

Abstract
Non-nucleoside Reverse Transcriptase Inhibitors (NNRTIs) are potent anti-HIV chemotherapeutics. Although there are FDA-approved NNRTIs, challenges such as the development of resistance have limited their utility. Here we describe the identification of novel NNRTIs through a combination of computational and experimental approaches. Based on the known plasticity of the NNRTI binding pocket (NNIBP), we adopted an ensemble-based virtual screening strategy: coupling receptor conformations from 10 x-ray crystal structures with 120 snapshots from a total of 480 ns of Molecular Dynamics (MD) trajectories. A screening library of 2,864 National Cancer Institute (NCI) compounds was built and docked against the ensembles in a hierarchical fashion. 16 diverse compounds were tested for their ability to block HIV infection in human tissue cultures using a luciferase-based reporter assay. 3 promising compounds were further characterized, using a HIV-1 RT based polymerase assay, to determine the specific mechanism of inhibition. We found that 2 of the 3 compounds inhibited the polymerase activity of RT (with potency similar to the positive control, the FDA-approved drug nevirapine). Through a computational approach, we were able to discover 2 compounds which inhibit HIV replication and block the activity of RT, thus offering the potential for optimization into mature inhibitors.
Keywords: HIV, reverse transcriptase, NNRTI, molecular dynamics, virtual screening
Introduction
HIV-1 Reverse Transcriptase (RT) is an important target of drugs that block the replication of Human Immunodeficiency Virus (HIV). HIV-1 reverse transcriptase (RT) catalyzes the conversion of the single-stranded RNA retroviral genome into a double-stranded linear DNA, which is then integrated into the human genome. Currently, there are two main classes of small molecule inhibitor that block this process: nucleoside RT inhibitors (NRTIs, which lack a 3′ OH and, when converted to the triphosphate form by cellular enzymes and incorporated into viral DNA, block DNA synthesis) and non-nucleoside RT inhibitors (NNRTIs, which act at an allosteric site and displace the end of viral DNA from the polymerase active site, blocking polymerization) (1). We have focused NNRTIs, because the allosteric binding pocket confers a higher degree of selectivity/specificity (to avoid harmful off-target activity) and because there is a large chemical space for drug design (2, 3). Although both major classes are used in combination as highly active antiviral therapies (HAART), HIV infections cannot be cured and infected patients have to follow these regimens for the duration of their lives; this places a considerable premium on developing inhibitors that have little or no long-term toxic effects.
Despite the approval of five NNRTIs by the FDA-(nevirapine, efavirenz, delavirdine, etravirine and rilpivirine), there is still a pressing need to discover novel NNRTI chemotypes, largely to mitigate the problems associated with the development of resistance. Nevirapine, the first FDA approved NNRTI, selects for resistance after one dose in a large number of infected patients (4). The hydrophobic NNRTI binding site is not evolutionarily conserved and many residues can be altered in ways that disrupt the binding of the first generation NNRTIs with only a very modest effect on the ability of the virus to replicate.
In this report, we describe the use of computational drug discovery techniques to identify new chemical “hits” that could be experimentally validated and potentially optimized to yield mature NNRTIs. Computational discovery of new NNRTIs has a relatively rich history, largely owed to the abundant structural database that is publically available in the Protein Databank (5). This work includes a variety of ligand-based and structure-based methodologies, including pharmacophore modeling, similarity optimization, molecular docking and free energy perturbation methods (3, 6–11) that can be used both for identifying hits and for lead optimization.
Because the NNRTI binding pocket is quite malleable, many of these computational studies have included sampling of the protein degrees of freedom through the use of multiple crystal structures, Monte Carlo sampling and pharmacophore models. Our approach used the dynamic structural information provided by molecular dynamics (MD) simulations, through a technique known as the relaxed complex scheme (RCS). This method uses multiple molecular dynamics snapshots to represent a dynamic ensemble of receptor coordinates for molecular docking (12, 13). The RCS has been used to identify hits for a number of molecular targets (14–17). This is an extension of our previous work on the elucidation of the inhibition mechanism of NNRTIs through MD simulations (18) to identify novel chemotypes that bind in the non-nucleoside binding pocket.
Methods and Materials
Virtual screening library
A library of 2,864 screening compounds that are available from the National Cancer Institute (NCI) was assembled using a combination of general chemical diversity and similarity to a selection of next-generation NNRTIs. The “diversity subset”, comprising 1,364 compounds, was taken directly from the preassembled NCI Diversity Set 2 (NCIDS-2: http://dtp.cancer.gov). The “similarity subset”, comprising 1,500 compounds, was built by querying the entire NCI repository for structures similar to a set of six leading NNRTIs currently under development by various companies (Figure 1). Similarity searching of the complete NCI screening library was carried out with Accelrys Discovery Studio, using a fingerprint-based method (http://www.accelrys.com). The aim of this combinatorial approach was to improve the probability of discovering active compounds, given the relatively small screening library. A library size of ~3,000 molecules was deemed tractable considering the computational demands of both the chosen docking algorithm and the multi-structure nature of ensemble-based docking.
Figure 1.
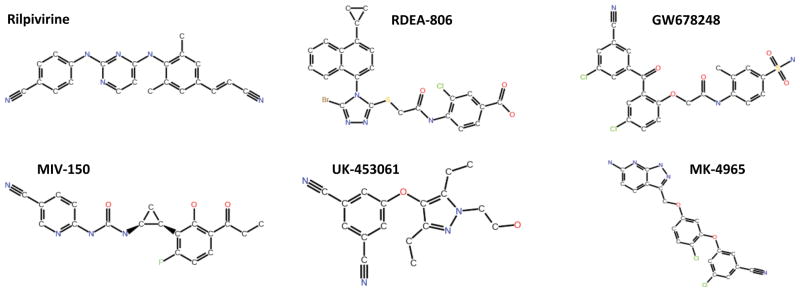
Six second-generation NNRTIs that were used to generate the “similarity subset” of the virtual screening library. Rilpivirine (Tibotec), RDEA-806 (Ardea), GW678248 (GSK), MIV-150 (Medivir/Chiron), UK-453061 (Pfizer) and MK-4965 (Merck).
Virtual screening workflow
A hierarchical scheme was used to identify a small number of the NCI compounds that could be tested experimentally for their ability to inhibit the polymerase activity of HIV-1 RT (Figure 2). In the first step, a set of 10 crystal structures of wild-type HIV-1 RT complexed with different NNRTIs was compiled. This crystallographic ensemble comprised the following PDB codes: 1VRT, 1HNV, 1RT1, 1VRU, 1FK9, 1EP4, 1BQM, 1KLM, 1RT4 and 2ZD1. These structures represent a structurally diverse collection of NNRTIs and consequently there is considerable variation in the conformation of the NNRTI Binding Pocket (NNIBP) (see Figure 3). The screening library was docked into each of the crystal structures and a mean binding energy was calculated for each compound across the ensemble. The library was then ranked by the mean binding score, to select for compounds that can adapt to, and bind to, diverse conformations of the NNIBP, as opposed to binding preferentially to a specific conformation. The top 150 compounds from this crystallographic screen (~5% of the screening library) were then moved forward to a secondary screen against conformations of the NNIBP extracted from MD simulations. This subset of the compounds represented a range of binding energies from −12.3 kcal/mol to −9.9 kcal/mol. Each compound was docked to 30 representative snapshots from each of four simulation systems. A weighted-mean binding energy was calculated for each compound in each simulation system, whereby the mean was weighted by the population size of the cluster from which the snapshot was extracted (12). Such a consensus scoring system gives more weight to compounds that bind well in more the common conformations of the binding site, as opposed to an unweighted mean which does not discriminate between common and rare conformers. A final re-scoring of the 150 compounds was achieved by taking the mean rank of each compound across all simulation systems.
Figure 2.

Overview of the virtual screening process used to select 16 compounds for experimental testing from a library of 2,864 compounds.
Figure 3.

Snapshots of the 10 crystal structures of HIV-1 RT used in the crystallographic ensemble, with the co-crystallized bound NNRTI. The NNIBP is depicted in molecular surface representation, colored by residue index. NNRTIs are shown in blue stick representation. Note the heterogeneity of the chemical structures that bind to this allosteric site.
Molecular docking
Virtual screening was carried out with components of Schrödinger Suite 2010. Receptor conformations (both crystallographic and from MD simulations) had all non-protein atoms eliminated prior to docking and were processed using the standard protein preparation workflow found within Maestro. All compounds were prepared with the LigPrep routine, including 3D conformer generation and protonation. Glide XP (Extra Precision) was used for all molecular docking, with only the lowest binding energy pose retained from each run (19). The GlideScore function was used to calculate ligand binding free energies.
Molecular dynamics (MD) simulations
Four different RT:drug complexes were simulated using the GROMACS simulation software, comprising the following NNRTIs: Nevirapine (NVP), 8-Cl TIBO (TBO), α-APA (APA) and UC-781 (UC1). For consistency, the starting protein coordinates for each system were taken from PDB code 1VRT (RT:Nevirapine complex). All MD was performed with GROMACS v.3.3.1 (20–22), using the GROMOS 53A6 force field (23, 24). Each protein-drug complex was bathed in explicit water, using a simple point charge (SPC) water model (25), yielding system sizes of ~160,000 atoms. A multi-copy simulation approach was adopted to enhance conformational sampling, whereby four independent trajectories of 30 ns were generated for each of the four systems. Further details of the simulation protocol have been described previously (18). Snapshots of the simulation systems can be seen in Figure 4.
Figure 4.

Snapshots of the MD simulations, comprising the complete HIV-1 RT heterodimer complexed with 4 diverse NNRTIs and bathed in a solvent of explicit water molecules and counter-ions to neutralize the net charge. HIV-1 RT shown in a ribbon representation, with the p66 and p51 subunits colored blue and red, respectively. A closeup view of the NNIBP, with the 4 NNRTIs superposed, in stick representation is shown in the inset.
In order to generate a tractable, representative set of diverse RT conformations from the MD simulations, a root-mean-square deviation (RMSD) based clustering was performed, using the “g_cluster” GROMACS tool. The aim of this step is to cluster MD snapshots with similar binding site conformations, so as to capture key movements in as few structures as possible. For each system, the four individual 30 ns trajectories were concatenated into a single 120 ns trajectory. Clustering was then performed on a set of 15 residues that form the NNIBP: Leu-100, Lys-101, Lys-103, Val-106, Val-179, Tyr-181, Tyr-188, Val-189, Gly-190, Phe-227, Trp-229, Leu-234, His-235, Pro-236 and Tyr-318. Using the motion of all heavy atoms in each binding site residue (backbone and sidechain), a RMSD cutoff was selected to allow for 30 clusters to be defined for each system. Each cluster centroid was then extracted from the MD trajectory as the representative of that cluster and used in virtual screening.
DNA constructs and virus production
The VSVg-pseudotyped HIV-1 vector was generated by transient transfection of human 293T cells (American Type Culture Collection No. CRL-11268), with the vector plasmid pNL4-3LucR+E− (26) and the VSVg glycoprotein plasmid pMD.G. The titer was determined by antibody staining for Gag (p24) expressing cells following infection of 293T cells. JM1186, an NL4-3 Nef+ IRES rluc vector encoding renilla luciferase was derived from NL4-3 Nef+ IRES eGFP vector (27) by replacement of the eGFP open reading frame with renilla luciferase (kindly provided by Sumit Chanda’s lab, Sanford-Burnham Medical Institute, La Jolla).
Tissue culture-based infectivity assays
For the initial screen of the ability of the compounds to block HIV-1 replication, ten thousand human 293T cells were plated in 80 μl DMEM + 10% FBS medium in each well of 96-well tissue culture plate. The next day, 10 μl of a solution of diluted compound was added to give a final concentration of 5 μM and the cells were incubated at 37°C for 1 hour. As a control, 5 μl of a nevirapine solution was added to give a final concentration of 10 μM and the cells incubated at 37°C for 1 hour. A 10 μl aliquot of medium containing the VSVg-pseudotyped HIV-1 vector (giving a multiplicity of infection of 0.15) was then added to each well. Twenty-four hours after the viral challenge, 100 μl of the Bright-Glo reagent (Promega, Madison, WI) was added to the medium to lyse the cells and provide the luciferin substrate for virus-encoded firefly luciferase, the level of luciferase was measure using a Topcount-HTS (PerkinElmer). After ten minutes, the luminescence associated with each sample was measured and used to quantify virus infectivity in each well. Cytotoxicity of the compounds was measured 24 hours after treatment of mock-infected cells by adding an equal volume of CellTiter-Glo (Promega, Madison, WI) and reading luminescence.
To determine the effect of the compounds on the expression of the HIV provirus, 293T cells were seeded, infected as described above, and passaged for 7 days to allow loss of most of the non-integrated forms of viral DNA. Ten thousand cells were then plated in each well of a 96-well tissue culture plate and the following day appropriate dilutions of each compound were added to give a final concentration of 5 μM and the cells were incubated at 37°C for 24 hours. HIV luciferase activity and cytotoxicity were determined as described above. In parallel plates, DNA was isolated and viral DNA was quantitated by qPCR as described in the next section.
For the assays employing the replication-competent HIV-1 vector JM1186, forty thousand human monocytic leukemia cells (THP-1) engineered to stably express the CD4 and CCR5 receptors were plated in each well. THP-1 cells were grown in RPMI 1640 with 10% FBS, Pen/Strep, 0.5 μg/ml and were differentiated by exposure to 5 ng/ml phorbol-12-myristate-13-acetate (PMA). Three days after activation, the test compounds were added to give a final concentration of 5 μM, as described above, and cells were incubated at 37°C for 1 hour prior to virus challenge. The cells were then mixed with 15 μl per well of the NL4-3 Nef+ IRES rluc virus vector (0.05 MOI) and spinoculated at 1200xg for 1 hour at 4°C. Plates were incubated at 37°C for 24 or 48 hours and viral infection was monitored by adding 100 μl per well of Renilla-Glo Luciferase Reagent (Promega) and measuring luciferase activity. Cell toxicity was assessed on a duplicate plate of samples by adding 100 μl Cell-Titer Glo Reagent. In parallel plates, DNA was isolated and viral DNA was quantitated by qPCR as described in the next section.
Real-time PCR quantification of viral DNA
Total cellular DNA was harvested 24 or 48 hours post infection from infected THP-1 cells using q-PCR lysis buffer (0.1M Tris-HCl pH8, 0.05M EDTA, 0.1M CaCl2, 1% Triton-X100, 1% SDS). Cells were washed with PBS, 50 μl of qPCR-lysis buffer was added to each well and the cells were dislodged from the plate by pipetting up/down five times. The cell lysis solution was added to a 96-well PCR plate and incubated for 58°C for 1 hour, followed by heat inactivation for 10 minutes at 95°C. Late HIV-1 reverse transcripts were quantified using real-time PCR and the ABI Prism 7900 Sequence Detection System (Applied Biosystems, Foster City, CA) with the following primers and probes: primers MH531, MH532, probe LRT-P (28). To normalize for the number of cellular DNA equivalents in the samples, a segment of the single-copy PBGD gene was amplified with primers PBGD1 (5′-AAGGGATTCACTCAGGCTCTTTC), PBGD2 (5′-GGCATGTTCAAGCTCCTTGG) and probe PBGD-P (5′-VIC-CCGGCAGATTGGAGAGAAAAGCCTGT-MGBNFQ).
HIV-1 RT enzyme assay
The purification of HIV-1 p66/p51 RT heterodimers has previously been described (29). The RT is from the B subtype HIV strain BH10 (Genebank HIVBH102). The NNRTI assay is similar to assays we have previously described (see above reference). Briefly, the −47 sequencing primer (5′- CGCCAGGGTTTTCCCAGTCACGAC-3′; New England Biolabs) was 5′ end-labeled with [γ-32P]ATP and T4 polynucleotide kinase. After purification, the labeled primer was annealed to single-stranded M13mp18 DNA (1.0 μg of DNA for each sample to be assayed) by heating and slow cooling. For each sample, 0.1 μg of wild-type RT (~17 nM final) added to the labeled primer template (~9.0 nM) in 25 mM Tris-Cl (pH 8.0), 75 mM KCl, 10.0 mM MgCl2, 100 μg of BSA per ml, and 10.0 mM CHAPS. The reaction mixture was supplemented with 0.5 μM (each) of dATP, dCTP, dGTP, and dTTP. The NNRTIs were added to a final concentration of 0, 0.0156, 0.0313, 0.0625, 0.125, 0.25,0.5, or 1.0 μM. The reactions were allowed to proceed at 37° for 60 min and were then halted by the addition of EDTA. The samples were precipitated by the addition of two volumes of ethanol, fractionated by electrophoresis on a 6.0% polyacrylamide gel, and the gel was autoradiographed. Phosphoimaging was used to determine the amount of signal in each lane. Primer extension products >90 nt in length were considered full length product. The percentage of the full length product was calculated for each reaction condition, then plotted. Reactions were done in duplicate.
Results and Discussion
Virtual screening
Using a primary screen based on the crystallographic ensemble of 10 RT structures, we were able to discard 95% of the compounds in the original library, retaining only the 150 compounds which docked in favorable poses across multiple receptor conformations. In a secondary screen, we re-ranked these compounds by their performance against an ensemble generated from a series of MD simulations. The highest ranked compounds thus represented molecules that were predicted to be able to adapt well to a highly dynamic binding site. The 50 highest re-ranked compounds from the secondary MD screen were chosen for additional testing. To reduce redundancy, the compounds were clustered into structurally similar groups, using Accelrys Discovery Studio (http://www.accelrys.com). A total of 20 compounds were picked, with the aim of selecting diverse chemotypes (see Figure 5). Of the final 20 compounds, 11 were derived from the similarity dataset and 9 from the diversity dataset. Only 16 of the 20 compounds were available, and these compounds were assayed using cell and enzyme-based methods.
Figure 5.

The 20 NCI compounds selected for experimental testing. Compounds are labeled according to the screening subset they came from: Diversity Set (D) and Similarity Set (S). Compounds marked with an asterisk were not available.
Cell-based assay
The effects of the compounds on HIV-1 replication in cultured cells was measured in a plate-based assay using a VSV g-pseudotyped HIV-1 vector (pNL4-3LucR+E−) encoding firefly luciferase (26). Human 293T cells were pretreated for 1 hr with 5 μM final concentration of the individual compounds and then challenged with the VSVg pseudotyped HIV-1 vector in the continued presence of that compound. 24 hours post-infection (hpi), the luciferase activity of each sample was determined. Of sixteen compounds tested under these conditions, three caused a 2-fold or greater decrease in luciferase activity compared to the DMSO control (Figure 6A). Moreover, at the 5 μM concentration used, none of the compounds showed any evidence of toxicity (Figure 6B). Because the initial screen was performed by scoring the expression of a firefly luciferase reporter gene from a VSVg-pseudotyped virus vector, it was possible that compounds NSC44556, NSC294378, NSC366102 inhibited either VSVg-mediated cell entry, an early step of HIV-1 replication, or firefly luciferase reporter activity. To differentiate among these possibilities, compounds NSC44556, NSC294378, NSC366102 were further tested in a THP-1 monocytic cell line engineered to stably express the cellular receptors and co-receptors that support virus entry. Nevirapine (NVP), a known NNRTI, was included as a positive control. THP-1 cells were pretreated for 1 hr with the compounds and infected with a replication-competent HIV-1 vector with a wild-type CCR5-tropic envelope glycoprotein. Since THP-1 cells express SAMHD1, a deoxynucleoside triphosphate triphosphohydrolase that reduces the available pool of deoxynucleotides present in the cytoplasm, the kinetics of HIV-1 are delayed compared to those in 293T cells (30). As seen in Figure 7A, luciferase levels were only slightly reduced at 24 hpi, even in cells treated with nevirapine. However, at 48 hpi, compound NSC366102 reduced luciferase levels by 85% compared to 75% and 55% for compounds NSC44556 and NSC294378, respectively.
Figure 6.

Effects of 16 compounds on HIV-1 infectivity. (A) The effects of the compounds (5 μM) on HIV-1 infectivity were determined by measuring firefly luciferase activity at 24 hours post-infection (hpi) in human 293T cells infected with a VSVg-pseudotyped HIV-1 vector encoding the luciferase reporter. DMSO was used as the control. (B) Cytotoxicity of the compounds was measured in mock-infected cells after treatment of the cells with the compounds for 24 hours. The experiments represent the mean of at least three independent experiments each performed in triplicate. The error bars represent the standard errors of the mean.
Figure 7.

Three compounds block an early step of HIV-1 replication. (A) Effect of three compounds (5 μM) in PMA differentiated THP-1 monocytes challenged with the NL4-3 Nef+ IRES rluc vector encoding renilla luciferase. Luciferase activity was measured at 24 or 48 hpi. Nevirapine (NVP, 10 μM) and DMSO were used as controls. Error bars show standard error mean from three independent experiments each performed in triplicate. (B) Effect of compounds on late HIV-1 reverse transcription products at 24 or 48 hpi. The values represent amounts of viral DNA relative to DMSO treated cell populations and were normalized to cellular GAPDH DNA levels. The data shown are the mean average values obtained from 3 independent experiments that were each performed in triplicate. The error bars represent the standard error.
To determine if compounds NSC44556, NSC294378, and NSC366102 affect viral DNA synthesis, a quantitative real-time PCR-amplification approach was used. Total DNA was isolated from cells at 24 and 48 hrs post infection and the amount of viral DNA synthesized was quantified with primers and a probe specific for late HIV-1 reverse transcription products (28). Under these conditions, compound NSC366102 reduced late RT products by 50% at 24 hpi and by 90% at 48 hpi (Figure 7B), suggesting that this compound hinders HIV-1 reverse transcription. Compound NSC44556 reduced late RT products by 36% and 69% at 24 and 48 hpi, respectively, while compound NSC294378 had only a modest effect (Figure 7B).
To further ensure that the compounds were affecting reverse transcription and not viral gene expression, the compounds were tested for their ability to affect the expression of previously established integrated proviral DNA. 293T cells were infected with the single-cycle pNL4-3LucR+E− virus and the cells were passaged for 7 days to reduce the amount of non-chromosomal forms of viral DNA. Compounds NSC44556, NSC294378, and NSC366102 were added to cells at a final concentration of 5 μM and 24 hours later the luciferase activity and the levels of viral DNA were measured. The compounds did not affect luciferase expression from integrated HIV-1 DNA (Figure 8).
Figure 8.

Compounds had no effect on gene expression from integrated HIV-1. 293T cells that contain an integrated vector DNA that carries and expresses firefly luciferase were treated with 5 μM of the compounds or 10 μM of NVP. Virus encoded firefly luciferase or late viral DNA was measured at 24 hpi. Error bars represent SEM of three independent luciferase or RT-PCR measurements.
Effects of the compounds on the polymerase activity of RT
Although the compounds were designed to bind to RT and were shown to inhibit HIV-1 DNA synthesis in cultured cells, this is not direct proof that they bind to, or directly affect, the polymerase activity of RT. The three compounds which showed promising results in the cell culture assays were tested for their effects on the polymerase activity of purified HIV-1 RT. One of the problems in assaying NNRTIs is that they bind to RT, blocking the chemical step of DNA synthesis. Unless the concentration of the compound is relatively high, there are two populations of RT: a non-bound, fully active RT set and a bound, inhibited RT set. If the reaction conditions are chosen such that the uninhibited (unliganded) RTs can freely bind to the template:primer and make relatively long DNA products, it can be difficult to accurately determine whether or not a subset of inhibited RTs is present in the reaction. To address this problem, we designed an in vitro assay so that the RTs in the assay repeatedly disassociate and reassociate with the same template:primer. Because the binding of an NNRTI does not impair the ability of RT to bind to a nucleic acid substrate (31, 32), both uninhibited and inhibited RTs will bind during the synthesis of individual DNAs. As the fraction of non-extending, NNRTI-bound RTs increases, the length of the DNA products will decrease. To accomplish this, we chose a long DNA template as the substrate (single-stranded, circular M13mp18 DNA), and a relatively low concentration of dNTPs (0.5 μM each dNTP), which will prevent the active RTs from making long products before they dissociate from the template. HIV-1 RT was present in the reactions at a final concentration of 17 nM and the reactions were allowed to proceed for 60 min at 37°C. Nevirapine was included as a positive control. As can be seen in Figure 9, adding NSC44556 and NSC366102 to the reactions generated inhibition curves that are similar to that obtained for Nevirapine. The amount of compound that would give a 50% reduction in the amount of the full length product was approximately 60 nM in both cases. These data show that these compounds directly inhibit the polymerase activity of HIV-1 RT. NSC294378 only had a slight effect on the polymerase activity of HIV-1 RT. Of the three compounds tested, NSC294378 had the smallest impact on HIV-1 replication in the in vivo experiments described above. Even if it does bind to the HIV-1 RT, it is the weakest of the compounds, which matches the data obtained with purified RT.
Figure 9.

Polymerase inhibition assay. As described in the Materials and Methods section, the three compounds that showed inhibitory activity in the cell-based assays were tested for their ability to inhibit the polymerase activity of HIV-1 RT. A radioactive primer annealed to a long template was extended by HIV-1 RT in the presence of varying concentrations of the compounds (the amount of DMSO was constant in all of the reactions), appropriate buffer, and 0.5 μM each dNTP. Nevirapine was included as a positive control for NNRTI inhibition. The reactions were allowed to proceed at 37° for 60 min and were then halted by the addition of EDTA. The samples were fractionated by electrophoresis on a 6.0% polyacrylamide gel, and the gel was autoradiographed. Phosphoimaging was used to determine the amount of signal in each lane. Primer extension products >90 nt in length were considered full length product. The percentage of the full length produced in each of the reaction conditions was calculated, then plotted. Reactions were done in duplicate.
Active compounds
The two compounds which caused RT-mediated inhibition of HIV replication have structural similarities to other compounds known to be active against RT. Upjohn laboratories identified and then detailed modifications of pyrimidine thioethers (33, 34). Bioisosteric replacement resulted in the clinical candidate PNU-142721, which potently inhibited wild-type HIV-1 RT and several RT mutants (35). More recently, difluoromethylbenzoxazole (DFMB) pyrimidine thioether derivatives were described that are potent inhibitors of wild-type RT and are moderately active against various mutants (36). NSC366102 contains a benzophenone, and compounds in this class can be potent and effective against a variety of RT mutants (37, 38). To the best of our knowledge, neither the pyrimidinone thoiether nor the benzophenone reported in this paper has been described previously as RT inhibitors.
Predicted poses of the two active compounds are shown in Figure 10, docked using the 1RT4 protein structure. Compounds were also docked into 1VRT, and similar orientations were obtained (results not shown). In the models, the pyrimidone of NSC44556 interacts with the backbone of lysine 101 and potentially with glutamine 138, whereas in published crystal structures of DFMB pyrimidine thioethers (2YKM, 2YKN) the pyrimidine near lysine 101 is slightly rotated towards valine 106 (36). It is interesting to note that a water molecule was co-crystallized in the binding pocket in both crystal forms, which may have affected the orientation of the pyrimidine. NSC366102 shows an intramolecular hydrogen bond between the carbonyl of the benzophenone with the secondary amine present in the linker. A similar orientation of the benzophenone is seen in the six benzophenone-RT co-crystal structures that have been determined (3DLE, 3DLG, 3DM2, 3DMJ, 3DOK, 3DOL), which also indicate an intramolecular hydrogen bond, but with an amide in the linker, as well as the C-ring pointing to the enzyme solvent interface (39). Solvent accessibility is indicated in the ligand interaction diagrams for the predicted poses (Figure 10). It has been suggested that the C-ring in benzophenone-based NNRTIs lies in the enzyme-solvent interface and that it could be optimized to gain specificity, improve pharmacokinetic properties, or to construct bifunctional inhibitors (3). Given that the two promising compounds we identified had very similar potency to the FDA-approved nevirapine, and that they also have similarities to other known inhibitors that have been shown to inhibit the replication of some important drug resistant viruses, characterization of the compounds against known RT mutants as a step towards potency optimization is warranted..
Figure 10.

Proposed binding modes of the 2 confirmed HIV-1 RT inhibitors that were active in enzyme and cell-based assays. Active compounds are shown in predicted poses based on docking into 1RT4 (top). Protein backbone is depicted as ribbons, and residues within 5 Å of the binding site are depicted as sticks. Intermolecular and intramolecular hydrogen bonding are denoted with black dashed lines. Two-dimensional ligand interaction diagrams (bottom) indicate predicted proximal residues for each of the inhibitors.
Conclusions
We set out to identify novel compounds that act as NNRTIs, using a combination of computational and experimental approaches that were based on previous molecular dynamics simulations. Taking into account the plasticity of the allosteric binding pocket and using a large collection of crystallographic data, we adopted an ensemble-based virtual screening strategy. Screening of 2,864 publically available compounds led to the testing of sixteen diverse small molecules and the identification of two compounds that block the replication of HIV in infected cells that inhibit polymerase activity of HIV-1 RT at a concentration of 60 nM.
Acknowledgments
We acknowledge the San Diego Supercomputing Center, the Center for Theoretical Biological Physics, and the National Biomedical Computational Resource for computational resources and the National Science Foundation, the National Institutes of Health and Howard Hughes Medical Institute for financial support. We also acknowledge the Nomis, Auen and Morris Foundations and the James B. Pendleton Charitable Trust for their support. This study was supported in part by the Intramural Research Program of the National Institutes of Health (NIH), National Cancer Institute, Center for Cancer Research. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
References
- 1.Hu WS, Hughes SH. Hiv-1 Reverse Transcription. Csh Perspect Med. 2012 Oct;2(10) doi: 10.1101/cshperspect.a006882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Das K, Arnold E, Hughes S. Nonnucleoside Reverse Transcriptase Inhibitors (NNRTIs) In: LeGrice S, Gotte M, editors. Human Immunodeficiency Virus Reverse Transcriptase. Springer; New York: 2013. pp. 123–39. [Google Scholar]
- 3.Zhan P, Chen X, Li D, Fang Z, De Clercq E, Liu X. HIV-1 NNRTIs: structural diversity, pharmacophore similarity, and implications for drug design. Med Res Rev. 2013 Jun;33( Suppl 1):E1–72. doi: 10.1002/med.20241. [DOI] [PubMed] [Google Scholar]
- 4.Palmer S, Boltz V, Martinson N, Maldarelli F, Gray G, McIntyre J, et al. Persistence of nevirapine-resistant HIV-1 in women after single-dose nevirapine therapy for prevention of maternal-to-fetal HIV-1 transmission. Proc Natl Acad Sci U S A. 2006 May 2;103(18):7094–9. doi: 10.1073/pnas.0602033103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, et al. The Protein Data Bank. Nucleic Acids Res. 2000 Jan;28(1):235–42. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barreiro G, Guimaraes CRW, Tubert-Brohman I, Lyons TM, Tirado-Rives J, Jorgensen WL. Search for non-nucleoside inhibitors of HIV-1 reverse transcriptase using chemical similarity, molecular docking, and MM-GB/SA scoring. J Chem Inf Model. 2007 Nov-Dec;47(6):2416–28. doi: 10.1021/ci700271z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nichols S, Domaoal R, Thakur V, Tirado-Rives J, Anderson K, Jorgensen W. Discovery of wild-type and Y181C mutant non-nucleoside HIV-1 reverse transcriptase inhibitors using virtual screening with multiple protein structures. J Chem Info Model. 2009 May;49(5):1272–9. doi: 10.1021/ci900068k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barreca ML, Rao A, De Luca L, Zappala M, Monforte AM, Maga G, et al. Computational strategies in discovering novel non-nucleoside inhibitors of HIV-1 RT. J Med Chem. 2005 May 5;48(9):3433–7. doi: 10.1021/jm049279a. [DOI] [PubMed] [Google Scholar]
- 9.Jorgensen WL, Ruiz-Caro J, Tirado-Rives J, Basavapathruni A, Anderson KS, Hamilton AD. Computer-aided design of non-nucleoside inhibitors of HIV-1 reverse transcriptase. Bioorg Med Chem Lett. 2006 Feb;16(3):663–7. doi: 10.1016/j.bmcl.2005.10.038. [DOI] [PubMed] [Google Scholar]
- 10.Bollini M, Domaoal RA, Thakur VV, Gallardo-Macias R, Spasov KA, Anderson KS, et al. Computationally-Guided Optimization of a Docking Hit to Yield Catechol Diethers as Potent Anti-HIV Agents. J Med Chem. 2011 Dec 22;54(24):8582–91. doi: 10.1021/jm201134m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ekkati AR, Bollini M, Domaoal RA, Spasov KA, Anderson KS, Jorgensen WL. Discovery of dimeric inhibitors by extension into the entrance channel of HIV-1 reverse transcriptase. Bioorg Med Chem Lett. 2012 Feb 15;22(4):1565–8. doi: 10.1016/j.bmcl.2011.12.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Amaro RE, Baron R, McCammon JA. An improved relaxed complex scheme for receptor flexibility in computer-aided drug design. J Comput Aid Mol Des [Article] 2008 Sep;22(9):693–705. doi: 10.1007/s10822-007-9159-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lin JH, Perryman AL, Schames JR, McCammon JA. The relaxed complex method: Accommodating receptor flexibility for drug design with an improved scoring scheme. Biopolymers. 2003 Jan;68(1):47–62. doi: 10.1002/bip.10218. [DOI] [PubMed] [Google Scholar]
- 14.Chan AH, Wereszczynski J, Amer BR, Yi SW, Jung ME, McCammon JA, et al. Discovery of Staphylococcus aureus Sortase A Inhibitors Using Virtual Screening and the Relaxed Complex Scheme. Chem Biol Drug Des. 2013 May 23; doi: 10.1111/cbdd.12167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cheng LS, Amaro RE, Xu D, Li WW, Arzberger PW, McCammon JA. Ensemble-based virtual screening reveals potential novel antiviral compounds for avian influenza neuraminidase. J Med Chem. 2008 Jul 10;51(13):3878–94. doi: 10.1021/jm8001197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Amaro RE, Schnaufer A, Interthal H, Hol W, Stuart KD, McCammon JA. Discovery of drug-like inhibitors of an essential RNA-editing ligase in Trypanosoma brucei. P Natl Acad Sci USA. 2008 Nov 11;105(45):17278–83. doi: 10.1073/pnas.0805820105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Durrant JD, Cao R, Gorfe AA, Zhu W, Li J, Sankovsky A, et al. Non-bisphosphonate inhibitors of isoprenoid biosynthesis identified via computer-aided drug design. Chem Biol Drug Des. 2011 Sep;78(3):323–32. doi: 10.1111/j.1747-0285.2011.01164.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ivetac A, McCammon JA. Elucidating the inhibition mechanism of HIV-1 non-nucleoside reverse transcriptase inhibitors through multicopy molecular dynamics simulations. J Mol Biol. 2009 May 8;388(3):644–58. doi: 10.1016/j.jmb.2009.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Friesner RA, Murphy RB, Repasky MP, Frye LL, Greenwood JR, Halgren TA, et al. Extra Precision Glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J Med Chem. 2006 Oct;49(21):6177–96. doi: 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
- 20.Berendsen HJC, Vanderspoel D, Vandrunen R. Gromacs - a Message-Passing Parallel Molecular-Dynamics Implementation. Comput Phys Commun. 1995 Sep;91(1–3):43–56. [Google Scholar]
- 21.Lindahl E, Hess B, Van Der Spoel D. GROMACS 3.0: a package for molecular simulation and trajectory analysis. J Mol Model. 2001;7(8):306–17. [Google Scholar]
- 22.Van der Spoel D, Lindahl E, Hess B, Groenhof G, Mark AE, Berendsen HJC. Gromacs: Fast, Flexible, and Free. J Comput Chem. 2005 Dec;26(16):1701–18. doi: 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
- 23.Scott WRP, Hunenberger PH, Tironi IG, Mark AE, Billeter SR, Fennen J, et al. The GROMOS biomolecular simulation program package. J Phys Chem A. 1999 May 13;103(19):3596–607. [Google Scholar]
- 24.Oostenbrink C, Villa A, Mark AE, Van Gunsteren WF. A biomolecular force field based on the free enthalpy of hydration and solvation: The GROMOS force-field parameter sets 53A5 and 53A6. J Comput Chem. 2004 Oct;25(13):1656–76. doi: 10.1002/jcc.20090. [DOI] [PubMed] [Google Scholar]
- 25.Hermans J, Berendsen HJC, Vangunsteren WF, Postma JPM. A Consistent Empirical Potential for Water-Protein Interactions. Biopolymers. 1984;23(8):1513–8. [Google Scholar]
- 26.Connor RI, Chen BK, Choe S, Landau NR. Vpr Is Required for Efficient Replication of Human-Immunodeficiency-Virus Type-1 in Mononuclear Phagocytes. Virology. 1995 Feb 1;206(2):935–44. doi: 10.1006/viro.1995.1016. [DOI] [PubMed] [Google Scholar]
- 27.Schindler M, Munch J, Kirchhoff F. Human immunodeficiency virus type 1 inhibits DNA damage-triggered apoptosis by a Nef-independent mechanism. J Virol. 2005 May;79(9):5489–98. doi: 10.1128/JVI.79.9.5489-5498.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Butler SL, Hansen MST, Bushman FD. A quantitative assay for HIV DNA integration in vivo. Nat Med. 2001 May;7(5):631–4. doi: 10.1038/87979. [DOI] [PubMed] [Google Scholar]
- 29.Boyer PL, Clark PK, Hughes SH. HIV-1 and HIV-2 Reverse Transcriptases: Different Mechanisms of Resistance to Nucleoside Reverse Transcriptase Inhibitors. J Virol. 2012 May;86(10):5885–94. doi: 10.1128/JVI.06597-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Goldstone DC, Ennis-Adeniran V, Hedden JJ, Groom HCT, Rice GI, Christodoulou E, et al. HIV-1 restriction factor SAMHD1 is a deoxynucleoside triphosphate triphosphohydrolase. Nature. 2011 Dec 15;480(7377):379–U134. doi: 10.1038/nature10623. [DOI] [PubMed] [Google Scholar]
- 31.Rittinger K, Divita G, Goody RS. Human-Immunodeficiency-Virus Reverse-Transcriptase Substrate-Induced Conformational-Changes and the Mechanism of Inhibition by Nonnucleoside Inhibitors. P Natl Acad Sci USA. 1995 Aug 15;92(17):8046–9. doi: 10.1073/pnas.92.17.8046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Spence RA, Kati WM, Anderson KS, Johnson KA. Mechanism of Inhibition of Hiv-1 Reverse-Transcriptase by Nonnucleoside Inhibitors. Science. 1995 Feb 17;267(5200):988–93. doi: 10.1126/science.7532321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Althaus IW, Chou KC, Lemay RJ, Franks KM, Deibel MR, Kezdy FJ, et al. The benzylthio-pyrimidine U-31,355, a potent inhibitor of HIV-1 reverse transcriptase. Biochem Pharmacol. 1996 Mar 22;51(6):743–50. doi: 10.1016/0006-2952(95)02390-9. [DOI] [PubMed] [Google Scholar]
- 34.Nugent RA, Schlachter ST, Murphy MJ, Cleek GJ, Poel TJ, Wishka DG, et al. Pyrimidine thioethers: A novel class of HIV-1 reverse transcriptase inhibitors with activity against BHAP-resistant HIV. J Med Chem. 1998 Sep 24;41(20):3793–803. doi: 10.1021/jm9800806. [DOI] [PubMed] [Google Scholar]
- 35.Wishka DG, Graber DR, Kopta LA, Olmsted RA, Friis JM, Hosley JD, et al. (−)-6-chloro-2-[(1-furo[2,3-c]pyridin-5-yl-ethyl)thio]-4-pyrimidinamine, PNU-142721, a new broad spectrum HIV-1 non-nucleoside reverse transcriptase inhibitor. J Med Chem. 1998 Apr 23;41(9):1357–60. doi: 10.1021/jm9801049. [DOI] [PubMed] [Google Scholar]
- 36.Boyer J, Arnoult E, Medebielle M, Guillemont J, Unge J, Jochmans D. Difluoromethylbenzoxazole Pyrimidine Thioether Derivatives: A Novel Class of Potent Non-Nucleoside HIV-1 Reverse Transcriptase Inhibitors. J Med Chem. 2011 Dec 8;54(23):7974–85. doi: 10.1021/jm200766b. [DOI] [PubMed] [Google Scholar]
- 37.Romines KR, Freeman GA, Schaller LT, Cowan JR, Gonzales SS, Tidwell JH, et al. Structure-activity relationship studies of novel benzophenones leading to the discovery of a potent, next generation HIV nonnucleoside reverse transcriptase inhibitor. J Med Chem. 2006 Jan 26;49(2):727–39. doi: 10.1021/jm050670l. [DOI] [PubMed] [Google Scholar]
- 38.Wyatt PG, Bethell RC, Cammack N, Charon D, Dodic N, Dumaitre B, et al. Benzophenone Derivatives - a Novel Series of Potent and Selective Inhibitors of Human-Immunodeficiency-Virus Type-1 Reverse-Transcriptase. J Med Chem. 1995 May 12;38(10):1657–65. doi: 10.1021/jm00010a010. [DOI] [PubMed] [Google Scholar]
- 39.Ren J, Chamberlain PP, Stamp A, Short SA, Weaver KL, Romines KR, et al. Structural basis for the improved drug resistance profile of new generation benzophenone non-nucleoside HIV-1 reverse transcriptase inhibitors. J Med Chem. 2008 Aug 28;51(16):5000–8. doi: 10.1021/jm8004493. [DOI] [PubMed] [Google Scholar]