

Abstract
Idiosyncratic drug hepatotoxicity is a hepatotoxicity subset that occurs in a very small fraction of human patients, is poorly predicted by standard preclinical models and in clinical trials, and frequently leads to post-approval drug failure. Animal models utilizing bacterial LPS co-administration to induce an inflammatory background and hepatocyte cell culture models utilizing cytokine mix co-treatment have successfully reproduced idiosyncratic hepatotoxicity signatures for certain drugs, but the hepatocyte signaling mechanisms governing these drug-cytokine toxicity synergizes are largely unclear. Here, we summarize our efforts to computationally model the signaling mechanisms regulating inflammatory cytokine-associated idiosyncratic drug hepatotoxicity. We collected a “cue-signal-response” (CSR) data compendium in cultured primary human hepatocytes treated with many combinations of idiosyncratic hepatotoxic drugs and inflammatory cytokine mixes (“cues”) and subjected this compendium to orthogonal partial-least squares regression (OPLSR) to computationally relate the measured intracellular phosphoprotein signals and hepatocellular death responses. This OPLSR model suggested that hepatocytes specify their cell death responses to toxic drug/cytokine conditions by integrating signals from four key pathways -- Akt, p70 S6K, ERK, and p38. An OPLSR model focused on data from these four signaling pathways demonstrated accurate predictions of idiosyncratic drug- and cytokine-induced hepatotoxicities in a second human hepatocyte donor, suggesting that hepatocytes from different individuals have shared network control mechanisms governing toxicity responses to diverse combinations of idiosyncratic hepatotoxicants and inflammatory cytokines.
I. Introduction
Hepatotoxicity is a major cause of failures of drug development and accurate assessment of human hepatotoxicity presents a significant challenge for the pharmaceutical industry [1]–[2]. Idiosyncratic drug hepatotoxicity is a hepatotoxicity subset that occurs in a very small fraction of human patients (~1 in 10,000) and accounts for ~10% of acute liver failure cases [3]. Idiosyncratic drug hepatotoxicity is poorly predicted by standard preclinical models and in clinical trials and frequently leads to post-approval drug failure [3]. LPS-administered rodent models [3]–[4] and cytokine co-treatment hepatocyte cell culture models [5] have been developed to reproduce idiosyncratic hepatotoxicities of some idiosyncratic hepatotoxicants. These models suggest that idiosyncratic drug hepatotoxicity can arise when mild drug-induced hepatocellular stresses synergize with inflammatory cytokine signaling to elicit hepatocellular death.
Multiple inflammatory cytokines (TNF, IL-1α, IFN-γ, and LPS itself) have been identified as contributing to inflammation-associated idiosyncratic drug hepatotoxicity in these animal and cell culture models. These cytokines stimulate a diversity of intracellular signaling pathways related to cell survival, stress, and apoptosis. Many of the complex signaling mechanisms activated by these individual cytokines are well-studied (reviewed in [6]–[8]). For example, TNF activates the MEK–ERK, IKK–NF-κB, JNK, and p38–HSP27 signaling pathways and the caspase cascade, whose integrated activities specificity hepatocyte responses to TNF in a context-sensitive manner.
Though, it is as of yet unclear how these cytokine-induced signals are integrated with drug-induced signals to elicit a synergistic cellular toxicity response. Of note, it has been demonstrated that the p38 signaling pathway contributes to the hepatotoxicity synergy between LPS-induced cytokine release and the idiosyncratic hepatotoxicant ranitidine in a mouse model [9]. Drug-induced hepatocellular stresses can induce activation of many of the signaling pathways stimulated by inflammatory cytokines and/or growth factors. For example, drug-induced reactive oxygen species (ROS) accumulation can activate the JNK and p38–HSP27 stress-response signaling pathways [6]. Consequently, we reasoned that further identification of the signaling mechanisms governing hepatocyte cell death responses to idiosyncratic hepatotoxicants and inflammatory cytokines would be benefited from a network-level examination of multiple intracellular signaling pathways.
II. Results
A. Collection of a drug and cytokine co-treament cue-signal-response phosphoproteomic data compendium
We collected a cue-signal-response (CSR) drug- and cytokine-induced hepatotoxicity data compendium in cultured primary human hepatocytes to examine the signaling mechanisms regulating inflammatory cytokine-associated idiosyncratic drug hepatotoxicity [10]. Human hepatocytes were treated with 66 different combinations of 11 “drug” conditions (six idiosyncratic hepatotoxicants, four corresponding “comparison” compounds, and a DMSO control) and six “cytokine” conditions (different mixes of TNF, IL-1α, IFN-γ, and LPS). Rigorously quantitative bead-based and cell death assays were utilized to allow for investigation of the quantitative relationships between signaling pathway activation and cell death measurements. Seventeen phosphoproteins, mechanistically associated with the MEK–ERK, mTOR–p70 S6K, Akt, IKK–NF-κB, JNK, p38–HSP27, STAT3, STAT6, cell cycle regulatory, and DNA damage signaling pathways, were measured at both early (0, 20 min) and delayed time-points (4, 24, and 48 hr) following drug and/or cytokine stimulation to capture a diversity of intracellular signaling pathways and dynamics. To quantify hepatotoxicity, hepatocyte LDH release was measured at 48 hr. This CSR phosphoproteomic compendium consisted of ~4500 (= 66 conditions × 17 phosphoproteins × 4 time-points) individual phosphoprotein signaling. In this CSR compendium, even the most positively and negatively correlated single-phosphoprotein signaling features were poorly predictive of the measured cell death [10]. These poorly predictive individual signaling relationships to the measured cell death responses suggested the need for a multivariate modeling approach to interpret the signal-response relationships present in this drug- and cytokine-induced hepatotoxicity data compendium.
B. Multipathway OPLSR model of the CSR data compendium identifies key molecular signals regulating drug- and cytokine-induced hepatotoxicity
To generate a multipathway model relating the observed signaling activities and cell death responses, we subjected the initial CSR data compendium to orthogonal partial-least squares regression (OPLSR), a data-driven modeling approach useful for suggesting relationships between intracellular signals and cell phenotypes without requiring a priori mechanistic knowledge [11]–[14]. OPLSR relates the measured phosphoprotein signaling data matrix (X) and the observed hepatocellular death responses (Y), each arrayed across all 66 treatment conditions, through a linear regression of the relationship Y = X·B, where B is a vector of regression coefficients that reflect how each phosphoprotein signal contributes to cell death [14]–[15]. An OPLSR model comprised of signaling time-course data from all 17 phosphoproteins demonstrated good model fitness (R2 = 0.92) for cross-validated predictions of the observed cell death responses for all 66 drug/cytokine co-treatment conditions [10].
Inspection of OPLSR model loadings and variable importance of projection (VIP) scores [16] identified four signaling pathways with phosphoproteins with highly informative model contributions at multiple time-points [10]. The model identified the MEK–ERK and p38–HSP27 signaling pathways as having significant pro-death contributions, and the Akt and mTOR–p70 S6K signaling pathways as having significant pro-survival contributions. In agreement with the model predictions, p38–HSP27 pathway is generally considered pro-apoptotic in hepatocytes due to its transcription regulation of effector caspases [17]. Counter to the model predictions, the MEK–ERK pathway is generally considered pro-survival hepatocytes through its activation of anti-apoptotic effectors [18]. Using small molecule inhibitors selected for their high efficacy and minimal toxicity, both MEK and p38 inhibition attenuated the drug-cytokine hepatotoxicity synergy for the idiosyncratic hepatotoxicant nortriptyline, confirming the OPLSR model interpretations that these pathways are pro-death mediators of inflammatory cytokine-associated idiosyncratic drug hepatotoxicity [10].
The importance of these four signaling pathways in the model suggested that many of the 17 phosphoproteins measured in the initial compendium were unnecessary for the model predictions and consequently the model could be reduced to a more interpretable set of protein signals. To reduce the complexity of the original model, phosphoproteins were removed from the model step-wise in order of the lowest VIP score. Model fitness was maintained until the top ~4–5 phosphoproteins remained (R2 = ~0.87–0.91) [10]. This emphasized that an equivalently predictive multipathway network model could be generated by focusing on representative phosphoproteins from four pathways -- MEK–ERK, Akt, mTOR–p70 S6K, and p38–HSP27 -- and that these representative phosphoproteins (e.g. p-ERK1/2, p-Akt, p-p70 S6K, and p-HSP27) could serve as a useful signaling network “gauge” [12], whose integrated activities accurately specify hepatocellular death responses to drug and/or cytokine stimulation.
C. A reduced multipathway OPLSR model accurately predicts hepatotoxicity signal-response relationships across human hepatocyte donors
To test the utility of this reduced multipathway OPLSR model, we collected a second drug- and cytokine-induced CSR hepatotoxicity data compendium containing just six phosphoprotein signals (p-MEK1, p-ERK1/2, p-Akt, p-p70 S6K, p-p38, and p-HSP27) from the four “network gauge” pathways (Fig. 1) [10]. This 6-phosphoprotein OPLSR model trained on data from donor #1 demonstrated reasonably accurate predictions of cell death responses for test data from conditions present in the training compendium (R2 = 0.56; Fig. 1). This demonstrated that the 6-phosphoprotein OPLSR model can accurately predict drug/cytokine responses across human hepatocyte donors.
Fig. 1.

OPLSR modeling demonstrates accurate predictions of drug- and cytokine-induced hepatotoxicity across human hepatocyte donors. Phosphoprotein signaling and response data from two human hepatocyte donors [10]. An OPLSR model was trained on the 66-condition (only a subset of 6 conditions shown here), 6-phosphoprotein CSR data compendium from donor #1. This OPLSR model generated quantitatively accurate predictions of cell death responses in donor #1 and donor #2, even though donor-specific signaling network activation profiles and cell death responses were observed under the same drug/cytokine treatments. Compare clarithromycin (CLA) ± cytokine mix across the two donors. The predictive accuracy of this OPLSR model suggests that a common-effector processing mechanism (f(x) = y) encompassing the integration of the survival and stress signaling network (x) yields quantitatively concerted cell death responses (y) to toxic drug/cytokine conditions exists and is shared between hepatocytes from different human donors. Data and schematic reproduced from [10].
The model’s ability to successfully predict hepatotoxicity signaling-response relationships across human hepatocyte donors for similar drug/cytokine conditions suggests that hepatocytes from multiple human donors share a “common effector” processing function [13]. In human hepatocytes from two different human donors, specific drug/cytokine treatment conditions (see clarithromycin + cytokine mix in donors #1 and 2 in Fig. 1) can elicit significantly different signaling network activation profiles and induce different levels of cell death but the trained OPLSR model can accurately predict the cell death responses from both signaling network activity profiles by considering all the time-dependent signaling activity variables across the six measured phosphoproteins.
III. Conclusions
Through data-driven modeling of phosphoproteomic data compendia, we demonstrated that synergistic induction of hepatocellular death by idiosyncratic hepatotoxicants and physiologically-relevant inflammatory cytokine co-stimuli in cultured primary human hepatocytes is governed by the integrated behaviors of multiple intracellular signaling pathways. OPLSR modeling identified an informative subset of four signaling pathways, two with model-assigned pro-survival functions -- Akt and mTOR–p70 S6K -- and two with model-assigned pro-death functions -- MEK–ERK and p38–HSP27. Together, these four signaling pathways represent a useful “network gauge” capturing the balance between survival and death signaling in drug- and cytokine-treated hepatocytes (Fig. 2). A simplified OPLSR model focused on these four signaling pathways demonstrated quantitatively accurate predictions of idiosyncratic drug- and cytokine-induced hepatotoxicities in a second human hepatocyte donor, but further confirmation in additional donors is warranted. This work demonstrates that hepatotoxicity can be thought of in terms of a “network toxicity”, in which the integrated behavior of multiple hepatocellular signaling pathways should be considered in evaluating the hepatotoxicity of a given treatment condition (Fig. 2). Further, these results demonstrate the utility of multipathway signaling network modeling of cellular behaviors in primary human hepatocyte cell culture models and offer promise for donor-specific predictions of hepatotoxicity responses to idiosyncratic hepatotoxicants from phosphoproteomic data.
Fig. 2.
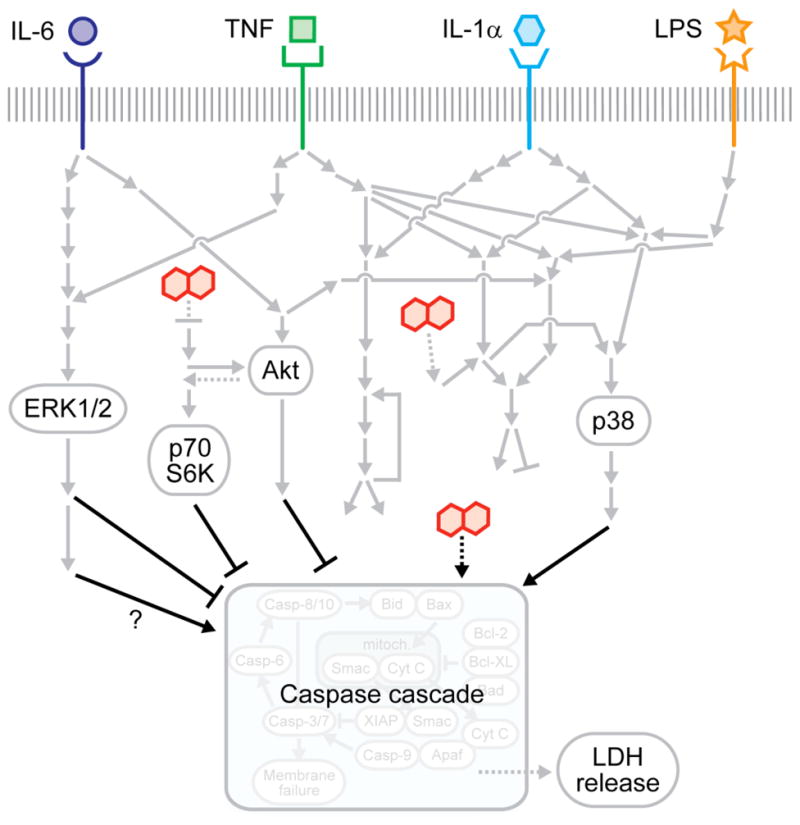
Inflammatory cytokine-associated idiosyncratic drug hepatotoxicity as a “network toxicity”. The multipathway modeling approach presented here suggests that an integration of multiple intracellular signaling pathway -- namely the MEK–ERK, mTOR–p70 S6K, Akt, and p38–HSP27 pathways -- activities is necessary for hepatocytes to specify death responses to hepatotoxic drug/cytokine co-treatment conditions. This provides motivation of the network-level consideration of multiple survival, stress, and apoptosis signaling pathways in evaluating the hepatotoxicity mechanisms of context-dependent hepatotoxic drugs. Schematic reproduced from [10].
Acknowledgments
This work was supported in part by a grant from Pfizer Inc., the Department of Defense Institute for Collaborative Biotechnologies (to D.A.L.), the MIT Center for Cell Decision Processes (NIH grant P50-GM68762; to D.A.L), the MIT Biotechnology Process Engineering Center (to L.G.G.), the MIT Center for Environmental Health Sciences (NIH grant U19ES011399; to L.G.G.), and a Whitaker Foundation Graduate Fellowship (to B.D.C.).
The authors thank Tachun Hang for technical assistance and Arthur Goldsipe, David de Graaf, Jinghai J. Xu, Bart Hendriks, and Peter Sorger for helpful discussions.
Contributor Information
Benjamin D. Cosgrove, Department of Biological Engineering, Massachusetts Institute of Technology, Cambridge, MA 02139 USA. He is now with the Molecular Imaging Program at Stanford, Stanford University School of Medicine, Stanford, CA 94305 USA
Leonidas G. Alexopoulos, Email: leo@mail.ntua.gr, Department of Systems Biology, Harvard Medical School, Boston, MA 02115 USA. He is now with the Department of Mechanical Engineering, National Technical University of Athens, Athens, Greece
Julio Saez-Rodriguez, Email: julio_saez@hms.harvard.edu, Department of Systems Biology, Harvard Medical School, Boston, MA 02115 USA.
Linda G. Griffith, Email: griff@mit.edu, Department of Biological Engineering, Massachusetts Institute of Technology, Cambridge, MA 02139 USA
Douglas A. Lauffenburger, Email: lauffen@mit.edu, Department of Biological Engineering, Massachusetts Institute of Technology, Cambridge, MA 02139 USA
References
- 1.Kaplowitz N. Drug Safety. 2001;24(7):483–490. doi: 10.2165/00002018-200124070-00001. [DOI] [PubMed] [Google Scholar]
- 2.Lee WM. N Eng J Med. 2003;349(5):474–485. doi: 10.1056/NEJMra021844. [DOI] [PubMed] [Google Scholar]
- 3.Kaplowitz N. Nat Rev Drug Discov. 2005;4(6):489–499. doi: 10.1038/nrd1750. [DOI] [PubMed] [Google Scholar]
- 4.Ganey GE, Luyendyk JP, Maddox JF, Roth RA. Chem Biol Interact. 2004;150(1):35–51. doi: 10.1016/j.cbi.2004.09.002. [DOI] [PubMed] [Google Scholar]
- 5.Cosgrove BD, et al. Toxicol Appl Pharmacology. 2009;237(3):317–330. doi: 10.1016/j.taap.2009.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schwabe RF, Brenner DA. Am J Physiol Gastrointest Liver Physiol. 2006;290(4):G583–G589. doi: 10.1152/ajpgi.00422.2005. [DOI] [PubMed] [Google Scholar]
- 7.Malhi H, Gores GJ. Gastroenterology. 2008;134(6):1641–1654. doi: 10.1053/j.gastro.2008.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tacke F, Luedde T, Trautwein C. Clin Rev Allergy Immunol. 2008;36(1):4–12. doi: 10.1007/s12016-008-8091-0. [DOI] [PubMed] [Google Scholar]
- 9.Deng X, et al. J Pharmacol Exp Ther. 2008;326(1):144–152. doi: 10.1124/jpet.108.137497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cosgrove BD, Alexopoulos LG, Hang T, Hendriks BS, Griffith LG, Sorger PK, Lauffenburger DA. submitted. [Google Scholar]
- 11.Janes KA, Yaffe MB. Nat Rev Mol Cell Biol. 2006;7(11):820–828. doi: 10.1038/nrm2041. [DOI] [PubMed] [Google Scholar]
- 12.Kumar N, Wolf-Yadlin A, White FM, Lauffenburger DA. PLoS Comput Biol. 2007;3(1):e4. doi: 10.1371/journal.pcbi.0030004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Miller-Jensen K, Janes KA, Brugge JS, Lauffenburger DA. Nature. 2007;448(7153):604–608. doi: 10.1038/nature06001. [DOI] [PubMed] [Google Scholar]
- 14.Geladi P, Kowalski B. Anal Chim Acta. 1986;185:1–17. [Google Scholar]
- 15.Bylesjo M, et al. J Chemometrics. 2006;20:341–351. [Google Scholar]
- 16.Wold S. Chemometrics and intelligent laboratory systems. 1994;23:149–161. [Google Scholar]
- 17.Ono K, Han J. Cell Signal. 2000;12(1):1–13. doi: 10.1016/s0898-6568(99)00071-6. [DOI] [PubMed] [Google Scholar]
- 18.McCubrey JA, et al. Curr Opin Investig Drugs. 2008;9(6):614–630. [PubMed] [Google Scholar]