

Abstract
Fiber strength is the key trait that determines fiber quality in cotton, and it is closely related to secondary cell wall synthesis. To understand the mechanism underlying fiber strength, we compared fiber transcriptomes from different G. barbadense chromosome introgression lines (CSILs) that had higher fiber strengths than their recipient, G. hirsutum acc. TM-1. A total of 18,288 differentially expressed genes (DEGs) were detected between CSIL-35431 and CSIL-31010, two CSILs with stronger fiber and TM-1 during secondary cell wall synthesis. Functional classification and enrichment analysis revealed that these DEGs were enriched for secondary cell wall biogenesis, glucuronoxylan biosynthesis, cellulose biosynthesis, sugar-mediated signaling pathways, and fatty acid biosynthesis. Pathway analysis showed that these DEGs participated in starch and sucrose metabolism (328 genes), glycolysis/gluconeogenesis (122 genes), phenylpropanoid biosynthesis (101 genes), and oxidative phosphorylation (87 genes), etc. Moreover, the expression of MYB- and NAC-type transcription factor genes were also dramatically different between the CSILs and TM-1. Being different to those of CSIL-31134, CSIL-35431 and CSIL-31010, there were many genes for fatty acid degradation and biosynthesis, and also for carbohydrate metabolism that were down-regulated in CSIL-35368. Metabolic pathway analysis in the CSILs showed that different pathways were changed, and some changes at the same developmental stage in some pathways. Our results extended our understanding that carbonhydrate metabolic pathway and secondary cell wall biosynthesis can affect the fiber strength and suggested more genes and/or pathways be related to complex fiber strength formation process.
Introduction
The cotton fiber is a terminally differentiated single cell derived from the epidermal cell of the developing ovule. After initiation, the fiber cell undergoes 1000- to 3000-fold elongation during its development. The development of cotton fibers involves four partially overlapping stages: initiation (−3 to +3 days post-anthesis; DPA), elongation and primary cell wall formation (3–23 DPA), secondary cell wall formation (16–40 DPA) and maturation (40–50 DPA) [1]–[6]. The most rapid period of fiber cell elongation begins around 10–16 DPA and continues to ∼20 DPA. Primary and secondary cell wall synthesis overlaps during the period of 16–25 DPA. During the secondary cell wall formation stage, the speed of cell elongation slows down and even stops.
Fiber strength is an important indicator of cotton fiber quality, and depends on formation of the secondary cell wall. Cellulose synthesis plays a predominant role in fiber cells, and cellulose accounts for >95% of the dry weight of the mature cotton fiber [3], [7]. Genome and EST sequencing have revealed that there are at least ten different CesA genes for cellulose synthase in Arabidopsis; CesA-like genes have also been reported in rice and barley [8]–[10]. In cotton (Gossypium raimondii), at least 15 cellulose synthase (CESA) sequences are required for cellulose synthesis [11]. A recent investigation in Arabidopsis thaliana using microarrays led to the identification of genes that are highly co-expressed with cellulose synthase genes and two mutants, irx8 and irx13, that have irregular xylem phenotypes, were also identified [12]. Sucrose synthase (Susy) is the enzyme that catalyzes the hydrolysis of sucrose to UDP-glucose that is then used as a substrate for cellulose synthesis. In cotton, the expression of Susy is higher at 16–32 DPA, and this enzyme plays a major role in partitioning carbon toward cellulose synthesis in the fiber [13]. SusC is another new sucrose synthase gene with a high level of expression during secondary cell wall synthesis [14]. Peroxide, mainly as H2O2, promotes cellulose synthesis as a signal of secondary cell wall synthesis [15], [16].
At present, many ovule- and fiber-specific cDNA libraries have been constructed and sequenced, and more than 268,000 expressed sequence tags (ESTs) from Gossypium are deposited in the NCBI database (http://www.ncbi.nlm.nih.gov). For genetic characterization of rapid cell elongation in cotton fibers, approximately 14,000 unique genes were assembled from 46,603 expressed sequence tags (ESTs) from developmentally-staged fiber cDNAs of a cultivated diploid species (G. arboreum L.). Eighty-one genes that were significantly up-regulated during secondary cell wall synthesis were found to be involved in cell wall biogenesis and energy/carbohydrate metabolism, which is consistent with the stage of cellulose synthesis during secondary cell wall modification in developing fibers [17]. Transcriptome profiling of the cotton fiber early in development by high-throughput tag-sequencing (Tag-seq) analysis using the Solexa Genome Analyzer reveals significant differential expression of genes in a fuzzless/lintless mutant [18]. High-throughput, genome-wide transcriptomic analysis of cotton under drought stress revealed a significant down-regulation of genes and pathways involved in fiber elongation, and an up-regulation of defense response genes [19]. More research have been processed in fiber initiation and elongation stage [20]–[24]. Saturated very-long-chain fatty acids (VLCFAs; C20:0–C30:0) exogenously applied in ovule culture medium significantly promoted fiber cell elongation in cotton (G. hirsutum L.) by activating ethylene biosynthesis [25], [26]. Previous investigations into cotton fiber development mainly focused on the elongation stage, and the number of genes reported from the later stages is quite small. Most of the genes up-regulated during secondary cell wall synthesis were related to cellulose synthesis, cell wall biosynthesis, and carbohydrate metabolism [17], [22], [27].
Chromosome segment introgression lines (CSILs) consist of a battery of near-isogenic lines that have been developed to cover the entire genomes of some crops, including tomato, rice, wheat, and cotton [28]–[31]. With the exception of a single, homozygous chromosome segment transferred from a donor parent, the remaining genome of each CSIL is the same as the recipient parent [31]. We used G. barbadense CSILs in the background of the standard genetic line of G. hirsutum, cv. TM-1, in order to understand the molecular mechanism behind superior quality fiber formation. Multi-point tests showed that three CSILs produced stronger fibers when compared to the recipient parent TM-1, but one CSIL produced weaker fibers. Using Solexa Genome sequencing, we analyzed transcriptome profiles from the CSILs and TM-1. We found that many genes were either up- or down-regulated at the stage of secondary cell wall synthesis, and that many metabolic pathways were altered in the CSILs.
Materials and Methods
Plant materials
G. hirsutum cv. TM-1, the genetic standard for Upland cotton, was obtained from the Southern Plains Agricultural Research Center, USDA-ARS, College Station/Texas, USA [32]. G. barbadense cv. Hai7124, an extra-long staple cotton that is widely grown in China, is descended from a selected individual in a study of inheritance of resistance to Verticillium dahlia [33], [34]. In this study, we identified three CSILs with stronger fiber or high fiber strength that carried different G. barbadense chromosome segment(s) in the recurrent parent TM-1. The detailed method of developing CSILs has been described previously [31]. We selected three CSILs, CSIL-35431, CSIL-31134, and CSIL-31010, in which the average fiber strength were 35.1, 34.73 and 34.28 cN/tex, respectively, significantly higher than TM-1, and also CSIL-35368 which had poorer fiber strength than TM-1(28.71 cN/tex) (Table S1). The introgressed G. barbadense chromosomal segments were different in the four lines [35]. Fiber samples were collected at 15, 20, and 25 DPA, frozen in liquid nitrogen, and stored at −70°C.
RNA isolation and evaluation
Total RNA was extracted from frozen tissue using an improved CTAB extraction protocol [36]. RNAs were evaluated for quality using RNA Pico Chips on an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA). All RNA samples were quantified and qualified with an RNA Integrity Number (RIN) >8, and 28S/18S rRNA band intensity (2∶1).
Library construction and sequencing
Digital gene expression libraries were constructed using the Illumina Gene Expression Sample Preparation Kit according to the manufacturer's instructions. We constructed and sequenced 14 libraries derived from immature fibers at 15, 20, and 25 DPA using the Solexa Genome Sequencing Analyzer system provided by BGI (Beijing Genomics Institute at Shenzhen, China), which gave 21 bp tags. The process was described in detail previously [18].
Data processing, statistical evaluation, and selection of differentially expressed genes (DEGs)
Raw data reads were filtered by the Illumina pipeline to produce clean data. All low-quality data, such as short tags (<21 nt) and singletons, were removed. A database of 21-base-long sequences was produced beginning with CATG using 37,505 reference genes from the diploid species G. raimondii (http://www.phytozome.net). The remaining high quality sequences were then mapped to this database; only a single mismatch was allowed, and more than one match was excluded. Gene expression levels were the summation of tags aligned to the different positions of the same gene. Expression levels are expressed as TPM, transcripts per million. To identify DEGs during fiber elongation, we compared pairs of DEG profiles from different libraries. Three fiber development periods for the four CSILs were compared with the same period for TM-1, and 11 comparisons were obtained. P- and Q-values were also calculated for every comparison [37]. DEGs were defined as FDR≤0.001 with an absolute value of |log2Ratio|≥1 to judge the significance of differences in transcript abundance.
Digital tag profiling analysis
DEG clustering in CSILs at different developmental stages were performed with Cluster3.0 (http://bonsai.hgc.jp/~mdehoon/software/cluster/software.htm). We also performed clustering with the ‘Self-organizing tree algorithm’ (SOTA, Multiple Array Viewer software, MeV 4.9.0) [38].
GO enrichment and KEGG (Kyoto Encyclopedia of Genes and Genomes) pathway analysis was done using BLAST2GO (http://www.blast2go.com/b2ghome). Mapman was also used to analyze metabolic pathway base on KEGG database [39].
Quantitative RT-PCR
Quantitative RT-PCR assays were performed on a 7500 Real-Time PCR system (Applied Biosystems, San Francisco, CA, USA). Reactions were performed in a final volume of 15 µL and contained 2 µL of diluted cDNA, 7.5 µL of 2× SYBR mix (Roche, Basel, Switzerland), and 200 nM of the forward and reverse primers. Primer lengths were designed to be between 18 and 24 nt using Beacon Designer 7, and PCR amplicon lengths were designed to be between 100 bp and 150 bp (Table S2). The thermal cycling conditions were 40 cycles of 95°C for 15 s, 60°C for 30 s, and 72°C for 30 s. All reactions were run in triplicate, and the cotton histone3 gene (ACC NO. AF024716) was used as an internal control for normalization of expression levels (F: 5′-GGTGGTGTGAAGAAGCCTCAT-3′, and R: 5′-AATTTCACGAACAAGCCTCTGGAA-3′). The relative gene expression levels were presented as 2−ΔCT.
Results
Statistical analysis of transcriptome data
The total number of sequence tags per library ranged from 7.0 to 8.5 million, and the number of distinct sequence tags was between 1.8 and 2.2 million. Approximately 50% of the clean tags were mapped to reference genes, and 60% of the reference genes were mapped with unambiguous tag (Table 1 and Table S3).
Table 1. The distribution of total and distinct tags.
| Summary | TM-1 | CSIL-35431 | CSIL-31010 | ||||||
| 15DPA | 20DPA | 25DPA | 15DPA | 20DPA | 25DPA | 15DPA | 20DPA | 25DPA | |
| Raw Data | 8374304 | 7231305 | 14267931 | 7461562 | 7389820 | 7298224 | 7471212 | 7007447 | 7203054 |
| Distinct Raw Tag | 389162 | 394430 | 494345 | 375999 | 371748 | 317147 | 283130 | 337525 | 386302 |
| Clean Tag | 8144920 | 7013147 | 14002534 | 7264426 | 7213791 | 7124575 | 7369012 | 6840101 | 7002766 |
| Distinct Clean Tag | 169700 | 177887 | 251173 | 182629 | 199257 | 148121 | 182184 | 172775 | 188639 |
| Unique Clean Tag Mapping to Gene | 3983215 | 3206328 | 6829327 | 3961228 | 3411249 | 3463314 | 3671014 | 3381955 | 3552566 |
| Total % of clean tag | 48.90% | 45.72% | 48.77% | 54.53% | 47.29% | 48.61% | 49.82% | 49.44% | 50.73% |
| Unique Distinct Clean Tag Mapping to Gene | 45380 | 40475 | 56172 | 53187 | 48453 | 41065 | 46720 | 45762 | 48147 |
| Total % of distinct tag | 26.74% | 22.75% | 22.36% | 29.12% | 24.32% | 27.72% | 25.64% | 26.49% | 25.52% |
| Unambiguous Tag-mapped Genes | 21498 | 20901 | 22594 | 21781 | 22325 | 20590 | 21900 | 22457 | 22811 |
| Percentage of reference genes | 57.32% | 55.73% | 60.24% | 58.07% | 59.53% | 54.90% | 58.39% | 59.88% | 60.82% |
Clean tags: tags after filtering dirty tags (low quality tags) from raw data.
Distinct tags: different kinds of tags.
Unambiguous tags: the clean tags after removing tags mapped to reference sequences from multiple genes.
To see whether the fiber transcriptomes at different developmental stages were different, the 23,237 genes which were expressed in at least three libraries at one stage (15 DPA, 20 DPA, or 25 DPA) were classified into six groups using the Multiple Array Viewer using TPM value (Figure 1A). Genes in Group 3 had higher expression levels at 15 DPA and 20 DPA than at the later stage (25 DPA). Genes in Group 4 had higher expression levels at 15 DPA than at either 20 DPA or 25 DPA. Genes in Group 5 showed the opposite expression pattern, with higher expression levels at 20 DPA and 25 DPA compared to 15 DPA. The other groups also showed distinct expression patterns (Figure 1A).
Figure 1. Statistical analysis of transcriptome data.

(A) SOTA clustering of the different genes using Log2(TPM). T, TM-1; A, CSIL-35431; B, CSIL-31010; C, CSIL-31134; D, CSIL-35368. 15, 15 DPA; 20, 20 DPA; 25, 25 DPA. (B) Distribution of functions of genes in different clusters. Yellow square indicated group 3, green square indicated group 4 and blue square indicated group 5. X-axis indicated different enriched process and Y-axis indicated number of hit-found genes in these processes.
Classification by gene function revealed that Group 3 is enriched in genes involved in protein catabolism, cell division, and cellulose biosynthesis, Group 4 is enriched in genes for cell morphogenesis, fatty acid biosynthesis, lipid transport, and wax biosynthesis, and Group 5 has more genes involved in glucose catabolism, response to chitin, and sucrose metabolism (Figure 1B). The unbalanced pattern of the expressed-gene functional distribution could possibly reflect some physiological events involved in secondary cell wall biosynthesis.
Cluster analysis of differentially expressed genes (DEGs) between and/or among CSILs
We specifically looked for DEGs in secondary cell wall fibers from 15 to 25 DPA, because previous studies have reported that the different sets of transcripts responsible for fiber secondary cell wall formation may be enriched at these stages of development [17], [22], [27]. Three fiber development periods for the four CSILs were compared with TM-1 at the same period. DEGs were defined as FDR≤0.001 with an absolute value of |log2Ratio|≥1. Analysis of the data indicated that many genes showed differential expression in the 11 comparison groups. The number of DEGs were about 6,000–8,000 in CSILs from 15 DPA to 25 DPA (Figure 2A). But the number of DEGs in CSIL-31010 at 20 DPA, CSIL-31010 at 25 DPA, and CSIL-31134 at 15 DPA, were 4,600, 10,106 and 2,060, respectively. We also found that the DEGs that were up-regulated or down-regulated were different in CSILs. There were ∼1,500–3,500 DEGs in common from 15 DPA to 25 DPA between CSIL-35431, CSIL-31010, and CSIL-35368 (Figure 2B).
Figure 2. Statistical of DEGs between CSILs and TM-1 at 15, 20 and 25 DPA.

(A) Up-regulated and down-regulated genes in different comparison. Red bar, up-regulated genes compared to TM-1; green bar, stand for down-regulated genes compared to TM-1, Blue square, total DEGs. CSILs included CSIL-35431, CSIL-3010, CSIL-31134, CSIL-35368 and TM-1. 15, 15DPA; 20, 20DPA; 25, 25DPA. (B) Common and special DEGs at 15 DPA, 20 DPA and 25 DPA.
To understand the mechanisms behind the changes in fiber strength observed in the CSILs, we also analyzed the common DEGs among CSIL-35431, CSIL-31010 and CSIL-31134 (Table S4). A total of 727 and 1796 common DEGs were selected at 15 and 20 DPA in three stronger fiber CSILs, respectively (Figure 3). More functional enrichment were shown at 15 DPA, including major CHO metabolism (carbohydrate), cell wall biosynthesis, amino acid metabolism and secondary metabolism (Figure 3E). Among these genes, 321 and 998 common upregulated DEGs between the same CSILs at 15 and 20 DPA were indentified, respectively (Figure 3). These common DEGs or processes maybe directly related to the fiber strength. However, these DEGs maybe function as downstream genes altered by the introgressed segments since these CSILs were inserted different G. barbadense segments in recipient TM-1.
Figure 3. Analysis of common and common upregulated DEGs among three stronger fiber CSILs.

(A, B, C, D) Common and common upregulated DEGs among three stronger fiber CSILs at 15 and 20 DPA. Common_up, common regulated DEGs. (B) Functional enrichment analysis of these DEGs using mapman software (Summary statistic type, wlcoxon). Colors from blue to red indicated that functions were enriched more significantly with smaller p-values.
To visualize the expression patterns of DEGs, we performed cluster analysis of 18,288 genes that were differentally expressed between CSIL-35431 and CSIL-31010 (Figure 4). These DEGs could be grouped into six clusters, designated G1–G6, based on their expression patterns. From 15 DPA to 20 DPA, the stages of fast fiber elongation and secondary cell wall deposition overlap, with the latter reaching a peak at around 20–25 DPA. We focused on clusters G1, G4, and G6 to conduct data analysis in order to identify genes that were either up-regulated or down-regulated during the secondary cell wall synthesis stage. Compared to the TM-1 control, 3,658 genes in cluster G1 were highly expressed at 15 and 20 DPA, 4,487 genes in G4 were highly expressed at 15 DPA, 20 DPA, and 25 DPA, 3,033 genes in G6 were highly expressed only at 25 DPA, and the other three groups showed various different expression patterns. Clustering results for 19,742 DEGs from the four CSILs showed five groups, indicating that the gene expression pattern in CSIL-31134 was distinct from the others at 15 DPA and 20 DPA, and that CSIL-35368 was similar to CSIL35431 and CSIL-31010 (Figure S1).
Figure 4. Heat map analysis of the expression of DEGs between CSILs and TM-1.
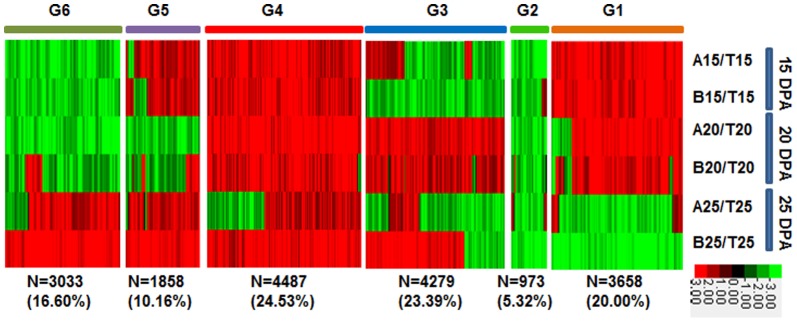
A, B and T indicated CSIL-35431, CSIL-31010 and TM-1, respectively. 15, 15DPA; 20, 20DPA; 25, 25DPA. Red color indicated up-regulated genes and green color indicated down-regulated genes. N = number of DEGs in different group.
Functional annotation by GO enrichment and KEGG analysis
To understand the mechanisms behind the changes in fiber strength observed in the CSILs, we analyzed DEG enrichment in the major functional GO categories of biological process, molecular function, and cellular component between CSIL-35431 and CSIL-31010. Based on the clustering results shown in Figure 4, G1 was enriched in genes for secondary cell wall biogenesis, glucuronoxylan biosynthesis, microtubule-based movement, and cellulose biosynthesis, G4 was enriched in genes for protein phosphorylation, response to chitin, and sugar-mediated signaling pathways, and G6 was enriched in fatty acid biosynthesis genes (Table 2). These data suggest that in the developmental stage of secondary cell wall deposition, DEGs were enriched for carbohydrate synthesis and cell wall formation.
Table 2. Enrichment analysis of gene ontologies from 15 to 25 DPA.
| Cluster | GO-ID | GO Ontology (Biological process) |
| G1 | GO:0010417 | glucuronoxylan biosynthetic process |
| 15DPA up-regulated | GO:0009834 | secondary cell wall biogenesis |
| 20DPA up-regulated | GO:0007018 | microtubule-based movement |
| 25DPA down-regulated | GO:0030244 | cellulose biosynthetic process |
| GO:0009753 | response to jasmonic acid stimulus | |
| G2 | GO:0015031 | protein transport |
| 15DPA down-regulated | GO:0015991 | ATP hydrolysis |
| 20DPA down-regulated | GO:0032544 | plastid translation |
| 25DPA down-regulated | GO:0016075 | rRNA catabolic process |
| GO:0006511 | ubiquitin catabolic process | |
| G3 | GO:0009734 | auxin mediated signaling pathway |
| 15DPA down-regulated | GO:0030259 | lipid glycosylation |
| 20DPA up-regulated | GO:0018106 | peptidyl-histidine phosphorylation |
| 25DPA down-regulated | GO:0009722 | detection of cytokinin stimulus |
| G4 | GO:0006468 | protein phosphorylation |
| 15DPA up-regulated | GO:0010200 | response to chitin |
| 20DPA up-regulated | GO:0006096 | glycolysis |
| 25DPA up-regulated | GO:0010182 | sugar mediated signaling pathway |
| GO:0009966 | regulation of signal transduction | |
| G5 | GO:0015884 | protein folding |
| 15DPA up-regulated | GO:0006886 | intracellular protein transport |
| 20DPA down-regulated | GO:0006122 | mitochondrial electron transport |
| 25DPA up-regulated | GO:0015914 | phospholipid transport |
| G6 | GO:0007267 | cell-cell signaling |
| 15DPA down-regulated | GO:0010025 | wax biosynthetic process |
| 20DPA down-regulated | GO:0006633 | fatty acid biosynthetic process |
| 25DPA up-regulated | GO:0006723 | hydrocarbon biosynthetic process |
G1–G6 according to Figure 3.
We applied the same GO analysis to the common DEGs at 15 DPA and 20 DPA in CSIL-35431 and CSIL-31010, respectively. These DEGs were enriched in genes for similar functional categories, such as cellular metabolic processes and carbohydrate metabolism, etc. We also found genes for some processes that were enriched only in CSIL35431 or CSIL-31010 (Figure S2).
Further GO analysis for CSIL-35368 and CSIL-31134 indicated that the DEGs in CSIL-35368 at 15 and 20 DPA were enriched in genes for lignin biosynthesis, secondary cell wall biogenesis, and response to chitin, which was similar to the enrichment found in CSIL-35431 and CSIL-31010. But at 15 and 20 DPA in the stronger fiber line CSIL-31134, GO enrichments were different from the other three lines, mainly in genes for ATP synthesis, proton transport, copper ion export, and oxidoreductase activity, but not in cell wall biosynthesis (Table S5).
Based on the results of GO analysis, we know that the secondary cell wall related biological process were impacted in the CSILs, but it is still not very clear how secondary cell wall biosynthesis was affected in the CSILs. Therefore, we performed pathway analysis on 18,288 DEGs in CSIL-35431 and CSIL-31010. The most highly enriched pathways found are listed in Table 3. KEGG analysis showed that the genes were enriched in pathways for starch and sucrose metabolism (328 genes), glycolysis/gluconeogenesis (122 genes), phenylpropanoid biosynthesis (101 genes), and oxidative phosphorylation (87 genes) (Table 3 and Figure S3). The regulation of some enzymes that catalyze sucrose, starch, and cellulose biosynthesis may have a direct or indirect impact on fiber quality. This could be especially true for sucrose and pectin metabolism, and many genes in these pathways were up-regulated. We also found that genes involved in phenylpropanoid and flavonoid biosynthetic processes were enriched in the CSILs.
Table 3. KEGG analysis of DEGs in CSIL-35431 and CSIL-31010.
| Pathway | DEGs with pathway annotation (2576) | References genes with pathway annotation(4601) | Ratio | DEGs distribution in each groups | |||||
| G1 | G2 | G3 | G4 | G5 | G6 | ||||
| Starch and sucrose metabolism | 328 | 563 | 58.26% | 87 | 15 | 54 | 89 | 31 | 52 |
| Purine metabolism | 251 | 459 | 54.68% | 35 | 20 | 53 | 65 | 30 | 48 |
| Phenylalanine metabolism | 140 | 261 | 53.64% | 29 | 5 | 26 | 47 | 12 | 21 |
| Amino sugar and nucleotide sugar metabolism | 137 | 211 | 64.93% | 47 | 5 | 23 | 36 | 12 | 14 |
| Pyrimidine metabolism | 128 | 227 | 56.39% | 15 | 13 | 26 | 37 | 15 | 22 |
| Glycolysis/Gluconeogenesis | 122 | 188 | 64.89% | 28 | 4 | 23 | 35 | 13 | 19 |
| T cell receptor signaling pathway | 115 | 212 | 54.25% | 28 | 4 | 23 | 34 | 11 | 15 |
| Pentose and glucuronate interconversions | 109 | 227 | 48.02% | 24 | 8 | 20 | 27 | 7 | 23 |
| Glycerolipid metabolism | 102 | 174 | 58.62% | 18 | 4 | 29 | 21 | 15 | 15 |
| Pyruvate metabolism | 102 | 182 | 56.04% | 16 | 8 | 18 | 29 | 12 | 19 |
| Phenylpropanoid biosynthesis | 101 | 220 | 45.91% | 26 | 6 | 17 | 28 | 8 | 16 |
| Galactose metabolism | 94 | 160 | 58.75% | 23 | 2 | 20 | 24 | 7 | 18 |
| Cysteine and methionine metabolism | 94 | 145 | 64.83% | 21 | 2 | 21 | 28 | 10 | 12 |
| Glycerophospholipid metabolism | 94 | 167 | 56.29% | 14 | 8 | 29 | 21 | 10 | 12 |
| Arginine and proline metabolism | 93 | 144 | 64.58% | 16 | 5 | 22 | 19 | 12 | 19 |
| Oxidative phosphorylation | 87 | 184 | 47.28% | 19 | 13 | 12 | 11 | 13 | 19 |
| Carbon fixation in photosynthetic organisms | 84 | 151 | 55.63% | 19 | 3 | 11 | 34 | 8 | 9 |
| Fatty acid degradation | 83 | 133 | 62.41% | 17 | 5 | 18 | 21 | 7 | 15 |
G1–G6 according to Figure 3.
Based on the cluster analysis of the weaker fiber line CSIL-35368, we hypothesized that changes in other biochemical pathways led to reduced fiber strength (Figure S1). Considering only those that were down-regulated in CSIL-35368, we found genes that participated in fatty acid degradation and biosynthesis, and also in carbohydrate metabolic pathways (Figure 2B and Figure S4).
Eight genes previously reported in the carbohydrate pathway were selected for quantitative RT-PCR. The expression patterns of these genes were consistent with the DEG data in TM-1 (Figure 5) and in the CSILs as well (Figure 6B and Figure 7B).
Figure 5. Quantitative RT–PCR validation of tag-mapped genes in TM-1.

These genes have been reported before, including 3 CesA genes (A,B,C) (homologous with AtCESA4, AtCESA7, AtCESA8, respectively), xyloglucan endotransglucosylase (D), beta -galactosidase (E), glycosyl hydrolase 9B7 (F), xylan alpha-glucuronosyltransferase 1, GUX1 (G), xylan alpha-glucuronosyltransferase 2, GUX2 (H).
Figure 6. Carbohydrate pathways that are differentially regulated during the secondary cell wall synthesis stage.

(A) Carbohydrate pathways. Genes up-regulated in CSIL-315431 and CSIL-31010 were selected to do heat map. ABAB indicated DEGs in CSIL-35431 at 15 DPA, CSIL-35431 at 20DPA, CSIL-3010 at 15DPA and CSIL-31010 at 20DPA, from left to right. Every square stand for one gene and every line stand for the same gene. Genes with red color expressed higher in CSILs than TM-1 and gray color stand for no difference. β-D-Fru, β-D-Fructose; α-D-Glu-1p, α-D-Glucose-1-phosphate; β-D-Fru-6p, β-D-Fructose-6-phosphate. (B) Quantitative RT–PCR validation of four CesA genes in CSILs and TM-1, Gorai.004G057400.1, Gorai.009G009700.1 and Gorai.011G037900.1 homologous with AtCESA4, AtCESA7 and AtCESA8, respectively.
Figure 7. NAC and MYB family genes involved in the regulation of secondary wall biosynthesis.

(A) 59 MYB family genes and 47 NAC family genes showed different expression level between CSILs and TM-1 at 15DPA, 20DPA and 25DPA. |Ratio|>2 and FDR<0.001. A, B, T indicated CSIL-35431, CSIL-31010 and TM-1. 15, 15DPA; 20, 20DPA; 25, 25DPA. (B) Quantitative RT–PCR validation of three transcription factors.
Carbohydrate metabolism in the secondary cell wall synthesis stage
Following the start of secondary cell wall formation, protein and carbohydrate metabolism genes involved in cell wall biosynthesis will be up-regulated [27]. We selected 72 DEGs associated with carbohydrate metabolism to investigate the mechanism of fiber development. These genes were related to pectin, sucrose, galactan, glucan, xyloglucan, and cellulose biosynthesis. We were interested in genes that are up-regulated in fiber cells at 15 DPA and 20 DPA, at the start of secondary cell wall formation. A heat map showing the different expression levels for these genes including cellulose synthase, sucrose synthase, pectin lyase, and other polysaccharides degradation in CSIL-35431 and CSIL-31010 is shown in Figure 6A. We found that the cellulose synthase genes were up-regulated in the CSILs at 15 DPA-25 DPA. It has been reported that cellulose biosynthesis predominates, and that many other metabolic pathways are down-regulated during secondary cell wall synthesis [27]. Moreover, we confirmed the expression patterns of cellulose synthase genes, annotated with the Arabidopsis genes AtCESA4, AtCESA7 and AtCESA8, using quantitative RT-PCR (Figure 4B). Proteins encoded by AtCESA4, 7, and 8 are specifically required to form a functional cellulose synthase complex (CSC) that is essential for secondary cell wall formation [40]–[42].
Transcription factors associated with secondary cell wall synthesis
Recent molecular and genetic studies have identified transcription factors that are involved in regulating secondary cell wall synthesis in Arabidopsis [43]–[45]. In our study, 97 MYB-type and 68 NAC-type transcription factors showed changes in expression between the CSILs and TM-1 (Table S6, Table S7). It was interesting that some NACs and MYBs were up-regulated in CSIL-35431 and CSIL-31010 during the secondary cell wall synthesis stage, especially at 15 DPA and 20 DPA. Defined as |log2Ratio|≥2, 59 MYB and 47 NAC transcription factors were selected for heat-map analysis (Figure 7A). Among these transcription factors, genes homologous with ATMYB2, ATMYB43, ATMYB73, ATNAC52, and ATNAC61 were expressed at higher levels in the CSILs. We confirmed that three transcription factors were up-regulated in CSILs from 15 DPA to 25 DPA (Figure 7B). In the MYB family, it has been reported that the expression of genes for MYB85, MYB52, MYB54, MYB69, MYB42, and MYB43 are developmentally associated with cells undergoing secondary wall thickening [45].
Different metabolic pathways associated with altered fiber strength
In order to investigate the mechanisms underlying changes in fiber strength, we analyzed several metabolic pathways including cell wall, lipids, minor CHO (carbohydrate) metabolism, and two secondary metabolite pathways. It is interesting that DEGs involved in cell wall proteins, cell wall pectin esterase, cell wall modification, cell wall cellulose synthesis, cell wall degradation/pectate lyases, lipid metabolism/FA synthesis, and lipid degradation showed distinct expression patterns or differential up/down-regulation at 20 DPA (Figure 8A). We found that up-regulated DEGs were similar to down-regulated DEGs both in CSIL-35431 and CSIL-35368. However, most of DEGs in CSIL-31010 were up-regulated at 20 DPA, while the opposite was true for DEGs in CSIL-31134, especially those genes involved in cell wall modification. In CSIL-31134, we also found a few genes in these metabolic pathways that were changed at 15 DPA except in cell wall modification, and in CSIL-31010, we found DEGs enriched in these metabolic pathways at 25 DPA (Figure S5). From the secondary metabolism results, we identified a few DEGs involved in flavonoid biosynthesis in CSIL-35431 and CSIL-31010 at 15 DPA. In contrast, more genes were up-regulated or down-regulated in CSIL-35368 at 15 DPA. It was obvious that DEGs from the phenylpropanoid pathways at 25 DPA were different from one another, and the expression pattern of DEGs in CSIL-31010 changed dramatically. Moreover, there were few genes that were up-regulated or down-regulated in CSIL-35368 at 25 DPA (Figure 7B). We assume that metabolic pathways in the CSILs at different developmental stages were changed in various ways as a result of the introgressed chromosmal segments from G. barbadense.
Figure 8. Metabolism analysis of DEGs in CSILs during the secondary cell wall biosynthesis stage.

(A) Motabolism overview in four CSILs at 20 DPA. (B) Secondary motabolism analysis in three CSILs at 15 DPA, 20 DPA and 25 DPA. 1, cell wall protein; 2, cell wall pectin esterases; 3, cell wall modification; 4, cell wall cellulose synthesis; 5, cell wall degradation/pectate lyases; 6, lipid metabolism/FA synthesis; 7, lipid degradation; 8, flavonoids; 9, phenylpropanoids/lignin biosynthesis. Blue square, down-regulated genes; Red square, up-regulated genes.
Discussion
G. hirsutum produces a high yield of cotton with moderate fiber strength. G. barbadense is characterized by a low yield, but with increased fiber fineness and strength. As a breeding target, we tried to combine the high yield of G. hirsutum with the superior fiber qualities of G. barbadense, and we also wanted to elucidate the molecular mechanism behind the formation of superior quality fibers. Fiber strength is an important indicator of the cotton fiber quality, and depends on the formation of the secondary cell wall. Genome-wide transcriptome profiling is effective at revealing significant genes and pathways involved in secondary cell wall formation. Transcriptome analysis showed that gene expression patterns and functional distribution were different during secondary cell wall biosynthesis.
Carbohydrate metabolism plays an important role in secondary cell wall synthesis
It is well known that the mature cotton fiber is composed of nearly pure cellulose, and that such a high level of cellulose synthesis requires an abundant supply of UDP-glucose [46], [47]. This means that a large amount of cellulose is required during the secondary cell wall synthesis stage. Functional classification and enrichment analysis showed that following the initiation of secondary cell wall synthesis, DEGs were enriched for secondary cell wall biogenesis, glucuronoxylan biological processes, and other carbohydrate metabolic pathways in the CSILs (Table 2). Focusing on carbohydrate metabolic pathways, it is obvious that the key intermediate in the multiple pathways is UDP-glucose, a substrate for cellulose synthesis. Our results showed that several CesA genes are expressed at higher levels during secondary cell wall synthesis than they are at earlier stages (Figure 6B). Ten AtCESA genes have been reported in Arabidopsis, and AtCESA4, 7, and 8 are specifically required to form the cellulose synthase complex (CSC) that is essential for secondary cell wall synthesis [40]–[42]. Similarly, three CESA isoforms have been identified during secondary cell wall synthesis in rice, maize, and Populus [10], [48], [49]. Also, many genes that participate in the degradation of poly- and oligo-saccharides were found to be up-regulated at 15 and 20 DPA, in order to produce more UDP-glucose for cellulose biosynthesis. Similarly, it has also been reported that during the secondary cell wall synthesis stage, certain metabolic pathways, including hydrolysis of fatty acids and non-cellulose poly- and oligo-saccharides, would be up-regulated [27]. Sucrose synthase (SuSy) has long been studied as a cytoplasmic enzyme in plant cells, where it serves to degrade sucrose and provide carbon for respiration and synthesis of cell wall polysaccharides and starch [50]. It has also been shown that genes associated with secondary cell wall biosynthesis are involved in sugar metabolism [51].
Multiple mechanisms affect fiber strength development
Except for carbohydrate metabolism, recent research has shown that transcription factors also affect fiber development during secondary cell wall biosynthesis. Several NAC- and MYB-type transcription factors were up-regulated in the CSILs compared to TM-1 from 15 DPA to 25 DPA, and these included cotton homologs of AtMYB2, AtMYB43, and AtNAC52 etc. (Figure 7A). The NAC-mediated transcriptional regulation of secondary wall biosynthesis is a conserved mechanism throughout vascular plants [44], [52]. SND2, a NAC transcription factor gene, regulates genes involved in secondary cell wall development in Arabidopsis fibers and increases fiber cell area in Eucalyptus [53]. A MYB75-associated protein complex is likely to be involved in modulating secondary cell wall biosynthesis in both the Arabidopsis inflorescence and stem [54]. It has also been found that the rice and maize MYB transcription factors, OsMYB46 and ZmMYB46, are functional orthologs of Arabidopsis MYB46/MYB83 and, when overexpressed in Arabidopsis, are able to activate the entire secondary wall biosynthetic program [55].
Several metabolic pathways were examined to determine the mechanism behind changes in fiber strength; these included cell wall, lipids, minor CHO metabolism, and two secondary metabolic pathways. Although results of the GO and KEGG analyses showed that CSIL-35431, CSIL-31010, and CSIL-35368 had similar patterns, fiber strength in these three lines were different. Our results support the hypothesis that different metabolic pathways can affect fiber strength, and the same pathway in the CSILs can be altered differentially at various times in development. DEGs in CSIL-31010 were up-regulated at 20 DPA, while the opposite was found for DEGs in CSIL-31134, especially those genes involved in cell wall modification. The expression levels of genes involved in flavonoid biosynthesis in the weak fiber line CSIL-35368 were changed dramatically at 15 DPA, but there were few genes changed at 25 DPA; this patter was the opposite of that in CSIL-35431 and CSIL-31010, lines with high quality fiber. We hypothesize that phenylpropanoid and flavonoid metabolism generally affected the fiber strength of CSIL-35368. Genes for phenylpropanoid and flavonoid biosynthesis showed significant enrichment and temporal differences in gene expression patterns which are associated with xylem formation [56]. It has been reported that expression levels of phenylpropanoid genes showed high correlations with specific fiber properties, supporting a role in determining fiber strength [57].
In conclusion, upland cotton CSILs carrying distinct G. barbadense chromosomal segments provide valuable material for research into fiber development. The G. barbadense chromosome segments resulted in different patterns of differentially expressed genes, and altered different metabolic pathways, mainly in carbohydrate metabolism. In addition, several transcription factor genes were found to be specifically up-regulated in the CSILs. Metabolic pathways involved in cell wall, lipid, phenylpropanoid, and flavonoid biosynthesis play a significant role during secondary cell wall formation, and are associated with the development of cotton fiber strength.
Supporting Information
Heat map of the expression of DEGs between 4 CSILs at 15–25 DPA.
(TIF)
Enrichment analysis of common DEGs at 15DPA and 20DPA in CSIL-35431 and CSIL-31010.
(TIF)
Heat map of DEGs participated in four metabolic pathways from 15 DPA to 25 DPA.
(TIF)
Pathway analysis of genes only down-regulated in CSIL-35368 from 15 DPA to 25 DPA.
(TIF)
Metabolism analysis of DEGs in CSILs at 15 DPA and 25 DPA.
(TIF)
Average fiber quality of 4 CSILs and TM-1.
(XLS)
Primer for quantitative RT-PCR.
(XLS)
Categorization and abundance of tags.
(XLS)
List of common DEGs among CSIL-35431, CSIL-31134 and CSIL-31010.
(XLS)
Enrichment analysis of gene ontologies in CSIL-35368 and CSIL-31010 at 15 DPA and 20 DPA.
(XLS)
Different expression level of 97 MYB transcription factors.
(XLS)
Different expression level of 68 NAC transcription factors.
(XLS)
Funding Statement
This work was financially supported in part by grants from the National Science Foundation of China (31330058) and the Priority Academic Program Development of Jiangsu Higher Education Institutions. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Basara AS, Malik CP (1984) Development of cotton fiber. Inter Rev Cyto 65–113. [Google Scholar]
- 2.Haigler TA, Jernstedt JA (1999) Molecular genetics of developing cotton fibers. In: AM Basra (Ed), Cotton Fibers. Hawthorne Press, New York, 231–267. [Google Scholar]
- 3. Kim HJ, Triplett BA (2001) Cotton fiber growth in planta and in vitro. Models for plant cell elongation and cell wall biogenesis. Plant Physiol 127: 1361–1366. [PMC free article] [PubMed] [Google Scholar]
- 4. Lee JJ, Hassan OS, Gao W, Wei NE, Kohel RJ, et al. (2006) Developmental and gene expression analyses of a cotton naked seed mutant. Planta 223: 418–432. [DOI] [PubMed] [Google Scholar]
- 5. Lee JJ, Woodward AW, Chen ZJ (2007) Gene expression changes and early events in cotton fibre development. Ann Bot 100: 1391–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wilkins TA, Arpat AB (2005) The cotton fiber transcriptome. Physiol Plant 124: 295–300. [Google Scholar]
- 7. Meinert MC, Delmer DP (1977) Changes in biochemical composition of the cell wall of the cotton fiber during development. Plant Physiol 59: 1088–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bolton JJ, Soliman KM, Wilkins TA, Jenkins JN (2009) Aberrant Expression of Critical Genes during Secondary Cell Wall Biogenesis in a Cotton Mutant, Ligon Lintless-1 (Li-1). Comp Funct Genom 659301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Richmond TA, Somerville CR (2000) The cellulose synthase superfamily. Plant Physiol 124: 495–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tanaka K, Murata K, Yamazaki M, Onosato K, Miyao A, et al. (2003) Three distinct rice cellulose synthase catalytic subunit genes required for cellulose synthesis in the secondary wall. Plant Physiol 133: 73–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Paterson AH, Wendel JF, Gundlach H, Guo H, Jenkins J, et al. (2012) Repeated polyploidization of Gossypium genomes and the evolution of spinnable cotton fibres. Nature 492: 423–427. [DOI] [PubMed] [Google Scholar]
- 12. Persson S, Wei H, Milne J, Page GP, Somerville CR (2005) Identification of genes required for cellulose synthesis by regression analysis of public microarray data sets. Proc Natl Acad Sci USA 102: 8633–8638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ruan YL, Chourey PS, Delmer DP, Perez-Grau L (1997) The Differential Expression of Sucrose Synthase in Relation to Diverse Patterns of Carbon Partitioning in Developing Cotton Seed. Plant Physiol 115: 375–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Brill E, van Thournout M, White RG, Llewellyn D, Campbell PM, et al. (2011) A novel isoform of sucrose synthase is targeted to the cell wall during secondary cell wall synthesis in cotton fiber. Plant Physiol 157: 40–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Potikha TS, Collins CC, Johnson DI, Delmer DP, Levine A (1999) The involvement of hydrogen peroxide in the differentiation of secondary walls in cotton fibers. Plant Physiol 119: 849–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yang YM, Xu CN, Wang BM, Jia JZ (2001) Effects of plant growth regulators on secondary wall thickening of cotton fibres. Plant Growth Regul 35: 233–237. [Google Scholar]
- 17. Arpat AB, Waugh M, Sullivan JP, Gonzales M, Frisch D, et al. (2004) Functional genomics of cell elongation in developing cotton fibers. Plant Mol Biol 54: 911–929. [DOI] [PubMed] [Google Scholar]
- 18. Wang QQ, Liu F, Chen XS, Ma XJ, Zeng HQ, et al. (2010) Transcriptome profiling of early developing cotton fiber by deep-sequencing reveals significantly differential expression of genes in a fuzzless/lintless mutant. Genomics 96: 369–376. [DOI] [PubMed] [Google Scholar]
- 19. Padmalatha KV, Dhandapani G, Kanakachari M, Kumar S, Dass A, et al. (2012) Genome-wide transcriptomic analysis of cotton under drought stress reveal significant down-regulation of genes and pathways involved in fibre elongation and up-regulation of defense responsive genes. Plant Mol Biol 78: 223–246. [DOI] [PubMed] [Google Scholar]
- 20. Chaudhary B, Hovav R, Rapp R, Verma N, Udall JA, et al. (2008) Global analysis of gene expression in cotton fibers from wild and domesticated Gossypium barbadense . Evol Dev 10: 567–582. [DOI] [PubMed] [Google Scholar]
- 21. Hovav R, Udall JA, Chaudhary B, Hovav E, Flagel L, et al. (2008) The evolution of spinnable cotton fiber entailed prolonged development and a novel metabolism. PLoS Genet 4: e25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hovav R, Udall JA, Hovav E, Rapp R, Flagel L, et al. (2008) A majority of cotton genes are expressed in single-celled fiber. Planta 227: 319–329. [DOI] [PubMed] [Google Scholar]
- 23. Ji SJ, Lu YC, Feng JX, Wei G, Li J, et al. (2003) Isolation and analyses of genes preferentially expressed during early cotton fiber development by subtractive PCR and cDNA array. Nucleic Acids Res 31: 2534–2543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Udall JA, Flagel LE, Cheung F, Woodward AW, Hovav R, et al. (2007) Spotted cotton oligonucleotide microarrays for gene expression analysis. BMC Genomics 8: 81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Qin YM, Hu CY, Pang Y, Kastaniotis AJ, Hiltunen JK, et al. (2007) Saturated very-long-chain fatty acids promote cotton fiber and Arabidopsis cell elongation by activating ethylene biosynthesis. Plant Cell 19: 3692–3704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Shi YH, Zhu SW, Mao XZ, Feng JX, Qin YM, et al. (2006) Transcriptome profiling, molecular biological, and physiological studies reveal a major role for ethylene in cotton fiber cell elongation. Plant Cell 18: 651–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gou JY, Wang LJ, Chen SP, Hu WL, Chen XY (2007) Gene expression and metabolite profiles of cotton fiber during cell elongation and secondary cell wall synthesis. Cell Res 17: 422–434. [DOI] [PubMed] [Google Scholar]
- 28. Eshed Y, Zamir D (1995) An introgression line population of Lycopersicon pennellii in the cultivated tomato enables the identification and fine mapping of yield-associated QTL. Genetics 141: 1147–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Liu S, Zhou R, Dong Y, Li P, Jia J (2006) Development, utilization of introgression lines using a synthetic wheat as donor. Theor Appl Genet 112: 1360–1373. [DOI] [PubMed] [Google Scholar]
- 30. Takai T, Nonoue Y, Yamamoto SI, Yamanouchi U, Matsubara K, et al. (2007) Development of chromosome segment substitution lines derived from backcross between indica donor rice cultivar ‘Nona bokra’ and japonica recipient cultivar ‘Koshihikari’. Breeding Sci 57: 257–261. [Google Scholar]
- 31. Wang P, Ding YZ, Lu QX, Guo WZ, Zhang TZ (2008) Development of Gossypium barbadense chromosome segment substitution lines in the genetic standard line TM-1 of Gossypium hirsutum . Chi Sci Bull 53: 1512–1517. [Google Scholar]
- 32. Kohel R, Richmond T, Lewis C (1970) Texas marker-1. Description of a genetic standard for Gossypium hirsutum L. Crop Sci 10: 670–671. [Google Scholar]
- 33. Pan J, Zhang T, Kuai B (1994) Studies on the inheritance of resistance to Verticillium dahliae in cotton. J Nanj Agric Univ 17. [Google Scholar]
- 34. Yang C, Guo W, Li G, Gao F, Lin S, et al. (2008) QTLs mapping for Verticillium wilt resistance at seedling and maturity stages in Gossypium barbadense L. Plant Sci 174: 290–298. [Google Scholar]
- 35. Wang P, Zhu Y, Song X, Cao Z, Ding Y, et al. (2012) Inheritance of long staple fiber quality traits of Gossypium barbadense in G. hirsutum background using CSILs. Theor Appl Genet 124: 1415–1428. [DOI] [PubMed] [Google Scholar]
- 36. Jiang JX, Zhang TZ (2003) Extraction of total RNA in cotton tissues with CTAB-acidic phenolic method. Cott Sci 15: 166–167. [Google Scholar]
- 37. Benjamini Y, Yekutieli D (2001) The control of the false discovery rate in multiple testing under dependency. Ann Stat 1165–1188. [Google Scholar]
- 38. Herrero J, Valencia A, Dopazo J (2001) A hierarchical unsupervised growing neural network for clustering gene expression patterns. Bioinformatics 17 (2) 126–136. [DOI] [PubMed] [Google Scholar]
- 39. Kanehisa M, Araki M, Goto S, Hattori M, Hirakawa M, et al. (2008) KEGG for linking genomes to life and the environment. Nucleic Acids Res 36: D480–D484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Taylor NG, Howells RM, Huttly AK, Vickers K, Turner SR (2003) Interactions among three distinct CesA proteins essential for cellulose synthesis. Proc Natl Acad Sci USA 100: 1450–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Taylor NG, Laurie S, Turner SR (2000) Multiple cellulose synthase catalytic subunits are required for cellulose synthesis in Arabidopsis . Plant Cell 12: 2529–2540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Taylor NG, Scheible WR, Cutler S, Somerville CR, Turner SR (1999) The irregular xylem3 locus of Arabidopsis encodes a cellulose synthase required for secondary cell wall synthesis. Plant Cell 11: 769–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Olsen AN, Ernst HA, Leggio LL, Skriver K (2005) NAC transcription factors: structurally distinct, functionally diverse. Trends Plant Sci 10: 79–87. [DOI] [PubMed] [Google Scholar]
- 44. Zhong R, Lee C, Ye ZH (2010) Functional characterization of poplar wood-associated NAC domain transcription factors. Plant Physiol 152: 1044–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zhong R, Lee C, Zhou J, McCarthy RL, Ye ZH (2008) A battery of transcription factors involved in the regulation of secondary cell wall biosynthesis in Arabidopsis . Plant Cell 20: 2763–2782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Delmer DP, Amor Y (1995) Cellulose biosynthesis. Plant Cell 7: 987–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Delmer DP, Haigler CH (2002) The regulation of metabolic flux to cellulose, a major sink for carbon in plants. Metab Eng 4: 22–28. [DOI] [PubMed] [Google Scholar]
- 48. Appenzeller L, Doblin M, Barreiro R, Wang HY, Niu XM, et al. (2004) Cellulose synthesis in maize: isolation and expression analysis of the cellulose synthase (CesA) gene family. Cellulose 11: 287–299. [Google Scholar]
- 49. Song DL, Shen JH, Li LG (2010) Characterization of cellulose synthase complexes in Populus xylem differentiation. New Phytol 187: 777–790. [DOI] [PubMed] [Google Scholar]
- 50. Amor Y, Haigler CH, Johnson S, Wainscott M, Delmer DP (1995) A membrane-associated form of sucrose synthase and its potential role in synthesis of cellulose and callose in plants. Proc Natl Acad Sci USA 92: 9353–9357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Hinchliffe DJ, Meredith WR, Yeater KM, Kim HJ, Woodward AW, et al. (2010) Near-isogenic cotton germplasm lines that differ in fiber-bundle strength have temporal differences in fiber gene expression patterns as revealed by comparative high-throughput profiling. Theor Appl Genet 120: 1347–1366. [DOI] [PubMed] [Google Scholar]
- 52. Zhong R, Lee C, Ye ZH (2010) Evolutionary conservation of the transcriptional network regulating secondary cell wall biosynthesis. Trends Plant Sci 15: 625–632. [DOI] [PubMed] [Google Scholar]
- 53. Hussey SG, Mizrachi E, Spokevicius AV, Bossinger G, Berger DK, et al. (2011) SND2, a NAC transcription factor gene, regulates genes involved in secondary cell wall development in Arabidopsis fibres and increases fibre cell area in Eucalyptus. BMC Plant Biol 11: 173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bhargava A, Ahad A, Wang S, Mansfield SD, Haughn GW, et al. (2013) The interacting MYB75 and KNAT7 transcription factors modulate secondary cell wall deposition both in stems and seed coat in Arabidopsis . Planta 237: 1199–1211. [DOI] [PubMed] [Google Scholar]
- 55. Zhong R, Lee C, McCarthy RL, Reeves CK, Jones EG, et al. (2011) Transcriptional activation of secondary wall biosynthesis by rice and maize NAC and MYB transcription factors. Plant Cell Physiol 52: 1856–1871. [DOI] [PubMed] [Google Scholar]
- 56. Brown DM, Zeef LA, Ellis J, Goodacre R, Turner SR (2005) Identification of novel genes in Arabidopsis involved in secondary cell wall formation using expression profiling and reverse genetics. Plant Cell 17: 2281–2295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Al-Ghazi Y, Bourot S, Arioli T, Dennis ES, Llewellyn DJ (2009) Transcript profiling during fiber development identifies pathways in secondary metabolism and cell wall structure that may contribute to cotton fiber quality. Plant Cell Physiol 50: 1364–1381. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Heat map of the expression of DEGs between 4 CSILs at 15–25 DPA.
(TIF)
Enrichment analysis of common DEGs at 15DPA and 20DPA in CSIL-35431 and CSIL-31010.
(TIF)
Heat map of DEGs participated in four metabolic pathways from 15 DPA to 25 DPA.
(TIF)
Pathway analysis of genes only down-regulated in CSIL-35368 from 15 DPA to 25 DPA.
(TIF)
Metabolism analysis of DEGs in CSILs at 15 DPA and 25 DPA.
(TIF)
Average fiber quality of 4 CSILs and TM-1.
(XLS)
Primer for quantitative RT-PCR.
(XLS)
Categorization and abundance of tags.
(XLS)
List of common DEGs among CSIL-35431, CSIL-31134 and CSIL-31010.
(XLS)
Enrichment analysis of gene ontologies in CSIL-35368 and CSIL-31010 at 15 DPA and 20 DPA.
(XLS)
Different expression level of 97 MYB transcription factors.
(XLS)
Different expression level of 68 NAC transcription factors.
(XLS)