to discern effects on solution antimicrobial efficacy, hemolytic activity and cytotoxicity. Monomers 3-((4-bromobutoxy)methyl)-3-methyloxetane (BBOx) and 3-((2-(2-methoxyethoxy)ethoxy)methyl)-3-methyloxetane (ME2Ox) were used to prepare precursor P[(BBOx)(ME2Ox)-50:50–4 kDa] copolyoxetane via cationic ring opening polymerization. The 1:1 copolymer composition and Mn (4 kDa) were confirmed by 1H NMR spectroscopy. After C–Br substitution by a series of tertiary amines, ionic liquid Cx-50 copolyoxetanes were obtained, where 50 is the mole percent of quaternary repeat units and “x” is quaternary alkyl chain length (2, 6, 8, 10, 12, 14, or 16 carbons). Modulated differential scanning calorimetry (MDSC) studies showed Tgs between −40 and −60 °C and melting endotherms for C14–50 and C16–50. Minimum inhibitory concentrations (MIC) were determined for Escherichia coli, Staphylococcus aureus, and Pseudomonas aeruginosa. A systematic dependence of MIC on alkyl chain length was found. The most effective antimicrobials were in the C6–50 to C12–50 range. C8–50 had better overall performance with MICs of 4 μg/mL, E. coli; 2 μg/mL, S. aureus; and 24 μg/mL, P. aeruginosa. At 5 × MIC, C8–50 effected >99% kill in 1 h against S. aureus, E. coli, and P. aeruginosa challenges of 108 cfu/mL; log reductions (1 h) were 7, 3, and 5, respectively. To provide additional insight into polycation interactions with bacterial membranes, a geometric model based on the dimensions of E. coli is described that provides an estimate of the maximum number of polycations that can chemisorb. Chain dimensions were estimated for polycation C8–50 with a molecular weight of 5 kDa. Considering the approximations for polycation chemisorption (PCC), it is surprising that a calculation based on geometric considerations gives a C8–50 concentration within a factor of 2 of the MIC, 4.0 (±1.2) μg/mL for E. coli. Cx-50 copolyoxetane cytotoxicity was low for human red blood cells, human dermal fibroblasts (HDF), and human foreskin fibroblasts (HFF). Selectivities for bacterial kill over cell lysis were among the highest ever reported for polycations indicating good prospects for biocompatibility.
Introduction
In connection with exploring amphiphilic soft blocks for modification of conventional polyurethanes, copolyoxetanes 1 were developed with quaternary and PEG-like ME2Ox side chains.3,4 Regardless of quaternary mole fraction, copolyoxetanes 1, designated C12-m (x = 12, m = mol %) are viscous, ionic liquids with low glass transition temperatures. With charged and uncharged segments, C12-m have structural characteristics related to naturally occurring antimicrobial peptides (AMPs) that inhibit the growth of bacteria at low concentrations.5,6 At ∼2 wt %, polyurethanes with ionic liquid soft blocks 1 were highly effective modifiers for contact kill coatings.3,7

Water solubility for C12-m led to an investigation that demonstrated high solution antimicrobial effectiveness for m = 14, 25, 42, 50, 60, 87, and 100 against E. coli, P. aeruginosa, and S. aureus.1 Minimum inhibitory concentration (MIC) assays define the antimicrobial concentration that precludes growth after overnight incubation.8−10 MIC tests showed that C12-m, with m = 40–60, had maximum biocidal activity. C12–43 had the lowest MIC for E. coli (4 μg/mL) and S. aureus (5.3 μg/mL), while C12–60 had the lowest MIC for P. aeruginosa (26 μg/mL). Kinetic assays with C12–43 at 5 × MIC gave >99% kill in 1 h for all the three bacterial strains with a starting inoculum ∼108 cfu/mL.
The highly effective biocidal activity of C12-m copolyoxetanes is of interest as, in contrast to conventional antibiotics, negligible buildup of resistance has been found for polycations. Even after 21 passages, no resistance was found for E. coli against a polymethacrylate polycation.11 A similar continued biocidal effectiveness was found for S. aureus against a phenylene ethynylene dication.12 This contrasting behavior may be due to a fundamental difference in killing mechanism for polycations (contact kill/membrane disruption) versus mechanisms based on metabolic processes developed over eons.13,14 That is, microbes possess/obtain antibiotic resistance genes as an evolutionary strategy for survival due to bacteria producing antimicrobials against other microbes. Resistant genes have therefore long been present, at least in microbes that produce antibiotics for self-protection. These antibiotic resistant genes can occur by mutation and be transferred from one bacterium to another and from one species to another by plasmids, transposons, and other subcellular elements.15
Previous research on polycations shows biocidal performance varies widely from nil (>2000 μg/mL) to levels rivaling commonly used antibiotics. For polycations, amphiphilic balance effecting biocidal effectiveness is often sensitive to what appears as a modest structural change. In one case, changing a butyl group in amine-functionalized methacrylates to a methyl group resulted in a 100× increase in MIC for E. coli.10 Another example is found for CSA-13 (10, Table 5), a molecular polycation, wherein a long alkyl chain resulted in a low MIC against E. coli, whereas without this chain, activity was lost.16,17 In view of this sensitivity, the effect of changing alkyl chain length on the antimicrobial properties of Cx-m copolyoxetanes 1 has been investigated. Based on prior work that showed highest antimicrobial effectiveness for C12-m, with m = 40–60,1 a series of Cx-50 copolyoxetanes was prepared (1, x = 2, 6, 8, 10, 12, 14, or 16 carbons). MIC assays were carried out to assess relative antimicrobial effectiveness. Kill kinetics were established for the most effective Cx-50 copolyoxetane, C8-50. Cytotoxicity for Cx-50 copolyoxetanes was examined via hemolytic assays and for toxicity to Human Foreskin Fibroblast (HFF) and Human Dermal Fibroblast (HDF) cell lines.
Table 5. Cx-50 Copolyoxetane Selectivities Calculated According to Eq 3a.
|
E. coli |
S. aureus |
P. aeruginosa |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| Cx-50 | RBC | HFF | HDF | RBC | HFF | HDF | RBC | HFF | HDF |
| C6-50 | 114 | 9000 | 9200 | 283 | 2200 | 2300 | 25 | 2000 | 2000 |
| C8-50 | 65 | 9300 | 9100 | 152 | 2100 | 2100 | 13 | 1800 | 1700 |
| C10-50 | 56 | 5700 | 5900 | 107 | 1100 | 11000 | 15 | 1500 | 1500 |
Selectivities for HFF and HDF are rounded to the nearest thousand.
By analogy with AMPs, the killing mechanism for polycations is thought to occur by ionic and hydrophobic interactions between the bacterial cellular membrane and multiple ammonium moieties.18 The mechanism for bacterial kill is not well understood but involves disruption of the phospholipid bilayer including ion diffusion through cell wall ion channels and a change in cell potential that results in cell death.18,19 In this connection, a geometric model has been developed for estimating polycation concentrations required for chemisorption on E. coli. Although major assumptions are made, the results are in fair agreement with MIC ranges observed experimentally.
Experimental Section
Materials
3-Bromomethyl-3-methyloxetane (BrOx) was generously provided by OMNOVA Solutions (Akron, OH). N,N-Dimethylethylamine (C2), N,N-dimethylhexylamine (C6), N,N-dimethyloctylamine (C8), N,N-dimethyldecylamine (C10), N,N-dimethyltetradecylamine (C14), and N,N-dimethylhexadecylamine (C16) were obtained from Aldrich. N,N-Dimethyldodecylamine (C12) was a gift from Lonza (Allendale, NJ). Methylene chloride (CH2Cl2) and tetrahydrofuran (THF) were obtained from Aldrich and dried by storing over 4 Å molecular sieves. Boron trifluoride diethyl etherate (BF3O(C2H5)2), tetrabutylammonium bromide (TBAB), 3-(hydroxymethyl)-3-methyloxetane, 1,4-dibromobutane, and sodium hydride (NaH) were also obtained from Aldrich and used as received. 1,4-Butanediol (BD) and 2-(2-methoxyethoxy)ethanol were purchased from Acros Chemicals and used as received. All M9 media components were purchased from Sigma Aldrich. Luria Broth (LB) was purchased from Fisher Scientific. Phosphate buffered saline (PBS, i.e., 10 mM phosphate, 138 mM NaCl, pH 7.4) was purchased from Sigma.
Synthesis and Characterization
Monomers 3-((4-bromobutoxy)methyl)-3-methyloxetane (BBOx) and 3-((2-(2-methoxyethoxy)ethoxy)methyl)-3-methyloxetane (ME2Ox) were synthesized and purified as described previously.1
P[(BBOx)(ME2Ox)-50:50-4]
This copolyoxetane intermediate was synthesized by cationic ring-opening polymerization in CH2Cl2.1 A higher ME2 monomer feed (55 mol % was used as BBOx is somewhat more reactive. For the designation P[(BBOx)(ME2Ox)-50:50-4], BBOx is the bromo-butoxymethyl side chain precursor to quat substitution, ME2Ox is the PEG-like side chain in 1, 50:50 is the mole ratio of repeat units, and 4 is the approximate molecular weight in kDa based on 1H NMR data and analyses are in Figure 1 and Supporting Information and GPC (Figure S3).
Figure 1.

1H NMR spectrum (400 MHz) after TFAA addition to P[BBOx-ME2Ox]. Assignments for α and β end group peaks are shown.
Cx-50 Copolyoxetanes
Safety Note
Dimethylalkylamines are toxic; C2 and C6 are hazardous and volatile. Reactions were carried out in a well-ventilated hood using appropriate personal safety equipment. C2-50 was made with caution using small quantities of dimethylethylamine. At the time this research was underway, dimethybutylamine (C4) was not commercially available.
P[(BBOx)(ME2Ox)-50:50-4] was quaternized with tertiary amines to obtain Cx-50 copolyoxetanes by a method described previously.1 The quantities of the precursor and the respective tertiary amines used for quaternization are provided in Table 1. After removing solvent and excess amine in vacuo (liquid nitrogen trap), Cx-50 copolyoxetanes were isolated as highly viscous liquids. 1H NMR data and analyses are in Supporting Information.
Table 1. Feed and Output Ratio for Quaternization of P[(BBOx)(ME2Ox)-50:50-4] and with Cx-50 Tg.
| x | feed, P[(BBOx)(ME2Ox)-50:50-4], g (mmol) | feed, Cx amine,a g (mmol) | ratio Cx/ME2Oxb | Tg (Tm), °C |
|---|---|---|---|---|
| 2 | 2.0 (0.35) | 0.6 (8.2) | 50/50 | –61 |
| 6 | 2.5 (0.44) | 1.42 (11.0) | 49.5/50 | –60 |
| 8 | 2.18 (0.38) | 1.55 (9.87) | 50.2/50 | –56 |
| 10 | 2.5 (0.44) | 1.75 (9.46) | 50.4/50 | –50 |
| 12 | 3 (0.53) | 4 (13.6) | 50/50 | –50 |
| 14 | 2 (0.035) | 2.17 (10.1) | 50.3/50 | –47 (−7) |
| 16 | 1.51 (0.27) | 1.84 (6.84) | 50.1/50 | –48 (7.2) |
Based on BBOx.
Ratio of Cx to ME2Ox from 1H NMR integration.
Instrumentation
1H NMR spectra were obtained using a Varian Mercury 300 MHz NMR spectrometer; FTIR spectra were acquired with a Magna-IR 760 spectrometer and Modulated Differential Scanning Calorimetry (MDSC) utilized a DSC Q1000 (TA Instruments).
Antimicrobial Testing
Bacterial Strains
Strains used for the biocidal tests were Escherichia coli DH5α, Pseudomonas aeruginosa PA01, and Staphylococcus aureus ATCC-25904. The cultures were streaked on Luria Agar plates and incubated overnight at 37 °C. A single colony from each strain was used to inoculate 6 mL of Luria Broth (LB), followed by overnight growth at 37 °C with vigorous shaking (225 rpm). A sample was diluted in fresh medium and incubated to 108–109 colony forming units per milliliter (cfu/mL) of the desired bacteria to generate a culture in the logarithmic growth phase.
Media
Several growth media were explored in order to avoid Cx-50 copolyoxetane precipitation. Luria Broth (LB) solutions were prepared with a range of salt concentrations (0.1–3 wt %). Although adequate bacterial growth was observed, the copolyoxetanes precipitated in media with ≥0.5 NaCl wt%. Subsequently, a formulation was developed using M9-mannitol medium that contained low salt and a small amount of mannitol. In the M9 mannitol medium containing 1 vol% LB, a density of 108 cells/mL was obtained overnight. E. coli and P. aeruginosa cultures grew well in the M9 mannitol media, while S. aureus had less growth. A modified Tryptic Soy Broth (TSB), a comparatively richer medium, proved suitable for S. aureus growth. The salt concentration in both media was similar (∼5 g/L). Although two different media formulations were used for the MIC tests, the extent of growth for all three bacterial strains was comparable. This mitigates a Cx-50 copolyoxetane salt effect for any particular strain.
MIC Assays
Minimum inhibitory concentrations for Cx-50 copolyoxetanes were determined by standard methods.9,10,20 Cx-50 copolyoxetane stock solutions were prepared in distilled water. Media (6 mL of M9 mannitol for E. coli and P. aeruginosa or TSB for S. aureus) were placed into Erlenmeyer flasks and 1:2 serial dilutions of Cx-50 stock solutions and 60 μL of the bacterial cultures (∼108 cfu/mL) were added sequentially to the flasks to establish an initial range for MICs. To determine MIC values with more precision, a series of tests was run above and below the initially established MIC with smaller concentration differences (1 μg/mL). Test mixtures were placed in shakers at 37 °C for 24 h. Triplicate determinations were carried out for each test series. Bacterial growth was examined by measuring solution optical density (λ = 600 nm) as well as visual examination of turbidity. This MIC assay is based on a change in optical density of 2 orders of magnitude and a visual observation of solution transparency.9,10,21
Kill Kinetics for C8-50 Copolyoxetane
Overall, C8-50 had the lowest MICs and was chosen for a study of kill kinetics. A 1:100 dilution of a bacterial suspension (106 cfu/mL) was prepared in Luria Broth (LB) and incubated at 37 °C until an optical density of ∼0.5 was attained (UV–vis). A 20 mL aliquot was centrifuged at 10000 rpm for 10 min, resulting in a bacterial pellet that was isolated and suspended in 20 mL of the respective growth medium used for the MIC tests (M9 mannitol for E. coli, P. aeruginosa, or TSB for S. aureus). The bacterial culture (107 cfu/mL) was divided in two; one served as a control, while C8-50 copolyoxetane was added to the other. At 0, 1, 2, and 3 h, 1 mL aliquots were taken from control and sample flasks and spun in a microcentrifuge at 1400 rpm for 3 min, resulting in a pellet. The supernatant was discarded and the pellet was resuspended in saline solution. This procedure was repeated twice to displace growth medium. A serial dilution was performed with 100 μL of the bacterial culture in saline. Aliquots of 100 μL were taken from each dilution and plated on Luria agar. After incubation at 37 °C for 24 h, the number of colonies was counted, compared with the control, and the percent kill was calculated.
Cytotoxicity
Human Cell Cytotoxity Assay
Cytotoxicity was tested using a CellTiter 96 Non-Radioactive Cell Proliferation Assay (Promega, Madison, WI). Human red blood cells (RBCs) were obtained from Innovative Research (Novi, MI) packed and transported on ice in a cooler (∼4 °C), refrigerated upon receipt, and used within one week. Immortalized human foreskin fibroblast cells and immortalized human dermal fibroblast cells were generously provided by Dr. Shawn Holt, Department of Pathology, and Dr. Kristoffer Valerie, Department of Radiation Oncology, both at Virginia Commonwealth University School of Medicine.
Human foreskin fibroblasts (HFF)/human dermal fibroblasts (HDF) were seeded in 96-well plates at a density of 1.5 × 103 cells/cm2. After incubation at 37 °C and 5% CO2 for 24 h, cells were exposed to different Cx-50 concentrations for 24 h followed by addition of tetrazolium dye solution and incubation for 4 h. During this period, living cells convert tetrazolium into a formazan product. A stop solution was added and the plate absorbance was read at 570 nm using a Biomate3 spectrophotometer (Thermo Electron Corporation). EC50 is determined by plotting absorbance vs concentration.22 From these plots, a maximum absorbance (plateau) was obtained. EC50 values were determined by locating the Cx-50 concentration corresponding to one-half of the maximum absorbance.
Hemolytic Activity Assay
One metric for defining hemolytic activity is HC50, the concentration of antimicrobial that kills 50% red blood cells (RBCs).12,20,23 A red blood cell lysis assay was developed following the procedure described by Palermo and Kuroda.10 RBCs (1 mL) were diluted with 9 mL of phosphate buffered saline, PBS, (i.e., 10 mM phosphate, 138 mM NaCl, pH 7.4, Sigma) and then centrifuged at 1000 rpm for 5 min. The resulting supernatant was removed using a pipet making sure not to disturb the RBCs and these steps were repeated two more times. The assay stock (90 μL) was mixed with each of the Cx-50 solutions (10 μL) using a sterile 96-well round-bottom polystyrene microplate to give a final solution of 3.3% v/v RBC. Based on a hemacytometer count, this was ∼108 red blood cells per mL.
PBS (10 μL) or Triton X-100 (10 μL, 1% v/v) were added to assay stock solutions as negative and positive hemolysis controls, respectively. Triton X-100, a nonionic surfactant is commonly used as a positive control in hemolysis assays.10 For Cx-50 copolyoxetane tests, 10 μL of stock solutions was added. The 96-well microtiter plate was secured in an orbital shaker at 37 °C and 250 rpm for 1 h and then centrifuged at 1000 rpm for 10 min. The resulting supernatant (10 μL) from each well was transferred to a new sterile 96-well round-bottom polystyrene microplate and diluted with PBS (90 μL).
Absorbance at 405 nm, characteristic of cell lysis,10 was determined using a Versamax EXT microplate reader (Molecular Devices, Sunnyvale, CA). Negligible absorbance was observed for the negative control (PBS) while the absorbance for the positive control was 3.9. The fraction of hemolysis is the absorbance for copolyoxetane treated wells divided by the average of readings from the positive control wells. Hemolysis was plotted as a function of Cx-50 concentration to obtain HC50, the copolyoxetane concentration that causes 50% hemolysis relative to the positive (Triton-X) control. Two separate trials were carried out in triplicate for each assay trial. Values reported are the average of the two trials.
Modeling
A geometric model was devised to estimate the number of polycations chemisorbed to a bacterium. This model is based on the dimensions of the bacterium, E. coli, and estimated chain dimensions of polycation C8-50 with a molecular weight of 5 kDa. E. coli dimensions are estimated from SEM images of bacteria grown in culture and adhered to a coverslip. These rod-shaped microbes are ∼0.5 μm in diameter and have a length of ∼5 μm.24,25 These dimensions are used for calculating total E. coli surface area.
The dimensions of polycation chains as globular random coils are calculated using the valence angle model.26 The diameter is estimated from root-mean-square (RMS) end-to-end distance, which is based principally on chain molecular weight.26 A single chain C8-50 molecular weight approximation is used with MW = 5 kDa giving a “diameter” of 2 nm. Details are provided in Supporting Information.
ChemDraw Ultra version 12.0.2.1076 was used to generate the image for C8-50 shown in Figure 6. Two MM2 energy minimizations were followed by an MM2 molecular dynamics using default parameters. The quaternary nitrogen was modeled as carbon; oxygen atoms are red; lone pairs are rose colored.
Figure 6.

ChemDraw3D structural representations for C8-50; the structure is an exactly alternating 16-mer end-capped with −CH3; MW = 4784 g/mol. Two MM2 energy minimizations were followed by an MM2 molecular dynamics using default parameters. The quaternary nitrogen was modeled as carbon; oxygen atoms are red; lone pairs are rose colored.
Results and Discussion
Previously we showed that increasing the linear charge density on C12-m copolyoxetanes 1 resulted in optimum biocidal activity at ∼50 mol%. Using MICs as a metric, an optimum range for biocidal efficiency was 40–60 mol % C12.1 To explore amphiphilic balance farther, the present study focuses on quaternary chain length keeping mol% quat constant. The compositions are designated Cx-50, where x = 2, 6, 8, 10, 12, 14, and 16. These copolyoxetanes were prepared via the intermediate P[(BBOx)(ME2Ox)-50:50–4], where polyol Mn ∼4 kDa. Thus, the mole percent alkylammonium bromide was in the middle of the range found most effective for C12. Preparation and characterization are briefly described followed by results from solution biocidal studies. A geometric model is then discussed for polycation chemisorption (PCC). Cytotoxicity was examined via hemolytic assays and for toxicity to HFF and HDF cell lines.
Copolyoxetane Preparation and Characterization
P[(BBOx)(ME2Ox)-50:50-4]
Cx-50 copolyoxetanes 1 were prepared in two steps as described previously.1 In brief, the intermediate P[(BBOx)(ME2Ox)-50:50-4] was prepared by ring-opening copolymerization using a BF3 catalyst and butane diol (BD) cocatalyst.27 Simultaneous addition of monomers gives copolyoxetanes with a mostly random architecture.281H NMR confirmed the 1:1 mol ratio of BBOx to ME2Ox (Figure S1). Assignments for chemical shifts are shown in Figure 1. Details for assignments and calculations are provided in Supporting Information, sections 1A and 1B.
Figure 1 shows the 1H NMR spectrum of P[BBOx-ME2Ox] after addition of trifluoroacetic anhydride (TFAA). Analysis of end group peaks provides Mn in two ways. First, a set of end group methylene peaks is associated with oxetane (α) and butoxy (β) groups (Figure 1, inset). The α/β integral ratio is 2.4, indicating that the BD cocatalyst competes for end group sites. Second, while the γ methyl end group peak is observed upfield from the main chain methyl peak for P[BBOx-ME2Ox] (Figure S2), it is downfield (0.85 ppm) from the main chain methyl peak after TFAA addition (Figure 1). Comparison of integrals for (α + β) and γ peaks, respectively, to main chain peaks gives 4.1 and 3.6 kDa for Mn (Table 2). Details for this calculation are provided in Supporting Information, section 1C.
Table 2. End Group Analysis for Determination of Mn.
| DPc |
Mn (kDa) |
GPCd (THF) |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| –CH3 | –CH2CH2– | αa | βa | α/ β | γb | α + β | γ | α + β | γ | Mn (kDa) | Mw (kDa) | PD | |
| shift/ppm (area, S) | 0.93 (15.98) | 1.6, 1.9 (10.29) | 4.26 (0.81) | 4.32 (0.34) | 2.4 | 0.85 (0.47) | 18 | 16 | 4.1 | 3.6 | 2.0 | 2.5 | 1.2 |
After addition of TFAA.
Neat P[(BBOx)-(ME2Ox)] copolyoxetane; Figure 1.
DP, degree of polymerization.
Excludes low MW peak assigned to cyclic tetramer.
Based on prior work, the inset in Figure 1 shows assignments for methylene groups adjacent to ME2Ox (α1) and BBOx (α2).28 The 1:1 ratio for these peaks confirms the 1:1 BBOx/ME2Ox ratio and supports our previous report showing that simultaneous addition of monomers results in random copolymers.28 Only one triplet is observed for β peaks as the intervening butoxy end group precludes differentiating butoxy end groups adjacent to oxetane (α) and butoxy (β) chain terminal groups.
A representative GPC for P[BBOx-ME2Ox] is shown in Figure S3. Oxetane ROP with BF3/BD catalysis results in cyclic byproducts.28−31 The sharp peak at longer retention volume is assigned to cyclic tetramer: MW calcd, 882; observed, 680 . The low molecular peak constitutes ∼12% of the total peak area. Considering polydispersity, GPC results, and end group analysis, the BBOx-ME2Ox copolyoxetane diols are designated P[(BBOx)(ME2Ox)-50:50-4], where 4 is molecular weight in kDa.
Cx-50 Copolyoxetanes
Quaternization with tertiary amines gave Cx-50 copolyoxetanes. The substitution reaction was carried out at 60 °C except for the reaction with C2 amine, which was carried out at 10 °C and utilized an ice jacketed condenser to contain the volatile amine. Complete substitution of C–Br to produce Cx-50 copolyoxetanes was established by 1H NMR spectroscopy (Table 1). Representative 1H NMR spectra for P[(BBOx)(ME2Ox)-50:50-4] and C8-50 are shown in Figure S1A and B, respectively. For C8-50 the absence of precursor peaks at 1.9 ppm (−CH2CH2CH2CH2Br) demonstrates complete quaternization. New peaks at 1.2–1.3 ppm (−CH2(CH2)6CH3 from C8) are characteristic for C8 substitution.
While it is clear that complete quaternization takes place, attempts to determine molecular weights on the quarternized copolyoxetanes was impeded by complex 1H NMR spectra after TFAA addition, suggesting chemical reaction. Several attempts to acquire GPC data were unsuccessful due to column adsorption. Quaternization adds 74–265 Da to the precursor P[(BBOx)(ME2Ox)-50:50-4] copolyoxetane molecular weight, which is a modest perturbation at most. To designate the quaternized copolyoxetanes, the designation Cx-50 is used based on the 50/50 BBOx-ME2Ox ratio in the precursor and the understanding that there may be a small cyclic polycation content.
Figure S4 shows FTIR spectra for P[(BBOx)(ME2Ox)-50:50-4] and C8-50 copolyoxetane. The C–Br peak appears at 650 cm–1 for the precursor but is absent for C8-50.
Cx-50 copolyoxetanes are viscous liquids at ambient temperature with Tgs ranging from −40 to −60 °C (Table 1, Figure 2). Tgs for C2 to C12 are 10–20 °C higher than P[(BBOx)(ME2Ox)-50:50-4], −71 °C, which is attributed to polar interactions of quaternary salt moieties. The low Cx-50 Tgs compare favorably with ionic liquid analogs reported by Ohno (−64 to −72 °C).32,33 The low Tgs for Cx-50 copolyoxetanes emphasize high molecular mobility due to a combination of ME2Ox side chains and ether functionality in the main chain and in the linkage to the alkyl side chain.
Figure 2.
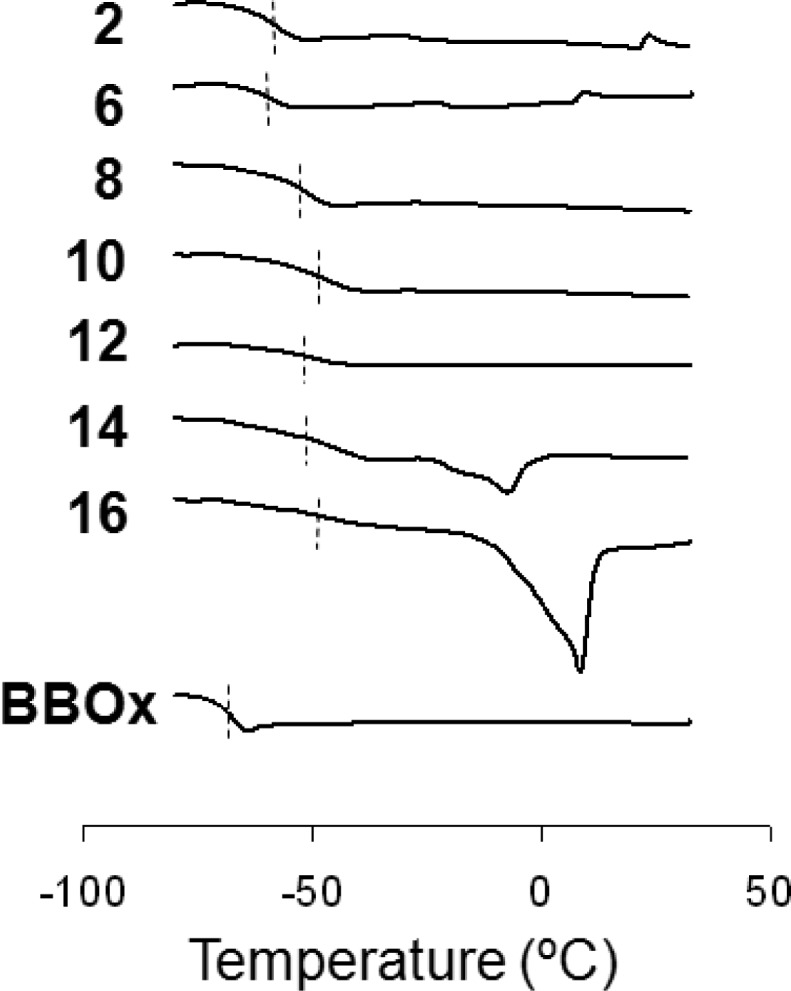
MDSC thermograms for Cx-50 and the intermediate P[(BBOx)(ME2Ox)-50:50-5.7]. Melting endotherms: C14-50, 5.5 J/g; C16-50, 22.4 J/g).
Tgs for C14-50 and C16-50 are broad, while endotherms at −7 and 10 °C are attributed to Tm for phases formed by C14 and C16 side chains (Table 1, Figure 2). Compared to poly(n-alkyl)acrylates, for which side chains with eight atoms crystallize,34 the chain length for onset of crystallization is x = 14 for Cx-50 copolyoxetanes. The longer side chain length required for C14-50 and C16-50 crystallization is ascribed to less favorable packing for copolyoxetanes that have one side chain per four main chain atoms compared to one side chain per two main chain atoms in polyacrylates.
Antimicrobial Tests
MIC Assays
Three pathogenic bacteria were the subject of minimum inhibitory concentration (MIC) tests: Gram(−) E. coli and P. aeruginosa and Gram(+) S. aureus (Table 3). MICs for DTAB1 are included for comparison with those previously reported by Seifert.2 This comparison shows that MICs vary depending on bacterial strain and details of laboratory test procedures. While interlaboratory test results may vary, intralaboratory tests and comparisons are generally reliable.
Table 3. Minimum Inhibitory Concentrations for Cx-50 Copolyoxetanesa.
| Cx-50 | E. coli | S. aureus | P. aeruginosa |
|---|---|---|---|
| C2-50 | >90 | >90 | >120 |
| C6-50 | 6.7 (±0.6) | 2.7 (±0.6) | 30.7 (±0.6) |
| C8-50 | 4.0 (±1.2) | 2.0 (±0) | 24.0 (±2.8) |
| C10-50 | 6.3 (±0.6) | 3.3 (±0.6) | 24.7 (±1.2) |
| C12-50 | 7.3 (±0.6) | 8.7 (±0.6) | 27.7 (±2.5) |
| C14-50 | 10.3 (±0.6) | 10.7 (±0.6) | 31.7 (±1.5) |
| C16-50 | 13.3 (±1.2) | 12.7 (±2.1) | 41.0 (±3.6) |
| DTABb | 17 (6) | 37 (6) | 103 (15) |
| DTABc | 77 | 19 | >154 |
The effect on MIC for changing the Cx-50 quaternary alkyl side chains is shown in Figure 3. The most effective antimicrobials are C6-50, C8-50, and C10-50, with C8-50 at the center of a shallow minimum. MICs for C8-50 are 4 μg/mL (E. coli), 2 μg/mL (S. aureus), and 24 μg/mL (P. aeruginosa). Overall, C8-50 is the most effective antimicrobial against the three bacterial strains tested.
Figure 3.
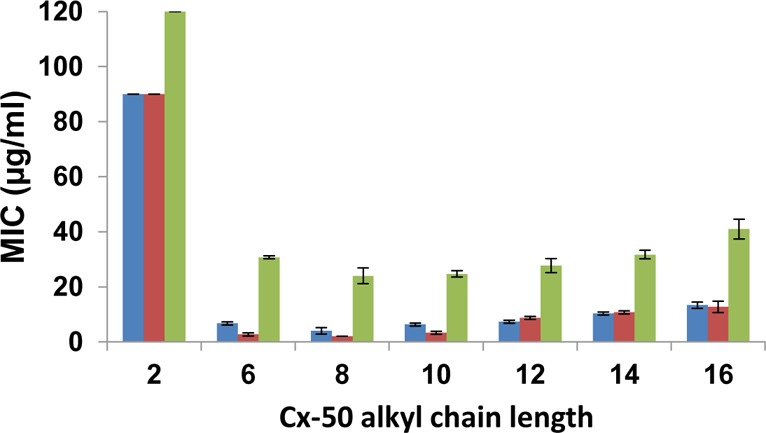
Minimum inhibitory concentrations as a function of quaternary chain length for Cx-50: blue, E. coli; red, S. aureus; green, P. aeruginosa.
Relatively high MIC values are observed for P. aeruginosa, which is the most resistant bacterium to all Cx-50 copolyoxetanes. C8-50 (24 μg/mL) and C10-50 (25 μg/mL) have similar MICs, which may be compared with 26 μg/mL for C12-60.1 The resistance of P. aeruginosa has been demonstrated for most antimicrobials.35 Additional lipopolysaccharides and other mechanisms are thought to decrease the interaction of antimicrobials with the membrane. Among all Gram(−) bacteria, P. aeruginosa has an overall outer cell membrane permeability that is 12–100× lower than E. coli.36−38
Depending on the bacterial strain, an increase in MIC of 4–45× is observed for C2-50 compared to C8-50 (Table 3). MICs for C2-50 are >90 μg/mL for Gram(−) E. coli and Gram(+) S. aureus and >120 μg/mL for P. aeruginosa (Figure 3). For C2-50 amphiphilic balance is clearly shifted in a way that precludes effective conjugation with bacterial membranes. The C4 amine required for C4-50 was not available when other Cx-50 compositions were prepared.
The MIC assay results in Table 3 and Figure 3 show that the Cx-50 copolyoxetanes are selective for E. coli and S. aureus compared to P. aeruginosa. Regular trends in antimicrobial effectiveness give evidence of internal consistency for MIC tests. However, variability of test results with different bacterial strains has been noted.2 In Table 3, MICs for dodecyltrimethyl ammonium bromide (DTAB) against E. coli 17 (6), S. aureus 37 (6), and P. aeruginosa 103 (15) in μg/mL are listed (these results were published previously).1 For DTAB against different strains of the same bacteria, Seifert reported 77, 19, and >125 μg/mL, respectively. Thus, intercomparisons of antimicrobial effectiveness are made with caution due to different bacterial strains and growth conditions and test protocols.
The results shown in Figure 3 may be compared with those for a series of amphiphilic polyoxanorbornenes with varying quaternary alkyl side chains (6, Table 5).39 With R = ethyl or butyl the MIC was 200 μg/mL for E. coli, while for hexyl and octyl analogs the MIC decreased to 12.5 and 4 μg/mL, respectively. For R = decyl, the MIC increased to 12.5 μg/mL. A similar trend was observed for Gram(+) B. subtilis. Considering the different main and side chain structures for 1 and polyoxanorbornenes 6, the trends are similar.
The relationships of MICs to structure are specific to polymer classes. Palermo noted that changing the butyl group in copolyacrylates 5 (Table 5) to a methyl group resulted in a 100 times increase in MIC for E. coli,10 while Savage found that a long alkyl chain for the molecular polycation CSA-13 was essential for a low MIC against E. coli.16 For some polymers, a “sweet spot” in amphiphilic balance that brings about low MICs is not easily attained or is dependent on bacterial strain. The lowest MIC reported by Sen for amphiphilic pyridinium methacrylate copolymers 4 (Table 5) against an E. coli challenge was 50 μg/mL for a butyl derivative, while a hexyl polymer was best against Gram(+) B. subtilis (30 μg/mL).23
Interestingly, antimicrobial effectiveness toward Gram(−) E. coli and P. aeruginosa and Gram(+) S. aureus differs little in the C6-50 to C10-50 compositional range (Figure 3). The uniformly high biocidal activity of C8-50 may portend low MICs for a broad range of pathogenic bacteria as there does not seem to be strain selectivity for polycations. Methacrylate copolymers displayed antimicrobial activity against drug-resistant S. aureus (MRSA) with the same level of activity as a drug susceptible laboratory strain.40
Killing Kinetics Assay
Although rapid bacterial kill is of clear importance, during a 20 year interval following the pioneering research of Ikeda in the early 1980s,41,42 little was published. Recent studies from the Kuroda laboratory on acrylate polycations have shed light on this subject11,40 Following Ikeda’s method, in view of antimicrobial effectiveness (Figure 3), C8-50 was selected for a kill kinetics assay.
Figure 4 shows bacterial kill over three hours for C8-50 at a concentration of 5× MIC. Three separate experiments were performed in triplicate yielding low standard deviations. Bacterial challenges were ∼108 cfu/mL Gram(−) E. coli and P. aeruginosa and Gram(+) S. aureus. Log reductions (s.d.) of 7 (0), 3.0 (0.2), and 4.5 (0.1) were obtained for S. aureus, E. coli, and P. aeruginosa, respectively, at the end of 1 h.
Figure 4.

Kill kinetics at 5 times MIC for C8–50: (A) E. coli; (B) P. aeruginosa; (C) S. aureus.
The kill kinetics study shows that the C8-50 affects a minimum log 2 kill of bacterial cells during the first hour. The MIC order for C8-50 MICs is S. aureus < E. coli ≪ P. aeruginosa, but the kill rate order is S. aureus ≫ P. aeruginosa > E. coli. Choosing 5× MIC resulted in a C8-50 concentration of 125 μg/mL against P. aeruginosa, while for E. coli the concentration was only 20 μg/mL. The higher C8-50 concentration against P. aeruginosa apparently accounts for the increased rate of kill. A more detailed study is required to elucidate details of concentration dependence.
The results in Figure 4 shows that at 20 μg/mL (5× MIC) 100% kill, that is, a 2 log reduction, was found for E. coli after 1 h followed by a 4 log reduction at 3 h. This rate may be compared with kill kinetics for PB27, a copolycation derived from poly(aminoethyl-co-butyl)methacrylate with 25 mol % butyl content (MW 2.7 kDa).11 PB27 has a MIC against E. coli of 16 μg/mL compared with 4 μg/mL for C8-50. The 2× MIC concentration used for kill kinetics for PB27 (32 μg/mL) was somewhat higher than that for C8-50 (20 μg/mL). In any event, PB27 affected a 3 log kill in 30 min, which is a more rapid than C8-50.
PB27 has a much higher MIC for S. aureus (250 μg/mL) compared to 2 μg/mL for C8-50. Despite the high 500 μg/mL for PB27 (2× MIC) used in a kill kinetics study, the 7 log/1 h kill rate for C8-50 is comparable. Looking forward, this result is important considering dose and potential side effects for polycation antimicrobials. However, several caveats must be applied including differences in bacterial strains, stages of growth, and growth media.
Kill kinetics for C8-50 may be compared with those obtained for the molecular polycation designated CSA-13.17 CSA-13 was tested at 10× MIC (10 μg/mL) against a clinical isolate of vancomycin-resistant S. aureus. CSA-13 affected a 3.8 log reduction/4 h and 4.8 log reduction/8 h.17,43 For these tests the bacterial challenges were 3.2 × 108 cfu/mL. This concentration is higher than the C8-50 challenge of ∼107 cfu/mL. By comparison, C8-50 at 5× MIC (10 μg/mL) effects a 7 log reduction against S. aureus (ATCC-25904) at 1 h, while CSA-13 affects a 3.2 log reduction in 3 h (Table S2).
Finally, in view of the limited development of heuristics for bacterial kill rate, the kill rates for antibiotics are noted. Ciprofloxacin, ofloxacin, sparfloxacin, and trovafloxacin were tested against two strains of S. aureus.44 For both ciprofloxacin and ofloxacin, at 8× MIC (4 μg/mL), a log reduction of <2.5 was observed at the end of 2 h for a starting inoculum of ∼106 cfu/mL.
Geometric Model for Polycation Bacterial Chemisorption
The killing mechanism for polycations including antimicrobial polypeptides (AMPs) is explained by ionic and hydrophobic interactions between the negatively charged bacterial cell membrane and multiple ammonium moieties.45 After or concurrently with polycation chemisorption, disruption of the phospholipid bilayer includes diffusion through the cell wall and binding to the cytoplasmic membrane. This leads to further changes due to osmotically driven release of cytoplasmic species such K+ and water incursion into the cell that result in cell death. This mechanism was described by Ikeda in the early 1980s.41,42,45 Subsequently, additional details and insights have been presented.17,19
Ikeda noted the greater release of K+ for polycations compared to single-charge quaternary moieties.42,45 With singly charged species, one mole of K+ salt may be released from the bacterium [B(nK+)nX–] per mole of ammonium (eq 1). For polycations the number of moles of K+ salt released can be as great as the number of charges m per polycation. Equation 2 illustrates a polycation with m charges releasing m × n equivalents of K+.
| 1 |
| 2 |
Several models have been proposed for polycation kill including carpet,46 barrel stave,47 and toroidal pore.48 Each of these models includes electrostatic interactions between polycations and anionic bacterial phospholipids leading to pore formation. While the amphiphilic nature of polycations is important to pore formation, the mechanism of interaction for initial inactivation is not well understood.49
For the carpet model, the polycation builds up on the bilayer bacterium surface. At a sufficiently high concentration, the polycation disturbs the lipid bilayer, induces the formation of micelles and produces pores.50 For the barrel stave model, α-helical peptides arrange into a bundle and insert into the bacterial membrane creating pores. Hydrophobic regions associate with lipids, while hydrophilic moieties create an inner pore.50 In the torodial pore mechanism, membrane disruption occurs as polycations insert individually.49 Polycations aggregate and interact with the lipid head groups resulting in continuous bending of the lipid monolayer. Both attached polycations and the lipid head groups create the lining of the pore.50
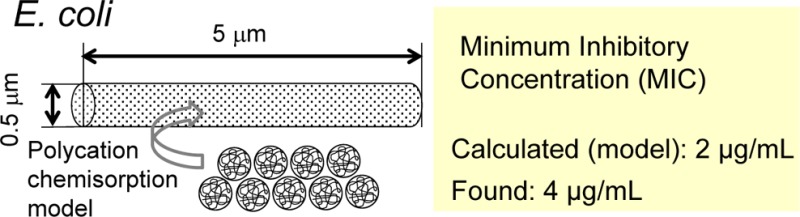
To provide additional insight into polycation interactions with bacterial membranes, a geometric model is proposed herein that provides an estimate of the maximum number of polycations that can chemisorb to a bacterium. This geometric model is based on the dimensions of the bacterium E. coli and chain dimensions estimated for polycation C8-50 with a molecular weight of 5 kDa.
E. coli dimensions are estimated from SEM images of bacteria grown in culture and adhered to a coverslip. These rod-shaped microbes are ∼0.5 μm in diameter and have a length of ∼5 μm.24 These dimensions are used for calculating the total E. coli surface area (Figure 5). The bacterium is assumed to be planktonic during a MIC test (1 bacterium per cfu).
Figure 5.
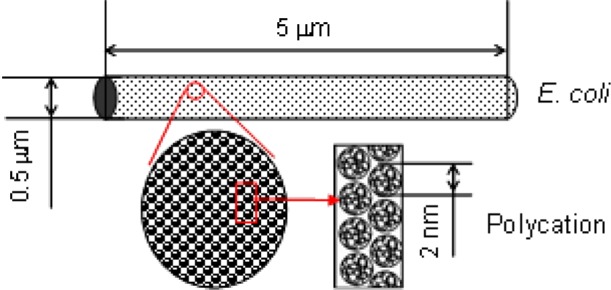
Geometric model for chemisorbed polycations: total surface area of E. coli, 8 μm2; polycation cross-section area, 3 nm2.
Figure 6 shows a ChemDraw3D structural representation for C8-50. An exactly alternating 16-mer, P[(ME2Ox)8(C8)8-50/50], MW 4784 Da, was chosen and end-capped with −CH3. An MM2 energy minimization was followed by an MM2 molecular dynamics simulation and a final energy minimization using default program parameters. The simulation yields a globular structure with ME2Ox, identified by red oxygen atoms, and C8 (gray) projecting from a polyoxetane core. The concentration of ME2Ox and C8 groups at the surface of the globular structure indicates ready availability for interaction with a bacterial membrane.
The dimensions of polycation chains as globular random coils may be calculated using the valence angle model.26 The diameter is estimated from root-mean-square (RMS) end-to-end distance, which is based principally on chain molecular weight.26 A single chain C8-50 was used with the molecular weight approximation MW = 5 kDa. The resulting RMS end-to-end distance gives a “diameter” of 2 nm (Figure 5). Because long side chains are not adequately taken into account by this model, the effective diameter is underestimated. Use of a more complicated model is not warranted due to polydispersity and chain expansion in solution due to side chain and solvent interactions. Using the calculated E. coli surface area, 2.6 × 106 polycation chains are required to cover the bacterium. The concentration of polycations in solution prior to chemisorption that corresponds to 2.6 × 106 C8-50 polycation chains is 2 μg/mL (details are provided in Supporting Information).
Considering the approximations for polycation chemisorption (PCC), it is surprising that a calculation based on geometric considerations gives a C8-50 concentration within a factor of 2 of the MIC, 4.0 (±1.2) μg/mL for E. coli (Figure 3, Table 2). With longer alky chain lengths, the MIC for Cx-50 copolyoxetanes increases regularly from C10-50 (6.3 μg/mL) to C16-50 (13.3 μg/mL). The MIC for C6-50 is 6.7 μg/mL, leaving C8-50 as the most effective antimicrobial against E. coli. Previously, Chakrabarty found MICs in the range 5–6.3 μg/mL for C12-m copolyoxetanes (x = 43, 50, 60, 87, 100) showing that biocidal effectiveness is somewhat more sensitive to alkyl chain length than mol % C12.1
Complete chemisorption depicted in Figure 5 is unlikely due to steric considerations. The calculated concentration for C8-50 chemisorption (∼2 μg/mL) is lower than the 4–7 μg/mL MIC range for C6-50–C12-50, which are most effective against E. coli. For complete kill, the minimum biocidal concentration MBC is typically a factor of 2–3 higher than MIC. The results from the PCC model are consistent with a sorption–desorption equilibrium in solution and that those polycations with optimum amphiphilic balance adsorb to the bacterial wall most strongly and effect maximum disruption at low concentrations.
For polycation C2-50 with a high MIC (>90 μg/mL for E. coli and S. aureus, >120 μg/mL P. aeruginosa) amphiphilic balance that includes not only charge but van der Waals interactions associated with the quaternary alkyl chain must result in inefficient chemisorption. Weak chemisorption lessens subsequent membrane disruption through pore formation or other mechanisms.40,51,52
The PCC calculation may be thought of as a calculation of polycation chemisorption based on the carpet model50 and is consistent with the thermodynamic driving force described by eq 2. The conceptual basis is related to layer-by-layer self-limiting deposition of polycations and polyanions.53,54 As such, the calculation asks “what if” complete chemisorption occurs. It seems unlikely that complete chemisorption and polysalt formation (eq 2) is required for pore formation and osmotic processes leading to cell membrane destabilization. MICs for Cn-50 polycations are consistently higher than that for PCC as are MICs for other polycations (Table 6). However, the uncertainties associated with MIC tests have been noted. At the least, PCC may offer a basis for additional experimentation.40
Table 6. Comparison of Cell Toxicity and Selectivity for Representative Polymeric and Molecular Polycationsa.
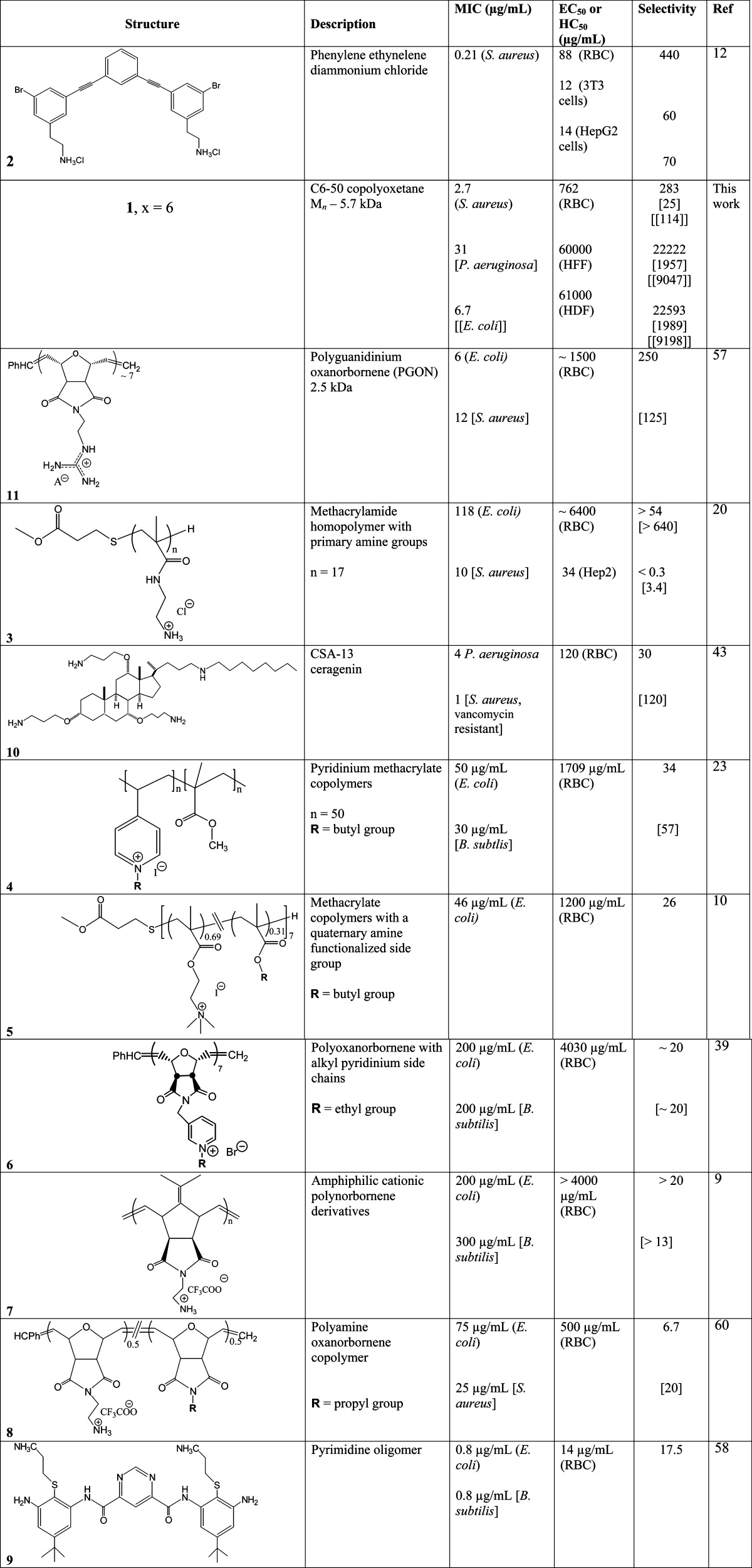
Selectivities use published MIC (μg/mL) and EC50 or HC50 (μg/mL) data. Where MICs for multiple bacteria are reported, S is reported in the sequence: no bracket, one bracket, two brackets. This is a notation that corresponds to the bacteria MICs.
Cytocompatibility
Cytotoxicity for HFF and HDF Cell Lines
Despite obvious differences, Cx-50 polycations may be regarded as analogs of naturally occurring antimicrobial polypeptides such as defensins and magainins.55 Thus, Cx-50 polycations are a class of synthetic mimics of antimicrobial peptides (SMAMPs). It has long been recognized that the antibacterial activity of polypeptides such as defensins is due to effects on microbial membranes. Defensin-like peptides, being positively charged, interact with negatively charged components of microbial membranes that include lipopolysaccharides (LPS) in Gram-negative bacteria, polysaccharides (teichoic acid) in Gram-positive bacteria, and phospholipids (phosphotidyl-glycerol).18 Since these membrane components are not found in mammalian cells, defensins in general appear to be “electrostatically specific” for prokaryotic cells.56
C6-50, C8-50, and C10-50 were tested for cytotoxicity toward mammalian cell lines as they were the most effective antimicrobials (Figure 3). Living cells convert tetrazolium dye into a formazan product. At a given time, absorbance at 570 nm for the dye provides a measure of cell viability. The concentration corresponding to 50% cell lysis, EC50, is determined from a plot of Cx-50 absorbance versus concentration.22 From these plots, a maximum absorbance (plateau) was obtained. EC50 was the Cx-50 concentration corresponding to one-half-maximum absorbance (Table 4).
Table 4. Cytotoxicity for Cx-50 Copolyoxetanes.
| EC50a (μg/mL) |
HC50 (μg/mL) | ||
|---|---|---|---|
| Cx-50 | HFF | HDF | RBC |
| C6-50 | 60000 | 61000 | 762 |
| C8-50 | 42000 | 41000 | 303 |
| C10-50 | 36000 | 37000 | 353 |
Rounded to the nearest thousand.
C6-50 has the least toxicity toward fibroblasts (60000 μg/mL for HFF and 61000 μg/mL for HDF). Toxicity for both cell lines is similar and increases with increasing length of the alkyl chain. Considering antimicrobial effectiveness (Figure 3) and this in vitro analysis of cytotoxicity, it appears that fibroblast cytotoxicity is more sensitive to quaternary chain length than is MIC.
Hemolytic Activity Assay
Percent lysis for human red blood cells (RBCs) versus Cx-50 concentration is shown in Figure 7. At the highest concentration for this test series (50 μg/mL), C6-50 and C12-50 have the lowest lysis (∼9%), while C8-50 affects 20% lysis. Lysis for C10-50 is intermediate. The modest trend for increasing lysis with increasing C6-50, C10-50, and C12-50 concentration indicates that the concentration affecting 50% human RBC lysis (HC50) is greater than 50 μg/mL. Even the poorest performer, C8-50 causes <25% lysis at 50 μg/mL.
Figure 7.

Percent RBC lysis vs Cx-50 concentration: (A) C6-50; (B) C8-50; (C) C10-50; (D) C12-50. Lines are meant to guide the eye.
To correlate with literature studies on antimicrobials, higher concentrations (to 5000 μg/mL) Cx-50 copolyoxetanes with x = 6, 8, and 10 were tested on RBCs (Figure 8). C6-50 was the least hemolytic with an HC50 of 762 μg/mL (Table 3). These hemolytic assays confirm the relatively benign nature of Cx-50 copolyoxetanes toward human RBCs (Figure 8).
Figure 8.

Percent RBC lysis as a function of Cx-50 concentration: (A) C6-50; (B) C8-50; (C) C10-50.
Selectivity
As a measure of the concentration required for lysing 50% human cells compared to the minimal concentration for inhibiting bacterial growth (MIC) selectivity, S, is defined by eq 3. High selectivities mean effective antimicrobial activity while minimizing effects on human cells. Selectivities provide an optimistic estimate of antimicrobial effectiveness, as the MIC is typically 3–4× lower than the minimal biocidal concentration, MBC.1 Nevertheless, selectivities provide a guide for potential in vivo applications of antimicrobials.
| 3 |
C6-50, C8-50, and C10-50 copolyoxetane selectivities for fibroblasts are generally high, while RBC selectivities are moderate (Table 5). Selectivities for human fibroblasts follow the EC50 trend with C6-50 and C8-50 being more selective compared to C10-50. For the tested strains of bacteria, RBC C6-50 selectivity was the highest overall and was included in Table 6 for comparison with polymer and molecular polycations. The highest C6-50 selectivity (283) was for S. aureus.
In detail, selectivities follow the order S. aureus (C6-50, 2300 to C10-50, 11000) > E. coli (C6-50, 9200 to C10-50, 5900) ≫ P. aeruginosa (C6-50, 2000 to C10-50, 1500). The lower selectivities for P. aeruginosa stem from MICs that are 5–6× higher than those for E. coli and S. aureus.
Comparative Antimicrobial Effectiveness and Cytotoxicity
The results for Cx-50 copolyoxetanes are compared with selected studies on molecular and macromolecular polycation antimicrobials in Table 6. The selected antimicrobials are optimum compositions from the cited references. The compositions follow an ordering that places those with highest selectivities toward the top of the table. Some judgment was exercised in this regard, as a given polycation may have variable selectivity depending on the tested organisms.
Phenylene ethynelene dication 2 has two primary alkylammonium functions per uncharged group, which is about double the linear charge density of Cx-50 copolyoxetanes (achieved with quaternary alkylammonium). An HC50 of 88 μg/mL against human RBCs was reported.12 For S. aureus dication 2 has a selectivity of 440, which is somewhat greater than that found for C6-50 (283). On the other hand, 2 has relatively low selectivity for other cell lines tested (60, 3T3; 70, HepG2). This comparison shows how radically different structures such as 2 (molecular aromatic, primary amine, 2:1 charge per neutral group, two C–Br bonds)12 and 1, C6-50, aliphatic, quat, 1:1 charge per neutral group) are both good antimicrobials with low cytotoxicity.
Polyguanidinium oxanorbornene (PGON) 11 was designated “non-membrane disrupting” due to a low RBC cytotoxicity, HC50 = 1500 μg/mL, and minimal vesicle–dye leakage.57 PGON MIC for E. coli is 6 μg/mL, which leads to a very good selectivity of 250 (Table 6). Higher MICs were found for other tested bacteria with correspondingly lower selectivities.
Like molecular polycation 2, 3 has primary alkylammonium functionality but is a low molecular weight polymethacrylamide (Table 6). Polymer 3 has an HC50 of 6400 μg/mL for human RBCs and an EC50 of 34 μg/mL for Hep2 cells.20 This polymer without alkyl side chains on N has the least toxicity toward mammalian cells, while copolymers with a butyl or hexyl pendant group exhibit high cytotoxicity. A copolymer version of 5 with 49 mol % cationic charge (designated 5a) has a comparable charge density and alkyl chain length to that of C6-50. However, 5a also has a low HC50 (<12 μg/mL) compared to 762 μg/mL for C6-50. The selectivity of 5a for human RBCs compared to E. coli and S. aureus is 0.15 compared to 114 (E. coli) and 283 (S. aureus) for C6-50. Once again our imperfect knowledge of polycation/cell interactions makes correlations difficult.
Pyridinium methacrylate copolymers 4 have a similar charge density compared to the Cx-50 copolyoxetanes.23 Copolymer 4 with butyl N-side chains has the least cytotoxicity toward human red blood cells (HC50 = 1709 μg/mL). However, relatively high MICs of 50 μg/mL (E. coli) and 30 μg/mL (B. subtilis) make the copolymer less selective (34 for E. coli and 57 for B. subtilis) toward human red blood cells.
Methacrylate copolymers 5 with quaternary amine functionalized side group exhibit high HC50 (1200 μg/mL).10 A high MIC (46 μg/mL) against a strain of E. coli leads to a low selectivity (26). Copolymer 5 with a 56 mol % quaternary ammonium charge has an HC50 of 67 μg/mL that gives a selectivity of 2, while the lowest selectivity (65) for the Cx-50 copolyoxetane series was for C8-50 (HC50 = 303 μg/mL). As above, subtle and as-yet not understood interactions for amphiphilic copolycation antimicrobials confound an easy interpretation of such comparisons.
Biocidal properties and cytotoxicity of polyoxanorbornenes 6 with alkyl pyridinium side chains gave an HC50 of 4030 μg/mL (R = ethyl), but again, selectivity was low (20) because of relatively high MICs for E. coli and B. subtilis.39 For R = hexyl, octyl, and decyl side chains, polyoxanorbornenes 6 with a 10 kDa molecular weight had lower HC50 values (202 μg/mL, <50 μg/mL, and <50 μg/mL, respectively) compared to the corresponding 3 kDa polymer. Amphiphilic cationic polynorbornenes 7 with a molecular weight of 1.6 kDa was the least hemolytic toward human red blood cells compared to its other structural analogs.9 Polymer 7 has an HC50 of >4000 μg/mL but rather poor antibacterial characteristics leads to low selectivities of >20 (E. coli) and >13 (B. subtilis).
Polyamine oxanorbornene copolymer 8 with a 50 mol % of cationic charge gave an HC50 of 500 μg/mL against human red blood cells and a selectivity of 7 (E. coli) and 20 (S. aureus).58 Primary amine functionalized pyrimidine oligomer 9 has good antimicrobial properties with low MIC but it is also toxic toward human red blood cells (HC50 = 14 μg/mL) which gave a selectivity of 17.5.58
An evaluation of the molecular polycation CSA-13 yielded a relatively low MIC (4 μg/mL) for P. aeruginosa (10, Table 5).59 However, CSA-13 has an HC50 of ∼120 μg/mL for RBCs giving a selectivity of 30.43
Conclusion
Previously we showed that increasing the linear charge density on C12-m copolyoxetanes 1 resulted in optimum biocidal activity at ∼50 mol %.1 In the present study, changing the quaternary alkyl chain length (x) for Cx-50 copolyoxetanes is found to influence biocidal activity. Optimum antimicrobial effectiveness is found for C8-50 (Figure 3), although unlike many other polycations and considering variables in biotesting, results for C6-50 and C12-50 were similar. In fact, it is remarkable that biocidal effectiveness is fairly insensitive to alkyl chain length in this range.
A geometric model provides an estimate of the maximum number of polycations that can chemisorb to E. coli. The PCC model is based on bacterial surface area and chain dimensions estimated for polycation C8-50 (5 kDa) and does not take into account polydispersity and chain expansion in solution due to side chain and solvent interactions. Considering approximations employed, it is surprising that the calculated C8-50 concentration required to form a monolayer (2 μg/mL) is within a factor of 2 of the MIC, 4.0 (±1.2) μg/mL for E. coli. This result suggests exploring the extent of polycation chemisorption in conjunction with kill kinetics to better understand polysalt formation described by eq 2 and the relationship to pore formation and osmotic processes leading to cell membrane destabilization due to stress.40a,b It is possible that the PEG/quat combination is synergistic with quaternary charge driving chemisorption and hydrated PEG providing a pathway for osmotically driven water and salt diffusion.
An investigation of in vitro cell compatibility has shown that the Cx-50 copolyoxetanes (x = 6, 8, and 10) have low toxicity toward human fibroblast cells. Selectivities for C6-50, C8-50, and C10-50 copolyoxetanes relative to MICs for E. coli and S. aureus are particularly high, while those relative to P. aeruginosa are moderate due to relatively high MICs. In particular, C6-50 exhibits low hemolysis of human red blood cells and has high selectivities for human cells compared to the tested bacteria. The water solubility of these polymers coupled with excellent antimicrobial behavior and cell compatibility makes these novel copolyoxetanes candidates for therapeutic applications.
Acknowledgments
The authors thank the National Science Foundation (DMR: Grants DMR-0207560, DMR-0802452, and DMR 1206259) for support of this research. D.E.O. thanks Public Health Service Grant AI-19146 from the National Institute of Allergy and Infectious Disease.
Supporting Information Available
Additional analytical details. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Chakrabarty S.; King A.; Kurt P.; Zhang W.; Ohman D. E.; Wood L. F.; Lovelace C.; Rao R.; Wynne K. J. Biomacromolecules 2011, 12, 757. [DOI] [PubMed] [Google Scholar]
- LaDow J. E.; Warnock D. C.; Hamill K. M.; Simmons K. L.; Davis R. W.; Schwantes C. R.; Flaherty D. C.; Willcox J. A. L.; Wilson-Henjum K.; Caran K. L.; Minbiole K. P. C.; Seifert K. Eur. J. Med. Chem. 2011, 46, 4219. [DOI] [PubMed] [Google Scholar]
- Kurt P.; Wood L.; Ohman D. E.; Wynne K. J. Langmuir 2007, 23, 4719. [DOI] [PubMed] [Google Scholar]
- Kurt P.; Wynne K. J. Macromolecules 2007, 40, 9537. [Google Scholar]
- Zasloff M. Proc. Natl. Acad. Sci. U.S.A. 1987, 84, 5449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zasloff M.; Martin B.; Chen H. C. Proc. Natl. Acad. Sci. U.S.A. 1988, 85, 910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurt P.; Gamble L. J.; Wynne K. J. Langmuir 2008, 24, 5816. [DOI] [PubMed] [Google Scholar]
- Albert M.; Feiertag P.; Hayn G.; Saf R.; Honig H. Biomacromolecules 2003, 4, 1811. [DOI] [PubMed] [Google Scholar]
- Ilker M. F.; Nusslein K.; Tew G. N.; Coughlin E. B. J. Am. Chem. Soc. 2004, 126, 15870. [DOI] [PubMed] [Google Scholar]
- Palermo E. F.; Kuroda K. Biomacromolecules 2009, 10, 1416. [DOI] [PubMed] [Google Scholar]
- Sovadinova I.; Palermo E. F.; Urban M.; Mpiga P.; Caputo G. A.; Kuroda K. Polymers 2011, 3, 1512. [Google Scholar]
- Tew G. N.; Clements D.; Tang H.; Arnt L.; Scott R. W. Biochim. Biophys. Acta, Biomembr. 2006, 1758, 1387. [DOI] [PubMed] [Google Scholar]
- Lebedeva M. N.Microbiology. 2nd ed; Meditsina, 1969.
- Todar K. G.http://textbookofbacteriology.net/.
- Mazel D.; Davies J. Cell. Mol. Life Sci. 1999, 56, 742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C.; Budge L. P.; Driscoll C. D.; Willardson B. M.; Allman G. W.; Savage P. B. J. Am. Chem. Soc. 1999, 121, 931. [Google Scholar]
- Lai X. Z.; Feng Y. S.; Pollard J.; Chin J. N.; Rybak M. J.; Bucki R.; Epand R. F.; Epand R. M.; Savage P. B. Acc. Chem. Res. 2008, 41, 1233. [DOI] [PubMed] [Google Scholar]
- Mor A. Drug Dev. Res. 2000, 50, 440. [Google Scholar]
- Tashiro T. Macromol. Mater. Eng. 2001, 286, 63. [Google Scholar]
- Palermo E. F.; Sovadinova I.; Kuroda K. Biomacromolecules 2009, 10, 3098. [DOI] [PubMed] [Google Scholar]
- Beckloff N.; Laube D.; Castro T.; Furgang D.; Park S.; Perlin D.; Clements D.; Tang H.; Scott R. W.; Tew G. N.; Diamond G. Antimicrob. Agents Chemother. 2007, 51, 4125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stratton T. R.; Rickus J. L.; Youngblood J. P. Biomacromolecules 2009, 10, 2550. [DOI] [PubMed] [Google Scholar]
- Sambhy V.; Peterson B. R.; Sen A. Angew. Chem., Int. Ed. 2008, 47, 1250. [DOI] [PubMed] [Google Scholar]
- NIAID, National Institute of Allergy and Infectious Diseases, National Institute of Health; http://www.niaid.nih.gov/SiteCollectionImages/topics/biodefenserelated/e_coli.jpg.
- Shu A.-C.; Wu C.-C.; Chen Y.-Y.; Peng H.-L.; Chang H.-Y.; Yew T.-R. Langmuir 2008, 24, 6796. [DOI] [PubMed] [Google Scholar]
- Chanda M.Advanced Polymer Chemistry; Marcel Dekker: New York, 2000. [Google Scholar]
- Malik A. A.; Archibald T. G.. Solvent-Free Process for the Preparation of Mono-Substituted Fluorinated Oxetane Monomers. Patent US6037483, 2000.
- Fujiwara T.; Makal U.; Wynne K. J. Macromolecules 2003, 36, 9383. [Google Scholar]
- Fujiwara T.; Wynne K. J. Macromolecules 2004, 37, 8491. [Google Scholar]
- Zhang W.; Henke D.; Presnall D.; Chakrabarty S.; Wang C.; Wynne K. J. Macromol. Chem. Phys. 2012, 213, 1225. [Google Scholar]
- Schulte B.; Dannenberg C. A.; Keul H.; Moller M. J. Polym. Sci., Part A: Polym. Chem. 2013, 51, 1243. [Google Scholar]
- Ohno H. Electrochim. Acta 2001, 46, 1407. [Google Scholar]
- Ohno H.; Yoshizawa M.; Ogihara W. Electrochim. Acta 2004, 50, 255. [Google Scholar]
- Yang F.; Wang X. H.; Zhou J. J.; Li L.; Zhang B. H. Acta Polym. Sin. 2008, 466. [Google Scholar]
- Kerr K. G.; Snelling A. M. J. Hosp. Infect. 2009, 73, 338. [DOI] [PubMed] [Google Scholar]
- Hancock R. E. W.; Bell A. Eur. J. Clin. Microbiol. Infect. Dis. 1988, 7, 713. [DOI] [PubMed] [Google Scholar]
- Hancock R. E. W. Clin. Infect. Dis. 1998, 27, S93. [DOI] [PubMed] [Google Scholar]
- Poole K. J. Antimicrob. Chemother. 2005, 56, 20. [DOI] [PubMed] [Google Scholar]
- Eren T.; Som A.; Rennie J. R.; Nelson C. F.; Urgina Y.; Nusslein K.; Coughlin E. B.; Tew G. N. Macromol. Chem. Phys. 2008, 209, 516. [Google Scholar]
- a Sovadinova I.; Palermo E. F.; Huang R.; Thoma L. M.; Kuroda K. Biomacromolecules 2011, 12, 260. [DOI] [PubMed] [Google Scholar]; b Hartmann M.; Berditsch M.; Hawecker J.; Ardakani M. F.; Gerthsen D.; Ulrich A. S. Antimicrob. Agents Chemother. 2010, 54, 3132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda T.; Yamaguchi H.; Tazuke S. Antimicrob. Agents Chemother. 1984, 26, 139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda T.; Hirayama H.; Yamaguchi H.; Tazuke S.; Watanabe M. Antimicrob. Agents Chemother. 1986, 30, 132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin J. N.; Rybak M. J.; Cheung C. M.; Savage P. B. Antimicrob. Agents Chemother. 2007, 51, 1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prenna M.; Montanari M. P.; Mingoia M.; Biavasco F.; Ripa S.; Varaldo P. E. Eur. J. Clin. Microbiol. Infect. Dis. 1998, 17, 816. [DOI] [PubMed] [Google Scholar]
- Ikeda T.; Hirayama H.; Suzuki K.; Yamaguchi H.; Tazuke S. Macromol Chem. Phys. 1986, 187, 333. [Google Scholar]
- Pouny Y.; Rapaport D.; Mor A.; Nicolas P.; Shai Y. Biochemistry 1992, 31, 12416. [DOI] [PubMed] [Google Scholar]
- Yang L.; Harroun T. A.; Weiss T. M.; Ding L.; Huang H. W. Biophys. J. 2001, 81, 1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuzaki K.; Murase O.; Fujii N.; Miyajima K. Biochemistry (Moscow) 1996, 35, 11361. [DOI] [PubMed] [Google Scholar]
- Som A.; Vemparala S.; Ivanov I.; Tew G. N. Biopolymers 2008, 90, 83. [DOI] [PubMed] [Google Scholar]
- Brogden K. A. Nat. Rev. Microbiol. 2005, 3, 238. [DOI] [PubMed] [Google Scholar]
- Brogden K. A. Nat. Rev. Microbiol. 2005, 3, 238. [DOI] [PubMed] [Google Scholar]
- Hale J. D.; Hancock R. E. Exp. Rev. Anti-Infect. Ther. 2007, 5, 951. [DOI] [PubMed] [Google Scholar]
- Fou A. C.; Rubner M. F. Macromolecules 1995, 28, 7115. [Google Scholar]
- Jaber J. A.; Schlenoff J. B. Langmuir 2007, 23, 896. [DOI] [PubMed] [Google Scholar]
- Tew G. N.; Liu D. H.; Chen B.; Doerksen R. J.; Kaplan J.; Carroll P. J.; Klein M. L.; DeGrado W. F. Proc. Natl. Acad. Sci. U.S.A. 2002, 99, 5110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raj P. A.; Dentino A. R. FEMS Microbiol. Lett. 2002, 206, 9. [DOI] [PubMed] [Google Scholar]
- Gabriel G. J.; Madkour A. E.; Dabkowski J. M.; Nelson C. F.; Nusslein K.; Tew G. N. Biomacromolecules 2008, 9, 2980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang H.; Doerksen R. J.; Jones T. V.; Klein M. L.; Tew G. N. Chem. Biol. 2006, 13, 427. [DOI] [PubMed] [Google Scholar]
- Chin J. N.; Jones R. N.; Sader H. S.; Savage P. B.; Rybak M. J. J. Antimicrob. Chemother. 2008, 61, 365. [DOI] [PubMed] [Google Scholar]
- Gabriel G. J.; Maegerlein J. A.; Nelson C. E.; Dabkowski J. M.; Eren T.; Nusslein K.; Tew G. N. Chem.–Eur. J. 2009, 15, 433. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.