

Summary
Stem-like glioma cells reside within a perivascular niche and display hallmark radiation resistance. Understanding of the mechanisms underlying these properties will be vital for the development of effective therapies. Here we show that the stem cell marker CD44 promotes cancer stem cell phenotypes and radiation resistance. In a mouse model of glioma, Cd44−/− and Cd44+/− animals showed improved survival compared to controls. The CD44 ligand Osteopontin shared a perivascular expression pattern with CD44 and promoted glioma stem cell-like phenotypes. These effects were mediated via the γ-secretase regulated intracellular domain of CD44, which promoted aggressive glioma growth in vivo and stem cell-like phenotypes via CBP/p300-dependent enhancement of HIF-2α activity. In human glioblastoma multiforme, expression of CD44 correlated with hypoxia-induced gene signatures and poor survival. Together, these data suggest that in the glioma perivascular niche, Osteopontin promotes stem cell-like properties and radiation resistance in adjacent tumor cells via activation of CD44 signaling.
Introduction
Despite intensive treatment with surgery, radiation and chemotherapy, glioblastoma multiforme (GBM) - the highest-grade glioma and most aggressive brain tumor - invariably recurs as an incurable lesion (Huse and Holland, 2010). Recurrence is tightly coupled to increased resistance to radiation and chemotherapy, hallmark features of stem-like glioma cells (Pietras, 2011). Stem-like glioma cells have been enriched experimentally based on expression of stem cell markers such as CD133 (Singh et al., 2003) and CD44 (Anido et al., 2010) or their ability to exclude Hoechst dye in the side population (SP) assay (Bleau et al., 2009), and are characterized by self-renewal ability, stem cell marker expression and resistance to radiation. Like stem cells in the normal brain subventricular zone (SVZ), stem-like glioma cells reside in a perivascular niche (PVN) thought to maintain the stem cell character of adjacent tumor cells (Calabrese et al., 2007). Indeed, we previously showed that nitric oxide from PVN endothelial cells activates Notch signaling in glioma cells, leading to increased stem cell characteristics (Charles et al., 2010). Thus, understanding how niche factors are involved in maintaining aggressive glioma cell phenotypes may help identifying novel potential targets for enhancing the efficacy of cancer therapeutics.
CD44, a glycoprotein transmembrane receptor, is a marker of stem cells from a variety of normal and neoplastic tissues (Zoller, 2011). As a receptor for extracellular matrix components such as hyaluronic acid (HA) and osteopontin (OPN), most described functions for CD44 are as an adhesion molecule. CD44-mediated adhesion is thought to be important, among other things, for stem cell homing to the niche, and indeed both HA and OPN have been described as components of stem cell niches (Haylock and Nilsson, 2005). Beyond adhesion, CD44 itself can act as an intracellular signaling molecule. The C-terminal intracellular domain (CD44ICD) initiates signaling by interacting with proteins like c-Src while membrane-bound (Bourguignon et al., 2001). In addition, CD44 is subject to proteolytic activation similar to that of Notch receptors: extracellular cleavage followed by γ-secretase-dependent release of CD44ICD (Murakami et al., 2003; Nagano et al., 2004; Nagano and Saya, 2004; Okamoto et al., 2001). Once released, CD44ICD localizes to both the cytoplasm and nucleus, however, the mechanisms underlying its signaling as well as its functions remain poorly understood.
In glioma, CD44 is expressed highly in the mesenchymal subtype of GBM (Phillips et al., 2006), and its expression has been used to enrich for stem-like cells (Anido et al., 2010). Here, we found that Cd44−/− and Cd44+/− mice survived longer than Cd44+/+ mice with PDGF-driven gliomas, indicating that CD44 actively contributes to aggressive glioma growth. We found that the CD44 ligand OPN shared a perivascular expression pattern with CD44 in PDGF-driven glioma, and OPN enhanced stemness of glioma cells including increasing the SP and radiation resistance. CD44ICD was sufficient to induce these stem cell characteristics in glioma cells, and potentiated aggressive tumor growth in PDGF-induced gliomas in vivo. We found that CD44ICD enhanced Hypoxia-Inducible Factor 2α (HIF-2α) function in a CBP/p300-dependent manner, and HIF-2α mediated downstream effects of CD44ICD on stemness. In human GBM, CD44 expression correlated with aggressive growth and poor survival in the proneural subtype, and CD44 expression was significantly correlated with hypoxia-induced gene signatures. Taken together, our data identify OPN as a stem cell-promoting extracellular factor in the GBM PVN and demonstrate that CD44 signaling via its intracellular domain promotes aggressive growth and stem cell characteristics by enhancing HIF-2α activity.
Results
Cd44 contributes to aggressive tumor growth in proneural GBM
Proneural GBM is characterized by elevated PDGFR signaling, and can be modeled by overexpressing PDGF in Nestin-expressing stem cells in the mouse brain. Specifically, we employed the RCAS/tv-a system (Holland et al., 1998), and infected Nestin-tv-a (Ntv-a) mice in a wildtype (generating a spectrum of low- to high grade gliomas) or Ink4a/Arf−/− (generating exclusively GBM) background with RCAS-PDGF. The resulting tumors displayed minimal detectable CD44 expression in the tumor bulk and high restricted expression in cells of the PVN (Fig. 1A). We then generated gliomas in Ntv-a mice crossed into a Cd44−/− and Cd44+/− background. The resulting gliomas were largely indistinguishable from Cd44+/+ tumors by hematoxylin/eosin (H/E) staining, but lacked any evidence of CD44 expression (Fig. 1B). Wildtype tumors were scored as either low- or high-grade gliomas based on histological features. Interestingly, in the Ntv-a Ink4a/Arf+/+ background, Cd44+/+ animals developed significantly more high-grade gliomas compared to Cd44+/− and Cd44−/− mice (Fig. 1C). In the Ntv-aInk4a/Arf−/− background, there was a significant survival difference between groups, with Cd44+/+ (n=10) tumors being the most aggressive, Cd44−/− (n=10) the least aggressive and Cd44+/− (n=16) tumors falling in between (Fig. 1D). These data support that CD44 actively contributes to tumor aggressiveness in proneural GBM.
Figure 1.

Cd44 contributes to aggressive growth in a mouse model of proneural GBM. A–B. Hematoxylin/Eosin and CD44 stainings by IHC of murine PDGFB-induced gliomas in a Cd44+/+ (A) and Cd44−/− (B) background. C. PDGFB-induced murine gliomas from Cd44+/+, Cd44+/− and Cd44−/− backgrounds were classified as low- or high-grade based on histological features. Data represent % of tumors within each background that were high- and low-grade. p<0.001. D. Kaplan-Meier plot of survival with tumors for Cd44+/+ (n=10), Cd44+/− (n=16) and Cd44−/− (n=10) Ntv-a mice injected with RCAS-PDGFB virus. *, p<0.05 **, p<0.01, ***, p<0.001 (Log-rank (Mantel-Cox) test).
The CD44 ligand OPN is restricted to the PVN and promotes stemness in proneural GBM
We next stained murine gliomas for CD44 and the CD44 ligand OPN by dual immunofluorescence and found perivascular co-localization of OPN and CD44 with little detectable expression in the tumor bulk (Fig. 2A), suggesting that OPN could bind CD44 in the proneural GBM PVN. Because of the PVN localization of OPN and CD44, and CD44 being a marker of stem-like cells, we asked whether activation of CD44 by OPN could induce the stem-like phenotype. Throughout the study, we chose two functional assays with clear clinical implications - the drug efflux-based SP assay that was previously demonstrated to enrich for stem-like tumor-initiating cells in human and murine glioma (Bleau et al., 2009; Golebiewska et al., 2011), and the colony formation assay following irradiation - in addition to expression of a panel of well-established stem cell markers (Nanog, Sox2, Oct4 and Id1), for a broad and relevant measure of stemness. First, culturing PDGF-induced glioma primary cultures (PIGPCs), human U251 and human T98G glioma cells in the presence of 400 ng/ml OPN for 48 h induced an increase in the SP (Fig. 2B). Conversely, treating PIGPCs with the IM7 anti-CD44 blocking antibody that blocks CD44-ligand interactions, including that of CD44 and OPN (Weiss et al., 2001), resulted in a marked decrease in the SP (Fig. 2C), suggesting that CD44 function is necessary for the SP phenotype. CD44 blocking similarly reduced the SP of primary human proneural GBM cells and primary murine GBM neurosphere cultures (Fig. 2C). Of note, Cd44 levels were significantly higher in sorted SP cells as compared to MP cells (Fig. S1A). Second, the stem cell markers Nanog, Sox2, Oct4 and Id1 were all upregulated in OPN-treated PIGPCs as well as primary human GBM cells, as shown by quantitative real-time PCR (qPCR) (Fig. 2D–E). Finally, PIGPCs treated with OPN formed more colonies than control cells in a colony formation assay following a single dose of 2 Gy irradiation (Fig. 2F). Together, these data suggest that OPN acts as a PVN factor to induce the stem-like state of PVN GBM cells. We next tested the tumor-initiating capacity of PIGPCs pre-treated or not with OPN in vitro prior to intracranial injection in recipient mice, and found no significant difference between groups in tumor formation or survival (Fig. S1B). These data are in line with recent advances separating stemness from tumor-initiating ability specifically in GBM (Barrett et al., 2012).
Figure 2.

OPN is expressed in the PVN and induces a stem-like phenotype in cultured glioma cells. A. Dual immunofluorescence of CD44 (green) and OPN (red) on DAPI (blue)-stained PDGFB-induced murine glioma. B. SP analysis of PIGPCs, U251 and T98G cells cultured with or without 400 ng/ml OPN as indicated for 48 h. Inset shows FTC-treated control sample. Right graphs show relative SP from at least three separate experiments. C. SP analysis of PIGPCs, primary human GBM cultures and primary murine neurosphere cultures treated with IgG control or anti-CD44 blocking antibodies for 72 h prior to analysis. Right graphs show relative SP from at least three separate experiments. D. qPCR data for relative mRNA expression of Nanog, Sox2, Oct4, and Id1 in PIGPCs treated with or without 400 ng/ml OPN for 24 h. Data represent average values of at least 3 independent experiments. Error bars represent SEM. *, p<0.05 **, p<0.01, Students t-test. E. qPCR data for relative mRNA expression of NANOG, SOX2, OCT4, and ID1 in primary human GBM cultures treated with or without 400 ng/ml OPN for 24 h. Data represent average values of at least 3 independent experiments. Error bars represent SEM. *, p<0.05, Students t-test. F. Fraction survival of PIGPCs treated with or without OPN at day 7 in a colony formation assay following irradiation (IR) compared to unirradiated controls (C). *, p<0.05, ns, not significant, Students t-test. G. qPCR data for relative mRNA expression of Nanog, Sox2, Oct4, and Id1 in PIGPCs treated with or without OPN for 24 h alone or in the presence of 10 μM MK-003 (GSI) as indicated. Data represent one representative experiment out of at least 3 independent experiments. Error bars represent SEM. *, p<0.05, ns, not significant, Students t-test. H. Western blot analysis of CD44 and Tubulin in U251 cells treated as indicated with 1000 ng/ml OPN or 200 μM TPA (positive control for CD44ICD) or 200 μM TPA with 10 μM MK-003 (GSI) (negative control for CD44ICD) as indicated. CD44EXT is CD44 cleaved extracellularly but still membrane bound. Right graph shows quantification of CD44ICD signal across three independent experiments. *, p<0.05, Students t-test. See also Fig. S1 and Fig. S7.
CD44 can be activated by proteolytic cleavage in a two-step process dependent on γ-secretase, resulting in the release of a CD44 intracellular domain (CD44ICD). Because of the well-documented inhibitory effects of γ-secretase inhibition on brain tumor stemness (at least partially mediated through inhibition of Notch) (Fan et al., 2006), we speculated that CD44ICD release may be involved downstream of OPN. To test this hypothesis, we treated PIGPCs with OPN in the presence of a γ-secretase-inhibitor (GSI; MK-003) or control, and measured expression of our panel of stem cell markers. Notably, γ-secretase-inhibition was sufficient to inhibit the effect of OPN on either marker in the panel (Fig. 2G), suggesting that γ-secretase-dependent release of CD44ICD could indeed be involved downstream of OPN-CD44. To test whether cleavage of CD44 is induced by OPN, we blotted for CD44 using an antibody recognizing the C-terminal end of the protein. Because this antibody recognizes human CD44, we utilized human U251 glioma cells. TPA, an activator of PKC and the best-described inducer of CD44ICD generation, was used to generate a positive control sample, and TPA-treatment in combination with γ-secretase inhibitor was used as a negative control. As expected, TPA induced a CD44 band slightly smaller than 15 kDa corresponding to CD44ICD (Fig. 2H). TPA in combination with γ-secretase inhibitor diminished this band, and instead resulted in accumulation of a band slightly larger than 15 kDa corresponding to CD44EXT (CD44 after extracellular cleavage, before CD44ICD release). OPN-treatment of U251 cells also resulted in increased CD44ICD release as compared to untreated control after 24 h (Fig. 2H - long exposure). Quantification of band intensities across several experiments revealed on average a 2-fold significant induction of CD44ICD by OPN treatment (Fig. 2H).
CD44ICD promotes aggressive growth in PDGF-induced GBM
An intracellular role for CD44 in glioma was supported by staining murine gliomas with an antibody detecting C-terminal CD44. Stainings revealed cells with CD44 expression in the nucleus (left panel, black arrow), membrane (middle panel, white arrow) and cytoplasm (right panel, gray arrow) (Fig. 3A), indicating that CD44 expression reflects the potential to be spontaneously activated in vivo. To study effects of CD44ICD release more carefully, we created a fusion protein with EGFP fused to the human CD44ICD N-terminus (EGFP-CD44ICD; Fig. 3B). When expressed in PIGPCs, EGFP-CD44ICD localized throughout the cell in both the cytoplasm and the nucleus (Fig. 3C). We inserted the EGFP-CD44ICD fusion gene into the RCAS vector and injected N-tva Ink4a/Arf−/− mice with RCAS-PDGF in combination with either RCAS-EGFP-CD44ICD or RCAS-GFP to study effects of CD44ICD in glioma in vivo. Tumors formed after co-injection of PDGFB with EGFP-CD44ICD contained a larger proportion of cells staining positive for intracellular/nuclear CD44 as detected by IHC with an antibody recognizing the C-terminal end and quantified using ImageJ (Fig. 3D, Fig. S2). Importantly, PDGFB/EGFP-CD44ICD-induced tumors were more aggressive as mice injected with PDGFB/GFP (n=8) survived significantly longer than those injected with PDGFB/EGFP-CD44ICD (n=6) (Fig. 3E). These data indicate that CD44ICD alone can contribute to aggressive tumor growth in glioma. Together with the finding that mice with Cd44−/− GBM survived longer than those with Cd44+/+ GBM (Fig. 1D), these data further establish CD44 as an active contributor to aggressive growth in PDGF-induced GBM.
Figure 3.

The CD44 intracellular domain (CD44ICD) promotes aggressive growth in a mouse model of proneural GBM. A. CD44 C-terminal staining by IHC on murine PDGFB-induced gliomas. Left panel shows nuclear staining (black arrow), middle panel shows membrane staining (white arrow) and right panel shows cytoplasmic staining (gray arrow). B. Schematic overview of the CD44 gene and the EGFPCD44ICD fusion construct. SP, signal peptide. ECD, extracellular domain. TM, transmembrane domain. ICD, intracellular domain. C. Expression of EGFP-CD44ICD in PIGPCs visualized by EGFP expression. Lower panels show nuclei by DAPI staining. D. CD44 C-terminal staining by IHC on murine gliomas induced by PDGFB in combination with RCAS-GFP or RCAS-CD44ICD as indicated. Graph shows quantification of CD44 staining, average values for all tumors. E. Kaplan-Meier plot of survival for mice injected with RCAS-PDGFB in combination with RCAS-GFP (n=8) or RCAS-CD44ICD (n=6) as indicated. p=0.02 (Log-rank (Mantel-Cox) test). See also Fig. S2.
CD44ICD promotes glioma stemness
To test whether CD44ICD alone could mimic the phenotype induced by OPN, we expressed EGFP-CD44ICD or control in a TetOn doxycycline-inducible system where CD44ICD expression could be rapidly controlled in T98G glioma cells, and subjected them to the same analyses to quantify stemness as in the OPN set of experiments. First, EGFP-CD44ICD expression resulted in a 4-fold increase in the SP as compared to EGFP-expressing controls (Fig. 4A), indicating that CD44ICD is sufficient to induce the SP phenotype in a subset of glioma cells. CD44ICD expression similarly increased the SP of PIGPCs (Fig. 4A). To test whether effects of CD44 blockade could be rescued by CD44ICD, we expressed CD44ICD stably in PIGPCs. Importantly, PIGPCs expressing EGFP-CD44ICD no longer underwent reduction of the size of SP by CD44 blocking antibodies as shown earlier (Fig. 4B), strongly arguing that CD44 blockade acts by inhibiting the release of CD44ICD. Second, we tested whether expression of CD44ICD could induce expression of our panel of stem cell markers. Indeed, Nanog, Sox2, Oct4 and Id1 expression was increased by CD44ICD expression in PIGPCs, primary human GBM cells and human U251 glioma cells (Fig. 4C–E). NANOG and SOX2 expression was significantly induced by EGFP-CD44ICD in T98G cells, whereas expression of OCT4 and ID1 was unaffected in this cell line (Fig. 4F). Finally, we tested the colony-forming ability of cells expressing EGFP-CD44ICD after a single dose of irradiation. After 1, 2 or 4 Gy irradiation, EGFP-CD44ICD-expressing T98G cells formed more colonies than controls (Fig. 4G), thus again mimicking the effect of OPN. Together, these data indicate that CD44ICD can drive parts of the stem cell phenotype in the PVN of PDGF-induced GBM.
Figure 4.

CD44ICD promotes a stem-like phenotype in glioma cells. A. SP analysis of T98G cells and PIGPCs expressing EGFP or EGFP-CD44ICD. Inset shows FTC-treated control sample. Right graphs show relative SP from at least three separate experiments. B. SP analysis of PIGPCs expressing EGFP or EGFP-CD44ICD treated with control or CD44 blocking antibodies for 72 h prior to analysis. Right graph shows relative SP from at least three separate experiments. C. qPCR data for relative mRNA expression of Nanog, Sox2, Oct4 and Id1 in PIGPCs expressing EGFP control or EGFP-CD44ICD. Data represent average values of at least 3 independent experiments. Error bars represent SEM. *, p<0.05, Students t-test. D. qPCR data for relative mRNA expression of NANOG, SOX2, OCT4 and ID1 in primary human GBM control cells or EGFP-CD44ICD-expressing cells. Data represent one representative experiment out of at least 3 independent experiments. Error bars represent SEM. *, p<0.05, Students t-test. E. qPCR data for relative mRNA expression of NANOG, SOX2, OCT4 and ID1 in U251 control or EGFP-CD44ICD-expressing cells. Data represent one representative experiment out of at least 3 independent experiments. Error bars represent SEM. *, p<0.05, Students t-test. F. qPCR data for relative mRNA expression of NANOG, SOX2, OCT4 and ID1 in T98G control or EGFP-CD44ICD-expressing cells. Data represent average values of at least 3 independent experiments. Error bars represent SEM. *, p<0.05, ns, not significant, Students t-test. G. Fraction survival of T98G control or EGFP-CD44ICD-expressing cells in a colony formation assay at day 7 following irradiation.
CD44ICD acts by enhancing Hypoxia-Inducible Factor 2α (HIF-2α) activity
Saya and colleagues demonstrated that CD44ICD could potentiate transactivation mediated by the transcriptional co-activator CBP/p300 (Okamoto et al., 2001). While CBP/p300 can potentiate transcription by a wide range of transcription factors, one such family of CBP/p300-dependent transcription factors known to regulate glioma stemness are the hypoxia-inducible factors (HIFs) (Li et al., 2009; Pietras et al., 2010; Semenza, 2007). We speculated that CD44ICD promoted stemness by enhancing HIF-dependent transcription, and transfected T98G cells expressing EGFP or EGFP-CD44ICD with a hypoxia responsive element (HRE)-luciferase reporter, in the presence of control or the hypoxia-mimetic 2,2-dipyridyl (DIP) to stabilize HIFs. DIP-treated EGFP-expressing cells displayed on average a 10-fold induction of HRE-luciferase as compared to control cells (Fig. 5A). Strikingly, EGFP-CD44ICD-expressing cells treated with DIP indeed had a 2–3 fold higher induction of HRE-luciferase as compared to DIP-treated control cells (Fig. 5A), thus supporting the hypothesis that CD44ICD potentiated HIF activation. Similarly, CD44ICD expression significantly enhanced HRE-activation even at normoxic conditions in primary human proneural GBM cells and U251 glioma cells (Fig. 5B–C). DIP stabilizes both HIF-1α and HIF-2α, so to elucidate whether this potentiation was dependent on HIF-1α, HIF-2α, or both, we overexpressed oxygen-insensitive versions of the HIFs (Yan et al., 2007) together with EGFP or EGFP-CD44ICD. In EGFP-expressing control cells, HIF-1α and HIF-2α both induced the HRE-luciferase reporter to a similar extent (Fig. 5D). Intriguingly however, CD44ICD potentiated induction of HRE-luciferase only together with HIF-2α, with no potentiation of HIF-1α-dependent HRE-activation in T98G cells (Fig. 5D). In U251 cells, in contrast, CD44ICD enhanced HRE activation both by HIF-1α and HIF-2α (Fig. 5E). However, knocking down HIF1A (Fig. S4A) in U251 cells by siRNA had no effect on the potentiated HRE activation by CD44ICD, while knockdown of HIF2A entirely diminished the difference between EGFP and CD44ICD-expressing cells (Fig. 5F), indicating that even in these cells, HIF2A was the primary HIF involved in CD44ICD signaling. Finally, genes of the stemness panel were induced by hypoxia in a HIF2A-dependent manner in U251 cells (Fig. S3). To test whether HIF-2α mediated the stemness-promoting effect of CD44ICD, we treated T98G TetOn-CD44ICD cells with control or two independent siRNAs targeting HIF2A (Fig. S4B), then cultured them with either DIP (to stabilize HIFs) or Dox (to induce CD44ICD expression). DIP treatment induced mRNA expression of the CD44ICD-induced stem cell markers SOX2 and NANOG (Fig. 5G). This induction was mediated by HIF-2α, as DIP did not induce these genes in HIF2A-knockdown cells (Fig. 5G). Importantly, SOX2- and NANOG-induction by CD44ICD (Fig. 4F) was also HIF-2α-dependent, as knockdown of HIF2A abrogated any effect of CD44ICD on these genes (Fig. 5G). Expression of the stem cell markers OCT4 and ID1, which were not induced by CD44ICD in T98G cells (Fig. 4F), was unaffected both by DIP- and Dox-treatment in these cells (Fig. 5G). We further tested expression of the well-described hypoxia-induced gene VEGF. As expected, VEGF was induced by DIP. However, this induction was not mediated by HIF-2α, as HIF2A knockdown did not affect VEGF-induction by DIP (Fig. 5H). In agreement with CD44ICD acting through HIF-2α, CD44ICD-induction did not affect VEGF expression (Fig. 5H).
Figure 5.

CD44ICD promotes stemness by enhancing HIF-2α activity. A. Relative HRE-luciferase normalized to Renilla for T98G cells expressing EGFP control or EGFP-CD44ICD, with or without 200 μM DIP for 24 h. Data represent mean values from at least three independent experiments. Error bars represent SEM. ***, p<0.001, ns, not significant, Students t-test. B. Relative HRE-luciferase normalized to Renilla for U251 cells expressing EGFP control or EGFP-CD44ICD, with or without 200 μM DIP for 24 h. Data represent one representative experiment out of at least three independent experiments. Error bars represent SEM. *, p<0.05, ***, p<0.001, Students t-test C. Relative HRE-luciferase normalized to Renilla for primary human GBM cells expressing EGFP control or EGFP-CD44ICD, with or without 200 μM DIP for 24 h. Data represent one representative experiment out of at least three independent experiments. Error bars represent SEM. *, p<0.05, ns, not significant, Students t-test. D-E. Relative HRE-luciferase normalized to Renilla for T98G (D) and U251 (E) cells expressing EGFP control or EGFP-CD44ICD co-expressed with pcDNA (empty vector), HIF-1α or HIF-2α as indicated. Data represent one representative experiment out of at least three separate experiments. Error bars represent SEM. *, p<0.05, ***, p<0.001, ns, not significant, Students t-test. F. Relative HRE-luciferase normalized to Renilla for U251 cells expressing EGFP or CD44ICD as indicated and transfected with control or siRNAs targeting HIF1A or HIF2A as indicated. *, p<0.05, ***, p<0.001, ns, not significant, Students t-test. G. qPCR data for relative mRNA expression of NANOG, SOX2, OCT4, and ID1 in T98G TetOn-CD44ICD cells treated with DIP (hypoxia) or Dox (CD44ICD) and transfected with control or HIF2A-targeting siRNAs. Data represent average values of at least 3 independent experiments. Error bars represent SEM. *, p<0.05, ns, not significant, Students t-test. H. qPCR data for relative mRNA expression of VEGF in T98G TetOn-CD44ICD cells treated with DIP (hypoxia) or Dox (CD44ICD) and transfected with control or HIF2A-targeting siRNAs. Data represent average values of at least 3 independent experiments. Error bars represent SEM. ns, not significant. I. SP analysis of T98G TetOn-CD44ICD cells treated with or without Dox (CD44ICD) and transfected with control or HIF2A-targeting siRNAs. Inset shows FTC-treated control sample. Graph shows relative SP from at least three separate experiments. See also Fig. S3 and S4.
We next asked whether the induction of SP by CD44ICD was also dependent on HIF-2α, and again treated cells transfected with siRNAs targeting HIF2A or control, with or without Dox. SP analysis revealed that while cells expressing non-targeting control siRNA induced the SP upon EGFP-CD44ICD expression, cells transfected with two independent siRNAs targeting HIF2A did not (Fig. 5I). Taken together, these data establish CD44ICD as an enhancer of HIF-2α function, and demonstrate that the stemness-promoting effects of CD44ICD are mediated by HIF-2α in these cells.
CD44ICD potentiates HIF-2α in a CBP/p300-dependent manner
To investigate the mechanism by which CD44ICD enhanced HIF-2α, we treated T98G TetOn-CD44ICD cells with DIP (to stabilize HIFs) and/or Dox (to induce CD44ICD expression) and measured HIF2A mRNA levels. Neither DIP nor Dox resulted in significantly altered HIF2A transcript levels (Fig. 6A and Fig. S5). On the other hand, T98G TetOn-CD44ICD cells expressed higher HIF-2α protein levels when treated with both DIP and Dox together as compared to DIP alone (Fig. 6B), suggesting that CD44ICD helps stabilize HIF-2α by inhibiting degradation rather than by increased transcription. As CD44ICD was previously demonstrated to potentiate CBP/p300-mediated transcription, we speculated that CD44ICD potentiation of HIF-2α was dependent on CBP and/or p300. To test this hypothesis, we transfected siRNAs targeting control, CBP or p300 (Fig. S4C–D), and expressed EGFP or CD44ICD in T98G cells and measured HRE-luciferase activity in the presence of DIP. Interestingly, in EGFP-expressing cells knockdown of CBP had no effect on HRE-luciferase activity, whereas p300 knockdown did significantly decrease HRE-luciferase signal (Fig. 6C). Strikingly however, both CBP and p300 siRNA significantly blunted the increase in HRE-luciferase activity induced by CD44ICD (Fig. 6C). Thus, CD44ICD potentiates HIF-2α signaling in a CBP/p300-dependent manner.
Figure 6.
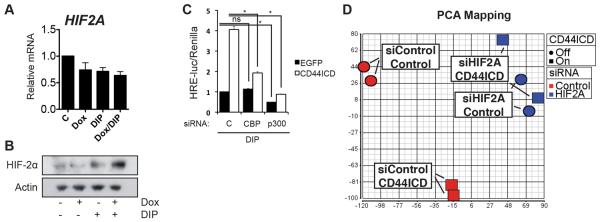
CD44ICD potentiates HIF-2α in a CBP/p300-dependent manner and HIF-2α accounts for global transcriptional effects of CD44ICD. A. QPCR analysis of HIF2A in T98G TetOn-CD44ICD cells treated with DIP (hypoxia) or Dox (CD44ICD) as indicated. Data represent average values of at least 3 independent experiments. Error bars represent SEM. B. Western blot analysis of HIF-2α and Actin in T98G TetOn-CD44ICD cells treated with DIP (hypoxia) or Dox (CD44ICD) as indicated. C. Relative HRE-luciferase normalized to Renilla for T98G cells expressing EGFP control or EGFP-CD44ICD, transfected with control or siRNAs targeting CBP or p300 as indicated. Data represent one representative experiment out of at least three separate experiments. Error bars represent SEM. *, p<0.05 ns, not significant, Students t-test. D. PCA mapping of T98G TetOn-CD44ICD cells treated with or without Dox and transfected with siRNA targeting HIF2A or control as indicated. Circles represent controls and squares represent Dox-treated samples. Red color indicates siControl-transfected, blue color indicates siHIF2A-transfected samples. See also Fig. S5.
We next asked to what extent HIF-2α mediated CD44ICD-induced gene expression changes globally, and performed microarray analysis on T98G TetOn-CD44ICD cells treated with or without Dox and transfected with either non-targeting control or HIF2A siRNA. Strikingly, principal component analysis (PCA) mapping showed that while control siRNA-transfected samples grouped separately based on CD44ICD expression, there was essentially no difference between groups in the HIF2A siRNA-transfected cells (Fig. 6D). These data suggest that the vast majority of CD44ICD-induced gene expression changes are mediated by HIF-2α.
CD44 is differentially expressed across GBM subtypes and correlates with short survival in proneural GBM
Finally, we stained a series of human GBM samples for CD44 protein expression by immunohistochemistry (IHC) and divided them according to their expression profile into the proneural, mesenchymal or classical GBM subtype (Phillips et al., 2006; Verhaak et al., 2010). Whereas mesenchymal and classical tumors expressed CD44 at high levels throughout the tumor, CD44 expression in proneural tumors was restricted to the PVN (Fig. 7A–C), similarly to CD44 expression in our murine PDGF-induced gliomas (Fig. 1A). We queried the dataset on human GBM from The Cancer Genome Atlas (TCGA) (2008) for CD44 gene expression, and found that patients with low levels of CD44 survived significantly longer than those with normal levels (Fig. 7D). Interestingly, although the difference in survival was modest, the observed correlation between CD44 expression and aggressive glioma growth held true only in the proneural group of GBM, with no survival difference between groups in other subtypes (Fig. 7D). We tested the hypothesis that CD44ICD expression shifted proneural GBM cells towards a mesenchymal phenotype. While some hallmark genes of the mesenchymal subtype like Stat3 were indeed induced by CD44ICD, others were not (Fig. S6), suggesting that CD44ICD is not sufficient to drive the mesenchymal phenotype.
Figure 7.

CD44 is differentially expressed across GBM subtypes and correlates with poor survival in proneural tumors. A-C. CD44 staining by IHC on human GBMs subdivided by their molecular subtype as indicated. D. Kaplan-Meier plots of human GBM specimens from the TCGA dataset (487 patients) based on low vs. normal gene expression of CD44, grouped by GBM subtype (128 proneural; 149 mesenchymal; 79 neural; 131 classical). P-values calculated by Log-rank (Mantel-Cox) test. E. GSEAs carried out on a CD44-correlated ranked gene list produced from the GBM TCGA dataset. Enrichment was observed for the indicated hypoxia-related gene sets (Winter_hypoxia_metagene NES=2.63, FDR q=0.000; Elvidge_hypoxia_up NES=2.54, FDR q=0.000; Harris_hypoxia NES=2.53, FDR q=0.000; Manalo_hypoxia_up NES=2.36, FDR q=0.000). See also Fig. S6.
To investigate the relation between CD44 and hypoxia-induced gene expression in GBM in vivo, we ranked genes in the TCGA dataset on GBM according to their correlation with CD44, then performed gene set enrichment analyses (GSEA) specifically for gene sets associated with hypoxia. CD44-correlated genes showed highly significant enrichment for four different published gene sets upregulated by hypoxia in independent systems (Fig. 7E), establishing a link between CD44 expression and hypoxia-induced gene expression in glioma consistent with our finding that CD44 signaling activates gene expression through HIF-2α in human GBM in vivo.
Discussion
Although CD44 was identified as a marker of mesenchymal GBM (Phillips et al., 2006), previous work paradoxically established that its expression can be used to isolate presumably rare stem-like glioma cells (Anido et al., 2010). Here, we confirmed high CD44 protein expression in mesenchymal GBM, found high expression throughout the tumor also in the classical subtype, but importantly, found restricted high expression in perivascular cells in proneural GBM. These findings suggest that the apparent paradox of CD44 can be resolved by subdividing gliomas into molecular subtypes, and open for the interpretation that CD44 expression highlight stem-like cells from proneural GBM while serving as a general marker of tumor cells in other subtypes. Bulk cells in proneural GBM have an oligodendrocytic phenotype, and CD44 does not seem to be generally expressed in this cell type. Stem-like tumor cells, on the other hand, have a more mesenchymal phenotype including expression of CD44, and reside primarily in the PVN. In other GBM subtypes, the tumor bulk consists of mesenchymal/astrocytic cells that naturally express CD44 (in fact, we previously identified CD44 as a marker of tumor-associated stromal astrocytes in proneural tumors (Katz et al., 2012)). In light of these expression patterns, it is intriguing that CD44 expression correlated with poor prognosis in GBM only of the proneural subtype. The correlation between aggressive tumor growth and CD44 expression appears causal in this tumor type, as Cd44−/− and Cd44+/− mice survived significantly longer than Cd44+/+ mice in a mouse model of proneural GBM, and as CD44ICD overexpression promoted aggressive growth in the same model.
The perivascular region is recognized as a niche for stem-like tumor cells in several brain tumors; cells surviving irradiation in medulloblastoma are almost exclusively perivascular, and stem cell markers appear enriched in the PVN (Calabrese et al., 2007; Hambardzumyan et al., 2008). It is expected that niche-derived factors actively maintain the stem-like state of adjacent tumor cells. For example, we have previously shown that endothelial cell-derived nitric oxide can activate Notch signaling and promote stemness in glioma cells (Charles et al., 2010). Here, we identify OPN as a factor expressed exclusively in the PVN in proneural GBM. OPN stimulated the SP, stem cell marker expression and radiation resistance in glioma cells, thus, OPN appears to be one niche-derived factor promoting the stem-like phenotype of PVN glioma cells. We recently identified SPP1 (encoding OPN) as the most upregulated gene in tumor-associated stromal astrocytes of the PVN as compared to normal brain astrocytes in a mouse model of proneural GBM (Katz et al., 2012). It is thus possible that stromal astrocytes in the PVN indirectly confer radiation-resistance and stemness on glioma cells through secretion of OPN, and may conceivably serve as non-transformed therapeutic targets. Tumor stemness is frequently viewed as a package comprising stem cell marker expression, drug/radiation resistance as well as increased tumorigenicity in vivo, factors that have indeed been shown to be co-regulated by niche factors like NO in the glioma PVN (Charles et al., 2010). While our study revealed increased stem cell marker expression, SP phenotype and radiation resistance, we found no increase in tumorigenicity of cells transiently pre-treated with OPN. These findings are in line with recent advances separating self-renewal and stemness from tumorigenicity specifically in glioma (Barrett et al., 2012), and point to the need of separate consideration of all these factors.
Release of the CD44ICD by γ-secretase in tumor cells is well described (Nagano and Saya, 2004; Okamoto et al., 2001), however, its consequences thus far are incompletely understood. OPN expression has been shown to correlate with intracellular CD44 in tumor material in vivo (Hsu et al., 2010), and we present data here demonstrating that OPN induced CD44ICD generation in human glioma cells. Furthermore, effects on some stem cell marker expression by OPN were abolished by γ-secretase inhibition, strongly supporting that OPN effects can be mediated by CD44ICD. Previous in vitro experiments have hinted at an active role for CD44ICD in tumor biology. For instance, CD44ICD was shown to promote neoplastic transformation of rat fibroblasts (Pelletier et al., 2006), and CD44ICD sustains proliferation of thyroid cancer cells (De Falco et al., 2012). Here, we show that expression of CD44ICD together with PDGF in a mouse model of proneural GBM resulted in more aggressive tumor growth and shorter survival as compared to control mice, thus identifying CD44 cleavage as a potential therapeutic target in human cancer. These data, to our knowledge, are the first demonstrating tumor-promoting effects of CD44ICD in an in vivo model of any tumor type.
CD44 was recently shown to increase stemness in colon cancer cells in a process dependent on the C-terminal end of CD44 (Su et al., 2011). We show here that CD44ICD is sufficient to induce some stemness and radiation resistance in a subset of glioma cells on its own. Mechanisms of CD44ICD signaling up to this point have been incompletely understood. Saya and colleagues described that transcription dependent on CBP/p300 was enhanced by CD44ICD expression (Okamoto et al., 2001). Our finding that CD44ICD enhances HIF-dependent transcription is consistent with a general enhancement of CBP/p300-dependent transcription, although further studies are needed to understand how specificity for either HIF is achieved. Recently, a CD44ICD responsive element (CIRE) was identified in the promoters of some genes activated by CD44ICD overexpression (Miletti-Gonzalez et al., 2012). Interestingly, such sequences were frequently found in genes also containing HREs. Experimental and TCGA data presented here support that CD44ICD induces genes with HREs in GBM, however, we demonstrate that CD44ICD activation of gene expression is dependent on HIF-2α, as HIF2A knockdown diminished any effects of CD44ICD on the specific target genes investigated here. It appears unlikely that binding of CD44ICD to CIREs is involved in this process, as CD44ICD could enhance HIF-2α activation of HREs in a reporter construct lacking any flanking CIREs. Furthermore, we did not find any CIREs in the promoter region of SOX2 (data not shown). Differences between CD44ICD-induced genes between cell lines, where applicable, were largely reflected in differences between hypoxia-induced genes, further supporting that CD44ICD signals mainly through this pathway (Fig. S7). Importantly, microarray analysis indicated that the vast majority of CD44ICD-mediated gene expression changes globally in our system were dependent on HIF2A, as there was no apparent difference between CD44ICD-expressing and control cells in PCA mapping when HIF2A was knocked down by siRNA. It should be noted, however, that at least under some experimental conditions, we found potentiation of HRE activation also by exogenous HIF-1α, suggesting that CD44ICD under certain circumstances may signal through both HIFs.
Our study supports recent findings indicating a crucial role for HIF-2α in maintaining glioma stemness (Bar et al., 2010; Li et al., 2009; Pietras et al., 2010; Soeda et al., 2009). Hypoxic conditions were first shown to induce dedifferentiation of tumor cells in neuroblastoma (Jogi et al., 2002). Similar findings have since supported this idea in other tumor forms and often implicated HIF-2α as a driver of this phenomenon (Keith and Simon, 2007; Mathieu et al., 2011; Pietras et al., 2010). Mechanisms involved include direct transcriptional activation or potentiation of activity of pluripotency factors by HIF-2α (Covello et al., 2006; Gordan et al., 2008). HIF-2α is further expressed at high levels in the PVN of brain and other neural tumors (Li et al., 2009; Pietras et al., 2008), a counter-intuitive finding supported by the seemingly aberrant stabilization of HIF-2α under normoxic conditions specifically in glioma stem-like cells (Li et al., 2009).
Finally, our findings open for the possibility that CD44 via its ICD plays a more active role than previously anticipated in stem cell biology in the wide variety of tissues and cancers where CD44 is regarded as a stem cell marker. Particularly, those cases where γ-secretase inhibition or HIF expression has profound effects on differentiation states, warrant more careful investigation into relative contributions of the Notch and CD44 pathways.
Experimental procedures
Generation of murine gliomas using RCAS/tv-a
Gliomas were induced in Nestin-tv-a (Ntv-a) mice by injecting RCAS-PDGFB-transfected DF-1 cells (ATCC) intracranially in the neonatal brain as previously described (Holland et al., 1998). Cd44+/− and Cd44−/− mice were generated by crossing Ntv-a Cd44+/+ mice with Cd44−/− mice (Jackson labs) and back-crossing at least 7 generations. Mice were monitored daily and euthanized upon symptoms of glioma (lethargy, enlarged skull, tilted head). All procedures were approved by the Institutional Animal Care and Use Commitee at Memorial Sloan-Kettering Cancer Center (protocol# 00-11-189).
Quantitative real-time PCR
RNA was isolated using QIAzol and the RNeasy mini kit (Qiagen), and cDNA synthesized using the Superscript III First strand synthesis kit (Invitrogen) according to manufacturers' recommendations. qPCR was performed using a 7900HT Fast Real Time PCR system and SYBR green (both Applied Biosystems), using primers listed in Supplemental Experimental Procedures. Relative gene expression was normalized to the expression of two housekeeping genes (UBC and SDHA) using the comparative Ct method (Vandesompele et al., 2002).
Side population, luciferase and colony formation assays
For SP, cells were re-suspended at 1×10^6 cells/ml then incubated at 37°C for 30' with or without Fumitremorgin C (FTC) (Sigma). Cells were then incubated for a further 90' with 5 μg/ml Hoechst 33342 with periodic shaking. Hoechst 33342 was excited at 407 nm using a trigon violet laser, and dual wavelength detection was performed using 450/40 (Hoechst 33342-Blue) and 695/40 (Hoechst 33342-Red) filters. For luciferase reporter assay, cells were co-transfected with HRE-luc ((Emerling et al., 2008); Addgene #26731) and pCMV-renilla (Promega), then analyzed for luciferase using the Dual-Luciferase Reporter Assay System (Promega) on a Veritas luminometer (Turner Biosystems). For colony formation assay, cells were plated at clonal density in 6-well plates, then irradiated. Colonies were fixed and manually counted at day 10 post-irradiation.
Plasmids
Human CD44ICD was amplified from pBabe-CD44S (Godar et al., 2008) (Addgene #19127), then subcloned into pEGFP-C1 (Clontech). The resulting fusion gene was further subcloned into RCAS-Y. The TetOn doxycycline-inducible cells were generated using the Retro-X TetOn system (Clontech). The HRE-luciferase plasmid was previously described (Emerling et al., 2008) (Addgene #26731). Oxygen-insensitive mutants of HIF1A and HIF2A (Yan et al., 2007) were obtained from Addgene (#18955 and #18956, respectively).
Affymetrix and TCGA data analysis
The TCGA dataset on GBM (540 patients) was analyzed using the cBio portal (http://cbioportal.org) with annotated TCGA data. CD44 and SPP1 expression one standard deviation lower than average was considered low. GSEA was performed using software and datasets available at http://www.broadinstitute.org/gsea/index.jsp. Hypoxia-related gene sets were previously described (Elvidge et al., 2006; Harris, 2002; Manalo et al., 2005; Winter et al., 2007). Affymetrix expression arrays (U133A) were performed by the MSKCC Genomics core facility, and data analyzed using Partek Genomics Suite. Data are available at Array Express (accession number E-MEXP-3914).
Statistical analysis
Statistical significance was calculated by indicated unpaired and two-sided tests using GraphPad/Prism.
Supplementary Material
Highlights
-
-
CD44 promotes aggressive glioma growth and cancer stem cell phenotypes
-
-
Osteopontin-CD44 signaling induces stemness and radiation resistance
-
-
The CD44 intracellular domain (CD44ICD) is sufficient to promote glioma growth
-
-
CD44ICD regulates HIF-2α via CBP/p300 to drive stemness and radiation resistance
Acknowledgements
We thank Madhu Menon at the MSKCC Flow Cytometry Core Facility, RARC staff, the Experimental Histopathology core facility at FHCRC, Jim Finney, Deby Kumasaka and Quanchao Zhang for skillful technical assistance, and members of the Holland lab for helpful discussions. We thank Dr. William G. Kaelin, Jr. (Dana Farber Cancer Institute), Dr. Navdeep Chandel (Northwestern University) and Dr. Robert Weinberg (Whitehead Institute for Biomedical Research) for plasmids. This work was supported by NIH grants U01 CA141502, U54 CA143798, U54CA163167 and RO1 CA94842. AP was supported by fellowships from the Swedish Research Council, the Tegger Foundation and the Swedish Childhood Cancer Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- The Cancer Genome Atlas Research Network Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–1068. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anido J, Saez-Borderias A, Gonzalez-Junca A, Rodon L, Folch G, Carmona MA, Prieto-Sanchez RM, Barba I, Martinez-Saez E, Prudkin L, et al. TGF-beta Receptor Inhibitors Target the CD44(high)/Id1(high) Glioma-Initiating Cell Population in Human Glioblastoma. Cancer Cell. 2010;18:655–668. doi: 10.1016/j.ccr.2010.10.023. [DOI] [PubMed] [Google Scholar]
- Bar EE, Lin A, Mahairaki V, Matsui W, Eberhart CG. Hypoxia increases the expression of stem-cell markers and promotes clonogenicity in glioblastoma neurospheres. Am J Pathol. 2010;177:1491–1502. doi: 10.2353/ajpath.2010.091021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett LE, Granot Z, Coker C, Iavarone A, Hambardzumyan D, Holland EC, Nam HS, Benezra R. Self-renewal does not predict tumor growth potential in mouse models of high-grade glioma. Cancer Cell. 2012;21:11–24. doi: 10.1016/j.ccr.2011.11.025. [DOI] [PubMed] [Google Scholar]
- Bleau AM, Hambardzumyan D, Ozawa T, Fomchenko EI, Huse JT, Brennan CW, Holland EC. PTEN/PI3K/Akt pathway regulates the side population phenotype and ABCG2 activity in glioma tumor stem-like cells. Cell Stem Cell. 2009;4:226–235. doi: 10.1016/j.stem.2009.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourguignon LY, Zhu H, Shao L, Chen YW. CD44 interaction with c-Src kinase promotes cortactin-mediated cytoskeleton function and hyaluronic acid-dependent ovarian tumor cell migration. J Biol Chem. 2001;276:7327–7336. doi: 10.1074/jbc.M006498200. [DOI] [PubMed] [Google Scholar]
- Calabrese C, Poppleton H, Kocak M, Hogg TL, Fuller C, Hamner B, Oh EY, Gaber MW, Finklestein D, Allen M, et al. A perivascular niche for brain tumor stem cells. Cancer Cell. 2007;11:69–82. doi: 10.1016/j.ccr.2006.11.020. [DOI] [PubMed] [Google Scholar]
- Charles N, Ozawa T, Squatrito M, Bleau AM, Brennan CW, Hambardzumyan D, Holland EC. Perivascular nitric oxide activates notch signaling and promotes stem-like character in PDGF-induced glioma cells. Cell Stem Cell. 2010;6:141–152. doi: 10.1016/j.stem.2010.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covello KL, Kehler J, Yu H, Gordan JD, Arsham AM, Hu CJ, Labosky PA, Simon MC, Keith B. HIF-2alpha regulates Oct-4: effects of hypoxia on stem cell function, embryonic development, and tumor growth. Genes Dev. 2006;20:557–570. doi: 10.1101/gad.1399906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Falco V, Tamburrino A, Ventre S, Castellone MD, Malek M, Manie SN, Santoro M. CD44 proteolysis increases CREB phosphorylation and sustains proliferation of thyroid cancer cells. Cancer Res. 2012;72:1449–1458. doi: 10.1158/0008-5472.CAN-11-3320. [DOI] [PubMed] [Google Scholar]
- Elvidge GP, Glenny L, Appelhoff RJ, Ratcliffe PJ, Ragoussis J, Gleadle JM. Concordant regulation of gene expression by hypoxia and 2-oxoglutarate-dependent dioxygenase inhibition: the role of HIF-1alpha, HIF-2alpha, and other pathways. J Biol Chem. 2006;281:15215–15226. doi: 10.1074/jbc.M511408200. [DOI] [PubMed] [Google Scholar]
- Emerling BM, Weinberg F, Liu JL, Mak TW, Chandel NS. PTEN regulates p300-dependent hypoxia-inducible factor 1 transcriptional activity through Forkhead transcription factor 3a (FOXO3a) Proc Natl Acad Sci U S A. 2008;105:2622–2627. doi: 10.1073/pnas.0706790105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan X, Matsui W, Khaki L, Stearns D, Chun J, Li YM, Eberhart CG. Notch pathway inhibition depletes stem-like cells and blocks engraftment in embryonal brain tumors. Cancer Res. 2006;66:7445–7452. doi: 10.1158/0008-5472.CAN-06-0858. [DOI] [PubMed] [Google Scholar]
- Godar S, Ince TA, Bell GW, Feldser D, Donaher JL, Bergh J, Liu A, Miu K, Watnick RS, Reinhardt F, et al. Growth-inhibitory and tumor- suppressive functions of p53 depend on its repression of CD44 expression. Cell. 2008;134:62–73. doi: 10.1016/j.cell.2008.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golebiewska A, Brons NH, Bjerkvig R, Niclou SP. Critical appraisal of the side population assay in stem cell and cancer stem cell research. Cell Stem Cell. 2011;8:136–147. doi: 10.1016/j.stem.2011.01.007. [DOI] [PubMed] [Google Scholar]
- Gordan JD, Lal P, Dondeti VR, Letrero R, Parekh KN, Oquendo CE, Greenberg RA, Flaherty KT, Rathmell WK, Keith B, et al. HIF-alpha effects on c-Myc distinguish two subtypes of sporadic VHL-deficient clear cell renal carcinoma. Cancer Cell. 2008;14:435–446. doi: 10.1016/j.ccr.2008.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hambardzumyan D, Becher OJ, Rosenblum MK, Pandolfi PP, Manova-Todorova K, Holland EC. PI3K pathway regulates survival of cancer stem cells residing in the perivascular niche following radiation in medulloblastoma in vivo. Genes Dev. 2008;22:436–448. doi: 10.1101/gad.1627008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris AL. Hypoxia--a key regulatory factor in tumour growth. Nat Rev Cancer. 2002;2:38–47. doi: 10.1038/nrc704. [DOI] [PubMed] [Google Scholar]
- Haylock DN, Nilsson SK. Stem cell regulation by the hematopoietic stem cell niche. Cell Cycle. 2005;4:1353–1355. doi: 10.4161/cc.4.10.2056. [DOI] [PubMed] [Google Scholar]
- Holland EC, Hively WP, DePinho RA, Varmus HE. A constitutively active epidermal growth factor receptor cooperates with disruption of G1 cell-cycle arrest pathways to induce glioma-like lesions in mice. Genes Dev. 1998;12:3675–3685. doi: 10.1101/gad.12.23.3675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu KH, Tsai HW, Lin PW, Hsu YS, Shan YS, Lu PJ. Clinical implication and mitotic effect of CD44 cleavage in relation to osteopontin/CD44 interaction and dysregulated cell cycle protein in gastrointestinal stromal tumor. Ann Surg Oncol. 2010;17:2199–2212. doi: 10.1245/s10434-010-0927-1. [DOI] [PubMed] [Google Scholar]
- Huse JT, Holland EC. Targeting brain cancer: advances in the molecular pathology of malignant glioma and medulloblastoma. Nat Rev Cancer. 2010;10:319–331. doi: 10.1038/nrc2818. [DOI] [PubMed] [Google Scholar]
- Jogi A, Ora I, Nilsson H, Lindeheim A, Makino Y, Poellinger L, Axelson H, Pahlman S. Hypoxia alters gene expression in human neuroblastoma cells toward an immature and neural crest-like phenotype. Proc Natl Acad Sci U S A. 2002;99:7021–7026. doi: 10.1073/pnas.102660199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz AM, Amankulor NM, Pitter K, Helmy K, Squatrito M, Holland EC. Astrocyte-specific expression patterns associated with the PDGF-induced glioma microenvironment. PLoS One. 2012;7:e32453. doi: 10.1371/journal.pone.0032453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keith B, Simon MC. Hypoxia-inducible factors, stem cells, and cancer. Cell. 2007;129:465–472. doi: 10.1016/j.cell.2007.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Bao S, Wu Q, Wang H, Eyler C, Sathornsumetee S, Shi Q, Cao Y, Lathia J, McLendon RE, et al. Hypoxia-inducible factors regulate tumorigenic capacity of glioma stem cells. Cancer Cell. 2009;15:501–513. doi: 10.1016/j.ccr.2009.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manalo DJ, Rowan A, Lavoie T, Natarajan L, Kelly BD, Ye SQ, Garcia JG, Semenza GL. Transcriptional regulation of vascular endothelial cell responses to hypoxia by HIF-1. Blood. 2005;105:659–669. doi: 10.1182/blood-2004-07-2958. [DOI] [PubMed] [Google Scholar]
- Mathieu J, Zhang Z, Zhou W, Wang AJ, Heddleston JM, Pinna CM, Hubaud A, Stadler B, Choi M, Bar M, et al. HIF induces human embryonic stem cell markers in cancer cells. Cancer Res. 2011;71:4640–4652. doi: 10.1158/0008-5472.CAN-10-3320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miletti-Gonzalez KE, Murphy K, Kumaran MN, Ravindranath AK, Wernyj RP, Kaur S, Miles GD, Lim E, Chan R, Chekmareva M, et al. Identification of function for CD44 intracytoplasmic domain (CD44-ICD): modulation of matrix metalloproteinase 9 (MMP-9) transcription via novel promoter response element. J Biol Chem. 2012;287:18995–19007. doi: 10.1074/jbc.M111.318774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami D, Okamoto I, Nagano O, Kawano Y, Tomita T, Iwatsubo T, De Strooper B, Yumoto E, Saya H. Presenilin-dependent gamma-secretase activity mediates the intramembranous cleavage of CD44. Oncogene. 2003;22:1511–1516. doi: 10.1038/sj.onc.1206298. [DOI] [PubMed] [Google Scholar]
- Nagano O, Murakami D, Hartmann D, De Strooper B, Saftig P, Iwatsubo T, Nakajima M, Shinohara M, Saya H. Cell-matrix interaction via CD44 is independently regulated by different metalloproteinases activated in response to extracellular Ca(2+) influx and PKC activation. J Cell Biol. 2004;165:893–902. doi: 10.1083/jcb.200310024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagano O, Saya H. Mechanism and biological significance of CD44 cleavage. Cancer Sci. 2004;95:930–935. doi: 10.1111/j.1349-7006.2004.tb03179.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto I, Kawano Y, Murakami D, Sasayama T, Araki N, Miki T, Wong AJ, Saya H. Proteolytic release of CD44 intracellular domain and its role in the CD44 signaling pathway. J Cell Biol. 2001;155:755–762. doi: 10.1083/jcb.200108159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelletier L, Guillaumot P, Freche B, Luquain C, Christiansen D, Brugiere S, Garin J, Manie SN. Gamma-secretase-dependent proteolysis of CD44 promotes neoplastic transformation of rat fibroblastic cells. Cancer Res. 2006;66:3681–3687. doi: 10.1158/0008-5472.CAN-05-3870. [DOI] [PubMed] [Google Scholar]
- Phillips HS, Kharbanda S, Chen R, Forrest WF, Soriano RH, Wu TD, Misra A, Nigro JM, Colman H, Soroceanu L, et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell. 2006;9:157–173. doi: 10.1016/j.ccr.2006.02.019. [DOI] [PubMed] [Google Scholar]
- Pietras A. Cancer stem cells in tumor heterogeneity. Adv Cancer Res. 2011;112:255–281. doi: 10.1016/B978-0-12-387688-1.00009-0. [DOI] [PubMed] [Google Scholar]
- Pietras A, Gisselsson D, Ora I, Noguera R, Beckman S, Navarro S, Pahlman S. High levels of HIF-2alpha highlight an immature neural crest-like neuroblastoma cell cohort located in a perivascular niche. J Pathol. 2008;214:482–488. doi: 10.1002/path.2304. [DOI] [PubMed] [Google Scholar]
- Pietras A, Johnsson AS, Pahlman S. The HIF-2alpha-driven pseudo-hypoxic phenotype in tumor aggressiveness, differentiation, and vascularization. Curr Top Microbiol Immunol. 2010;345:1–20. doi: 10.1007/82_2010_72. [DOI] [PubMed] [Google Scholar]
- Semenza GL. Hypoxia-inducible factor 1 (HIF-1) pathway. Sci STKE. 2007;2007 doi: 10.1126/stke.4072007cm8. cm8. [DOI] [PubMed] [Google Scholar]
- Singh SK, Clarke ID, Terasaki M, Bonn VE, Hawkins C, Squire J, Dirks PB. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003;63:5821–5828. [PubMed] [Google Scholar]
- Soeda A, Park M, Lee D, Mintz A, Androutsellis-Theotokis A, McKay RD, Engh J, Iwama T, Kunisada T, Kassam AB, et al. Hypoxia promotes expansion of the CD133-positive glioma stem cells through activation of HIF-1alpha. Oncogene. 2009;28:3949–3959. doi: 10.1038/onc.2009.252. [DOI] [PubMed] [Google Scholar]
- Su YJ, Lai HM, Chang YW, Chen GY, Lee JL. Direct reprogramming of stem cell properties in colon cancer cells by CD44. Embo J. 2011;30:3186–3199. doi: 10.1038/emboj.2011.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, Speleman F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002;3 doi: 10.1186/gb-2002-3-7-research0034. RESEARCH0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, Miller CR, Ding L, Golub T, Mesirov JP, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17:98–110. doi: 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss JM, Renkl AC, Maier CS, Kimmig M, Liaw L, Ahrens T, Kon S, Maeda M, Hotta H, Uede T, et al. Osteopontin is involved in the initiation of cutaneous contact hypersensitivity by inducing Langerhans and dendritic cell migration to lymph nodes. J Exp Med. 2001;194:1219–1229. doi: 10.1084/jem.194.9.1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter SC, Buffa FM, Silva P, Miller C, Valentine HR, Turley H, Shah KA, Cox GJ, Corbridge RJ, Homer JJ, et al. Relation of a hypoxia metagene derived from head and neck cancer to prognosis of multiple cancers. Cancer Res. 2007;67:3441–3449. doi: 10.1158/0008-5472.CAN-06-3322. [DOI] [PubMed] [Google Scholar]
- Yan Q, Bartz S, Mao M, Li L, Kaelin WG., Jr. The hypoxia-inducible factor 2alpha N-terminal and C-terminal transactivation domains cooperate to promote renal tumorigenesis in vivo. Mol Cell Biol. 2007;27:2092–2102. doi: 10.1128/MCB.01514-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoller M. CD44: can a cancer-initiating cell profit from an abundantly expressed molecule? Nat Rev Cancer. 2011;11:254–267. doi: 10.1038/nrc3023. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.