

Abstract
Conventional chemotherapy of ovarian cancer often fails because of initiation of drug resistance and/or side effects and trace of untouched remaining cancerous cells. This highlights an urgent need for advanced targeted therapies for effective remediation of the disease using a cytotoxic agent with immunomodulatory effects, such as shikonin (SHK). Based on preliminary experiments, we found SHK to be profoundly toxic in ovarian epithelial cancer cells (OVCAR-5 and ID8 cells) as well as in normal ovarian IOSE-398 cells, endothelial MS1 cells, and lymphocytes. To limit its cytotoxic impact solely to tumor cells within the tumor microenvironment (TME), we aimed to engineer SHK as polymeric nanoparticles (NPs) with targeting moiety toward tumor microvasculature. To this end, using single/double emulsion solvent evaporation/diffusion technique with sonication, we formulated biodegradable NPs of poly(lactic-co-glycolic acid) (PLGA) loaded with SHK. The surface of NPs was further decorated with solubilizing agent polyethylene glycol (PEG) and tumor endothelial marker 1 (TEM1)/endosialin-targeting antibody (Ab) through carbodiimide/N-hydroxysuccinimide chemistry. Having characterized the physicochemical and morphological properties of NPs, we studied their drug-release profiles using various kinetic models. The biological impact of NPs was also evaluated in tumor-associated endothelial MS1 cells, primary lymphocytes, and epithelial ovarian cancer OVCAR-5 cells. Based on particle size analysis and electron microscopy, the engineered NPs showed a smooth spherical shape with size range of 120 to 250 nm and zeta potential value of −30 to −40 mV. Drug entrapment efficiency was ~80%–90%, which was reduced to ~50%–60% upon surface decoration with PEG and Ab. The liberation of SHK from NPs showed a sustained-release profile that was best fitted with Wagner log-probability model. Fluorescence microscopy and flow cytometry analysis showed active interaction of Ab-armed NPs with TEM1-positive MS1 cells, but not with TEM1-negative MS1 cells. While exposure of the PEGylated NPs for 2 hours was not toxic to lymphocytes, long-term exposure of the Ab-armed and PEGylated NPs was significantly toxic to TEM1-positive MS1 cells and OVCAR-5 cells. Based on these findings, we propose SHK-loaded Ab-armed PEGylated PLGA NPs as a novel nanomedicine for targeted therapy of solid tumors.
Keywords: nanomedicine, nanoparticle, ovarian cancer, shikonin, targeted therapy, tumor vasculature
Introduction
The survival rate of ovarian cancer patients is often low, particularly in patients with recurrent malignancies. After conventional chemotherapy, high percentages of patients are inevitably prone to profound toxicity in normal cells/tissue and drug resistance.1–3 Further, the malignancy may relapse, complicating the second course of chemotherapy; advanced nanomedicines have been shown to be more effective than conventional chemotherapy.4,5 Battles with cancer can be substantially improved if cytotoxicity is combined with anticancer immunosurveillance. This can be accomplished by implementing advanced innocuous nanomedicines with the ability to deliver cytotoxic and immunomodulatory agents to the tumor microenvironment (TME), induce toxic impacts solely within the cancerous cells, and activate the immune system.
Of various anticancer agents, very few show such characteristics; however, among numerous natural products examined for anticancer and immunomodulatory effects, shikonin (SHK) and alkannin derivatives (R-enantiomer and S-enantiomer, respectively) appear to provide such pharmacological effects.6–9 SHK- and alkannin-derivatives have previously been produced through bioprocess engineering of Echium italicum L,10,11 and these compounds showed substantial inhibitory effects in various cancerous cell lines (Barar et al, unpublished data, 2014). Despite having immunomodulatory effects,9 SHK, as a highly liposoluble naphthoquinone pigment, can inhibit pyruvate kinase M2 activity,12,13 upregulate p53 activity and downregulate cyclin-dependent protein kinase 4 activity,14 upregulate BCL2-associated X protein (BAX) and downregulate B-cell lymphoma 2 (BCL2),6 and inhibit extracellular signal-regulated kinases (ERK) and protein kinase B pathways.15 All these studies confirm that SHK can induce significant cytotoxicity, while its cytotoxic impacts are not specific to cancer cells. Hence, like any other cytotoxic agent, SHK needs to be delivered solely to the cancer cells, preferably using targeted nanomedicines.
Smart targeted nanoparticles (NPs) have been shown to accumulate within the TME through both passive (ie, enhanced permeation and retention [EPR] effect) and active (ie, targeting a cancer marker molecule) mechanisms, resulting in enhanced effects, mainly in cancerous cells.16–18 Given the fact that biodegradable polymeric NPs elicit negligible side effects, they have been widely investigated for efficient and safe delivery of encapsulated/entrapped chemotherapeutics to the target cells.17 Of these, poly(lactic-co-glycolic acid) (PLGA) is the most studied polymer;19–21 it is also a safe biocompatible polymer in comparison with synthetic polymers/lipids used as delivery systems.22–24 PLGA polymer has previously been used for delivery of insulin21 and doxorubicin (Adriamycin).19 In the current work, we aimed to engineer SHK-loaded, surface-modified PLGA NPs (eg, SHK-loaded, polyethylene glycol [PEG]ylated, and antitumor endothelial marker 1 [TEM1] antibody [Ab]/single-chain variable fragment [scFv]-armed PLGA NPs) and investigate their biological effects in tumor endothelial TEM1-positive MS1 cells, TEM1-negative MS1 cells, epithelial (OVCAR-5 and ID8) cells, and primary isolated lymphocytes.
Materials and methods
Materials
SHK, fluorescein (FSN), Tween 80, Pluronic® F-68, NH2-PEG3000-COOH, NH2-PEG3000-OH, sulforhodamine B (SRB), 2-(N-morpholino)ethanesulfonic acid (MES), ethylenedi-aminetetraacetic acid (EDTA), 4′,6-diamidino-2-phenylindole (DAPI), phalloidin–tetramethylrhodamine B isothiocyanate (phalloidin-TRITC), acid-terminated (50:50) poly(D,L-lactide-co-glycolide) (24,000–38,000 and 38,000–54,000 Mw), N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC), N-hydroxysulfosuccinimide sodium salt (NHS), dichloromethane, chloroform, dimethyl sulfoxide, O-(2-aminoethyl)PEG 3000, N,N-diisopropylethylamine p-toluenesulfonate salt, methoxypolyethylene glycol amine, polyvinyl alcohol (Mowiol® 4–88), Trypan Blue, Triton™ X-100, and staurosporine were purchased from Sigma-Aldrich (St Louis, MO, USA). Roswell Park Memorial Institute medium (RPMI) 1640 (GlutaMAX™), fetal bovine serum, penicillin G, streptomycin, Dulbecco’s Modified Eagle’s Medium, and L-glutamine were purchased from Invitrogen (Life Technologies, Carlsbad, CA, USA). All cell lines were from American Type Culture Collection (ATCC) (Manassas, VA, USA) and human TEM1-positive MS1 cells were developed in our laboratory as reported previously.25 All tissue culture dishes and Coomassie dye were purchased from Thermo Fisher Scientific (Waltham, MA, USA). Anti-TEM1 (H-63) Ab was from Santa Cruz Biotechnology, Inc (Santa Cruz, CA, USA). Anti-TEM1 scFvs were produced in our laboratory (courtesy of Dr Aizhi Zhao). The secondary streptavidin-allophycocyanin (APC) tagged Ab and fixable viability dye eFluor 506 were purchased from eBioscience (San Diego, CA, USA). Annexin V-EnzoGold was purchased from Enzo Life Sciences (Farmingdale, NY, USA). All other chemicals not listed were purchased in the highest purity available.
Formulation of NPs
For formulation of PLGA NPs, we capitalized on a single emulsion-solvent evaporation technique. Briefly, SHK (15 mg/mL) and FSN (5 mg/mL) were dissolved in 3 mL of oil phase (chloroform or ethyl acetate) containing 3% PLGA and surfactant (either 2%–4% Pluronic F68 or 2%–5% polyvinyl alcohol). The oil phase was mixed with 10mL of the water phase (ie, double-distilled water containing 1% Tween 80) over an ice bath under sonication at 40 W output for 4 minutes (2×2 minutes) using a probe sonicator (VCX 130; Sonics and Materials, Inc, Newtown, CT, USA). Then, the oil phase was eliminated by evaporation under reduced pressure (150–200 mbar) using a rotary evaporator (30 rpm) (Hei-VAP Advantage ML/G5B; Heidolph North America, Grove Village, IL, USA). Drug-free PLGA NPs were formulated using the same procedure. The effects of formulation variables on the NP characteristics were examined. The formulated NPs were resuspended in 1 mL trehalose (250 mM), used as cryoprotectant (to prevent lyophilization-induced aggregation), supplemented with 0.5% Tween 80, and lyophilized for later use.
Particle size and zeta potential analyses
To determine the size and zeta potential of NPs, 1 mg/mL suspension of the formulated NPs was prepared in double-distilled water. Then, 20 μL of each sample was diluted to 1 mL and, after brief sonication, the size and zeta potential of the NPs were analyzed using a Zetasizer Nano-ZS (Malvern Instruments, Malvern, UK).
Transmission (TEM) and scanning (SEM) electron microscopy
For morphological characterization, the formulated NPs were evaluated by TEM and SEM. For TEM analysis, a sample of NPs (0.5 mg/mL) was suspended in water and sonicated for 30 seconds. One drop of this suspension was placed over a carbon-coated copper TEM grid (150–400 mesh; Ted Pella, Inc, Redding, CA, USA) and allowed to dry. Images were visualized at 120 KeV at indicated magnifications on a Tecnai 12 microscope (FEI, Hillsboro, OR, USA) equipped with a Gatan, Inc (Pleasanton, CA, USA) 896 2.2.1 US1000 camera. For SEM analysis, PLGA NPs were applied to mica and allowed to air dry. Then, the samples were imaged at 5–10 KeV on a JEOL JSM-7500F microscope (JEOL USA, Inc, Peabody, MA, USA) at designated magnifications. The NPs were sputtered with gold in order to improve their conductivity and placed on a copper stub before the acquisition of SEM images.
Drug content (DC%), drug entrapment (DE%), drug loading (DL%), and yielding (YE%) efficiencies
Both SHK and FSN contents of the NPs were estimated by ultraviolet (UV) spectrophotometric method. Briefly, a designated amount of lyophilized SHK-loaded NPs was dissolved in acetonitrile (1 mg/mL) and kept on a shaker at 37°C for 72 hours for complete release of the entrapped drug. Then, samples were removed and centrifuged at 10,000× g for 5 minutes at 4°C to extract the drug present in the solution. The supernatant (700 μL) was collected, and the absorbance of the supernatant was measured by a UV spectrophotometer, (Genesys 6; Thermo Fisher Scientific) at a wavelength of 520 nm. As shown in Table 1, we calculated DC%, DE%, DL%, and yielding efficiency (YE%) for all formulations.
Table 1.
DC%, DE%, DL%, and process YE% of shikonin-loaded NPs before and after modification
| Formulation characteristics | Plain NPs | PEGylated NPs | Ab-armed NPs |
|---|---|---|---|
| 12.8 | 10.4 | 7.5 | |
| 87.0 | 65.0 | 42.0 | |
| 14.0 | 11.8 | 8.6 | |
| 85.0 | 75.0 | 60.0 |
Abbreviations: Ab, antibody; DC%, drug content efficiency; DE%, drug entrapment efficiency; Df, free drug; DL%, drug loading efficiency; Dt, total drug; NP, nanoparticle; PEG, polyethylene glycol; Pt, total polymer; YE%, yielding efficiency.
Release kinetics analysis
In vitro release kinetics of SHK from PLGA NPs was determined using phosphate-buffered saline (PBS) release buffer (137 mM NaCl, 2.7 mM KCl, 4.3 mM Na2HPO4, 1.47 mM KH2PO4) at various pHs (pH 4.4, 5.4, 6.4, and 7.4) at 37°C. The NPs (25 mg) were dispersed in 5 mL of the PBS buffer and divided into equal aliquots (1 mL each). These tubes were kept on a shaker at 37°C and 150 rpm. At designated time intervals (1, 2, 4, 8, 12, 24, 48, and 72 hours), these tubes were taken from the shaker and centrifuged at 9,000× g at 4°C for 5 minutes. Then, the supernatant was removed to estimate the amount of released drug using the Genesys 6 UV spectrophotometer, at a wavelength of 520 nm (SHK) and 490 nm (FSN). The release data were analyzed using various kinetics models (Table 2), as described previously.26
Table 2.
The kinetics models used to fit the release data
| Kinetics model | Equation | Coefficient of determination (R2)
|
|||
|---|---|---|---|---|---|
| pH 4.4 | pH 5.4 | pH 6.4 | pH 7.4 | ||
| Zero order | F=k0t | 0.777 | 0.788 | 0.762 | 0.728 |
| First order | Ln(1−F) = −kf t | 0.901 | 0.922 | 0.915 | 0.945 |
| Higuchi | 0.918 | 0.926 | 0.910 | 0.884 | |
| Power law | LnF = Lnkp + pLnt | 0.927 | 0.933 | 0.915 | 0.910 |
| Square root of mass | 0.652 | 0.650 | 0.608 | 0.520 | |
| Hixson–Crowell | 0.805 | 0.820 | 0.799 | 0.796 | |
| Three seconds root of mass | 0.710 | 0.723 | 0.693 | 0.663 | |
| Weibull | Ln[−Ln(1−F)] = −βLn td + βLn t | 0.961 | 0.970 | 0.960 | 0.974 |
| Linear probability | Z = Z0 +qt | 0.778 | 0.790 | 0.763 | 0.727 |
| Wagner log-probability | Z′ = Z′0 +q′Lnt | 0.997 | 0.998 | 0.997 | 0.993 |
Notes: Parameters of models were obtained by linear regression. F represents fraction of drug released up to time t. The k0, kf, kH, p, kP, k1/3,k1/2, k2/3, td, β, Z0, Z′0, q, and q′ are parameters of the models. Z and Z′ denote probits of fraction of drug released at any time. Ln: natural logarithm.
PEGylation and Ab conjugation
We implemented one-/two-step PEGylation and Ab conjugation approaches for surface modifications.27 For one-step PEGylation and Ab conjugation, a designated amount (10 mg) of acid-terminated PLGA NPs was resuspended in 2–4 mL MES (pH 6) and activated with 250 μL EDC (1 mg/mL) and 250 μL NHS (1 mg/mL) for 2 hours at room temperature. After washing with 50 mM sucrose, pH 8 (2×) by centrifu-gation at 8,500× g (4°C) for 5 minutes, the activated NPs were resuspended in PBS and incubated with 250 μL NH2-PEG3000-OH and 100 μL Ab (at a final molar ratio of 10:1) for 2 hours at room temperature (or at 4°C overnight). For two-step conjugation, the same process was performed separately for each of PEGylation and Ab conjugation (PEG:Ab/ScFv ratio 10:1). The modified NPs were washed twice using 50 mM sucrose (pH 8) by centrifugation at 8,500× g (4°C) for 5 minutes to remove the unconjugated molecules. The supernatant was collected to calculate the conjugation efficiency using UV spectrophotometry analysis, and the NPs were lyophilized for further studies. Figure 1 shows a schematic representation of PEGylation and Ab conjugation.
Figure 1.

Schematic illustration of PEGylation and antibody conjugation.
Notes: (A) Activation of acid-terminated PLGA NPs by EDC and NHS. (B) One-step PEGylation and antibody conjugation. (C) Two-step PEGylation and antibody conjugation.
Abbreviations: Ab, antibody; EDC, N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride; FSN, fluorescein; NHS, N-hydroxysulfosuccinimide sodium salt; NP, nanoparticle; PEG, polyethylene glycol; PLGA, poly(lactic-co-glycolic acid); SHK, shikonin; scFv, single-chain variable fragment.
Determination of Ab conjugation efficiency
For conjugation efficiency, Bradford method, using Coomassie dye, was employed. Briefly, 300 μL of Coomassie plus reagent was added to Ab-conjugated NPs (10 mg dispersed NPs in 200 μL PBS pH 7.4) and incubated for 10–15 minutes. Then, the absorbance was measured at 595 nm using a microplate reader (EL800; BioTek, Winooski, VT, USA). The results were compared to a standard curve of bovine serum albumin (BSA) solution using a designated concentration range (10 μg/mL to 1,000 μg/mL).
Cell culture
Cell lines were cultured onto well-plates using RPMI 1640 (GlutaMAX™) supplemented with 10% fetal bovine serum, 100 units/mL penicillin G, and 100 μg/mL streptomycin. Lymphocytes were further supplemented with 20 U/mL of interleukin (IL)-2.
The cultured cells were kept in a humidified incubator with 5% CO2 at 37°C during cultivation and experiments. The primary lymphocyte isolation was performed by the Human Immunology Core (HIC) facility at the University of Pennsylvania, Philadelphia, PA, USA. The lymphocytes were kept in 24-well plates at a seeding density of 5×105 cells/well and 20 U/mL of IL-2 was supplemented in the media to help their proliferation before the assay.
Fluorescence microscopy
Fluorescence microscopy was used for evaluation of the cellular interaction of SHK-loaded PLGA NPs and the cellular impact of free SHK.
For the cellular interaction of SHK-loaded PLGA NPs armed with anti-TEM1 Ab/scFv, briefly, TEM1-positive and TEM1-negative MS1 cells were seeded onto the coverslip and, at 40%–50% confluency, were treated with a designated amount of NPs for 1–2 hours. The cellular association/interaction of NPs was terminated by transferring the plate onto ice. Then, cells were washed four times with ice-cold PBS (1x) to remove any NP remains and concurrently fixed and permeabilized (using 4% paraformaldehyde with 0.1% Triton™ X-100 prepared in PBS) for 20 minutes at room temperature. After washing three times with PBS, the nonspecific binding sites were blocked with 1% BSA prepared in PBS for 30 minutes’ incubation at room temperature. After removal of BSA and washing three times with PBS, the cells were stained with phalloidin-TRITC solution for 40 minutes at room temperature then washed three times with PBS, and the nucleus was stained by DAPI. The coverslips were inverted onto microscope slides and sealed with nail polish. Cell morphology and characteristics were monitored using a fluorescence microscope (ECLIPSE Ti; Nikon Corporation, Tokyo, Japan) equipped with a charge-coupled device camera (Photometrics CoolSnap HQ2; Roper Industries, Inc, Sarasota, FL, USA) and NIS-Elements AR software (v 3.2; Nikon Corporation).
A similar approach was undertaken to examine the morphological changes of the OVCAR-5 cells treated with free SHK or SHK-loaded NPs. For the cellular impact of free SHK, the cultivated cells in coverslip, at 40%–50% confluency, were treated with a designated amount of free SHK (5.0,10.0, or 20.0 μM). After 24 hours, the cells were subjected to the fluorescence microscopy analysis as described above.
Cell migration assay
To address the impacts of SHK on cell proliferation and migration, we used a simple cell scratch assay. Briefly, the confluent OVCAR-5 cells cultivated in six-well plates were scraped in a straight line to create a scratch in the middle of the well using a 200 μL pipette tip. After washing four times with PBS, the scratched cells were exposed to designated concentrations of free SHK or SHK-loaded NPs. The morphological changes over a designated time period were evaluated using an ECLIPSE Ti light microscope for 16 hours post-treatment.
Cytotoxicity evaluation
The bioactivity of free SHK and SHK-loaded PLGA NPs was evaluated by SRB assay, as described previously.28 Briefly, cells were cultured at a seeding density of 3.0×104 cells/cm2 onto 96-well plates. After 24 hours, the cells were treated with designated concentrations of SHK or equivalent SHK-loaded NPs for 24 and 48 hours. To study the impact of targeting moiety on cytotoxicity, the treatments were removed after 2 hours of initial exposure and then cells were allowed to grow for 24 or 48 hours. At the end of incubation, the treated cells were fixed using 10% trichloroacetic acid for 60 minutes at 4°C. After washing four times with water, the plates were left to dry and then stained with SRB staining solution (0.057% w/v in 1% v/v acetic acid solution) for 30 minutes at room temperature. The cells were washed four times with 1% acetic acid solution. After drying, the cells were solubilized with 200 μL of 10 mM Tris base solution (pH 10.5) on a shaker for 30 minutes. The absorbance was measured using an EL800 microplate reader at 570 nm. The cell viability was calculated as percentage of untreated control cells. Trypan Blue exclusion assay was used to assess the cytotoxicity potential of SHK-loaded PLGA NPs in Jurkat cell line.
Flow cytometry analysis
To detect any transition from apoptosis to necrosis, we implemented the flow cytometry assay using phospholipid-binding annexin V protein, which binds to phosphatidylserine with a high affinity in the presence of Ca2+ and is considered the main sign for occurrence of early apoptosis.29 Briefly, the cultivated cells in a six-well plate were treated with various concentrations of SHK (as free drug or NP formulation). Twelve hours post-treatment, the treated and untreated cells were gently trypsinized and centrifuged at 200× g for 5 minutes. Then, 1.0×106 cells were washed with 5.0 mL ice-cold PBS by centrifugation at 200× g for 5 minutes and resuspended in 500 μL of binding buffer (1x), containing 5 μL of apoptosis detection reagent (Annexin V-EnzoGold) and 5 μL of necrosis detection reagent, and incubated for 15 minutes at room temperature. The samples were then analyzed by flow cytometry using the FL2 channel for the apoptosis detection reagent and the FL3 channel for the necrosis detection reagent. The treated samples were compared with the positive control cells (ie, by UV irradiation for 15 minutes for MS1 cells and for 30 minutes for OVCAR-5) and the untreated control cells. Flow cyto metry assay was also performed to study the internalization of Ab-/scFv-armed NPs by TEM1-positive MS1 cells and TEM1-negative MS1 cells. For uptake study, cells were treated with free SHK or SHK-loaded NPs for 2 hours, and then the plates were transferred onto ice and the internalization of NPs was analyzed. Besides, the expression of TEM1 in MS1 cells was also validated and quantified by flow cytometry analysis.
DNA fragmentation analysis
DNA fragmentation analysis was performed in the ovarian cancer OVCAR-5 cells. For DNA fragmentation, cells were cultured overnight and then treated with different concentrations of SHK. After 24 hours, the cells were trypsinized and 2.0×106 cells were resuspended in 500 μL Tris–EDTA buffer (10 mM Tris-HCl pH 7.4, 1 mM EDTA) containing 0.2% Triton™ X-100. After vortexing for 1 minute, 100 μL of ice-cold 5M NaCl was added to the sample, which was vortexed for a further 1 minute to remove the DNA from associated proteins. Then, 700 μL of ice-cold isopropanol was added to the sample, which was vortexed for 1 minute and kept overnight at −20°C. DNA was then recovered by centrifugation at 16,000× g at 4°C for 10 minutes. DNA pellets were washed twice with 700 μL ice-cold 85% ethanol by centrifugation at 16,000× g at 4°C for 10 minutes. Then, 150 μL of Tris–EDTA buffer was added to each sample and the samples incubated at 37°C overnight. DNA samples were then mixed with loading buffer and incubated at 65°C for 10 minutes prior to electrophoresis using 1.2% agarose gel containing ethidium bromide (0.5 mg/mL) and Tris–acetate–EDTA buffer (1x) and visualized using a gel documentation system.
Statistical analysis
Data were statistically analyzed using t-test or one-way analysis of variance. A P-value less than 0.05 was used to show statistical significance.
Results
Size, zeta potential, and morphology analyses
Based upon formulation methods and components, the size of the engineered PLGA NPs varied from 120 to 250 nm. Figure 2 represents the size distribution and zeta potential of the engineered PLGA NPs.
Figure 2.

Size distribution and zeta potential of PLGA NPs.
Notes: (A) Size and (B) zeta potential of unloaded PLGA NPs. (C) Size and (D) zeta potential of shikonin-loaded PLGA NPs. Red arrow represents diameter Z-average.
Abbreviations: NPs, nanoparticles; PLGA, poly(lactic-co-glycolic acid).
The zeta potential of SHK-loaded NPs was approximately −35 mV. The TEM and SEM micrographs displayed the engineered PLGA NPs with a spherical shape and smooth surface. Figure 3 represents the TEM and SEM micrographs of the SHK-loaded PLGA NPs.
Figure 3.

TEM and SEM micrographs of the shikonin-loaded PLGA NPs.
Notes: (A) TEM image of PLGA NPs. (B) Maximized TEM image of a single PLGA NP. (C) Maximized SEM image of a single PLGA NP.
Abbreviations: NP, nanoparticle; PLGA, poly(lactic-co-glycolic acid); SEM, scanning electron microscopy; TEM, transmission electron microscopy.
DE% and release kinetics
Table 1 shows the DC%, DE%, DL%, and YE% of SHK-loaded NPs before and after modification. After PEGylation and Ab/scFv conjugation, these values were significantly reduced. FSN content was similar to SHK (data not shown).
Figure 4 demonstrates the release of SHK from PLGA NPs. Table 2 represents various kinetics models used to fit the SHK release profile. As shown in Table 2 and Figure 4 (inset), SHK release profile was best fit with Wagner log-probability. The pattern of drug liberation revealed that 20% of drug was released within the first 2 hours, while 80% of drug was released after 48 hours. The release of SHK from NPs appeared to be somewhat pH-dependent, with highest release at pH 7.4.
Figure 4.

The release profile of shikonin from poly(lactic-co-glycolic acid) nanoparticles.
Note: The inset shows the release data fit with the Wagner log-probability.
Abbreviation: Ln t, natural logarithm of time.
PEGylation and conjugation efficiency
After formulation, PLGA NPs were further solubilized through PEGylation and conjugated with either anti-TEM1 Ab or scFv Ab fragment (Figure 1). One-step PEGylation and Ab conjugation resulted in NPs with higher drug content, while the PEGylation and Ab conjugation efficiency appeared to be somewhat variable. The drug content of the two-step PEGylation and Ab conjugation was lower than that of the one-step approach, while the PEGylation and Ab conjugation efficiency appeared to be consistent. Bradford Coomassie dye analysis revealed conjugation efficiency values of ~15% and ~20% for anti-TEM1 Ab and scFv Ab fragment, respectively.
Fluorescence microscopy
Figure 5 represents the active targeting of TEM1 by Ab-armed PLGA NPs in TEM1-positive MS1 cells. Both Ab-armed and scFv-armed PLGA NPs markedly interacted with the TEM1-positive MS1 cells, but not the TEM1-negative MS1 cells.
Figure 5.

Fluorescence and transmission light microscopy of anti-TEM1 Ab-/scFv-armed fluorescein- and shikonin-loaded PLGA NPs in endothelial MS1 cells.
Notes: (A) Untreated TEM1-negative MS1 cells. (B) Anti-TEM1 Ab-armed NPs treated TEM1-negative MS1 cells. (C) Anti-TEM1 scFv-armed NPs treated TEM1-negative MS1 cells. (D) Untreated TEM1-positive MS1 cells. (E) Anti-TEM1 Ab-armed NPs treated TEM1-positive MS1 cells. (F) Anti-TEM1 scFv-armed NPs treated TEM1-positive MS1 cells. Green color represents fluorescein-loaded PLGA NPs. (G–I), respectively, represent light microscopy, fluorescence microscopy, and light microscopy–fluorescence microscopy superimposed images of anti-TEM1 scFv-armed NPs treated TEM1-positive MS1 cells. Phalloidin–tetramethylrhodamine B isothiocyanate was used to stain the cytoplasmic F-actin (red). 4′,6-diamidino-2-phenylindole was used to stain the nucleus (blue).
Abbreviations: Ab, antibody; NPs, nanoparticles; PLGA, poly(lactic-co-glycolic acid); scFv, single-chain variable fragment; TEM1, tumor endothelial marker 1.
We designed Ab-armed PLGA NPs loaded with SHK to target tumor endothelial cells through TEM1 antigen, by which the NPs can be accumulated within the TME and taken up by epithelial ovarian cancer cells through both passive and active targeting mechanisms.
Figure 6 represents the fluorescence microscopy images of free SHK-treated OVCAR-5 cells. Marked morphological changes in cytoplasm together with shrinkage of nucleus were observed in the epithelial ovarian cancer OVCAR-5 cells treated with free SHK in a dose-dependent manner. Both free SHK and SHK-loaded NPs imposed similar morphological changes in OVCAR-5 cells as well as in ID8 cells.
Figure 6.
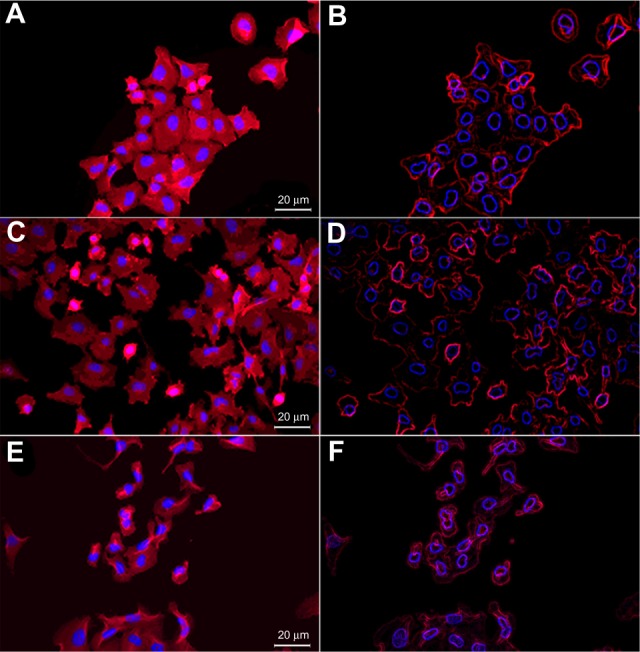
Fluorescence microscopy (FM) images of free shikonin (SHK) effects in OVCAR-5 cells.
Notes: (A) FM image of the untreated cells. (B) Stylized image of the untreated cells. (C) FM image of the treated cells with 5.0 μM SHK. (D) Stylized image of the treated cells with 10.0 μM SHK. (E) FM image of the treated cells with 20 μM SHK. (F) Stylized image of the treated cells with 20.0 μM SHK.
Cell migration assay
Figure 7 represents the migration of OVCAR-5 cells in the presence of SHK at the concentrations of 0.5 μM (Figure 7C), 2.5 μM (Figure 7D), and 5.0 μM (Figure 7E). The untreated cells (Figure 7B) managed to fill the gap within 6 hours post-scratching as compared to that of time 0 (Figure 7A); however, SHK significantly reduced the growth and migration of the treated cells: the higher the SHK concentration, the greater the inhibition of cell growth and migration (Figure 7F).
Figure 7.

Effect of SHK on migration of ovarian cancer OVCAR-5 cells.
Notes: Cells were scratched and imaged at time 0 (A) and 6 hours post-scratching (B–E). (B) Untreated control cells. (C) Cells treated with 0.5 μM SHK. (D) Cells treated with 2.5 μM SHK. (E) Cells treated with 5.0 μM SHK. (F) Cell migration analysis of treated cells in comparison with the scratched cells at time 0.
Abbreviations: SHK, shikonin; UT, untreated.
Interaction of Ab-armed NPs with TEM1-positive MS1 cells
To determine the specific binding of formulated NPs and targeting efficiency, the TEM1-positive MS1 cells were quantified using flow cytometry. Based on our flow cytometry analysis, over 90% of the transfected MS1 cells showed TEM1 expression. Accordingly, the TEM1-positive MS1 cells showed significantly higher uptake of the Ab-/scFv-armed PLGA NPs compared to the TEM1-negative MS1 cells (data not shown).
Cellular toxicity
To evaluate the cytotoxic effect of SHK, we first examined the toxicity impacts of the free SHK in the normal and cancerous cells using SRB assay. We observed that free SHK affected viability of normal ovarian IOSE-398 cells (Figure 8A), Jurkat cells (Figure 8B), ovarian cancer ID8 cells (Figure 8C), and OVCAR-5 cells (Figure 8D) in a concentration-dependent manner. To avoid the nonspecific cytotoxic impacts of free SHK, we aimed at engineering SHK-loaded PEGylated PLGA NPs. Figure 9 represents the cytotoxic effects of free SHK, free PLGA NPs, SHK-loaded PLGA NPs, and SHK-loaded PEGylated PLGA NPs in primary T-cells. The PEGylation process significantly reduced the cytotoxic effects of SHK.
Figure 8.

Cytotoxic effects of SHK.
Notes: (A) Free SHK-treated ovarian normal cells. (B) Free SHK-treated Jurkat cells. (C) Free SHK-treated ovarian cancer ID8 cells. (D) Free SHK-treated ovarian cancer OVCAR-5 cells. DOX was used as positive control. An asterisk represents statistical significance (P<0.05).
Abbreviations: DOX, doxorubicin; SHK, shikonin; UT, untreated.
Figure 9.
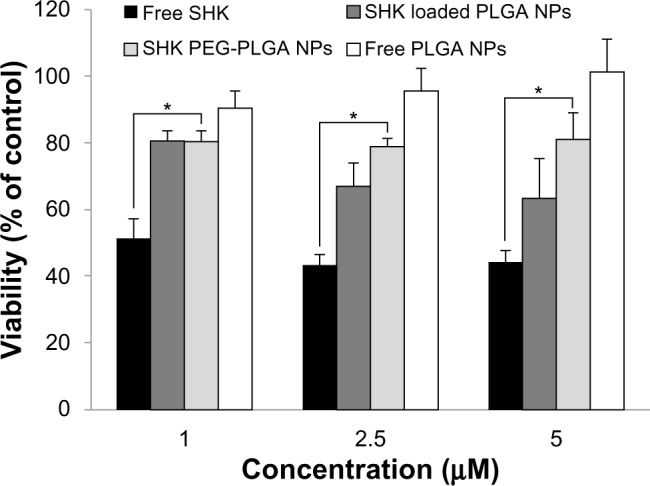
The cytotoxic effects of free SHK, free PLGA NPs, SHK-loaded PLGA NPs, and SHK-loaded PEGylated PLGA NPs in primary T-cells.
Notes: T-cells were exposed to SHK for 2 hours and then sulforhodamine B cytoto xicity assay was conducted. *Statistical significance (P<0.05).
Abbreviations: NPs, nanoparticles; PLGA, poly(lactic-co-glycolic acid); SHK, shikonin; PEG, polyethylene glycol.
Next, the TEM1-positive MS1 cells and TEM1-negative MS1 cells were treated with Ab-/scFv-armed NPs and unarmed NPs loaded with SHK. Figure 10 represents the cytotoxic effects of Ab-/scFv-armed PEGylated PLGA NPs loaded with SHK 2.5 μM (Figure 10A) or 5 μM (Figure 10B) in TEM1-positive MS1 cells and TEM1-negative MS1 cells after 24 hours. Similar impacts were seen at 48 hours post-treatment (data not shown). The Ab-/scFv-armed PEGy-lated PLGA NPs loaded with SHK induced significantly (P<0.05) greater cytotoxicity in TEM1-positive MS1 cells, but not in TEM1-negative MS1 cells.
Figure 10.

The cytotoxicity of anti-TEM1 Ab-/scFv-armed PEGylated PLGA NPs loaded with SHK in TEM1-positive and -negative MS1 endothelial cells.
Notes: The cultured cells were exposed to treatments for 2 hours and then subjected to either SRB cytotoxicity assay (A and B) or flow cytometry apoptosis/necrosis analysis (C–F). The SRB cytotoxicity assay was performed after 24 hours in cells treated with NPs containing SHK 2.5 μM (A) or 5.0 μM (B). The flow cytometry apoptosis/necrosis analysis was conducted after 6 hours. (C) Untreated control cells. (D) Free SHK-treated cells. (E) SHK-loaded Ab-armed PEGylated PLGA NP-treated cells. (F) SHK-loaded scFv-armed PEGylated PLGA NP-treated cells. The concentration of SHK was set at 5 μM in all treatments. *Statistical significance (P<0.05).
Abbreviations: Ab, antibody; NPs, nanoparticles; PEG, polyethylene glycol; PLGA, poly(lactic-co-glycolic acid); scFv, single-chain variable fragment; SHK, shikonin; SRB, sulforhodamine B; TEM1, tumor endothelial marker 1; UT, untreated.
To revalidate the SRB results and reveal the stage of apoptosis/necrosis, we further performed flow cytometry analysis. We found that free SHK or SHK-loaded NPs were able to induce necroptosis (a programmed necrosis) (Figure 10C–F), while the scFv-armed PEGylated PLGA NPs loaded with SHK induced greater cytotoxicity in TEM1-positive MS1 cells (Figure 10F).
We assumed that the NPs could be accumulated within the TME through both passive and active targeting mechanisms. Thus, to assess the final end point of the engineered NPs, their cytotoxicity was examined in the ovarian epithelial OVCAR-5 cells. We witnessed that the growth of the OVCAR-5 cells was significantly inhibited upon treatment with SHK in a dose-dependent manner. The cells treated with 10.0 μM SHK showed over 80% and 90% inhibition after 24 (Figure 11A) and 48 (Figure 11B) hours, respectively.
Figure 11.
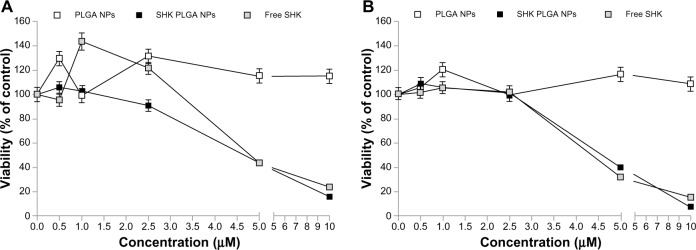
The cytotoxic effects of free SHK or SHK-loaded NPs in ovarian cancer OVCAR-5 cells.
Notes: Cells were treated with free SHK or SHK-loaded NPs. Cytotoxicity analyses were conducted 24 (A) and 48 (B) hours post-treatment.
Abbreviations: NPs, nanoparticles; PLGA, poly(lactic-co-glycolic acid); SHK, shikonin.
DNA fragmentation analyses
To evaluate the impact of SHK on DNA integrity, presented in Figure 12, we also performed DNA defragmentation analysis in OVCAR-5 cells treated with various concentrations of SHK (2.5, 5.0, and 10.0 μM) for 24 hours. We found that SHK could induce significant DNA fragmentation in a dose-dependent manner.
Figure 12.

Effect of shikonin on DNA defragmentation in OVCAR-5 cells.
Notes: Lanes 1–4 represent untreated control cells and shikonin (2.5, 5.0, and 10.0 μM)-treated cells, respectively.
Abbreviation: MWL, molecular weight ladder.
Discussion
Ovarian cancer, as the most common type of gynecologic malignancy, is one of the leading causes of cancer deaths among women. After conventional chemotherapy, most ovarian cancer patients show relapse and become drug resistant. Although cancer chemotherapy has been accepted as an effective treatment modality for ovarian cancer, this approach is often associated with inadvertent intrinsic side effects, mainly because of the cytotoxic nature of most anticancer chemotherapeutic agents. To overcome such problems, a large number of investigators have attempted to deliver chemotherapeutics exclusively to the target cancerous cells within the TME. As the tumor microvasculature displays a disordered fenestrated endothelial architecture, the nanomedicines can be accumulated within the TME based upon the EPR effect (so-called “passive targeting”). Such nanomedicines can be further armed with homing devices to exploit “active targeting” mechanisms as well.30 So far, various drug delivery systems have been used in engineering nanoscale formulations for delivery of cytotoxic agents. In comparison with toxic non-biodegradable polymer-/lipid-based drug delivery systems,22,31–34 biodegradable polymers such as PLGA appear to be a safer biocompatible system for delivery of anticancer agents.17,19,21 In the current study, we capitalized on the engineering of Ab-armed PEGylated PLGA NPs for delivery of SHK, which is known as cytostatic, cytotoxic and immunomodulatory secondary metabolite in a concentration-dependent manner.10,11 SHK can effectively activate both receptor- and mitochondria-mediated apoptosis7 and circumvent cancer drug resistance by induction of a necroptotic death.35 In order to induce specific cytotoxic impacts solely in cancer cells within the TME, we aimed to harness both passive and active targeting mechanisms by engineering SHK-loaded PLGA NPs armed with TEM1-targeting Ab/scFv.
Depending on the composition and method used (ie, polymer and surfactant type and concentration, sonication), as described previously,36 we found that the engineered PLGA NPs showed a size range of 150–250 nm (Figure 2) and a smooth surface (Figure 3). Among several formulations examined, PLGA (50:50 with Mw of 38,000–54,000 [3% w/v]) and Pluronic® F68 (3% w/v) provided NPs at a size range of ~200 nm with a sound distribution pattern and high DE% (~90%).
The release of SHK from PLGA NPs was studied at different pHs to reveal the pattern of releases within the vesicular machineries of the target cells as well as the TME, in which the pH is acidic. We found slower release at lower pH (Figure 4), which indicates sustained release of SHK in acidic conditions, resulting in longer presence of drug molecules at the TME. To disclose the drug release kinetics, the release data were fit using several models (Table 2), as described previously.26 The release data were best fit with Wagner log-probability and, to some extent, with Weibull models (Table 2), which is in consensus with a previous report on the release of lipophilic drugs from polymeric NPs.26 We assumed that these NPs can reach the target tumor site within 2 hours,37,38 during which period the liberation of SHK (~20%) can favor the cancer therapy objectives by activating the immune system.9 Once these NPs reached into the TME by EPR effect, they could attach to the tumor endothelial cells through homing devices (targeting TEM1) and accumulate within the TME where SHK is released (>80%) within 24–48 hours. Such accumulation of SHK within the TME can induce maximum cytotoxic effects. It should be noted that the plain PLGA NPs are inherently prone to opsonization and reticuloendothelial system clearance.39,40 To avoid such problems, the PLGA NPs have been PEGylated to become invisible to the immune system and to enhance the EPR effect.39,41 For active and specific targeting of cancer cells in the current study, the PLGA NPs were PEGylated and further conjugated with anti-TEM1 Ab/scFv using EDC/NHS chemistry (Figure 1). We found that a 20:1 molar ratio of homing device:PEG provided NPs with significant potential to bind to the TEM1 that is overexpressed in tumor vasculature endothelia. The anti-TEM1 Ab/scFv-armed PEGylated PLGA NPs were significantly able to bind the TEM1-positive MS1 cells, but not the TEM1-negative MS1 cells (Figure 5). We found that the binding potential of the scFv-armed NPs was significantly greater than the Ab-armed NPs, perhaps due to the smaller size and mass of the scFv that allow a higher level of conjugation compared to the whole Ab. It should be stated that the surface decoration of the PLGA NPs was associated with some drug loss (Table 1), which was taken into account for cytotoxicity studies. The two-step post-formulation surface modification processes of PEGylation and Ab/scFv conjugation resulted in greater loss of SHK.
Since the main objective of the study was to specifically deliver SHK to, and investigate its impacts in the target cells, we first compared the morphological changes of the OVCAR-5 cells treated with SHK using fluorescence microscopy. Morphological evaluation showed that SHK induced profound morphological changes in ovarian cancer OVCAR-5 cells (Figure 6). We found that the SHK-treated cells showed somewhat lower phalloidin staining, especially regarding the formation of lamellipodial ruffling.42 This emphasizes the impact of SHK on cell motility and migration capacity, which was further confirmed by cell migration assay (Figure 7). Based on morphological changes, it seems SHK can induce chromatin condensation and nuclear shrinkage. These findings are in consensus with reported effects of SHK on C6 glioma cells;43 nonetheless, the genomic and/or epigenomic impacts of SHK are yet to be fully investigated. Huang et al, in 2013, demonstrated that SHK can elicit cell death mainly via necroptosis mechanism,43 which is in full agreement with our results in both OVCAR-5 and MS1 cells.
The SRB cytotoxic assay revealed that free SHK can induce toxicity in the ovarian cancer OVCAR-5 cells as well as in the normal ovarian IOSE-398 cells and Jurkat cells (Figure 8). Such nonspecific toxicity further highlighted the inevitability of specific delivery of SHK to the TME through passive and active targeting mechanisms in order to diminish the nonspecific side effects. Thus, to exploit both passive and active targeting potentials, we engineered SHK-loaded PEGylated PLGA NPs decorated with anti-TEM1 Ab/scFv. TEM1 has been reported to be expressed by the endothelial cells of ovarian tumor vasculature25,44–47 and other malignancies such as brain and abdominal tumors.48,49 Based on the release profile, less than 20% of drug can be released within 2 hours, during which the engineered NPs can safely pass the blood stream and reach the target site with negligible inadvertent detrimental impacts on blood-circulating immune system cells. To this end, the toxic effects of SHK-loaded NPs were tested in primary lymphocytes and it was found that the SHK-loaded PEGylated NPs were significantly safer than non-PEGylated NPs (Figure 9). To evaluate the effect of anti-TEM1 Ab/scFv-armed SHK-loaded PLGA NPs, we used TEM1-negative MS1 cells and human TEM1-transfected MS1 cells. In accordance with the fluorescence microscopy results (Figure 5), we found that the anti-TEM1 Ab/scFv-armed SHK-loaded PLGA NPs could induce significantly greater toxicity in TEM1-positive MS1 endothelial cells (Figure 10). These findings may verify our primary assumption that the active targeting of TEM1 could enhance the EPR effect through binding to the TEM1-expressing tumor endothelial cells during circulation in blood. We speculate that, once attached to the tumor vasculature, these NPs are entrapped and accumulated solely within the tumor site. Our findings confirmed that decoration of PLGA NPs with anti-TEM1 scFv provides higher antigen-binding capacity as compared to the PLGA NPs conjugated with anti-TEM1 Ab (Figures 5 and 10). The results of the SRB toxicity assay were further confirmed using flow cytometry apoptosis/necrosis assay (Figure 10E and F). Our results are in accordance with previous reports showing that SHK can induce both apoptosis and necrosis in cancer cells.50–52 Further, given that annexin V is not cell permeable, the binding of externalized phosphatidylserine (PS) is selective for early apoptotic cells, hence we aimed to discriminate the stage of apoptosis that occurred in the cells treated with free SHK or SHK-loaded NPs. Using TEM1-positive MS1 cells (with 95% expression of TEM1) and TEM1-negative MS1 cells, we found that Ab- or scFv-armed PLGA NPs loaded with SHK were able to induce significantly (P<0.05) greater cytotoxic impacts in TEM1-positive MS1 cells than in TEM1-negative MS1 cells (Figure 10A and B). Further, the impact of unarmed NPs encapsulating SHK was greater than that of the free SHK, showing the potential of passive targeting. Our preliminary confocal microscopy showed that the engineered SHK-loaded NPs can be taken up by the epithelial cancerous cells (data not shown) and induce profound toxicity in a dose-dependent manner as shown for the OVCAR-5 cells (Figure 11). Based on the flow cytometry analysis, SHK-treated cells showed both apoptosis and necrosis after 6 hours (Figure 10C–F), while they became mainly necrotic after 24 hours, which was further confirmed by DNA fragmentation assay (Figure 12). In fact, SHK is a naturally occurring naphthoquinone with profound inhibitory effects on cancer cell proliferation both in vivo and in vitro.6,53 Chen et al reported in 2012 that SHK can induce generation of reactive oxygen species, depletion of glutathione, disruption of mitochondrial transmembrane potential, upregulation of p53, cleavage of poly(adenosine diphosphate [ADP]-ribose) polymerase, catalase downregu-lation, super oxide dismutases (SOD1) upregulation, BCL2 downregulation, and BAX upregulation in U87MG glioma cells.6 In human glioma cells, SHK-induced necroptosis appears to occur through multiple pathways, while it has also been shown to inhibit the glycolytic path in cancers through inhibiting pyruvate kinase-M2 (PKM2).13 Similarly, skin epidermal JB6 P+ cells treated with SHK showed marked induction of apoptosis through suppression of PKM2 activation, and decreased level of lactate as a glycolysis marker, thus mitochondrial dysfunction.12 In summary, SHK can induce necroptosis through multiple pathways, such as by inhibiting the activity of PKM2. Thus, if SHK is specifically delivered to cancer cells using targeted nanomedicines, it can result in significant suppression of dysregulated molecular events within the TME.
Conclusion
SHK is able to induce marked cytotoxic impacts in ovarian cancer cells. However, similar to other cytotoxic agents, such as doxorubicin, its toxic impact is nonspecific, resulting in inadvertent detrimental impact on normal cells. Based on our findings, we propose biodegradable SHK-loaded PLGA NPs PEGylated and armed with anti-TEM1 scFv as a new targeted nanomedicine for ovarian cancer therapy as well as for other solid tumors.
Acknowledgments
The authors are thankful to Dr Andrew Tsourkas, Dr Aizhi Zhao, Dr John Lambris, Euihye Jung, Steve Santoro, and Thomas Garrabrant (University of Pennsylvania, Philadelphia, PA, USA) for their technical support. This work was supported by NIH R01CA156695, SPORE in Ovarian Cancer Pilot P50 CA083638-14, and the Honorable Tina Brozman Foundation.
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Guddati AK. Ovarian cancer stem cells: elusive targets for chemotherapy. Med Oncol. 2012;29(5):3400–3408. doi: 10.1007/s12032-012-0252-6. [DOI] [PubMed] [Google Scholar]
- 2.Rocconi RP, Sullivan P, Long B, et al. Treatment of chemotherapy-induced anemia in ovarian cancer patients: does the use of erythropoiesis-stimulating agents worsen survival? Int J Gynecol Cancer. 2012;22(5):786–791. doi: 10.1097/IGC.0b013e31825104f4. [DOI] [PubMed] [Google Scholar]
- 3.Bookman MA. First-line chemotherapy in epithelial ovarian cancer. Clin Obstet Gynecol. 2012;55(1):96–113. doi: 10.1097/GRF.0b013e31824b45da. [DOI] [PubMed] [Google Scholar]
- 4.Meier W, du Bois A, Reuss A, et al. Topotecan versus treosulfan, an alkylating agent, in patients with epithelial ovarian cancer and relapse within 12 months following 1st-line platinum/paclitaxel chemotherapy. A prospectively randomized phase III trial by the Arbeitsgemeinschaft Gynaekologische Onkologie Ovarian Cancer Study Group (AGO-OVAR) Gynecol Oncol. 2009;114(2):199–205. doi: 10.1016/j.ygyno.2009.04.026. [DOI] [PubMed] [Google Scholar]
- 5.Ferrero JM, Weber B, Geay JF, et al. Second-line chemotherapy with pegylated liposomal doxorubicin and carboplatin is highly effective in patients with advanced ovarian cancer in late relapse: a GINECO phase II trial. Ann Oncol. 2007;18(2):263–268. doi: 10.1093/annonc/mdl376. [DOI] [PubMed] [Google Scholar]
- 6.Chen CH, Lin ML, Ong PL, Yang JT. Novel multiple apoptotic mechanism of shikonin in human glioma cells. Ann Surg Oncol. 2012;19(9):3097–3106. doi: 10.1245/s10434-012-2324-4. [DOI] [PubMed] [Google Scholar]
- 7.Chen HM, Wang PH, Chen SS, et al. Shikonin induces immunogenic cell death in tumor cells and enhances dendritic cell-based cancer vaccine. Cancer Immunol Immunother. 2012;61(11):1989–2002. doi: 10.1007/s00262-012-1258-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wiench B, Eichhorn T, Paulsen M, Efferth T. Shikonin directly targets mitochondria and causes mitochondrial dysfunction in cancer cells. Evid Based Complement Alternat Med. 2012;2012:726025. doi: 10.1155/2012/726025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Long S, GuangZhi Y, BaoJie G, et al. Shikonin derivatives protect immune organs from damage and promote immune responses in vivo in tumour-bearing mice. Phytother Res. 2012;26(1):26–33. doi: 10.1002/ptr.3503. [DOI] [PubMed] [Google Scholar]
- 10.Zare Kh, Khosrowshahli M, Nazemiyeh H, Movafeghi A, Azar AM, Omidi Y. Callus culture of Echium italicum L. towards production of a shikonin derivative. Nat Prod Res. 2011;25(16):1480–1487. doi: 10.1080/14786410902804857. [DOI] [PubMed] [Google Scholar]
- 11.Zare K, Nazemiyeh H, Movafeghi A, et al. Bioprocess engineering of Echium italicum L: induction of shikonin and alkannin derivatives by two-liquid-phase suspension cultures. Plant Cell Tissue Organ Cult. 2010;100(2):157–164. [Google Scholar]
- 12.Li W, Liu J, Zhao Y. PKM2 inhibitor shikonin suppresses TPA-induced mitochondrial malfunction and proliferation of skin epidermal JB6 cells. Mol Carcinog. 2012 Dec 19; doi: 10.1002/mc.21988. Epub. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen J, Xie J, Jiang Z, Wang B, Wang Y, Hu X. Shikonin and its analogs inhibit cancer cell glycolysis by targeting tumor pyruvate kinase-M2. Oncogene. 2011;30(42):4297–4306. doi: 10.1038/onc.2011.137. [DOI] [PubMed] [Google Scholar]
- 14.Wu Z, Wu L, Li L, Tashiro S, Onodera S, Ikejima T. p53-mediated cell cycle arrest and apoptosis induced by shikonin via a caspase-9-dependent mechanism in human malignant melanoma A375-S2 cells. J Pharmacol Sci. 2004;94(2):166–176. doi: 10.1254/jphs.94.166. [DOI] [PubMed] [Google Scholar]
- 15.Nam KN, Son MS, Park JH, Lee EH. Shikonins attenuate microglial inflammatory responses by inhibition of ERK, Akt, and NF-kappaB: neuroprotective implications. Neuropharmacology. 2008;55(5):819–825. doi: 10.1016/j.neuropharm.2008.06.065. [DOI] [PubMed] [Google Scholar]
- 16.Danhier F, Feron O, Préat V. To exploit the tumor microenvironment: passive and active tumor targeting of nanocarriers for anti-cancer drug delivery. J Control Release. 2010;148(2):135–146. doi: 10.1016/j.jconrel.2010.08.027. [DOI] [PubMed] [Google Scholar]
- 17.Omidi Y, Davaran S. Impacts of biodegradable polymers: towards biomedical applications. In: Sharma SK, Mudhoo A, Clark JH, editors. A Handbook of Applied Biopolymer Technology: Synthesis, Degradation and Applications. London: Royal Society of Chemistry; 2011. pp. 388–418. [Google Scholar]
- 18.Omidi Y. Smart multifunctional theranostics: simultaneous diagnosis and therapy of cancer. Bioimpacts. 2011;1(3):145–147. doi: 10.5681/bi.2011.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mooguee M, Omidi Y, Davaran S. Synthesis and in vitro release of adriamycin from star-shaped poly(lactide-co-glycolide) nano- and microparticles. J Pharm Sci. 2010;99(8):3389–3397. doi: 10.1002/jps.22106. [DOI] [PubMed] [Google Scholar]
- 20.Thamake SI, Raut SL, Gryczynski Z, Ranjan AP, Vishwanatha JK. Alendronate coated poly-lactic-co-glycolic acid (PLGA) nanoparticles for active targeting of metastatic breast cancer. Biomaterials. 2012;33(29):7164–7173. doi: 10.1016/j.biomaterials.2012.06.026. [DOI] [PubMed] [Google Scholar]
- 21.Davaran S, Omidi Y, Rashidi MR, et al. Preparation and in vitro evaluation of linear and star-branched PLGA nanoparticles for insulin delivery. J Bioact Compat Polym. 2008;23(2):115–131. [Google Scholar]
- 22.Omidi Y, Barar J, Heidari HR, Ahmadian S, Yazdi HA, Akhtar S. Microarray analysis of the toxicogenomics and the genotoxic potential of a cationic lipid-based gene delivery nanosystem in human alveolar epithelial a549 cells. Toxicol Mech Methods. 2008;18(4):369–378. doi: 10.1080/15376510801891286. [DOI] [PubMed] [Google Scholar]
- 23.Omidi Y, Barar J. Induction of human alveolar epithelial cell growth factor receptors by dendrimeric nanostructures. Int J Toxicol. 2009;28(2):113–122. doi: 10.1177/1091581809335177. [DOI] [PubMed] [Google Scholar]
- 24.Kafil V, Omidi Y. Cytotoxic impacts of linear and branched polyethylenimine nanostructures in A431 cells. Bioimpacts. 2011;1(1):23–30. doi: 10.5681/bi.2011.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhao A, Nunez-Cruz S, Li C, Coukos G, Siegel DL, Scholler N. Rapid isolation of high-affinity human antibodies against the tumor vascular marker Endosialin/TEM1, using a paired yeast-display/secretory scFv library platform. J Immunol Methods. 2011;363(2):221–232. doi: 10.1016/j.jim.2010.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Barzegar-Jalali M, Adibkia K, Valizadeh H, et al. Kinetic analysis of drug release from nanoparticles. J Pharm Pharm Sci. 2008;11(1):167–177. doi: 10.18433/j3d59t. [DOI] [PubMed] [Google Scholar]
- 27.Barar J, Omidi Y. Surface modified multifunctional nanomedi-cines for simultaneous imaging and therapy of cancer. Bioimpacts. 2014;4(1):3–14. doi: 10.5681/bi.2014.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vichai V, Kirtikara K. Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat Protoc. 2006;1(3):1112–1116. doi: 10.1038/nprot.2006.179. [DOI] [PubMed] [Google Scholar]
- 29.Vermes I, Haanen C, Steffens-Nakken H, Reutelingsperger C. A novel assay for apoptosis. Flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein labelled Annexin V. J Immunol Methods. 1995;184(1):39–51. doi: 10.1016/0022-1759(95)00072-i. [DOI] [PubMed] [Google Scholar]
- 30.Barar J, Omidi Y. Translational approaches towards cancer gene therapy: Hurdles and hopes. Bioimpacts. 2012;2(3):127–143. doi: 10.5681/bi.2012.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Barar J, Omidi Y. Intrinsic bio-signature of gene delivery nanocarriers may impair gene therapy goals. Bioimpacts. 2013;3(3):105–109. doi: 10.5681/bi.2013.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Omidi Y, Hollins AJ, Drayton RM, Akhtar S. Polypropylenimine dendrimer-induced gene expression changes: the effect of complexation with DNA, dendrimer generation and cell type. J Drug Target. 2005;13(7):431–443. doi: 10.1080/10611860500418881. [DOI] [PubMed] [Google Scholar]
- 33.Omidi Y, Hollins AJ, Benboubetra M, Drayton R, Benter IF, Akhtari S. Toxicogenomics of non-viral vectors for gene therapy: A microarray study of lipofectin- and oligofectamine-induced gene expression changes in human epithelial cells. J Drug Target. 2003;11(6):311–323. doi: 10.1080/10611860310001636908. [DOI] [PubMed] [Google Scholar]
- 34.Barar J, Hamzehei H, Mortazavi Tabatabaei SA, SE H-A, Omidi Y. Genomic signature and toxicogenomics comparison of polycationic gene delivery nanosystems in human alveolar epithelial A549 cells. Daru J Pharm sci. 2009;17(3):139–147. [Google Scholar]
- 35.Andreopoulou E, Gaiotti D, Kim E, et al. Pegylated liposomal doxorubi-cin HCL (PLD; Caelyx/Doxil): experience with long-term maintenance in responding patients with recurrent epithelial ovarian cancer. Ann Oncol. 2007;18(4):716–721. doi: 10.1093/annonc/mdl484. [DOI] [PubMed] [Google Scholar]
- 36.Adibkia K, Barzegar Jalali M, Nokhodchi A, et al. A review on the methods of preparation of pharmaceutical nanoparticles. Pharm Sci. 2010;15(4):303–314. [Google Scholar]
- 37.Snehalatha M, Venugopal K, Saha RN, Babbar AK, Sharma RK. Etoposide loaded PLGA and PCL nanoparticles II: biodistribution and pharmacokinetics after radiolabeling with Tc-99m. Drug Deliv. 2008;15(5):277–287. doi: 10.1080/10717540802006500. [DOI] [PubMed] [Google Scholar]
- 38.Avgoustakis K, Beletsi A, Panagi Z, et al. Effect of copolymer composition on the physicochemical characteristics, in vitro stability, and biodistribution of PLGA-mPEG nanoparticles. Int J Pharm. 2003;259(1–2):115–127. doi: 10.1016/s0378-5173(03)00224-2. [DOI] [PubMed] [Google Scholar]
- 39.Amoozgar Z, Park J, Lin Q, Yeo Y. Low molecular-weight chitosan as a pH-sensitive stealth coating for tumor-specific drug delivery. Mol Pharm. 2012;9(5):1262–1270. doi: 10.1021/mp2005615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yadav KS, Chuttani K, Mishra AK, Sawant KK. Effect of size on the biodistribution and blood clearance of etoposide-loaded PLGA nanoparticles. PDA J Pharm Sci Technol. 2011;65(2):131–139. [PubMed] [Google Scholar]
- 41.Hamidi M, Azadi A, Rafiei P. Pharmacokinetic consequences of pegylation. Drug Deliv. 2006;13(6):399–409. doi: 10.1080/10717540600814402. [DOI] [PubMed] [Google Scholar]
- 42.Vitorino P, Meyer T. Modular control of endothelial sheet migration. Genes Dev. 2008;22(23):3268–3281. doi: 10.1101/gad.1725808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Huang C, Luo Y, Zhao J, et al. Shikonin kills glioma cells through necroptosis mediated by RIP-1. PLoS One. 2013;8(6):e66326. doi: 10.1371/journal.pone.0066326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Conejo-Garcia JR, Buckanovich RJ, Benencia F, et al. Vascular leukocytes contribute to tumor vascularization. Blood. 2005;105(2):679–681. doi: 10.1182/blood-2004-05-1906. [DOI] [PubMed] [Google Scholar]
- 45.Nanda A, St Croix B. Tumor endothelial markers: new targets for cancer therapy. Curr Opin Oncol. 2004;16(1):44–49. doi: 10.1097/00001622-200401000-00009. [DOI] [PubMed] [Google Scholar]
- 46.Christian S, Winkler R, Helfrich I, et al. Endosialin (Tem1) is a marker of tumor-associated myofibroblasts and tumor vessel-associated mural cells. Am J Pathol. 2008;172(2):486–494. doi: 10.2353/ajpath.2008.070623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bagley RG, Honma N, Weber W, et al. Endosialin/TEM 1/CD248 is a pericyte marker of embryonic and tumor neovascularization. Microvasc Res. 2008;76(3):180–188. doi: 10.1016/j.mvr.2008.07.008. [DOI] [PubMed] [Google Scholar]
- 48.Carson-Walter EB, Winans BN, Whiteman MC, et al. Characterization of TEM1/endosialin in human and murine brain tumors. BMC Cancer. 2009;9:417. doi: 10.1186/1471-2407-9-417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nanda A, Karim B, Peng Z, et al. Tumor endothelial marker 1 (Tem1) functions in the growth and progression of abdominal tumors. Proc Natl Acad Sci USA. 2006;103(9):3351–3356. doi: 10.1073/pnas.0511306103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Han W, Xie J, Li L, Liu Z, Hu X. Necrostatin-1 reverts shikonin-induced necroptosis to apoptosis. Apoptosis. 2009;14(5):674–686. doi: 10.1007/s10495-009-0334-x. [DOI] [PubMed] [Google Scholar]
- 51.Xuan Y, Hu X. Naturally-occurring shikonin analogues – a class of necroptotic inducers that circumvent cancer drug resistance. Cancer Lett. 2009;274(2):233–242. doi: 10.1016/j.canlet.2008.09.029. [DOI] [PubMed] [Google Scholar]
- 52.Han W, Li L, Qiu S, et al. Shikonin circumvents cancer drug resistance by induction of a necroptotic death. Mol Cancer Ther. 2007;6(5):1641–1649. doi: 10.1158/1535-7163.MCT-06-0511. [DOI] [PubMed] [Google Scholar]
- 53.Chen X, Yang L, Oppenheim JJ, Howard MZ. Cellular pharmacology studies of shikonin derivatives. Phytother Res. 2002;16(3):199–209. doi: 10.1002/ptr.1100. [DOI] [PubMed] [Google Scholar]