

Abstract
One of the primary mechanisms through which a cell exerts control over its metabolic state is by modulating expression levels of its enzyme-coding genes. However, the changes at the level of enzyme expression allow only indirect control over metabolite levels, for two main reasons. First, at the level of individual reactions, metabolite levels are non-linearly dependent on enzyme abundances as per the reaction kinetics mechanisms. Secondly, specific metabolite pools are tightly interlinked with the rest of the metabolic network through their production and consumption reactions. While the role of reaction kinetics in metabolite concentration control is well studied at the level of individual reactions, the contribution of network connectivity has remained relatively unclear. Here we report a modeling framework that integrates both reaction kinetics and network connectivity constraints for describing the interplay between metabolite concentrations and mRNA levels. We used this framework to investigate correlations between the gene expression and the metabolite concentration changes in Saccharomyces cerevisiae during its metabolic cycle, as well as in response to three fundamentally different biological perturbations, namely gene knockout, nutrient shock and nutrient change. While the kinetic constraints applied at the level of individual reactions were found to be poor descriptors of the mRNA-metabolite relationship, their use in the context of the network enabled us to correlate changes in the expression of enzyme-coding genes to the alterations in metabolite levels. Our results highlight the key contribution of metabolic network connectivity in mediating cellular control over metabolite levels, and have implications towards bridging the gap between genotype and metabolic phenotype.
Author Summary
Regulation of metabolic activity in response to environmental and genetic perturbations is fundamental to the growth and maintenance of all cells. A primary regulatory process used by cells to control the activity of their metabolic network is the alteration in the expression of enzyme-coding genes. How these alterations regulate metabolite concentrations is an important question in the quest towards unraveling the genotype-phenotype relationship. The link between the expression levels of enzymes and metabolite concentrations is governed by the kinetics of individual reactions, which in turn are interlinked with each other due to the complex connectivity structure of metabolic networks. Although the enzyme-metabolite relationship is relatively well studied at the level of individual reactions, our understanding of the regulation of metabolite levels in complex networks has remained incomplete. In this study, we show that the constraints imposed by the network connectivity are key determinants of the relationship between gene expression and metabolite concentration changes. Our results provide mechanistic insight into the function of complex metabolic networks and have implications for health and biotechnological applications.
Introduction
Cellular metabolic networks provide basic biochemical building blocks and a thermodynamically favorable environment for growth and maintenance. Due to this crucial role of metabolism, cells have evolved various mechanisms to regulate metabolic reactions in response to genetic and environmental changes. Metabolic reactions can be regulated either by modulating the availability of the corresponding enzymes, e.g. through altered transcription and/or translation, or, by modulating the enzyme activities through post-translational modifications or through binding of small molecules. Our knowledge of the landscape of transcriptional, translational and post-translational regulation of metabolism is expanding with the increasing availability of datasets that provide genome-wide views of the abundance and interactions between mRNAs, proteins and metabolites [1]–[6]. Although the relative contribution of each of these regulatory layers is still unclear and is likely to be context dependent, it has long been clear that the adjustments in the cellular metabolic phenotype (i.e., rates of reactions, or fluxes, and metabolite levels) often involve changes at the level of gene expression [7]–[9]. For example, previous studies have shown that the gene expression changes in metabolic networks are centered on metabolites that are crucial for adjusting the network state in response to specific perturbations [10], [11]. Despite successful outcomes of these and other studies suggesting a strong link between transcriptional regulation and changes in metabolite levels [7], [10], [12], [13], the relationship between the two has remained elusive.
The task of developing models for describing the relationship between gene expression and metabolite concentrations is challenging due to the multiple layers of regulation involved in between (Figure 1A). Several of the regulatory mechanisms involved, such as translational control or allosteric regulation, are currently poorly understood at the scale of the whole network. Measurement of protein abundances or enzyme activities is also currently difficult to perform at the network scale and in complex systems such as human tissues. Thus, in the absence of data for intermediate molecular players, a detailed investigation of the link between gene expression and metabolite levels has both a fundamental and a practical appeal. In particular, it is of interest to estimate the degree to which the changes at the level of gene expression affect changes in metabolite concentration and to uncover the underlying mechanisms determining their relationship. In this study, we explore the hitherto poorly understood role of network connectivity constraints in controlling metabolite concentrations in a eukaryotic model organism, Saccharomyces cerevisiae. We postulate that two primary mechanisms will largely determine the association between the changes in mRNA and metabolite levels: reaction kinetics (which are non-linear by nature [14]–[16]) and the mass balance constraints imposed by the network, i.e. the balance between production and consumption of metabolites. Although we here focus on mRNA levels due to genome-wide coverage of the available transcriptomics datasets, the proposed model can also be readily applied to enzyme abundance or activity data.
Figure 1. From gene expression to metabolite levels.

A) Metabolite levels are only indirectly affected by changes in gene expression levels, through changes in the corresponding enzyme abundances. Metabolites, in turn, can provide feedback to the regulatory network controlling enzyme abundance/activity. B) Transcript-metabolite relationships are usually many-to-one. We discarded transcripts with insignificant changes (P≤0.05) (grey circles). For the remaining transcripts we combined the corresponding fold changes to derive gene-expression scores for reactions and thereby for consumption or production of metabolites (see Main text). C) Schematic workflow used for the proposed concentration change coupling analysis (CoCCoA). In the first step, physiological measurements from growth experiments are used to constrain the genome-scale metabolic model (Methods). Subsequent flux simulations (Methods) help in identifying the directionality and range of fluxes under the conditions being compared. Next, by using the comparative transcriptome data, fold changes at the individual gene-expression level are mapped on to the reactions in the network (panel B). CoCCoA integrates the mapped gene-expression data with the network topology by using a model formulation derived from the MM reaction kinetics mechanism and mass balance constraints (main text). The main output from the algorithm is a measure of transcriptional control over metabolite levels, or the CoCCoA scores, which are tested for correlation with the experimentally measured metabolite concentration changes. D) Schematic representation of three different CoCCoA models with varying degrees of network connectivity constraints.
The role of reaction kinetics in controlling metabolite concentration has been previously examined mostly from the perspective of the isolated reaction-metabolite pairs. With such a reaction-centric approach, a previous study on yeast metabolism was able to partially explain changes in the intracellular metabolite levels when using protein abundance as a measure of enzyme availability [3]. However, no correlation was observed in the same study when using gene expression data instead of protein abundances. One possible reason for the lack of strong correlation between gene expression and metabolite levels when looking at the isolated enzyme-metabolite pairs is that the large connectivity inherent to metabolic networks is not taken into account. A large fraction of intra-cellular metabolites participate in multiple reactions. For example, over 25% of the yeast metabolites participate in more than three reactions [17]. Consequently, abundance of an enzyme catalyzing a particular reaction cannot completely determine the concentrations of the participating metabolites or the rate of the reaction. Indeed, correlations between mRNA and fluxes, and even between enzyme activities and fluxes, have been often found to be poor [18]–[21]. Approaches accounting for the network connectivity of metabolites have been successful in linking gene expression to metabolites in an empirical or qualitative manner [10], [12], [22]–[24], but have achieved only a limited success on the quantitative front. Advantages of both reaction-centered kinetics approaches and network topology-based approaches can be combined in network kinetic models that include detailed kinetics of all involved reactions [25]–[28]. However, application of kinetic models to large metabolic networks is difficult due to their reliance on a large number of parameters. Such parameters are either currently unavailable, or their estimation requires comprehensive measurements of intra-cellular states of interest (e.g. metabolite concentrations, enzyme abundances, and fluxes) in the vicinity of the perturbation to be modeled.
In this study, we propose a steady-state model of the transcriptional control of metabolite concentrations. Our model integrates reaction kinetics and metabolic network connectivity constraints without requiring the knowledge of kinetic parameters. In essence, the model uses mass balance constraints to bridge the individual reaction kinetic constraints with those of the other reactions in the network. The resulting equations provide a log-linear relationship between the fold-change in the concentration of a given metabolite to the fold-changes in the expression of its neighboring genes, as well as topologically more distant genes.
Results
By analogy to flux coupling analysis [29], which describes how steady-state fluxes are linked with each other, we termed our approach Concentration Change Coupling Analysis (CoCCoA). Starting with a classical reaction kinetics model, which treats each reaction as an isolated system consisting of a single enzyme and its substrate, we developed a network kinetics approach by accounting for the interactions between different reactions through their shared metabolites. As there is currently a lack of information on in vivo enzyme kinetics mechanisms at the network-scale, we used the single-substrate Michaelis-Menten (MM) kinetics for all reactions. In essence, MM kinetics describes the flux or reaction rate V as a function of three parameters: i) concentration of the substrate, S; ii) maximum capacity of the enzyme pool, V max; and iii) a parameter reflecting the enzyme's kinetic properties, KM (Figure 1A). The central idea of CoCCoA is to use mass balance constraints on the flux term V to link single-reaction kinetics to the other reactions in the network. We considered MM kinetics in the fold-change space, which allowed us to eliminate the need to know the KM values. For each metabolite, CoCCoA provides an overall transcriptional change score (CoCCoA score) according to the CoCCoA equations, which are developed in the subsequent sub-sections. To assess the proportion of variance in metabolite changes that can be attributed to transcriptional regulation, we compared the calculated CoCCoA scores with the experimentally measured metabolite concentration changes. The overall workflow used is depicted in Figure 1B.
The first step in our analysis is to calculate a representative transcriptional fold-change for each reaction. As the yeast metabolic network consists of several reactions that are each governed by multiple proteins, we classified all reactions into three types: i) reactions catalyzed by a single enzyme, ii) reactions catalyzed by two or more isoenzymes, and iii) reactions catalyzed by enzyme complexes. We then applied the following rules to calculate the representative fold-changes for all reactions: in the case of isoenzymes, we averaged the fold changes of the related transcripts, while in the case of complexes, we picked the transcript with the lowest fold change (Figure 1C). We used only significantly changed transcripts (P-value≤0.05) in the presented analysis. Relaxation of this filtering criterion did not change the overall results (Figure S1).
Experimental datasets and the metabolic network
We used four published experimental datasets for evaluating the proposed CoCCoA models. These case studies included three pairwise comparisons – one genetic [3] and two environmental perturbations [23], [30] – and a time-course dataset obtained during the yeast metabolic cycle [9], [31]. In all pair-wise comparison studies, both gene expression and metabolite concentration data were obtained from the same experiment. In the case of the metabolic cycle data, although the sampling for transcriptome and metabolome was performed in two separate experiments, the experimental setups were identical and the sampling was performed at comparable time-points spanning all phases of the metabolic cycle. While the metabolic cycle is fundamentally a non-steady-state phenomenon, the observed transcript oscillation period of about 300 minute means that a reasonable degree of pseudo-steady-state can be assumed for applying our model. In the case of the three pairwise comparison studies, the correlations between CoCCoA scores and metabolite concentrations provided a perturbation-centered perspective wherein the responses of different metabolites were analyzed jointly. The metabolic cycle case study allowed us to additionally evaluate the gene expression-metabolite relationship from a metabolite-centered perspective, wherein the response of each metabolite was assessed individually for its conformity to the proposed model.
A genome-scale metabolic reconstruction of S. cerevisiae [17] was used to obtain the metabolite-reaction-gene connectivity information and to estimate the reaction directionalities (Methods). For each case study, we used the experimental measurements of exchange fluxes (uptake and secretion rates of metabolites) to constrain and simulate a flux balance model. Accordingly, we removed all blocked reactions and reactions for which the flux directions could not be unambiguously assigned. We also excluded the reactions for which the predicted flux directions did not agree between the two conditions being compared.
Depending on the extent to which the network connectivity information is included in the calculations, we term the CoCCoA models as 0th degree, 1st degree, 2nd degree, and so on (Figure 1D). 0th degree CoCCoA relies on the enzyme kinetics alone and thus considers only the consumption reaction(s) of any given metabolite. 1st degree CoCCoA additionally considers the production of the metabolite by using mass balance constraints. 2nd degree CoCCoA further expands the degree of network connectivity accounted for in the model by including the producing reactions of the precursors of the metabolite in question. Alternatively, the 2nd and higher degree models can also be expanded on both the consumption and production sides of the metabolite as described in the following sub-sections (also see Text S1).
0th degree concentration change coupling analysis
We consider metabolite concentration changes relative to a reference condition that can be arbitrarily chosen from the conditions pertaining to the experiment under investigation. Assuming that the enzyme properties (represented by KM) remain unchanged in the experiment, by using MM kinetics one obtains (Text S1):
| (1) |
Where * denotes the reference condition. The relative nature of this formulation allows circumventing the problem of the lack of availability of in vivo KM values. Furthermore, by assuming that 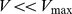 &
&
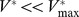 , and that the ratio
, and that the ratio 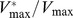 can be approximated by the gene expression ratio, equation (1) simplifies to a log-linear relationship (equation (2), Text S1). Both of these assumptions are critically examined in the next sub-section. The model represented by equation (2) is hereby termed 0th degree coupling, meaning that the metabolite S is not considered to be directly coupled to any other metabolite and is connected only to the enzyme that uses it as a substrate (Figure 1D).
can be approximated by the gene expression ratio, equation (1) simplifies to a log-linear relationship (equation (2), Text S1). Both of these assumptions are critically examined in the next sub-section. The model represented by equation (2) is hereby termed 0th degree coupling, meaning that the metabolite S is not considered to be directly coupled to any other metabolite and is connected only to the enzyme that uses it as a substrate (Figure 1D).
| (2) |
Evaluation of non-saturation and mRNA-protein correlation assumptions
The first assumption used in deriving equation (2) implies that the enzyme is not saturated. The opposite situation, i.e. an enzyme approaching saturation, is not amenable for establishing the metabolite-gene expression relationship (or metabolite-enzyme abundance relationship in general), as the reaction velocity will then be only a weak function of the substrate concentration. Recent studies have shown that in vivo concentrations for several metabolites, especially from central carbon metabolism, are close to the corresponding KM values [32]. At these concentrations, reaction rates V are close to half of the Vmax. Although the assumption of V≪Vmax is not strictly applicable in this flux regime, numerical simulations showed modest errors (around 20%) due to this approximation (Figure S2). Moreover, if the saturation level does not change drastically between the two conditions being compared, the error remains close to zero (Figure S2). Given the advantage that this approximation brings, namely elimination of the need for knowing the in vivo kinetic parameters, the cost of the approximation error appears to be acceptable.
The second major assumption is that the fold-change in mRNA level can be used as a proxy for the fold-change in enzyme abundance and ultimately for the fold-change in Vmax. Critical examination of this assumption is of particular importance as the role of translational efficiency and post-translational modifications in regulating metabolic enzymes is becoming increasingly evident [18], [33]–[36]. We examined our assumption by analyzing published experimental data for S. cerevisiae where genome-wide mRNA and protein fold changes were simultaneously measured. In support of the assumption, the correlations between the mRNA and the protein fold changes corresponding to the metabolic genes were found to be both significant and strong (Dataset 1–3 [37], [38], R2 = 0.77, P = 0.04; R2 = 0.66, P = 0.0365, R2 = 0.76, P = 0.0036; dataset 4 [39], R2 = 0.4, P = 0.296; dataset 5 [40], R2 = 0.57, P = 0.0681; dataset 6 [41], R2 = 0.43, P = 0.0001) (Figure 2A). We note that these correlations involving only metabolic genes are stronger than the correlations calculated by including the non-metabolic genes (Figure 2A, Figure S3). As mRNA and protein levels have recently been demonstrated to be in good agreement in mammalian systems as well [42], we expect that the assumption of proportionality between gene expression and protein abundance fold changes will be valid in a broad range of organisms.
Figure 2. Changes in mRNA and protein abundances are strongly correlated for metabolic genes.
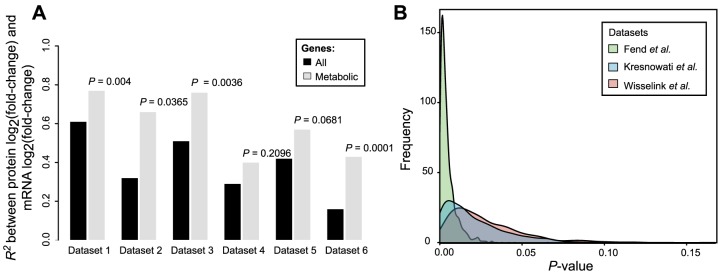
A) Shown are the coefficients of determination for mRNA fold-change – protein fold-change correlations; black: with all proteins measured in each of the datasets, gray: only with metabolic proteins. P-values denoting the significance of the improved correlations for metabolic genes were estimated based on random sampling from the set of measurements including all proteins. B) Distributions of P-values obtained for 1st degree CoCCoA analysis while accounting for the variability in the correlations between the mRNA and protein fold-changes.
Evaluation of 0th degree concentration change coupling analysis
Under the condition of flux homeostasis, i.e. no flux change between the two conditions being compared, the metabolite concentration ratio in equation (2) becomes dependent only on the transcript change. The resulting 0th degree CoCCoA model is equivalent to the analysis of the transcript/protein-metabolite relationship reported by Sauer and co-workers [3]. According to this model, we observed a significant correlation between transcriptional and metabolite changes in the glucose pulse case study (r = 0.81, P = 0.048). In the other two pairwise comparison case studies, the 0th degree model failed to correlate with the experimentally observed metabolite changes (Figure 3B). In case of the metabolic cycle dataset, around 31% of the measured metabolites showed significant correlations (FDR 10%) (Figure 3C, Figure S1). In all four case studies investigated here, flux homeostasis cannot be assumed as the growth rate as well as the substrate uptake and product secretion rates were affected by the corresponding perturbations. Our attempts to obtain reliable flux estimates by using flux balance analysis were not fruitful since only a limited number of physiological measurements were available to constrain the model, resulting in a high degree of uncertainty in the flux estimates. Thus, in the absence of reliable intra-cellular flux estimates, the 0th degree model was found to be insufficient for relating gene expression changes to metabolite levels.
Figure 3. Metabolite concentration changes are correlated with CoCCoA scores.

A) Equations illustrating the calculations of CoCCoA scores of different degrees (Main text, Figure S5). B) Correlations between experimentally measured metabolite concentration changes and CoCCoA scores for the three pairwise comparison datasets [3], [23], [30]. Note that the number of data points that can be tested varies for each coupling degree as the number of genes included in the analysis changes with the degree of the CoCCoA equation (see main text). Metabolites marked in gray could not be included in the analysis as the directions of the fluxes linked to them were altered between the growths on two different carbon sources [30]. C) Pearson correlation coefficients comparing experimentally measured metabolite concentration changes and CoCCoA scores for the metabolic cycle study [9]. D) Overlap between the significantly expressed genes in the three pairwise comparison case studies. Only genes that were used for the calculation of CoCCoA scores are included. E) Number of genes used in the CoCCoA score calculations increase with the increasing model degree. 3PG = 3-Phospho-D-glycerate; G6P = Glucose 6-phosphate; F26P = Fructose 2,6-bisphosphate; F16P = Fructose 1,6-bisphosphate; FUM = Fumarate; CIT = Citrate; MAL = Malate; AKG = alpha-Ketoglutarate; T6P = Trehalose 6-phosphate; PEP = Phosphoenolpyruvate; S7P = Sedoheptulose 7-phosphate; RBU5P = Ribulose 5-phosphate; R5P = Ribose 5-phosphate; DHAP = dihydroxyacetone phosphate; M6P = Mannose 6-phosphate.
1st degree concentration change coupling analysis
At steady state, the sum of fluxes producing a particular metabolite must be equal to the sum of fluxes through the reactions that use it as a substrate. For a metabolite with a single production reaction and a single consumption reaction, the steady state assumption combined with equation (2) leads to equation (3) (Supplementary Text S1).
| (3) |
Tprod and Tcons denote expression levels of the genes corresponding to the enzymes producing and consuming S, respectively. R refers to the concentration of the metabolite that is the precursor of S. The relation described by equation (3) implies a coupling between the concentration changes in R and S, and is here defined as 1st degree coupling. In comparison to the 0th degree coupling, the flux term ln(V/V*) is eliminated in the 1st degree coupling equation and is replaced by two new terms, ln(Tprod/Tprod*) and ln(R/R*). Equation (3) brings a new network perspective to enzyme kinetics, whereby gene expression and metabolite concentration changes in the adjacent reactions are linked through the mass balance constraint. Each metabolite pool is thus linked to the reactions consuming it as well as on the reactions producing it (Figure 3A). When multiple reactions are consuming (or producing) the same metabolite S, the consumption (or production) term can be approximated by the geometric mean of the transcript ratios of all the consumption (or production) reactions (Supplementary Text S1).
To evaluate the 1st degree model, we compared the experimentally measured metabolite concentration ratios with the 1st degree CoCCoA scores based on the transcript fold changes – the first two terms on the right-hand side of equation (3). We note that, although the strict application of our model requires the use of the ln(R/R*) term, these measurements are often not available. Moreover, a model that is completely independent of the metabolite concentration data will likely be of more practical value. Omitting the ln(R/R*) term equates to assuming that the preceding metabolite's concentration does not change between the two conditions; an alternative to omitting this term is explored below, in the 2nd degree CoCCoA model, in which the ln(R/R*) term is estimated by use of the 1st degree CoCCoA model. The effect of omitting the ln(R/R*) term on CoCCoA scores is further discussed in the later sub-section “Post-transcriptional regulation”.
Comparisons of the 1st degree CoCCoA scores to metabolite concentrations yielded significant correlations in the two environmental perturbation case studies (r = 0.88, P = 0.0099 and r = −0.96, P = 0.038), and a reasonably good correlation (r = −0.61, P = 0.06) for the genetic perturbation case study (Figure 3B, Figure S1). For the metabolic cycle case study, around 25% of the measured metabolites showed significant correlations (FDR 10%) (Figure 3C, Figure S1). Although our model suggests positive correlation between CoCCoA scores and metabolite changes, we observed negative correlations in the cases of two of the pairwise comparisons and for some of the metabolites in the metabolic cycle case study. The possible reasons underlying this discrepancy are discussed in the subsequent sub-section “Negative correlations in CoCCoA”. We maintain that the negative slopes do not invalidate the significance of the observed correlations, but rather hint at the existence of unaccounted parameters/mechanisms leading to the reversal of slope in some cases.
The number of transcripts that can be used for the calculation of the CoCCoA scores typically increases as more distant reaction nodes in the network are included with the increasing CoCCoA degree. Consequently, the number of metabolites that could be assigned CoCCoA scores varied between the coupling degrees. For example, in the C-source change study [30] (Figure 3B), only 4 metabolites have significant transcript changes corresponding to their consuming reactions and hence only these could be compared against the experimental data for the 0th degree analysis. In contrast, 2nd degree CoCCoA scores could be calculated for 7 metabolites.
Network propagation of concentration control
In a similar manner as going from the 0th to the 1st degree coupling, the CoCCoA equations can be further extended to include more distant nodes in the metabolic network. By replacing the concentration ratio in the right-hand side of Equation 3 (i.e. R/R*) with the 1st degree CoCCoA relationship for the corresponding precursor metabolite (in this case, R), we obtained the 2nd degree coupling relationship. This 2nd degree model accounts for the gene expression changes corresponding to the precursor's production reactions (Figure 3A) (Text S1). In all case studies, the 2nd degree correlations remained as strong as for the 1st degree. This result is notable since the 2nd degree coupling score includes expression data from the genes that are further away from the metabolites in question. With further extension of the CoCCoA model in a similar manner, we observed significant correlations up to the 6th degree coupling (P≤0.05, Fendt et al. case study, Figure S4).
To gain further insight into the metabolite concentration control at different network distances, we examined this problem from a metabolite-centric perspective by taking advantage of the broad metabolite coverage of the metabolic cycle case study. First, we extended the higher degree CoCCoA formulation so as to include information from all intermediate reaction steps up to the desired degree (Text S1). For example, the calculation of the 3rd degree CoCCoA score includes fold changes from the genes associated with the reactions involving all metabolites that are three steps upstream or downstream from the metabolite in question. The inclusion of genes within a desired network distance can either be restricted to the consumption or the production side of the metabolite, or both included simultaneously. This formulation also allowed us to include, if available, measured concentrations of the neighboring metabolites within the desired distance of a given metabolite, and thereby to assess the effect of changes in neighboring metabolites over its concentration. The algorithm used for calculating CoCCoA scores using this formulation is described in Text S1. In brief, this algorithm first enumerates all paths starting from the metabolite of interest to identify genes that are within a given network distance. Next, it uses graph topology-based heuristics to weight and incorporate the expression fold-changes corresponding to these genes into the CoCCoA equations by using mass balance considerations. Following this, we evaluated the ability of these CoCCoA scores to explain the concentration changes observed during the metabolic cycle. Overall, positive correlations are apparent for most of the metabolites (∼66%, Figure 4A,C). For the long distance scores, a slightly lower number of metabolites showed positive correlations. The contrast between the close and distant neighbors, however, should be interpreted in light of the highly connected nature of the metabolic network. The numbers of genes that are included in the calculation of CoCCoA scores already reach a plateau at the 4th degree (Figure 4B), and thus, even relatively modest distances can mean inclusion of a very large fraction of the network. Consequently, the CoCCoA scores for a given metabolite will become ‘diluted’ due to the noise stemming from the inclusion of gene expression changes pertaining to the reactions that are only indirectly affecting the metabolite of interest. These results from the metabolic cycle case study, together with the results from the pairwise comparison studies, suggest that the close neighbors in the metabolic network exert the majority of the control over metabolite concentrations.
Figure 4. CoCCoA models link the changes in gene expression and metabolite concentrations during yeast metabolic cycle.

A) Pearson correlation coefficients comparing the experimentally observed metabolite concentration changes and CoCCoA model scores. For a given metabolite, 16 different CoCCoA scores were computed, accounting for a network distance of up to 4, both upstream and downstream of the metabolite in question. Scores were divided into two sets: close (distance ≤2) and distant (distance >2). Shown values are the median correlation coefficients in each set. B) CoCCoA models account for a large fraction of genes in the metabolic network at relatively short distances. C) Correlations between the experimentally observed metabolite concentration changes and CoCCoA model scores improve with the inclusion of the concentrations of the precursors and/or products of the metabolite in question. D) Example correlations between the CoCCoA model scores and the metabolite concentration changes. Different shapes of data points mark different stages of the yeast metabolic cycle. † - Metabolites with previous evidence for post-translational regulation of at least one of their neighboring enzymes [34].
The correlations between the CoCCoA scores and the metabolite concentration changes were further strengthened when the experimentally determined fold-changes in the concentration of the upstream and/or downstream metabolites were also used in the calculation (Figure 4C). This improvement further supports the CoCCoA theory, as the inclusion of concentration changes for the upstream/downstream metabolites stems from the joint mass balance and kinetic considerations, e.g. as illustrated in equation (3).
Limitations of CoCCoA formulation
CoCCoA is not applicable in the case of perturbations that are likely to drastically affect the kinetic properties (KM values) of several enzymes in the network, or if the metabolite concentrations are considerably above the corresponding KM values (saturated enzymes). Furthermore, the CoCCoA model needs to exclude reactions for which the flux directionality is ambiguous, and it assumes that the flux directions do not change for the rest of the reactions. Post-translational regulatory mechanisms, which can affect the kinetic parameters, are also not included in the current CoCCoA formulation, as sufficient data are not available to enable their modeling. The latter is perhaps the most restrictive limitation of our model. Post-translational regulation is known to play a crucial role in the yeast central metabolism, wherein several enzymes are controlled by allosteric binding of small molecules [35], [43], [44] and/or through post-translational modifications such as phosphorylation [33]. Together, these various assumptions and limitations can lead to poor or no correlations. In the case of the pairwise comparisons, poor correlations can also result from the pooling of metabolites with positive and negative correlation with CoCCoA scores into a single plot.
According to the CoCCoA model, all examined correlations would be expected to be positive. Among the pairwise comparison case studies, positive correlations were observed only for the glucose pulse study (Figure 3B). In the case of the metabolic cycle study, a significant majority of the metabolites (∼66%, P = 2.9×10−8, exact binomial test) showed positive correlations (Figure 4A,C). Flux regulation due to allosteric binding by small molecules and post-translational modifications are likely to be the major factors underlying this discrepancy between the expected positive slopes and the observed negative slopes for the remaining 34% metabolites. The possible causes and implications of negative correlation are discussed in the subsequent sub-section “Negative correlations in CoCCoA”.
Robustness of CoCCoA towards differences in gene expression and protein abundance fold changes
We found that the enzyme-coding genes in yeast exhibit significantly stronger correlations between mRNA and protein fold-changes than do the non-metabolic genes (Figure 2A, Figure S3). The slopes of these correlations were, however, different across different datasets examined. To evaluate the robustness of CoCCoA towards this variation, we re-performed 1st degree CoCCoA analysis multiple times (1000 simulations), adjusting the transcript ratios in each simulation by a randomly sampled correction factor to account for the expected difference between the mRNA and protein fold changes. The sampling space for the correction factors was estimated based on the variance in the slopes of linear regression lines between mRNA and protein abundance fold changes across different datasets (Figure S3, Text S1). We then examined the number of simulations in which the correlation between the 1st degree CoCCoA scores and the metabolite fold changes remained significant. For all three pairwise comparison case studies, the correlations remained significant (P≤0.05) in a large fraction of these simulations (99%, 86% and 91%) (Figure 2B).
Discussion
Network constraints over metabolite concentration changes
The correlation between metabolites concentrations and transcript fold changes becomes evident only when including network connectivity constraints (1st and higher degree CoCCoA). Thus, inclusion of gene expression changes associated with the both upstream and downstream reactions was critical for explaining metabolite concentration changes. For all three pairwise comparison case studies, CoCCoA models explained more than 60% of the variation in metabolite changes based on gene expression. For the metabolic cycle case study, CoCCoA could explain variation in about 33% of the measured metabolites, with correlation coefficients as strong as 0.90.
The use of a genome-scale metabolic model was crucial in CoCCoA analysis in order to capture the large connectivity inherent to metabolic networks. Even for a sparsely connected metabolite such as D-Ribose 5-phosphate, for which the 0th degree score accounted for only 4 transcripts, the 2nd degree CoCCoA score accounted for transcriptional information from 47 genes in the Fendt et al. study [3] (Figure 3B). With the increasing degree of CoCCoA equations, larger numbers of genes become part of the CoCCoA score (Figure 3E, Figure 4B). We also observed that, in general, the inclusion of new genes when moving from the 1st to the 2nd degree CoCCoA maintains the significance of the correlation (Figure 3B, Figure 3C). This observation implies strong co-regulation of genes that are linked through common substrates/products. Indeed, co-expression of metabolic genes at short network distances has been observed in earlier studies [45]. The CoCCoA theory suggests that homeostasis of metabolite concentrations is one of the objectives of such topology-oriented co-regulation in metabolic networks.
Post-transcriptional regulation
There are several layers of regulation that the CoCCoA model does not take into account: translational and post-translational regulation, and the kinetic effects of neighboring metabolites' concentrations. With regard to translational regulation, we observed strong correlations between transcript and protein fold changes (Figure 2A, Figure S3), and the CoCCoA results were found to be relatively robust in light of the known variability (Figure 2B). However, it remains to be seen whether these correlations extend through post-translational regulation to enzyme activity. Increasing availability of the genome-wide protein phosphorylation/acetylation data may aid in addressing this question. The CoCCoA framework can be used with enzyme abundance or enzyme activity fold changes in place of transcript fold changes. Using this information could lead to more accuracy in CoCCoA scores, and could furthermore aid in identifying the relative contribution of the different regulatory layers in controlling metabolite concentrations.
The effects of neighboring metabolites' concentrations could be examined thanks to the large coverage of metabolite measurements in the metabolic cycle dataset. The power of CoCCoA in explaining variance in metabolite concentration changes was substantially improved following the inclusion of data for the neighboring metabolites, representing ln(R/R*) terms in the CoCCoA equations (2.54 fold increase in the median correlation coefficient; P = 6.6×10−8, Wilcox test; Figure 4C). In accordance with the CoCCoA theory, this improvement indicates that a metabolite's neighbors in the metabolic network play an important role in determining its level.
CoCCoA is applicable to perturbations of different nature
Formulation of CoCCoA in a relative manner, i.e. in the fold-change space, allowed us to circumvent the problem of the unavailability of in vivo kinetic parameters. A major advantage following this relative formulation is that the CoCCoA models do not need any parameter fitting. Indeed, the applicability of CoCCoA was found to be quite broad in terms of the perturbation or experimental design underlying the data. The four datasets considered in this study represent three different biological perturbations, namely gene knockout [3], nutrient pulse [23], change in carbon source [30], as well as a fundamental rhythmic phenomenon associated with the cell cycle [9], [31]. These case studies also span two distinct cultivation types, batch [3] and chemostat [9], [23], [30], [31]. We also verified the differences in the nature of these perturbations at the level of gene expression changes: the three pairwise comparison studies were found to have only a small overlap in terms of the significantly responding genes (Figure 3D). Additionally, the CoCCoA model was found to be applicable over a broad range of concentration changes displayed by different metabolites during the yeast metabolic cycle (Figure 4D).
Negative correlations in CoCCoA
Intriguingly, several of the observed negative correlations were found to be not only significant but also quite strong, with R2 values as high as 0.86 (Figure 4). These negative correlations are indicative of the mechanisms that are unaccounted for in the CoCCoA model and/or highlight cases in which the assumptions of the model do not apply. We observed that the number of positive correlations in the metabolic cycle case study increased considerably when using the concentration change data from the neighboring metabolites (the ln(R/R*) term in the right-hand side of the 1st degree CoCCoA equation) (Figure 4C). This observation suggests that the kinetic effect due to changes in the neighboring metabolites is an important factor contributing to negative correlations. The results from the analysis of the metabolic cycle data also hint that the negative slopes might be characteristic to certain metabolites, for example, those for which the producing/consuming enzymes are regulated predominantly and/or prevalently at the post-translational level. Indeed, we found that the metabolites with poor or negative correlations in the metabolic cycle case study are enriched in the metabolites with previous evidence for post-translational regulation of at least one of their neighboring enzymes (metabolites marked with † in Figure 4A, data for post-translationally regulated enzymes from [34], P = 0.0006). In these cases, the post-translational regulation may be counteracting the transcriptional change. Post-translational regulation of an enzyme can change its KM value and can thereby directly affect CoCCoA scores. Consider, for example, 1st degree CoCCoA score. When changes in KM values are included in the 1st degree CoCCoA formula, the score becomes: 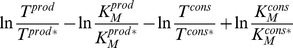 . If the post-translational modifications counteract the transcriptional changes, the KM ratios in this new score will partially cancel out or even override the transcript ratios. When assuming constant KM values, the discrepancy between the transcript and KM ratios in some cases might be sufficiently large to result in CoCCoA scores with opposite sign. On the other hand, the inconsistency between the directions of transcriptional and post-translational regulation (or post-transcriptional regulation in general) implies non-optimal regulation and is unlikely to be a general mechanism used by the cell. However, non-optimal regulation is a possibility for certain enzymes, with two plausible biological explanations: i) the highly non-linear scenario of regulation (resulting from the concerted action of reaction kinetics, incl. allosteric regulation, and mass balance and thermodynamic constraints) can mean that the cell needs to make some locally non-optimal choices in order to achieve a global optimality in regulating the overall metabolism (for example, to take advantage of the distinct time-scales at which post-translational and transcriptional regulations act); ii) the observed behavior is both locally and globally non-optimal in case of certain perturbations. The second scenario would imply that the perturbations in question are unknown or new to the cells in the evolutionary sense.
. If the post-translational modifications counteract the transcriptional changes, the KM ratios in this new score will partially cancel out or even override the transcript ratios. When assuming constant KM values, the discrepancy between the transcript and KM ratios in some cases might be sufficiently large to result in CoCCoA scores with opposite sign. On the other hand, the inconsistency between the directions of transcriptional and post-translational regulation (or post-transcriptional regulation in general) implies non-optimal regulation and is unlikely to be a general mechanism used by the cell. However, non-optimal regulation is a possibility for certain enzymes, with two plausible biological explanations: i) the highly non-linear scenario of regulation (resulting from the concerted action of reaction kinetics, incl. allosteric regulation, and mass balance and thermodynamic constraints) can mean that the cell needs to make some locally non-optimal choices in order to achieve a global optimality in regulating the overall metabolism (for example, to take advantage of the distinct time-scales at which post-translational and transcriptional regulations act); ii) the observed behavior is both locally and globally non-optimal in case of certain perturbations. The second scenario would imply that the perturbations in question are unknown or new to the cells in the evolutionary sense.
In addition to the unaccounted post-transcriptional regulation, the simplifying assumptions of constant flux directions and flux split ratios may also be contributing to the observed negative correlations. A wrongly considered flux direction for a reaction would mean that the corresponding fold change in the expression level would be treated in the opposite direction. Similarly, moderate changes in the flux split ratio can also cause a sign reversal in the CoCCoA score if one of the fluxes is significantly lower than the other(s). The interaction between the various missing/simplifying factors in our model can further amplify the difference between the CoCCoA scores and actual concentration changes. How these interactions lead to the reversal of correlation while retaining statistical significance is yet unclear. Further investigation into the mechanisms underlying these intriguing negative correlations would require network-wide in vivo measurements of fluxes, metabolite concentrations, protein abundances and functional post-translational modifications. Nevertheless, we note that our model revealed significant correlations in several cases, including perturbations of very different nature. We also note that, in the case of metabolic cycle dataset, significantly more positive correlations were observed than negative (P = 2.9e-08, exact binomial test). Together, the statistical and mechanistic considerations suggest that the CoCCoA model captures considerable mechanistic essence of the complex processes governing metabolite levels.
Overall, our model-guided analysis highlighted the role of metabolic network connectivity in modulating metabolite concentration changes and revealed much stronger correlations between gene expression and metabolite levels than previously appreciated. The proposed model can be extended to include translational and post-translational regulation as the data becomes available. From this perspective, we see CoCCoA as a framework with a strong mechanistic yet parameter-free basis, rather than a general relationship. We anticipate that, due to its parameter-free nature, the CoCCoA framework will be widely applicable for modeling metabolite level changes in large metabolic networks.
Methods
Experimental datasets
Four different experimental studies reporting gene expression and metabolite concentration measurements for the yeast S. cerevisiae were used. The first study, Fendt et al. [3], includes a comparison between wild type yeast and a mutant strain lacking GCR2, a transcription factor responsible for activation of glycolytic genes [46]. In the second study, Kresnowati et al. [23], yeast cultures were grown in carbon-limited chemostat cultures and subjected to a step change in glucose concentration. In the third study, Wisselink et al. [30], an evolutionarily engineered strain was grown on either glucose or arabinose as the sole carbon source. In the fourth study, oscillating chemostat cultures were sampled covering different phases of the metabolic cycle [9]. A summary of the growth conditions and descriptions of datasets from all case studies is provided in Table S1. Metabolite data was used as available in the original studies; a significance cut-off α = 10% was chosen to control for the type 1 error. For the metabolic cycle data, we considered metabolites with the periodicity P-value≤0.05 as reported in the original study [31]. As the datasets used did not distinguish between the cytosolic and mitochondrial concentration of metabolites, we regarded all metabolite concentrations as cytosolic. Since 2-phosphoglycerate and 3-phosphoglycerate are usually indistinguishable in the MS measurements, we considered only 3-phosphoglycerate gene neighbors and excluded 2-phospoglycerate.
Metabolic network and flux variability analysis
A genome-scale reconstruction of Saccharomyces cerevisiae metabolic network by Forster et al. [17] was used to map metabolite-reaction-gene connectivity. For each of the case studies, the functional reaction directions of reversible reactions were estimated by using flux variability analysis [47]. For this purpose, the model was constrained with the physiological data obtained from the publications reporting the used datasets (Table S2, Table S3, Table S4, and Table S5) [3]. Linear programming problems were solved using the glpk solver (http://www.gnu.org/software/glpk/) accessed through a C library.
Transcription data analysis
Preprocessing of the Affymetrix CEL files was carried out with the statistical software environment R/Bioconductor (www.bioconductor.org). Probe intensities were corrected for background by using robust multi-array average method (RMA) [48] using only perfect-match probes, and normalization was performed using the quantiles algorithm. Gene expression intensity values were calculated from the perfect-match probes with median polish summarization method [49]. Significance of the differential expression was calculated by using the empirical Bayes test as implemented in the limma package [50].
Statistical analysis
Pearson correlation coefficients between log2 metabolite fold changes and CoCCoA scores were calculated with the statistical software R (www.r-project.org) using the function cor.test(). Metabolite changes were used as dependent variables and CoCCoA scores as independent. P-values for the null hypothesis of no correlation (regression slope = 0) were estimated by using the same function. In addition, we performed a permutation test by shuffling gene labels before calculating CoCCoA scores. The originally paired data was randomly permuted without replacement 1000 times. For each permutation, a correlation coefficient was calculated and the P-value was estimated as a fraction of squared correlation coefficients that were larger than in the case of the original paired data. The results were similar to those estimated with the cor.test() function.
Code used for CoCCoA score calculations
Supporting Information
Correlations between experimentally measured metabolite concentration changes and CoCCoA scores based on gene expression fold changes. A) Facet columns correspond to three different pairwise comparison datasets used in our work; rows represent different CoCCoA models. B) Heatmap of Pearson correlation coefficients assessing the applicability of CoCCoA models to the metabolic cycle study. In the present figure, the significance thresholds for the transcript and metabolite fold changes are relaxed in comparison to the data shown in Figure 3B,C in the main text.
(PNG)
{kind=link}
Estimates of error in predicting metabolite concentration changes with MM kinetics when assuming V≪Vmax (see main text for the motivation behind the use of this assumption). A. Error (Z-axis) as a function of V/Vmax and V*/Vmax *. The error function is shown in supplementary Text S1 (equation 11). B. Error estimates around the points where V/Vmax = V*/Vmax *, representing a situation in which the enzyme saturation levels remain unchanged in the perturbed condition. C. 2-D projection of the plot in A, where the errors are represented with different colors.
(PNG)
{kind=link}
Correlation between protein abundance changes and the corresponding mRNA abundance changes is stronger for metabolic proteins. A, D, G, J, M, P) Correlation including all proteins measured in different datasets. B, E, H, K, N, Q) Correlations including only metabolic proteins (as per genome-scale metabolic model by [17]). C, F, I, L, O, R) Histogram of 10,000 different correlation coefficients obtained for randomly chosen protein-transcript pairs (number of chosen pairs for each correlation being equal to the number of metabolic proteins measured in the corresponding dataset). Blue area denotes the number of random correlations that were higher than those obtained for the correlation based on the actual data. Each row of plots represents a different dataset (from top to bottom), 1, 2, 3 –[38], [51]; 4 –[41]; 5 –[40]; 6 –[41].
(PNG)
{kind=link}
Coefficients of determination for the correlations between experimentally measured metabolite concentration changes and CoCCoA scores corresponding to different degrees. The significance of correlations was assessed against correlations obtained with random permutations of gene labels.
(PNG)
{kind=link}
Example CoCCoA score calculations. Shown is the case of fumarate in the Fendt et al. case study.
(PNG)
{kind=link}
Summary of the growth conditions from the three pairwise comparison case studies used in our analysis.
(DOCX)
Physiological data from the pairwise comparison case study 1.
(DOCX)
Physiological data from the pairwise comparison case study 2.
(DOCX)
Physiological data from the pairwise comparison case study 3.
(DOCX)
Reaction directions used for the case studies 1 and 2.
(DOCX)
Supporting text.
(DOCX)
Acknowledgments
We are grateful to T. Çakir and A. R. Brochado for feedback on the manuscript. We thank B. Klaus for discussion on the statistical tests.
Funding Statement
The study was partially funded by Novozymes A/S (http://www.novozymes.com/) through PhD study grant to AZ. SS was funded by European Molecular Biology Laboratory. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Gallego O, Betts MJ, Gvozdenovic-Jeremic J, Maeda K, Matetzki C, et al. (2010) A systematic screen for protein-lipid interactions in Saccharomyces cerevisiae. Mol Syst Biol 6: 430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Costenoble R, Picotti P, Reiter L, Stallmach R, Heinemann M, et al. (2011) Comprehensive quantitative analysis of central carbon and amino-acid metabolism in Saccharomyces cerevisiae under multiple conditions by targeted proteomics. Mol Syst Biol 7: 464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Fendt SM, Buescher JM, Rudroff F, Picotti P, Zamboni N, et al. (2010) Tradeoff between enzyme and metabolite efficiency maintains metabolic homeostasis upon perturbations in enzyme capacity. Mol Syst Biol 6: 356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Li X, Gianoulis TA, Yip KY, Gerstein M, Snyder M (2010) Extensive in vivo metabolite-protein interactions revealed by large-scale systematic analyses. Cell 143: 639–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Oliveira AP, Patil KR, Nielsen J (2008) Architecture of transcriptional regulatory circuits is knitted over the topology of bio-molecular interaction networks. BMC Syst Biol 2: 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wang Q, Zhang Y, Yang C, Xiong H, Lin Y, et al. (2010) Acetylation of metabolic enzymes coordinates carbon source utilization and metabolic flux. Science 327: 1004–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Murray DB, Beckmann M, Kitano H (2007) Regulation of yeast oscillatory dynamics. Proc Natl Acad Sci U S A 104: 2241–2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tai SL, Boer VM, Daran-Lapujade P, Walsh MC, de Winde JH, et al. (2005) Two-dimensional transcriptome analysis in chemostat cultures. Combinatorial effects of oxygen availability and macronutrient limitation in Saccharomyces cerevisiae. J Biol Chem 280: 437–447. [DOI] [PubMed] [Google Scholar]
- 9. Tu BP, Kudlicki A, Rowicka M, McKnight SL (2005) Logic of the yeast metabolic cycle: temporal compartmentalization of cellular processes. Science 310: 1152–1158. [DOI] [PubMed] [Google Scholar]
- 10. Patil KR, Nielsen J (2005) Uncovering transcriptional regulation of metabolism by using metabolic network topology. Proc Natl Acad Sci U S A 102: 2685–2689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zelezniak A, Pers TH, Soares S, Patti ME, Patil KR (2010) Metabolic network topology reveals transcriptional regulatory signatures of type 2 diabetes. PLoS Comput Biol 6: e1000729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bradley PH, Brauer MJ, Rabinowitz JD, Troyanskaya OG (2009) Coordinated concentration changes of transcripts and metabolites in Saccharomyces cerevisiae. PLoS Comput Biol 5: e1000270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Urbanczyk-Wochniak E, Luedemann A, Kopka J, Selbig J, Roessner-Tunali U, et al. (2003) Parallel analysis of transcript and metabolic profiles: a new approach in systems biology. EMBO Rep 4: 989–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Michaelis L, Menten M (1913) Die kinetik der invertinwirkung. Biochem Z 49. [Google Scholar]
- 15. Briggs GE, Haldane JB (1925) A Note on the Kinetics of Enzyme Action. Biochem J 19: 338–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Van Slyke DD, Cullen GE (1914) THE MODE OF ACTION OF UREASE AND OF ENZYMES IN GENERAL. J Biol Chem 19: 141–180. [Google Scholar]
- 17. Forster J, Famili I, Fu P, Palsson BO, Nielsen J (2003) Genome-scale reconstruction of the Saccharomyces cerevisiae metabolic network. Genome Res 13: 244–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Daran-Lapujade P, Rossell S, van Gulik WM, Luttik MA, de Groot MJ, et al. (2007) The fluxes through glycolytic enzymes in Saccharomyces cerevisiae are predominantly regulated at posttranscriptional levels. Proc Natl Acad Sci U S A 104: 15753–15758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yang C, Hua Q, Shimizu K (2002) Integration of the information from gene expression and metabolic fluxes for the analysis of the regulatory mechanisms in Synechocystis. Appl Microbiol Biotechnol 58: 813–822. [DOI] [PubMed] [Google Scholar]
- 20. Rossell S, van der Weijden CC, Kruckeberg AL, Bakker BM, Westerhoff HV (2005) Hierarchical and metabolic regulation of glucose influx in starved Saccharomyces cerevisiae. FEMS Yeast Res 5: 611–619. [DOI] [PubMed] [Google Scholar]
- 21. Rossell S, van der Weijden CC, Lindenbergh A, van Tuijl A, Francke C, et al. (2006) Unraveling the complexity of flux regulation: a new method demonstrated for nutrient starvation in Saccharomyces cerevisiae. Proc Natl Acad Sci U S A 103: 2166–2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cakir T, Patil KR, Onsan Z, Ulgen KO, Kirdar B, et al. (2006) Integration of metabolome data with metabolic networks reveals reporter reactions. Mol Syst Biol 2: 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kresnowati MT, van Winden WA, Almering MJ, ten Pierick A, Ras C, et al. (2006) When transcriptome meets metabolome: fast cellular responses of yeast to sudden relief of glucose limitation. Mol Syst Biol 2: 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Moxley JF, Jewett MC, Antoniewicz MR, Villas-Boas SG, Alper H, et al. (2009) Linking high-resolution metabolic flux phenotypes and transcriptional regulation in yeast modulated by the global regulator Gcn4p. Proc Natl Acad Sci U S A 106: 6477–6482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Klipp E, Nordlander B, Kruger R, Gennemark P, Hohmann S (2005) Integrative model of the response of yeast to osmotic shock. Nat Biotechnol 23: 975–982. [DOI] [PubMed] [Google Scholar]
- 26. Westerhoff HV, Chen YD (1984) How do enzyme activities control metabolite concentrations? An additional theorem in the theory of metabolic control. Eur J Biochem 142: 425–430. [DOI] [PubMed] [Google Scholar]
- 27. Westerhoff HV, Groen AK, Wanders RJ (1984) Modern theories of metabolic control and their applications (review). Biosci Rep 4: 1–22. [DOI] [PubMed] [Google Scholar]
- 28. Cleland WW (1989) The kinetics of enzyme-catalyzed reactions with two or more substrates or products. I. Nomenclature and rate equations. 1963. Biochim Biophys Acta 1000: 213–220. [PubMed] [Google Scholar]
- 29. Burgard AP, Nikolaev EV, Schilling CH, Maranas CD (2004) Flux coupling analysis of genome-scale metabolic network reconstructions. Genome Res 14: 301–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wisselink HW, Cipollina C, Oud B, Crimi B, Heijnen JJ, et al. (2010) Metabolome, transcriptome and metabolic flux analysis of arabinose fermentation by engineered Saccharomyces cerevisiae. Metab Eng 12: 537–551. [DOI] [PubMed] [Google Scholar]
- 31. Tu BP, Mohler RE, Liu JC, Dombek KM, Young ET, et al. (2007) Cyclic changes in metabolic state during the life of a yeast cell. Proc Natl Acad Sci U S A 104: 16886–16891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bennett BD, Kimball EH, Gao M, Osterhout R, Van Dien SJ, et al. (2009) Absolute metabolite concentrations and implied enzyme active site occupancy in Escherichia coli. Nat Chem Biol 5: 593–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Oliveira AP, Ludwig C, Picotti P, Kogadeeva M, Aebersold R, et al. (2012) Regulation of yeast central metabolism by enzyme phosphorylation. Mol Syst Biol 8: 623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Oliveira AP, Sauer U (2012) The importance of post-translational modifications in regulating Saccharomyces cerevisiae metabolism. FEMS Yeast Res 12: 104–117. [DOI] [PubMed] [Google Scholar]
- 35. Metallo CM, Vander Heiden MG (2010) Metabolism strikes back: metabolic flux regulates cell signaling. Genes Dev 24: 2717–2722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ptacek J, Devgan G, Michaud G, Zhu H, Zhu X, et al. (2005) Global analysis of protein phosphorylation in yeast. Nature 438: 679–684. [DOI] [PubMed] [Google Scholar]
- 37. Usaite R, Jewett MC, Oliveira AP, Yates JR 3rd, Olsson L, et al. (2009) Reconstruction of the yeast Snf1 kinase regulatory network reveals its role as a global energy regulator. Mol Syst Biol 5: 319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Usaite R, Wohlschlegel J, Venable JD, Park SK, Nielsen J, et al. (2008) Characterization of global yeast quantitative proteome data generated from the wild-type and glucose repression saccharomyces cerevisiae strains: the comparison of two quantitative methods. J Proteome Res 7: 266–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Washburn MP, Koller A, Oshiro G, Ulaszek RR, Plouffe D, et al. (2003) Protein pathway and complex clustering of correlated mRNA and protein expression analyses in Saccharomyces cerevisiae. Proc Natl Acad Sci U S A 100: 3107–3112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ideker T, Thorsson V, Ranish JA, Christmas R, Buhler J, et al. (2001) Integrated genomic and proteomic analyses of a systematically perturbed metabolic network. Science 292: 929–934. [DOI] [PubMed] [Google Scholar]
- 41. Griffin TJ, Gygi SP, Ideker T, Rist B, Eng J, et al. (2002) Complementary profiling of gene expression at the transcriptome and proteome levels in Saccharomyces cerevisiae. Mol Cell Proteomics 1: 323–333. [DOI] [PubMed] [Google Scholar]
- 42. Schwanhausser B, Busse D, Li N, Dittmar G, Schuchhardt J, et al. (2011) Global quantification of mammalian gene expression control. Nature 473: 337–342. [DOI] [PubMed] [Google Scholar]
- 43. Braus GH (1991) Aromatic amino acid biosynthesis in the yeast Saccharomyces cerevisiae: a model system for the regulation of a eukaryotic biosynthetic pathway. Microbiol Rev 55: 349–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Luttik MA, Vuralhan Z, Suir E, Braus GH, Pronk JT, et al. (2008) Alleviation of feedback inhibition in Saccharomyces cerevisiae aromatic amino acid biosynthesis: quantification of metabolic impact. Metab Eng 10: 141–153. [DOI] [PubMed] [Google Scholar]
- 45. Kharchenko P, Church GM, Vitkup D (2005) Expression dynamics of a cellular metabolic network. Mol Syst Biol 1: 2005 0016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Uemura H, Jigami Y (1992) Role of GCR2 in transcriptional activation of yeast glycolytic genes. Mol Cell Biol 12: 3834–3842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Mahadevan R, Schilling CH (2003) The effects of alternate optimal solutions in constraint-based genome-scale metabolic models. Metab Eng 5: 264–276. [DOI] [PubMed] [Google Scholar]
- 48. Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, et al. (2003) Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 4: 249–264. [DOI] [PubMed] [Google Scholar]
- 49. Irizarry RA, Bolstad BM, Collin F, Cope LM, Hobbs B, et al. (2003) Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res 31: e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Smyth GK (2004) Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol 3: Article3. [DOI] [PubMed] [Google Scholar]
- 51. Usaite R, Nielsen J, Olsson L (2008) Physiological characterization of glucose repression in the strains with SNF1 and SNF4 genes deleted. J Biotechnol 133: 73–81. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Correlations between experimentally measured metabolite concentration changes and CoCCoA scores based on gene expression fold changes. A) Facet columns correspond to three different pairwise comparison datasets used in our work; rows represent different CoCCoA models. B) Heatmap of Pearson correlation coefficients assessing the applicability of CoCCoA models to the metabolic cycle study. In the present figure, the significance thresholds for the transcript and metabolite fold changes are relaxed in comparison to the data shown in Figure 3B,C in the main text.
(PNG)
Estimates of error in predicting metabolite concentration changes with MM kinetics when assuming V≪Vmax (see main text for the motivation behind the use of this assumption). A. Error (Z-axis) as a function of V/Vmax and V*/Vmax *. The error function is shown in supplementary Text S1 (equation 11). B. Error estimates around the points where V/Vmax = V*/Vmax *, representing a situation in which the enzyme saturation levels remain unchanged in the perturbed condition. C. 2-D projection of the plot in A, where the errors are represented with different colors.
(PNG)
Correlation between protein abundance changes and the corresponding mRNA abundance changes is stronger for metabolic proteins. A, D, G, J, M, P) Correlation including all proteins measured in different datasets. B, E, H, K, N, Q) Correlations including only metabolic proteins (as per genome-scale metabolic model by [17]). C, F, I, L, O, R) Histogram of 10,000 different correlation coefficients obtained for randomly chosen protein-transcript pairs (number of chosen pairs for each correlation being equal to the number of metabolic proteins measured in the corresponding dataset). Blue area denotes the number of random correlations that were higher than those obtained for the correlation based on the actual data. Each row of plots represents a different dataset (from top to bottom), 1, 2, 3 –[38], [51]; 4 –[41]; 5 –[40]; 6 –[41].
(PNG)
Coefficients of determination for the correlations between experimentally measured metabolite concentration changes and CoCCoA scores corresponding to different degrees. The significance of correlations was assessed against correlations obtained with random permutations of gene labels.
(PNG)
Example CoCCoA score calculations. Shown is the case of fumarate in the Fendt et al. case study.
(PNG)
Summary of the growth conditions from the three pairwise comparison case studies used in our analysis.
(DOCX)
Physiological data from the pairwise comparison case study 1.
(DOCX)
Physiological data from the pairwise comparison case study 2.
(DOCX)
Physiological data from the pairwise comparison case study 3.
(DOCX)
Reaction directions used for the case studies 1 and 2.
(DOCX)
Supporting text.
(DOCX)