

Abstract
Sirtuin1 (SIRT1) regulates central metabolic functions such as lipogenesis, protein synthesis, gluconeogenesis and bile acid (BA) homeostasis through deacetylation. Here, we describe that SIRT1 tightly controls the regenerative response of the liver.
We performed partial hepatectomy (PH) to transgenic mice that overexpress SIRT1 (SIRT). SIRT mice showed increased mortality, impaired hepatocyte proliferation, BA accumulation and profuse liver injury after surgery. The damaging phenotype in SIRT mice correlated with impaired FXR activity due to persistent deacetylation and lower protein expression that led to decreased FXR-target gene expression; SHP, BSEP and increased Cyp7A1. Next, we convincingly show that 24-norUrsodeoxycholic acid (NorUDCA) attenuates SIRT protein expression, increases the acetylation of FXR and neighboring histones, restores trimethylation of H3K4 and H3K9 and increases miR34a expression, thus re-establishing BA homeostasis. Consequently, NorUDCA restored liver regeneration in SIRT mice, which showed increased survival and hepatocyte proliferation. Furthermore, a Leucine-enriched diet restored mTOR activation, acetylation of FXR and histones, leading to an overall lower BA production through SHP-inhibition of Cyp7A1 and higher transport (BSEP) and detoxification (Sult2a1) leading to an improved liver regeneration. Finally, we found that human HCC samples have increased presence of SIRT1, which correlated with absence of FXR suggesting its oncogenic potential.
Conclusions
Overall, we define SIRT1 as a key regulator of the regenerative response in the liver through post-transcriptional modifications that regulate the activity of FXR, histones and mTOR. Moreover, our data suggest that SIRT1 contributes to liver tumorigenesis through dysregulation of BA homeostasis by persistent FXR deacetylation.
Keywords: Sirtuin1, FXR, Bile Acids, mTOR, deacetylation, liver regeneration
INTRODUCTION
Sirtuin 1 (SIRT1) is a class III NAD+-dependent histone deacetylase that tightly regulates lipid, glucose and bile acids (BA) metabolism (1, 2). SIRT1 is activated in situations of low energy availability and links nutritional status with metabolic homeostasis (1). SIRT1 regulates AMPK by deacetylation of LKB1 (1), which are involved in tissue repair processes (3). Contrarily to SIRT1, mTOR is activated in high-energy conditions and controls cell growth and proliferation (4). mTORC1 promotes protein synthesis by activating the S6 ribosomal protein and this axis is essential to regulate cell cycle during liver regeneration after partial hepatectomy (PH) (5). BA are also essential for the regeneration of the liver after PH (6) although, when present in excess, BA can be toxic and promote hepatocyte death (7). Therefore, a fine regulation of BA metabolism is essential to preserve liver homeostasis and a proper response to injury. The orphan nuclear receptor (NR) Farnesoid X receptor (FXR; NR1H4) is the master regulator of BA, lipid and glucose metabolism (8). SIRT1 directly modulates FXR activity by deacetylation of this NR and neighboring histones that strictly control target gene transcription (9, 10). In the present work we describe that modulation of SIRT1 is essential for the regenerative response in the liver through mechanisms involving the regulation of i) FXR and ii) mTOR signaling pathways by dynamic acetylation/deacetylation, which overall maintain BA homeostasis.
BA-mediated toxicity is also involved in the pathogenesis of cirrhosis from diverse etiologies (11, 12). The role of SIRT during tumorigenesis remains controversial, as both pro- and anti-oncogenic properties have been described (13, 14). Here, we show that increased SIRT1 expression correlated with a low presence of FXR in human HCC samples, supporting the significance of SIRT1 deacetylase activity in regulating BA homeostasis and the response to liver injury.
Overall, our data defines SIRT1 as a key regulator of the regenerative response of the liver, controlling BA homeostasis, protein synthesis and cell proliferation through deacetylation of FXR and histones and regulation of mTOR. Importantly, our results underscore the implication of mTOR in regulating BA metabolism as a negative regulator of SIRT1.
MATERIALS AND METHODS
Experimental procedures in animals
SIRT1 overexpressing animals (SIRT) were generated as described (14) in a C57/Bl6J background. Males from 8–12 weeks of age were treated and used according to the guidelines of the National Academy of Sciences (National Institutes of Health publication 86-23, revised 1985). SIRT mice were fed with a diet enriched in 3% Leucine or a diet containing 0.5% w/w 24-norursodeoxycholic acid (15, 16) for 2 or 3 weeks respectively before 2/3 partial hepatectomy (PH).
Materials
Recombinant TNF was obtained from Peprotech. Deoxycholic acid (DCA) was purchased from Sigma-Aldrich.
Bile acid determination
Bile acids were extracted from liver samples, and analyzed by high performance liquid chromatography-tandem mass spectrometry as previously reported (17).
Analysis of gene expression
mRNA was extracted from snap-frozen livers using TriZol Reagent (Invitrogen). Quantitative RT-PCR (qPCR) was performed using SYBR Green reagent (Quantas biosciences) in a MyiQ single color Real-time PCR detection system (Biorad). Gene expression was normalized with GAPDH and shown in times vs WT expression before PH (basal). Primers can be provided upon request.
(Additional M&M are provided as supplemental material).
RESULTS
Overexpression of SIRT1 impairs hepatocyte proliferation after PH
Liver damage leads to a regenerative response that restores organ mass and function (18). During liver regeneration, the expression of SIRT1 was downregulated at the initial stages to recover later at 48h after PH (Fig. 1A), highlighting the regulation of SIRT1 throughout the regenerative response.
Fig. 1. Overexpression of SIRT1 impairs liver regeneration in mice.

(A) qPCR analysis of SIRT expression in WT animals at different timepoints after PH. (B) Kaplan Meier curve depicting survival of SIRT and WT mice after PH. (C) IHC using an anti-BrdU Ab (red) and stained with DAPI (blue) on liver sections after PH. (D) Analysis of Cyclin D1, Cyclin E and Cyclin A mRNA expression by qPCR. (E) IHC performed in liver sections using a phistone H3 Ab (green) vs DAPI+ nuclei (blue) (F) pmTORC1, pS6 and pAMPK levels in liver extracts analyzed by western blot (WB) analysis (Values are mean ± SD. Survival curve: n = 20 animals; *P <0.05, **P <0.01, ***P<0.001 [WT vs SIRT]).
To define the role of SIRT1 during liver regeneration, we used transgenic mice that moderately overexpress SIRT1 (SIRT) (Suppl. Fig. 1A, B). SIRT mice showed poor prognosis after PH as 40% of SIRT mice died within the first 48h after resection (Fig. 1B). Hepatocyte proliferation was significantly attenuated in SIRT mice as shown by BrdU staining (Fig. 1C and Suppl. Fig. 1C) and analysis of cyclin D1, E and A2 mRNA expression after PH (Fig. 1D). Additionally, the mitotic response was significantly impaired in SIRT mice 72h after PH as shown by p-histone H3S10 IHC (Fig. 1E and Suppl. Fig. 1D). Finally, we found impaired activation of the mTOR pathway in SIRT mice before and after PH (Fig. 1F), in agreement with the recently described inhibitory effect of SIRT1 over mTORC1 (19). WT mice showed basal p-mTOR and p-S6 expression that was upregulated 6h after PH (Fig. 1F) whereas SIRT mice showed low basal p-S6 levels that were unaffected after PH. SIRT1 activates AMPK (1). Accordingly, AMPK was basally phosphorylated in SIRT mice compared to WT animals (Fig. 1F, Suppl. Fig. 1E). PH triggered phophorylation of AMPK in WT mice whereas in SIRT animals pAMPK expression remained unchanged after resection (Fig. 1F, Suppl. Fig. 1E). Furthermore, we found that cytosolic shuttling of HuR, a pAMPK target protein, was attenuated in SIRT mice after PH (Suppl. Fig. 1F). HuR stabilizes the mRNA of diverse molecules involved in cell proliferation after PH (20). Accordingly, MAT2A (Suppl. Fig. 1F, G) and NOS2 (Suppl. Fig. 1G) expression were attenuated in SIRT mice. Overall, these data suggest that persistent phosphorylation of AMPK renders it non-functional in SIRT mice, further contributing to impairment of liver regeneration.
Overexpression of SIRT1 impairs FXR-mediated signaling leading to BA accumulation and liver injury after PH
BA-signaling is essential for liver regeneration (6) whereas excessive presence of BA is toxic for hepatocytes, promoting apoptosis and necrosis (7). Histological analysis of livers from SIRT mice showed no parenchymal damage and appeared similar to WT animals prior to PH (Fig. 2A; left panels). BA pool size in untreated SIRT livers was similar to WTs (Fig 2B) but its composition was shifted towards secondary BA (Suppl. Table 1). The poor prognosis of SIRT mice within the first 48h after PH was associated with the presence of bile infarcts, evidenced by profuse presence of necrotic areas and inflammatory cell infiltration in the liver (Fig. 2A; right panels). In line with bile infarcts, we found a significant increase of total BA levels, especially conjugated, in SIRT mice 48h after PH (Fig. 2B). BA metabolism is tightly controlled by FXR as it activates SHP, leading to inhibition of Cyp7a1. In SIRT mice, we found that FXR was strongly deacetylated at basal conditions (Fig. 2C, Suppl Fig. 2A), which correlated with lower protein expression (Fig. 2C, Suppl Fig. 2A, B). Lower acetylation of FXR was associated with downregulation of SHP and increased Cyp7A1 expression, indicating impaired FXR-signaling (Fig. 2D). Finally, expression of the canalicular export pump; BSEP and detoxification-related enzyme Sult2a1 were significantly downregulated in SIRT mice (Fig. 2D). Interestingly, BA metabolism-related gene expression in SIRT mice resembles what was found in WT animals after 8h of fasting, characterized by BA synthesis and attenuated transport (Suppl. Fig. 3A). These data indicates that overexpression of SIRT1 mimics a ‘fasting like’ status despite the fed-status of SIRT mice. In accordance, we found low glucose levels in serum from SIRT mice at basal conditions (Suppl. Fig. 3B). SIRT1 regulates glucose metabolism through PGC1α-gluconeogenesis (21). In SIRT mice, deacetylation of PGC1α correlated with lower levels of total protein (Suppl. Fig. 3C), suggesting its degradation (22). Accordingly, PEPCK and PDK4 were significantly downregulated in SIRT mice (Suppl. Fig. 3D). Overall, these data suggest that ‘fasting-like’ status characterized by dysregulation of gluconeogenesis and BA metabolism may contribute to the poor regenerative capacity of the liver in SIRT mice after PH.
Fig. 2. Overexpression of SIRT1 leads to altered BA and glucose homeostasis due to dysregulation of FXR and PGC1α.
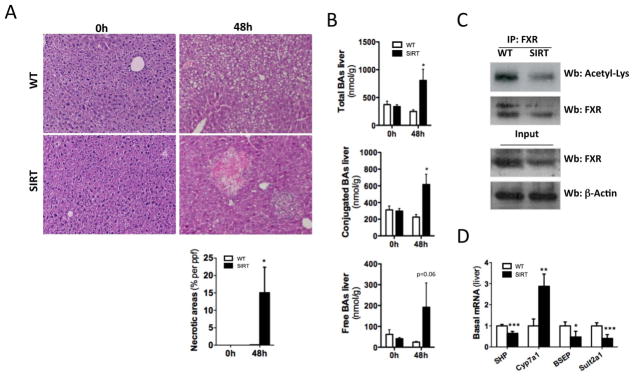
(A) H&E staining on liver sections at 0h and 48h after PH (B) Quantification of bile acids (BA) in livers from WT and SIRT mice before and 48h after PH. (C) Immunoprecipitation (IP) using a FXR Ab and WB analysis using Acetylated Lysine. WB of nuclear extracts FXR and β-Actin (input). (D) Analysis of SHP, Cyp7a1, BSEP and Sult2a1 mRNA expression by qPCR in livers at basal conditions. (Values are mean ± SD. n = 5 animals/time point; *P <0.05, **P <0.01, ***P<0.001 [WT vs SIRT]).
Overexpression of SIRT1 sensitizes the liver and hepatocytes to BA-induced injury
At high concentrations, certain species of BA induce toxicity and cell death (7). Accordingly, BA accumulation in the regenerating liver of SIRT mice correlated with profuse hepatocyte death 48h after PH (Fig. 3A). Next, we performed in vitro experiments to better define the impact of BA in SIRT1-overexpressing hepatocytes. We observed increased apoptosis, oxidative stress and pJNK in isolated SIRT hepatocytes when cultured in serum starvation conditions (Fig 3B–D), supporting the essential role of SIRT1 in protecting the cell against energetic stress. Furthermore, deoxycholic acid (DCA) promoted profuse death of SIRT hepatocytes (Fig. 3B). Additionally, ROS was further increased 4h and 6h after DCA treatment in SIRT hepatocytes (Fig. 3C). Basal phosphorylation of JNK in SIRT hepatocytes was further sustained after DCA, whereas transient and milder activation of JNK was found in WT cells after treatment (Fig. 3D). Inhibition of JNK-signaling with SP600125 significantly reduced DCA-induced hepatocyte death and ROS formation in SIRT hepatocytes (Fig. 3E), supporting the role of JNK in mediating BA-cell damage (7). Overall, these data point out that SIRT1 overexpression sensitizes hepatocytes to BA-death.
Fig. 3. Overexpression of SIRT1 sensitizes the liver and hepatocytes to BA-induced damage.

(A) TUNEL assay on liver sections 48h after PH. (B) TUNEL assay on primary hepatocytes stimulated with DCA (100μM) for 24h (C) Primary hepatocytes incubated with DCFH-DA (Red) before and 6h after stimulation with DCA. (D) WB of whole protein extracts using pJNK Ab. (E) Inhibition of pJNK with SP600125 protected SIRT hepatocytes from DCA-induced ROS and apoptosis as shown by DCFH-DA and TUNEL assay. (n = 5 animals/time point; In vitro experiments were performed three times, each in duplicate).
NorUDCA restores hepatocyte proliferation after PH in SIRT mice
NorUDCA has strong beneficial effects against sclerosing cholangitis and diet-induced NASH by improving bile flow and hydrophilicity and detoxification of BA (15, 16). We fed SIRT mice with a diet enriched in NorUDCA for 3 weeks, after which PH was performed. Acetylation and protein expression of FXR (Fig. 4A, Suppl. Fig 4A, B) were restored in NorUDCA/SIRT mice and SHP mRNA levels increased (Fig. 4B). Interestingly, we found elevated Cyp7A1 mRNA levels in NorUDCA/SIRT mice (Fig 4B), while protein expression was lower in these animals compared to SIRT mice (Suppl. Fig 4C). As expected, NorUDCA increased expression of bile export pumps and detoxification-related molecules in SIRT mice (Fig. 4B). Importantly, NorUDCA caused only mild restoration of acetylation of PGC1α (Suppl. Fig. 5A) and no significant differences were observed in total protein levels and PGC1α target gene expression (data not shown), suggesting a certain specificity of NorUDCA over FXR-signaling.
Fig. 4. NorUDCA restores FXR-signaling and promotes liver regeneration in SIRT mice after PH.

(A) IP using a FXR Ab and further WB analysis using Acetylated Lysine Ab of liver extracts at basal conditions. WB analysis of nuclear extracts using FXR and β-Actin Abs (lower panel). (B) Analysis of BA metabolism-related gene expression by qPCR in livers (C) Quantification of BA in livers from SIRT and NorUDCA/SIRT mice before and 48h after PH. (D) H&E staining, TUNEL assay and BrdU staining on liver sections 48h after PH (E) IHC using an anti-SIRT1 Ab (F) WB analysis of nuclear protein extracts using H3K4me3 and H3K9me3 Ab. (Values are mean ± SD. n = 5 animals/time point; *P <0.05, **P <0.01, ***P<0.001 [SIRT vs NorUDCA/SIRT]).
Feeding SIRT mice with NorUDCA shifted the basal free BA composition, as 90% of the unconjugated BA content consisted of NorUDCA. After PH, the overall free BA pool size was reduced in NorUDCA/SIRT compared to SIRT mice (Fig 4C). The conjugated BA pool was significantly reduced before and after PH in NorUDCA/SIRT mice (Fig. 4C). Total BA pool was basally higher in NorUDCA/SIRT mice consisting mainly of this modified BA. After PH, NorUDCA treatment reduced the total BA pool (Fig. 4C), supporting the well-described impact of NorUDCA on promoting BA efflux and detoxification (15). In line with the shift towards a hydrophilic non-toxic BA pool (mainly consisting of NorUDCA), we observed an absence of necrotic areas, parenchymal degeneration and hepatocyte cell death in NorUDCA/SIRT mice (Fig. 4D, Suppl. Fig. 6A). BrdU analysis and qPCR of cyclins confirmed that NorUDCA restored hepatocyte proliferation 48h after PH in SIRT mice (Fig. 4D, Suppl. Fig. 6B). mTORC1/pS6 signaling was not restored upon NorUDCA feeding (Suppl. Fig. 6C), suggesting a potential specificity over FXR-signaling. Importantly, we found that miR34a expression was low in SIRT mice but restored upon NorUDCA feeding (Suppl. Fig. 6D). Accordingly, NorUDCA decreased SIRT1 protein expression in SIRT mice (Fig 4E, Suppl. Fig. 6E). Moreover, SIRT mice exhibited increased presence of the trimethylated forms of both H3K4 and H3K9 basally, whereas NorUDCA decreased their expression back to WT levels (Fig. 4F). Restoration of acetylation by NorUDCA correlated with recovery of mRNA expression of FXR in NorUDCA/SIRT mice (Suppl. Fig. 6F). Finally, all NorUDCA/SIRT mice survived after PH, confirming the beneficial impact of NorUDCA during the regenerative response of the liver in the context of SIRT1 overexpression (Suppl. Fig. 6G).
Overall, our data indicates that the beneficial effects of NorUDCA during the regenerative process are, at least in part, mediated by the restoration of the functionality of FXR through mechanisms involving transcriptional and translational regulation, leading to the recovery of FXR mRNA, protein and dynamic acetylation.
Recovery of mTOR-signaling restores BA homeostasis and liver regeneration in SIRT mice
mTORC1 is directly activated by amino acids (Aa) through RagGTPases (4) and leucine is the most effective Aa in inducing this process (23). In order to restore mTOR signaling, we fed SIRT mice with a 3% leucine-rich diet (Leu) for 2 weeks after which PH was performed. Leu successfully restored mTORC1 signaling to levels comparable to those found in WT animals; basal S6-phosphorylation was found in Leu/SIRT mice and further increased at 6h and 24h after PH (Suppl. Fig. 7A, B) and AMPK activation was also restored (Suppl. Fig. 7A, B). In Leu/SIRT mice, recovery of mTORC1 signaling led to a significant improvement in survival after PH, as the mortality rate was only 20% (Suppl. Fig. 7C), and to normalization of hepatocyte proliferation (Fig. 5A, Suppl. Fig. 7D, E).
Fig. 5. Leucine enriched diet recovers mTOR signaling and restores liver regeneration in SIRT mice.

(A) IHC using an anti-BrdU Ab (Red) and stained with DAPI (blue) showing restored proliferation in Leu/SIRT mice. (B) H&E staining of liver sections 48h after PH (20x) (C) Bile acids were measured in livers 48h after PH (D) IP using a FXR Ab and further WB analysis using Acetylated Lysine Ab of liver extracts at basal conditions. WB analysis of nuclear extracts using FXR and β-Actin Abs (lower panel). (E) IHC using an anti-SIRT Ab showing normalization of SIRT levels in Leu/SIRT livers. (F) IHC showing aberrant tri-methylation of H3K4 and H3K9 in SIRT mice, which was reverted by Leu (Values are mean ± SD. n = 5 animals/time point; *P <0.05, **P <0.01, ***P<0.001 [SIRT vs Leu/SIRT]).
Interestingly, H&E staining revealed significant improvement of the liver parenchyma in Leu/SIRT mice 48h after PH, and no signs of bile infarcts were detected (Fig. 5B). Leu reduced the accumulation of conjugated and total bile acids at 48h after PH in SIRT mice (Fig. 5C) and restored FXR acetylation and total protein levels (Fig. 5D, Suppl. Fig. 7F–H), which correlated with increased SHP and BSEP expression and Cyp7a1 downregulation (Suppl. Fig. 7I). Interestingly, Leu lowered SIRT1 protein expression in SIRT mice (Fig. 5E, Suppl. Fig. 7J) and restored the expression of H3K4 and H3K9 to WT levels (Fig. 5F). Finally, Leu attenuated the increased apoptosis found in isolated SIRT hepatocytes when cultured in serum starvation conditions (Suppl. Fig. 7K). Overall, these data indicate that restoration of the mTORC1 signaling pathway circumvents the detrimental impact of SIRT1 overexpression on hepatocyte proliferation and BA accumulation. The potential mechanisms underlying these effects rely on the impact of mTOR on SIRT1-regulated acetylation of FXR and neighboring histones. Also, our data points to a link between mTORC1 and regulation of BA metabolism.
SIRT1 is overexpressed in HCC patients
The role of SIRT during tumorigenesis remains controversial (13, 14). We examined the expression of SIRT1 in a cohort of human HCC from different etiologies. SIRT1 mRNA expression was increased in HCC samples (Suppl. Fig 8A). A more detailed study of different HCC samples by IHC and further quantification revealed that SIRT1 was significantly overexpressed in samples derived from HCV and ASH (Fig. 6A). Interestingly, FXR expression was significantly downregulated in HCC human samples; both from an HCV and ASH etiology (Fig. 6B). Overall, these data support the implication of SIRT during liver tumorigenesis in a context of BA metabolism misregulation and suggests that this oncogenic role might be linked to the ability of SIRT1 to regulate FXR through deacetylation.
Fig. 6. SIRT1 is highly expressed in HCC human samples.

(A) IHC using a SIRT1 and FXR (B) antibody in human HCC samples from patients with HCV (n=10) and ASH (n=10) etiology. Quantitative assessments of IHC staining were performed using FRIDA image analysis software. Expressed in % of positive staining per area *P <0.05, **P <0.01, ***P<0.001 [Control vs HCC]).
DISCUSSION
SIRT1 is essential for cell metabolism control as, in situations of low energy availability (low ATP), it promotes catalytic processes to restore cell-energy homeostasis (1, 2). Here, we describe that SIRT1 overexpression has a detrimental impact on liver regeneration as poor survival and impaired proliferation were found after PH. In apparent contradiction, recent work showed that reduced SIRT1 expression in elder mice correlated with impaired liver regeneration, which was restored after normalization of SIRT levels in these aged mice (24). Interestingly, both Jin et al. and our work emphasize the critical relevance of the fine-tuned regulation of SIRT deacetylase activity for the regeneration of the liver, as either deficiency or excess have detrimental impact on this process. Here we show that SIRT overexpressing mice have persistent deacetylase activity that profoundly alters key metabolic responses such as i) BA metabolism and ii) protein synthesis, highlighting the role of SIRT1 in controlling the regenerative response of the liver.
FXR is the master regulator of BA metabolism. FXR activates SHP that represses the expression of Cyp7A1; the BA synthesis rate-limiting enzyme (8). Additionally, essential BA transporters like BSEP are positively regulated by FXR. Little is known regarding the mechanisms by which FXR regulates gene expression although post-transcriptional modifications (PTM) seem to play a key role. PTM occur at two levels; modifying the NR and its cofactors, and/or modifying histones at the promoters of the NR-target genes (reviewed in (25)). FXR is tightly regulated by p300- and SIRT1-mediated acetylation/deacetylation. Whereas acetylation is generally related to gene activation, Kemper et al. showed that acetylation of FXR by p300 leads to lower transactivation potential. Accordingly, deacetylation of FXR by SIRT1 allows interaction with RXR, leading to DNA binding or to its degradation by ubiquitination (9). Interestingly, the FXR-target gene SHP is differently regulated as histone deacetylation, by SIRT1, of its promoter contributes to silence transcription (25). This underlines that the complex mechanism by which SIRT1 controls BA metabolism through deacetylation must be a dynamic process. In addition to the ostensible deacetylation of FXR and low transcriptional capacity, we found aberrant methylation of histones in SIRT mice, as both H3K4 and H3K9 were tri-methylated. The histone methyltransferase Set9 competes with histone deacetylases to methylate H3 on the lysine 4. H3K4me3 inhibits the methyltransferase Suv39H1, which (tri-)methylates H3K9. This regulatory mechanism avoids simultaneous methylation of H3K4me3 and H3K9me3 as they have generally opposing activities; activation and repression of gene transcription respectively. Sir2, the SIRT1 analogue in yeast promotes trimethylation of H3K4 and H3K9 (26, 27). H3K4m3 relates with FXR activation of target gene expression such as BSEP (28) whereas acetylation of H3K9 by p300 is essential for SHP-mediated activation of its transcriptional activity (25).
Overall, we propose that SIRT1 modulates BA metabolism through two mechanisms; i) PTM of NR (FXR) and its cofactors (SHP) that repress target gene transcription (CYp7a1, BSEP) and ii) PTM of histones permitting methylation through deacetylation. Both of these mechanisms are altered when SIRT1 is overexpressed leading to dysregulated FXR activity and aberrant methylation of H3K4 and H3K9.
SIRT mice show no signs of liver injury at basal conditions and have comparable levels of liver BA than WT. However, a closer analysis shows that misregulation of FXR-related BA metabolism found basally correlates with a shift towards enhanced presence of secondary BA in SIRT livers. These data suggest that the role of the intestine in bile acid homeostasis might be affected in SIRT mice. Although it is out of the scope of this work, the unbalance towards secondary BA in SIRT mice, despite having a similar BA pool size to WT animals, may indicate enhanced intestine elimination rate due to reduced re-uptake by the ileum. Also, consistently with enhanced intestinal elimination, our data points to a role of intestinal bacteria in changing the BA pool composition in SIRT mice.
Previous work showed that BA homeostasis is essential for liver regeneration as FXR−/− mice show impaired response after PH (6). Accordingly, our data suggests that persistent deacetylation of FXR by SIRT1 contributes to defective liver regeneration through misregulation of BA synthesis, transport and detoxification. Consequently, accumulation of BA in regenerating livers of SIRT mice may cause severe necrosis and tissue damage supporting the toxicity of BA when present in excess (7). We show that BA-mediated liver injury was attenuated and liver regeneration was fully restored in NorUDCA/SIRT animals. Our data not only confirms what was previously described regarding the therapeutic role of NorUDCA during liver damage (15, 16) but also provides new insights in the mechanisms by which NorUDCA exerts its beneficial effects. Our results suggest that NorUDCA controls the acetylation status of the cell by regulating the expression of SIRT1. Thus, in agreement with the known regulation of SIRT1 by miR34a (29), we found that NorUDCA recovered the expression of this microRNA attenuating SIRT1. Overall, our data suggest that regulation of FXR by NorUDCA undergoes through transcriptional and translational mechanisms, involving restoration of mRNA and protein expression, and by regulating the acetylation status in the cell through SIRT1.
Furthermore, we found that persistent deacetylation by SIRT1 promotes a ‘fasting-like’ status in the body characterized by increased BA synthesis through Cyp7a1. During fasting, interaction of SIRT1 and FXR increases whereas it is reduced upon re-feeding and in presence of BA; the natural FXR ligands (8). During fasting SIRT1 regulates FXR through various mechanisms; repressing gene transcription by deacetylating histones or direct deacetylation of FXR and further ubiquitination and proteosomal degradation, which may explain the lower protein levels found in SIRT mice.
Fasting promotes activation of PGC1α by SIRT1-mediated deacetylation in order to increase gluconeogenesis and blood glucose levels. In SIRT mice, we found that low acetylation of PGC1α correlated with impaired gluconeogenesis, which may be explained by persistent deacetylation by SIRT1 and further degradation by ubiquitination (30). Overall, our data regarding the regulation of FXR and PGC1α further underlines the importance of SIRT1 as a regulator of the fine-tuning of the acetylation/deacetylation process, which seems essential to orchestrate a proper response to tissue injury.
SIRT1 regulates the energy status of the cell through activation of AMPK/LKB1 (1, 4, 19). Accordingly, SIRT mice showed phosphorylation of AMPK at basal conditions that was not further activated after PH, contrarily to what we observed in WT animals. These results suggest that persistent phosphorylation of AMPK renders it non-functional in SIRT mice as we found downregulation of AMPK-target molecules like HuR, MAT2A and NOS2. These data support the key role of LKB1/AMPK to mediate cell growth during liver regeneration (3).
We describe that mTOR signaling was attenuated in SIRT mice throughout the regenerative response, supporting the role of SIRT1 as a negative regulator of mTOR (19). Interestingly, we provide evidences of a feedback regulation of SIRT1 by mTOR as Leu attenuated SIRT1 expression in SIRT mice. Our data links the previously described regulation of SIRT1-deacetylase activity by mTOR-mediated phosphorylation (31) with the degradation of SIRT by phosphorylation (32). Leu restored mTORC1 signaling that correlated with reduced BA-accumulation and liver injury, pointing to a link between mTOR and BA metabolism. Accordingly, mTOR activation by Leu restored FXR acetylation and its target gene expression in SIRT mice and attenuated methylation of H3K4 and H3K9 reaching comparable WT liver levels. We propose that the direct regulation of SIRT1 by mTORC1 influences histone and protein acetylation, which may explain the potential of mTOR to regulate histone acetylation (33)
It is worth to note that the beneficial effects of Leu and NorUDCA seem directly related to counteracting the detrimental effects of SIRT overexpression. Thus, WT animals fed with Leu showed attenuated hepatocyte proliferation (Suppl. Fig. 9A) whereas NorUDCA/WT mice showed comparable number of proliferating hepatocytes than WT mice after PH (Suppl. Fig. 9B).
PTM such as chromatin alterations by acetylation/deacetylation are involved in carcinogenesis. The role of SIRT1 during tumorigenesis remains controversial as it has both pro- and anti-carcinogenic effects (34). Here we confirm that SIRT1 is highly expressed in human HCC samples, regardless of etiology; HCV or ASH. Based on this fact, we propose that the tumorigenic characteristic of SIRT1 relies on its role as a regulator of BA metabolism as both HCV- and ASH-cirrhosis are characterized by dysregulated BA metabolism (11, 12). This might explain the apparent contradiction with a previous study (14) where the protection against inflammation and ROS exerted by SIRT1 overexpression seemed sufficient to circumvent tumor development. Moreover, we convincingly show that SIRT1 overexpression leads to dysregulation of FXR; a well know feature of metabolic disease and tumor development, as deletion of FXR leads to HCC development (35). Previous work related hyperacetylation of FXR with cancer, metabolic- and aging-related diseases. Importantly, we propose that overexpression of SIRT1 correlates with an absence of FXR expression in tumors potentially due to hypoacetylation and further degradation of FXR. These compelling data evidence the inverse correlation between SIRT1 and FXR during tumorigenesis and points to the oncogenic role of SIRT1.
Overall, our data underscore the significance of maintaining the deacetylation activity of SIRT1 as a dynamic process in the liver during the regenerative response after injury. Also, our work highlights the need to propose the cautious use of SIRT1-activating drugs, as persistent SIRT1 activity may have detrimental effects on the liver in response to injury. Importantly, our findings underline the potential use of NorUDCA- and mTORC1-activators as therapeutic tools in the context of an aberrant overexpression of SIRT1, to counteract liver metabolic diseases involving dysregulated BA homeostasis and/or the induction of a regenerative response after injury.
Supplementary Material
Acknowledgments
This work was supported by grants from the Instituto de Salud Carlos III; FIS, PS12/00402 (to N.B.), NIH AT-1576 (to M.L.M.-C., and J.M.M.), ETORTEK-2010 (to M.L.M.-C), Educación Gobierno Vasco 2011 (to M.L.M.-C), PI11/01588 (to M.L.M.-C). Plan Nacional SAF2011-29851 (to JMM), Plan Nacional SAF2010-15517 (to JJ.G.-M). Basque Government IT-336-10 (to PA) and UFI 11/20 (to PA). N.B. is funded by the Program Ramon y Cajal (Ministry of Economy and Competitiveness, Spain). Ciberehd is funded by the Instituto de Salud Carlos III. We want to thank Imanol Zubiete-Franco for editing the manuscript.
Footnotes
Conflict of Interest Statement: The authors declare that they have no competing financial interests. The Medical University of Graz has filed a patent on the medical use of norUDCA and Michael Trauner is listed as co-inventor.
References
- 1.Ruderman NB, Xu XJ, Nelson L, Cacicedo JM, Saha AK, Lan F, Ido Y. AMPK and SIRT1: a long-standing partnership? American journal of physiology Endocrinology and metabolism. 2010;298:E751–760. doi: 10.1152/ajpendo.00745.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schug TT, Li X. Sirtuin 1 in lipid metabolism and obesity. Annals of medicine. 2011;43:198–211. doi: 10.3109/07853890.2010.547211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vazquez-Chantada M, Ariz U, Varela-Rey M, Embade N, Martinez-Lopez N, Fernandez-Ramos D, Gomez-Santos L, et al. Evidence for LKB1/AMP-activated protein kinase/endothelial nitric oxide synthase cascade regulated by hepatocyte growth factor, S-adenosylmethionine, and nitric oxide in hepatocyte proliferation. Hepatology. 2009;49:608–617. doi: 10.1002/hep.22660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sengupta S, Peterson TR, Sabatini DM. Regulation of the mTOR complex 1 pathway by nutrients, growth factors, and stress. Molecular cell. 2010;40:310–322. doi: 10.1016/j.molcel.2010.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Espeillac C, Mitchell C, Celton-Morizur S, Chauvin C, Koka V, Gillet C, Albrecht JH, et al. S6 kinase 1 is required for rapamycin-sensitive liver proliferation after mouse hepatectomy. The Journal of clinical investigation. 2011;121:2821–2832. doi: 10.1172/JCI44203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Huang W, Ma K, Zhang J, Qatanani M, Cuvillier J, Liu J, Dong B, et al. Nuclear receptor-dependent bile acid signaling is required for normal liver regeneration. Science. 2006;312:233–236. doi: 10.1126/science.1121435. [DOI] [PubMed] [Google Scholar]
- 7.Higuchi H, Bronk SF, Takikawa Y, Werneburg N, Takimoto R, El-Deiry W, Gores GJ. The bile acid glycochenodeoxycholate induces trail-receptor 2/DR5 expression and apoptosis. The Journal of biological chemistry. 2001;276:38610–38618. doi: 10.1074/jbc.M105300200. [DOI] [PubMed] [Google Scholar]
- 8.Modica S, Gadaleta RM, Moschetta A. Deciphering the nuclear bile acid receptor FXR paradigm. Nuclear receptor signaling. 2010;8:e005. doi: 10.1621/nrs.08005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kemper JK, Xiao Z, Ponugoti B, Miao J, Fang S, Kanamaluru D, Tsang S, et al. FXR acetylation is normally dynamically regulated by p300 and SIRT1 but constitutively elevated in metabolic disease states. Cell metabolism. 2009;10:392–404. doi: 10.1016/j.cmet.2009.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fang S, Tsang S, Jones R, Ponugoti B, Yoon H, Wu SY, Chiang CM, et al. The p300 acetylase is critical for ligand-activated farnesoid X receptor (FXR) induction of SHP. The Journal of biological chemistry. 2008;283:35086–35095. doi: 10.1074/jbc.M803531200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Trinchet JC, Gerhardt MF, Balkau B, Munz C, Poupon RE. Serum bile acids and cholestasis in alcoholic hepatitis. Relationship with usual liver tests and histological features. Journal of hepatology. 1994;21:235–240. doi: 10.1016/s0168-8278(05)80401-5. [DOI] [PubMed] [Google Scholar]
- 12.Shlomai A, Halfon P, Goldiner I, Zelber-Sagi S, Halpern Z, Oren R, Bruck R. Serum bile acid levels as a predictor for the severity of liver fibrosis in patients with chronic hepatitis C. Journal of viral hepatitis. 2013;20:95–102. doi: 10.1111/j.1365-2893.2012.01628.x. [DOI] [PubMed] [Google Scholar]
- 13.Jang KY, Noh SJ, Lehwald N, Tao GZ, Bellovin DI, Park HS, Moon WS, et al. SIRT1 and c-Myc promote liver tumor cell survival and predict poor survival of human hepatocellular carcinomas. PloS one. 2012;7:e45119. doi: 10.1371/journal.pone.0045119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Herranz D, Munoz-Martin M, Canamero M, Mulero F, Martinez-Pastor B, Fernandez-Capetillo O, Serrano M. Sirt1 improves healthy ageing and protects from metabolic syndrome-associated cancer. Nature communications. 2010;1:3. doi: 10.1038/ncomms1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fickert P, Wagner M, Marschall HU, Fuchsbichler A, Zollner G, Tsybrovskyy O, Zatloukal K, et al. 24-norUrsodeoxycholic acid is superior to ursodeoxycholic acid in the treatment of sclerosing cholangitis in Mdr2 (Abcb4) knockout mice. Gastroenterology. 2006;130:465–481. doi: 10.1053/j.gastro.2005.10.018. [DOI] [PubMed] [Google Scholar]
- 16.Beraza N, Malato Y, Vander Borght S, Liedtke C, Wasmuth HE, Dreano M, de Vos R, et al. Pharmacological IKK2 inhibition blocks liver steatosis and initiation of non-alcoholic steatohepatitis. Gut. 2008;57:655–663. doi: 10.1136/gut.2007.134288. [DOI] [PubMed] [Google Scholar]
- 17.Nytofte NS, Serrano MA, Monte MJ, Gonzalez-Sanchez E, Tumer Z, Ladefoged K, Briz O, et al. A homozygous nonsense mutation (c. 214C->A) in the biliverdin reductase alpha gene (BLVRA) results in accumulation of biliverdin during episodes of cholestasis. Journal of medical genetics. 2011;48:219–225. doi: 10.1136/jmg.2009.074567. [DOI] [PubMed] [Google Scholar]
- 18.Fausto N, Campbell JS, Riehle KJ. Liver regeneration. Hepatology. 2006;43:S45–53. doi: 10.1002/hep.20969. [DOI] [PubMed] [Google Scholar]
- 19.Ghosh HS, McBurney M, Robbins PD. SIRT1 negatively regulates the mammalian target of rapamycin. PloS one. 2010;5:e9199. doi: 10.1371/journal.pone.0009199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Varela-Rey M, Beraza N, Lu SC, Mato JM, Martinez-Chantar ML. Role of AMP-activated protein kinase in the control of hepatocyte priming and proliferation during liver regeneration. Experimental biology and medicine. 2011;236:402–408. doi: 10.1258/ebm.2011.010352. [DOI] [PubMed] [Google Scholar]
- 21.Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, Puigserver P. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature. 2005;434:113–118. doi: 10.1038/nature03354. [DOI] [PubMed] [Google Scholar]
- 22.Trausch-Azar J, Leone TC, Kelly DP, Schwartz AL. Ubiquitin proteasome-dependent degradation of the transcriptional coactivator PGC-1{alpha} via the N-terminal pathway. The Journal of biological chemistry. 2010;285:40192–40200. doi: 10.1074/jbc.M110.131615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Laplante M, Sabatini DM. mTOR signaling at a glance. Journal of cell science. 2009;122:3589–3594. doi: 10.1242/jcs.051011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jin J, Iakova P, Jiang Y, Medrano EE, Timchenko NA. The reduction of SIRT1 in livers of old mice leads to impaired body homeostasis and to inhibition of liver proliferation. Hepatology. 2011;54:989–998. doi: 10.1002/hep.24471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kemper JK. Regulation of FXR transcriptional activity in health and disease: Emerging roles of FXR cofactors and post-translational modifications. Biochimica et biophysica acta. 2011;1812:842–850. doi: 10.1016/j.bbadis.2010.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Guillemette B, Drogaris P, Lin HH, Armstrong H, Hiragami-Hamada K, Imhof A, Bonneil E, et al. H3 lysine 4 is acetylated at active gene promoters and is regulated by H3 lysine 4 methylation. PLoS genetics. 2011;7:e1001354. doi: 10.1371/journal.pgen.1001354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Imai S, Armstrong CM, Kaeberlein M, Guarente L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature. 2000;403:795–800. doi: 10.1038/35001622. [DOI] [PubMed] [Google Scholar]
- 28.Ananthanarayanan M, Li Y, Surapureddi S, Balasubramaniyan N, Ahn J, Goldstein JA, Suchy FJ. Histone H3K4 trimethylation by MLL3 as part of ASCOM complex is critical for NR activation of bile acid transporter genes and is downregulated in cholestasis. American journal of physiology Gastrointestinal and liver physiology. 2011;300:G771–781. doi: 10.1152/ajpgi.00499.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Choi SE, Fu T, Seok S, Kim DH, Yu E, Lee KW, Kang Y, et al. Elevated microRNA-34a in obesity reduces NAD levels and SIRT1 activity by directly targeting NAMPT. Aging cell. 2013 doi: 10.1111/acel.12135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Anderson RM, Barger JL, Edwards MG, Braun KH, O’Connor CE, Prolla TA, Weindruch R. Dynamic regulation of PGC-1alpha localization and turnover implicates mitochondrial adaptation in calorie restriction and the stress response. Aging cell. 2008;7:101–111. doi: 10.1111/j.1474-9726.2007.00357.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Back JH, Rezvani HR, Zhu Y, Guyonnet-Duperat V, Athar M, Ratner D, Kim AL. Cancer cell survival following DNA damage-mediated premature senescence is regulated by mammalian target of rapamycin (mTOR)-dependent Inhibition of sirtuin 1. The Journal of biological chemistry. 2011;286:19100–19108. doi: 10.1074/jbc.M111.240598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gao Z, Zhang J, Kheterpal I, Kennedy N, Davis RJ, Ye J. Sirtuin 1 (SIRT1) protein degradation in response to persistent c-Jun N-terminal kinase 1 (JNK1) activation contributes to hepatic steatosis in obesity. The Journal of biological chemistry. 2011;286:22227–22234. doi: 10.1074/jbc.M111.228874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen H, Fan M, Pfeffer LM, Laribee RN. The histone H3 lysine 56 acetylation pathway is regulated by target of rapamycin (TOR) signaling and functions directly in ribosomal RNA biogenesis. Nucleic acids research. 2012;40:6534–6546. doi: 10.1093/nar/gks345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Song NY, Surh YJ. Janus-faced role of SIRT1 in tumorigenesis. Annals of the New York Academy of Sciences. 2012;1271:10–19. doi: 10.1111/j.1749-6632.2012.06762.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang F, Huang X, Yi T, Yen Y, Moore DD, Huang W. Spontaneous development of liver tumors in the absence of the bile acid receptor farnesoid X receptor. Cancer research. 2007;67:863–867. doi: 10.1158/0008-5472.CAN-06-1078. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.