

Abstract
Background
Copy number variations (CNVs) are structural genetic mutations consisting of segmental gains or losses in DNA sequence. Although CNVs contribute substantially to genomic variation, few genetic and imaging studies report association of CNVs with alcohol dependence (AD). Our purpose is to find evidence of this association across ethnic populations and genders. This work is the first AD-CNV study across ethnic groups and the first to include the African American population.
Methods
This study considers two CNV datasets, one for discovery (2,345 samples) and the other for validation (239 samples), both including subjects with AD and healthy controls of European and African ancestry. Our analysis assesses the association between AD and CNV losses across ethnic groups and gender by examining the effect of overall losses across the whole genome, collective losses within individual cytogenetic bands and specific losses in CNV regions.
Results
Results from the discovery dataset showed an association between CNV losses within 16q12.2 and AD diagnosis (p = 4.53x10−3). An overlapping CNV region from the validation dataset exhibited the same direction of effect with respect to AD (p = 0.051). This CNV region affects the genes CES1p1 and CES1, which are members of the carboxylesterase (CES) family. The enzyme encoded by CES1 is a major liver enzyme that typically catalyzes the decomposition of ester into alcohol and carboxylic acid and is involved in drug or xenobiotics, fatty acid and cholesterol metabolisms. In addition, the most significantly associated CNV region was located at 9p21.2 (p = 1.9×10−3) in our discovery dataset. Although not observed in the validation dataset, probably due to small sample size, this result might hold potential connection to AD given its connection with neuronal death. In contrast, we did not find any association between AD and the overall total losses or the collective losses within individual cytogenetic bands.
Conclusions
Overall, our study provides evidence that the specific CNVs at 16q12.2 contribute to the development of alcoholism in African American and European American populations.
Keywords: CNV, alcoholism, addiction, European Americans, African Americans
Introduction
Alcohol dependence (AD) is a disease marked by the abuse of alcohol consumption with harmful consequences for the patient and his surroundings. The heritability of this disease, estimated at 32–73% (Heath et al., 1997), places the genetic risk factor as one of the most important contributors of AD prognosis. Many studies confirm this genetic influence by reporting significant association between AD and single nucleotide polymorphisms (SNPs), variations in one DNA nucleotide base pair. Examples of these findings include SNPs located within GABA receptor genes (Song et al., 2003, Dick and Foroud, 2003, Reich et al., 1998) and alcohol metabolic genes (Boccia et al., 2007).
Although most genetic studies on AD used SNPs, there is an emerging interest in a different source of genetic variant known as copy number variations (CNVs), which in contrast to SNPs encompass multiple nucleotides of DNA code. CNVs are structural variations in DNA sequence consisting of excess (gain) or deficiency (loss) of sections of DNA sequence, which contribute substantially to genomic variation (Sebat et al., 2004, Iafrate et al., 2004). Several studies have reported strong evidence on the association between CNVs and various diseases. For instance, one study reported that losses affecting the CCL3L1 gene are associated with enhanced HIV susceptibility (Gonzalez et al., 2005). A study by Guilamtre et al. showed associations between Autism and losses located at 22q11, spanning the PRODH and DGCR6 genes (Guilmatre et al., 2012). Losses at the C4 gene have shown to increase Systemic Lupus Erythematous susceptibility (Yang et al., 2007). Capuzzo et al. have reported a link between gains located at 7p11.2, affecting the EGFR gene, and a better response of survival rate to lung cancer (Cappuzzo et al., 2005). Wilson et al. have distinguished several gains encompassing the GLUR7 and AKAP5 genes only in mental disorder patients (Wilson et al., 2006). Patients with schizophrenia (Stone et al., 2008) exhibit an increased rate of rare CNVs across the whole genome. All these studies present evidence of the CNV impact in human disease susceptibility.
Up to date, few studies have centered on the association between CNVs and AD (Bae et al., 2011, Lin et al., 2012, Liu et al., 2011, Boutte et al., 2012). Bae et al. (Bae et al., 2011) reported losses at 20q13.33 as a protective factor against the risk for AD using a dataset consisting of 1,138 Koreans. Lin et al. (Lin et al., 2012) showed associations of CNV dosage and AD in two CNV regions located at 6q14.1 and 5q13.2. The study by Lin et al. utilized a public dataset consisting of 2,488 European Americans (EAs), which is part of the Study of Addiction: Genetics and Environment (SAGE) (Bierut et al., 2010), available through NCBI dbGaP (http://www.ncbi.nlm.nih.gov/gap). Their results suggest that AD risk and CNV dosage has a negative relationship at 5q13.2 and a positive relationship at 6q14.1. In addition, our research group has utilized magnetic resonance imaging (MRI) data of the brain as a mediator between CNVs and AD, to enhance the detection power within relatively small samples (Liu et al., 2011),(Boutte et al., 2012). Losses identified at 22q13.1 were associated with functional responses in the precuneus to alcohol cues in binge drinkers. CNVs at 11q14.2 were marginally associated with brain volume variations as well as hazardous drinking behavior. All these studies demonstrate that CNVs have the potential to influence the risks for AD.
Collaboration studies, such as SAGE, often consist on multi-ethnic samples. This ethnic variety may confound further analysis since differences in genetic variations among diverse ethnic populations are substantial. Evolutionist theories and empirical evidence lend support to this suggestion in particular to CNVs. The out-of-Africa theory (Vigilant et al., 1991) suggests that higher number of CNVs should be expected in African descendants compared to non-African descendants (Pinto et al., 2007). Not surprisingly, some CNV studies have reported evidence of significant differences between EA and African American (AA) populations. For instance, a study on psychometric intelligence shows a larger effect of the length of rare losses in European descendants compared to African descendants (Yeo et al., 2012). Another study shows that significantly more subjects in EA (75%) carry a loss affecting GSTM1 gene than in AA (25%) (Huang et al., 2009). These reports suggest that ethnicity should be carefully considered in CNV association studies.
Previous AD-CNV association researches focus on EA or Korean individuals, yet no former study contemplates more than one ethnicity disregarding the substantial difference and similarity in genetic susceptibility between populations. A common disease such as AD might hold biological pathways affected by genetic mutations common to multiple populations besides ethnic differences. This motivates us to search for evidence of CNV associations with alcohol dependence across ethnic populations and genders. Our work is the first AD-CNV association study across ethnic groups and the first to include AA population by leveraging multi-ethnic samples within the collaboration study, SAGE. This study takes advantage of all samples available in SAGE and focuses on losses exhibiting similar effects for both EAs and AAs. Our analysis ponders three different types of CNV ensembles: overall losses across the whole genome, collective losses within individual cytogenetic bands and specific losses in individual CNV regions.
Materials and Methods
Participants
Our study included a discovery dataset and an independent validation dataset. The discovery dataset came from the public genetic database known as dbGaP, specifically the SAGE project (Bierut et al., 2010). Three large complementary studies individually ascertained for alcohol dependence form the SAGE project. These include the Collaborative Study on the Genetics of Alcoholism (Reich et al., 1998, Foroud et al., 2000), the Collaborative Genetic Study of Nicotine Dependence (Bierut et al., 2007, Saccone et al., 2007), and the Family Study of Cocaine Dependence (Bierut et al., 2008). Overall, the SAGE dataset consists of 4,032 subjects where cases are subjects identified as having a lifetime diagnosis of DSM-IV alcohol dependence and control subjects do not report a history of drinking with significant AD symptoms and do not have a lifetime diagnosis of drug dependence. For our study, we retained 2,345 out of 4,032 SAGE samples following a quality control procedure further explained in the Genotyping and CNV calls section. In summary, we excluded all cell line samples, bad CNV quality samples, first-degree relatives and subjects tagged as “Other” (an intermediate score of AD). Table 1 provides complete demographic information.
Table 1.
Demographic table of 2,345 CNV participants in the discovery dataset.
| Healthy Control (1320) | Alcohol Dependent (1025) | Total (2345) | ||
|---|---|---|---|---|
| Race | EA | 981 | 756 | 1737 |
| AA | 339 | 269 | 608 | |
| Gender | Female | 434 | 621 | 1055 |
| Male | 886 | 404 | 1290 | |
| Age | Mean ± SD | 39.5 ± 9.34 | 39.2 ± 8.90 | 39.33 ± 9.10 |
The validation dataset consisted of 239 European descendant samples (Hispanic and non-Hispanic) collected at the Mind Research Network during a genetic study on alcoholism (Claus et al., 2011, Liu et al., 2011). Recruited subjects only included people with a minimum of five binge-drinking episodes in the past month. Binge drinking means five or more drinks per episode for men, and four or more drinks for women. Diagnostic interviews confirmed that subjects are free of severe brain injury, brain-related medical problems, and symptoms of psychosis. The alcohol dependence scale (ADS) (Skinner and Allen, 1982) determined AD severity of every subject. Subjects with a total ADS score greater or equal than 14 were classified as AD and the rest as non-AD (controls) (Saunders et al., 1993), resulting in 106 AD cases and 133 controls. See Table 2 for the demographic information.
Table 2.
Demographic table of 239 participants in the validation dataset.
| Healthy Control (133) | Alcohol Dependent (106) | Total (239) | ||
|---|---|---|---|---|
| Ethnicity | Hispanic | 39 | 50 | 89 |
| Non-Hispanic | 94 | 56 | 150 | |
| Gender | Female | 36 | 39 | 75 |
| Male | 98 | 66 | 164 | |
| Age | Mean ± SD | 29.3 ± 9.02 | 35.06 ± 9.6 | 31.9 ± 9.72 |
Genotyping and CNV Calls
The discovery dataset DNA was extracted from whole blood or cell line and genotyped at the Center for Inherited Disease Research (CIDR) using the Illumina Human 1 M-duo bead chip. The chip spans 1,049,008 loci. CNVs were detected based on the intensity value of log-R-ratio and Beta allele frequency of each locus using the processing pipeline proposed by Chen et al. (Chen et al., 2011). This pipeline was designed to report a CNV call only if the two independent detection algorithms, Hidden Markov Models (PennCNV) (Wang et al., 2007) and Circular Binary Segmentation (Olshen et al., 2004), agree with each other. As a quality control in the detection pipeline, we excluded CNVs smaller than 500 base pairs (bp)and subjects with an excessive total number of CNVs (mean + 3 standard deviations), which helps in reducing the number of false positives and improves the reliability of CNV calls. A two-sample t-test on the number of losses for samples from whole blood and cell lines suggested that significantly more CNV calls for cell line samples (p = 2.4x10−12) were observed. This strongly indicated that cell lines induce additional CNV calls as reported by previous studies (Reddy et al., 1994). Thus, we excluded 1,431 cell line samples from our analysis.
Despite advances in CNV discovery techniques, CNV detection from SNP microarrays still presents low reliability with a considerable high probability of false calls, especially in the case of CNV gains (Zhang et al., 2011). We have evaluated a set of 22 CNVs called by the approach implemented in this study (Chen et al., 2011) using TaqMan PCR (Liu et al., 2012), and 93% of the losses but none of the gains were validated. Additionally, many studies have confirmed that losses are more likely to have deleterious effects than gains (Yeo et al., 2012, Liu et al., 2012, Liu et al., 2011, Stefansson et al., 2008). Thus, we choose to investigate only losses and their association with AD. Overall, we identified 36,813 losses from 2,345 samples (15.7 losses per subject). We did not excluded CNVs at non-coding regions of the genome since the literature suggests that a majority of associated variants fall outside coding regions (Manolio et al., 2009).
We define overlapping CNVs from different samples as CNVs that share the same loci or are three or less markers apart from each other. Then, a CNV region (CNVR) is a chromosome location with CNVs observed in our samples with a frequency larger than 1%, and the boundary of a CNVR is set to be the extreme positions of the overlapping CNVs. There were 378 individual CNVRs encompassing losses with a frequency larger than 1%.
For the validation dataset, DNA was extracted from saliva and genotyped by Illumina Human 1 M-duo bead chips. Losses were detected using the same pipeline as for the discovery dataset. A total of 4,083 losses were identified from 239 samples (17.08 losses per subject) and 333 individual CNV regions had losses with a frequency larger than 1%.
Genome-wide Association Analyses
Total Loss burden test
We first explored the difference between cases and controls in terms of the overall loss burden, measured by the total number of losses per subject. We applied an N-way ANOVA to test the main effect of the overall loss burden on diagnosis while controlling for race (AA/EA) and gender including interaction terms.
Cytoband and CNVR Test
The overall loss burden across genome provides a gross measure of CNVs. Thus, we explored the collective effect of all CNVs within each of 811 cytogenetic bands, given that nucleotides within each cytoband share similar GC contents, and genes within each cytoband share similar expression breadth (Lercher et al., 2003). Studying cytoband genetic effects, an intermediate level between genomic and individual genetic variant, strikes to find a balance between overall genomic burden and specific genetic effect. We group all CNV calls in each cytoband according to NCBI36/hg18 genome map.
Four categorical variables were considered in this step: the presence of losses in a cytoband or CNVR, AD diagnosis, race and gender. We used the Cochran–Mantel–Haenszel (CMH) test to assess repeated effects of losses on AD across populations and gender. Specifically, we built four 2-by-2 contingency tables to test the association between the presence of losses and AD diagnosis, while stratifying the samples into four groups (AA males, AA females, EA males and EA females). This test was conducted for each cytoband and CNVR, where the null hypothesis stated that there were no consistent association between losses and AD diagnosis across AA males, AA females, EA males and EA females.
Validation test of Loss association with AD
Given that we focused on CNV effects common to AA and EA populations in the discovery dataset, we expected to replicate the findings in the European descendant only validation dataset. First, we tested the validity of the effect of overall losses by applying a two-sample t-test to the loss burden on AD. Second, we tested the effect observed in associated (p < 0.01) cytobands and/or CNVRs from the discovery dataset. For this purpose, we used CNVRs in the validation dataset that overlapped with those identified in the discovery dataset. We assessed if the AD effect in the validation dataset was of the same direction as that observed in the discovery dataset. Finally, we reported the observed level of significance using the Fisher’s exact test in 2 by 2 contingency tables, one for each region, where the presence of losses and diagnosis are the nominal variables. Since the validation dataset is small (239 samples), it is not recommendable to further stratify samples to use the CMH test, as applied to the discovery dataset. Because of the small number of samples, we relaxed the significance threshold to 0.1 in the validation dataset.
Replication of regions previously reported as associated with AD
We expected to observe some degree of similarity between our results and those reported from other CNV studies of AD cohorts. Since our analysis shared part of EA samples with the study by Lin et al. (Lin et al., 2012), we evaluated their reported CNVRs located at 5q13.2 (Chromosome 5: 68,921,426–70,412,247) and 6q14.1 (Chromosome 6: 79,034,386–79,090,197) against AD. Additionally, we evaluated the CNVR at 20q13.33 (Chromosome 20: 61,195,302–61,195,978) reported for Korean population (Bae et al., 2011). First, we identified CNVRs in our discovery dataset overlapping with those regions previously reported. Then, we compared the direction of effect and significance of our analysis with their results.
Results
Total Loss burden discovery
The ANOVA test on the number of losses suggested no significant association between the overall loss burden and AD (p=0.33) or gender (p=0.06). On the other hand, a significant group difference was observed in terms of race (p=1.25x10−164), where the mean number of losses is higher in AAs compared to EAs (21 vs. 14 losses per subject), as shown in Table 3.
Table 3.
Mean number of losses for race and diagnosis.
| Diagnosis (p=0.33) | ||
|---|---|---|
| Race (p=1.25x10−164) | Healthy Control (n=1320) | Alcohol Dependent (n= 1025) |
| EA (n=1737) | 14.08 ± 4.61 | 13.62 ± 4.45 |
| AA (n = 608) | 20.63 ± 6.82 | 21.23 ± 6.35 |
Cytoband region discovery
We tested 727 cytobands, excluding 84 with no losses, and did not observe any significant effect from the collective losses within individual cytobands on AD after Bonferroni correction. However, we found five cytobands associated with AD with an uncorrected significance level of p<0.01 (6q12, 9p21.2, 11q14.1, 3p14.1 and 6p22.1). See Figure 1 for a graphical representation of the distribution of CNVs for each of these cytobands. Given the limited incidence per CNVR, which implies low power of detection, we considered these regions as potential candidates for AD association and further validated their effects using the validation dataset.
Figure 1.
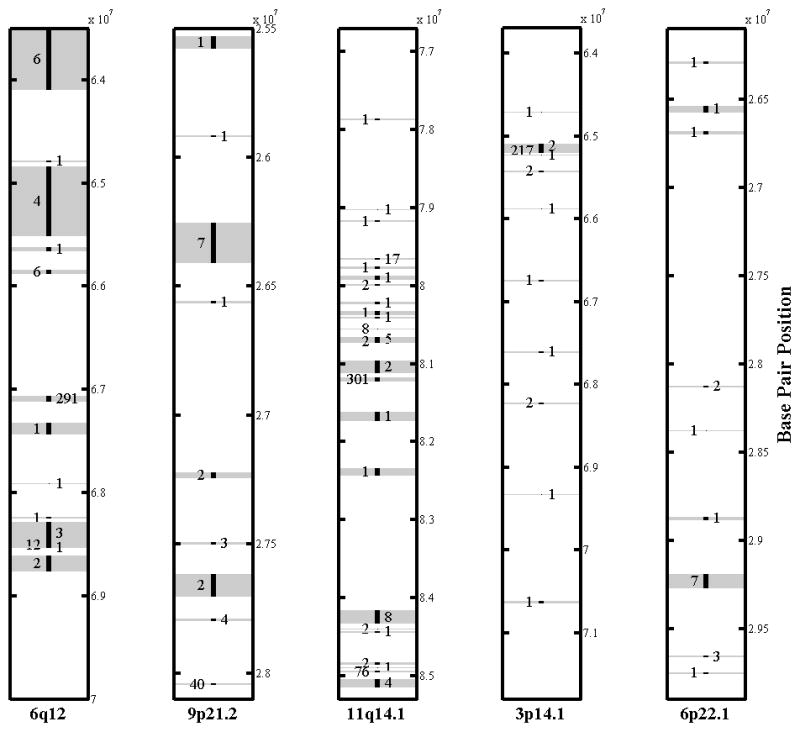
CNVR discovery
None of the CNVRs exhibited significant AD associations after Bonferroni correction. Similarly to the cytoband region discovery, we identified six CNVRs with p<0.01. These CNVRs are located at 9p21.2, 6q12, 3p14.1, 16q12.2, 11q14.1 and 2p25.1, ordered by observed significance level. The CNVR with the lowest p-value, located at 9p21.2 (Chromosome 9: 28,039,518–28,043,556), spanned 4Kbp affecting the LINGO2 gene. This CNVR showed an AD effect with a p-value of 1.9×10−3 where we consistently observed a lower frequency of subjects with losses in controls compared to patients for EA males (OR 6, 95% CI: 1.89–19.5), EA females (OR 1.71, 95% CI: 0.45–6.51), AA males (OR 1.33, 95% CI: 0.29–6.06) and AA females (OR 2.62, 95% CI: 0.52–13.215). The breakpoints of CNVs at 9p21.2 CNVR and another CNVR of 16q12.2 are plotted in Figure 2, where CNVR 16q12.2 carries the validated effect on AD reported in the following.
Figure 2.

Validation results
Overall Loss burden validation
The two sample t-test on the validation dataset suggested that the overall loss burden did not significantly differ between cases and controls (p = 0.93) as suggested by the discovery dataset.
Cytoband region validation
Among the five cytobands implicated as AD-related in the discovery dataset, none showed similar effect direction in the validation dataset.
CNVR validation
The discovery dataset suggested six AD-related CNVRs, among which four overlapped with CNVRs in the validation dataset (3p14.1, 6q12, 11q14.1 and 16q12.2). We did not observe any CNVs at 9p21.2 (Chromosome 9: 28,039,518–28,043,556) which presented the most significant association with AD in the discovery data. Only a CNVR, located at Chromosome 16: 54,362,680–54,377,001 in the validation dataset fell inside the region (Chromosome 16: 54,342,070–54,403,172) identified in the discovery dataset and affected genes CES1p1 and CES1. This CNVR reported the same direction of effect on AD as in the discovery. In the validation dataset, AD patients exhibited a lower frequency of losses compared to controls (OR 0.16, 90% CI: 0.02–0.93) and the Fisher’s exact test reported a p-value of 0.051. Since the validation sample includes Hispanic and non-Hispanic (all European descendant), we conducted an additional logistic regression test, controlling for two top principal components from genome-wide SNP data (possible population structure), age and gender. It leaded to the same result that AD patients are 89% less likely to carry a loss at 16q12.2 (p=0.06). In the discovery dataset, AD patients also showed a lower frequency of losses compared to controls (p=4.53×10−3). This was consistently observed in EA males (OR 0.47, 95% CI: 0.25–0.87), EA females (OR 0.65, 95% CI: 0.37–1.16), and AA females (OR 0.21, 95% CI: 0.02–1.93). The observed difference for AA males (OR 1.06, 95% CI: 0.37–3) reported the opposite direction but with a very small effect, close to no effect.
Replication of CNVRs previously reported associated with AD
We tested three CNVRs located at 5q13.2, 6q14.1 and 20q13.33 previously suggested for their association with AD. The first region, located at 5q13.2, exhibited the same direction of effect for EAs (p = 0.09) in our discovery dataset as that reported by Lin et al. (Lin et al., 2012). Extending the analysis to include AAs, we observed a significance of 0.43 from the CMH test of four subgroups. We found lower ratio of subjects with losses in AD patients compared to controls for the sets of EA males (OR 0.76, 95% CI: 0.4–1.4), EA females (OR 0.56, 95% CI: 0.27–1.17) and AA females (OR 0.72, 95% CI: 0.19–2.74). However, the direction of effect was the opposite for AA males (OR 3.3, 95% CI: 1.09–10.28). The validation dataset showed the same direction of association as reported by Lin et al. where a lower ratio of subjects with losses was observed in AD patients compared to controls (OR 0.83, 90% CI: 0.28–2.45).
The second AD associated CNVR, located at 6q14.1, reported opposite direction of effect to that suggested by Lin et al. for EAs (Lin et al., 2012), noticeable lower ratio of loss incidence and significance of 0.26 for EAs. Extending the analysis to including AAs, we observed a significance of 0.15 from the CMH test of four subgroups. In detail, EA males did not report losses, EA females reported only two losses in AD patients, and AD patients had higher ratio of subjects with losses compared to controls in AA males (OR 2.17, 95% CI: 0.65–7.3) and AA females (OR 1.27, 95% CI: 0.53–3.09). The validation dataset showed almost no difference between the control and case groups (OR 0.97, 90% CI: 0.44–2.1). Note that Lin’s test at this region mainly reflected the large effect of gains that cannot be studied in our data.
The last AD associated region, located at 20q13.33 suggested by Bae et al. for Koreans (Bae et al., 2011), had no overlapping CNVRs in the discovery nor in the validation dataset.
Discussion
This CNV association study constitutes a step forward in better understanding how CNVs affect the risk of developing AD across different ethnic populations and genders. To the best of our knowledge, our work is the first CNV-AD association study to take advantage of a large multi-ethnic dataset. The main results of our analysis suggest that losses located at 16q12.2 (Chromosome 16: 54,342,070–54,403,172) have a protective effect, while losses at 9p21.2 (Chromosome 9: 28,039,518–28,043,556) may have (further validation is needed) a deleterious effect on the risk for AD, regardless of race or gender as summarized in Table 4. The effect of losses at 16q12.2 can potentially come from reduced duplications of this region, since regions with low copy repeats (region-specific repeat sequences) or segmental duplications are susceptible to genomic rearrangements resulting in CNVs (Lee and Lupski, 2006). Thus, more losses at 16q12.2 in healthy controls may reflect less of duplication of this region, which may impose a protective factor on the risk to AD. In addition, we report our analysis for the CNVR at 5q13.2 (Chromosome 5: 68,921,426–70,412,247), reported by Lin et al. (Lin et al., 2012), showing the same direction of effect for EA but a different pattern for AA population.
Table 4.
Associations of CNV Losses and Alcohol Dependence
| European American | African American | P-value | Genes | ||||
|---|---|---|---|---|---|---|---|
|
| |||||||
| CNVR | Female | Male | Female | Male | |||
| 16q12.2 (61.1Kbp, 14 markers) | case/control% | 4.49%/9.07% | 6%/8.88% | 5.21%/4.92% | 0.67%/3.07% | 4.5×10−3 | CES1, CES1P1 |
| Odds ratio | 0.47 | 0.65 | 1.06 | 0.21 | |||
| Chr16: 54,342,070–54,403,172 | 95% CI | (0.25–0.87) | (0.37–1.16) | (0.37–3) | (0.02–1.93) | ||
|
| |||||||
| 9p21.2* (4Kbp, 4 markers) | case/control% | 3.4%/0.58% | 1.72%/1.01% | 2.6%/1.97% | 3.82%/1.49% | 1.9×10−3 | LING02 |
| Odds ratio | 6 | 1.71 | 1.33 | 2.62 | |||
| Chr9: 28,039,518–28,043,556 | 95% CI | (1.89–19.5) | (0.45–6.51) | (0.29–6.06) | (0.52–13.22) | ||
Not validated. No overlapping region was found in the validation dataset.
After no effect from overall total losses or the collective losses within individual cytogenetic bands was observed, we constrained our study to common CNVR (>1%), following the common disease-common variant hypothesis. About 12% of American adults have had an alcohol dependence problem at some time in their life (Hasin et al., 2007). For such a common disorder, we considered that common CNVRs may have a higher impact on AD risk than rare CNVRs, which instead are more likely associated with rare disorders such as schizophrenia or autism (Sebat et al., 2007, Stone et al., 2008). For completeness, we conducted a 2-way ANOVA test on ethnicity and overall rare CNV losses and found no significant difference of rare CNVs between cases and controls (p = 0.24), suggesting that rare CNVs may less likely contribute to AD. Therefore, we focused on validating the hypothesis of Common Disease-Common Variant (Hemminki et al., 2008) for AD.
Although the analysis of CNVRs reports no significant association with AD after Bonferroni correction, two individual CNV regions may still hold potential biological functional impact. The CNVR at 9p21.2 holds the highest observed significance in the discovery dataset (p=1.9×10−3), but since no CNVR at the same region was observed in the validation dataset (possibly due to the small sample size), we cannot perform validation test. Nevertheless, the consistency observed in four subgroups of the discovery samples merited further discussion. The other promising CNVR is at 16q12.2 and presents the same effect on AD in both the discovery and validation datasets. In contrast to the lack of effect from cytobands and the total burden of losses, these results may indicate that AD is more attributable to specific local variations instead of extensive variations of the genome or chromosomes. Furthermore, we conducted a power test for this region showing that we have a power of 0.724, probability to detect such effect with Type I error of 0.05 uncorrected for multiple comparisons. Even though the power of detection under multiple comparisons is low, the CNVR at 16q12.2 does not pass multiple comparison correction, the results are promising because of the agreement with the validation tests.
The CNVR at 16q12.2 spans the two genes CES1p1 and CES1. Both belonging to the carboxylesterase (CES) gene family which is responsible for catalyzing the hydrolysis of various xenobiotics, such as cocaine and heroin, and endogenous substrates with ester (Imai et al., 2006). Gene CES1p1 does not encode a protein, but is a pseudogene and shares a similar DNA sequence with CES1. The enzyme CES1, encoded by gene CES1, is a major liver enzyme that typically catalyzes the decomposition of ester into alcohol and carboxylic acid. It is involved in xenobiotic (Redinbo et al., 2003), fatty acid and cholesterol (Jernås et al., 2009) metabolisms. Mutations within the 16q12.2 cytoband or the CES1 gene have been connected with cerebral cortical malformations (Piao et al., 2002), amyotrophic lateral sclerosis (Abalkhail et al., 2003), anti-tuberculosis drug-induced hepatotoxicity (Wu et al., 2012) and obesity (Friedrichsen et al., 2013). Even though we do not know the correspondent pathway, the association of this CNVR with AD is promising given the fact that the spanned genes have a direct relationship with liver and hydrolysis functions involving the alcohol compound.
The CNVR at 9p21.2 exhibits the strongest association with AD in our discovery analysis (p = 1.9x10−3). This CNVR affects LINGO2 gene. The function of LINGO2 has not been well investigated and is currently unknown (Wu et al., 2011). However, a high degree of similarity exists between LINGO1 and LINGO2 (Vilariño-Güell et al., 2010), members of the leucine-rich repeat (LRR) family. LINGO1 expression increases after neuronal damage and decreases during axonal sprouting (Ji et al., 2006). This fact suggests that LINGO1 is a regulator of neuronal death. Therefore, disruption of this gene might have implications in neurological diseases proposing an interesting potential connection with AD, but further validation is required.
Given our multi-ethnic framework, the replication of results from previous studies is important because it extends the CNV association analysis to AAs. In the case of the EA population, we observed the consistent effect of CNV losses at 5q13.2 in our dataset, compared with what Lin et al. reported. However, the CMH test for EAs suggested no significance dependence (p = 0.09). AA males also exhibit similar effects from losses at 5q13.2, but females exhibit a strong opposite direction of effect. This constitutes evidence that AD morbidity may also vary according to combinations of ethnicity and gender. The noticeable difference in our results for 6q14.1 may be due to the exclusion of cell line samples and emphases on losses only in our analysis. The CNVR at 20q13.33 reported by Bae et al. (Bae et al., 2011) do not overlap with any region in our datasets, which may reflect the population specificity and/or the different genotyping platforms. A limitation of our study was the two AD classification criteria in the discovery and validation datasets. However, a pair of studies suggests the high degree of similarity and consistency of ADS and DSM-IV (Saxon et al., 2007, Hasin, 2003), supporting consistent results in our study using any of these criteria. Overall, our study provides evidence that the presence of losses has a protective effect against the risk for AD in the CNVR at 16q12.2 and likely a deleterious effect at 9p21.2, which is common to both EA and AA populations. Moreover, the implicated gene CES1 has functional coherence with the disease of our interest. These findings suggest that local individual CNVR, not the global or cytoband collective CNVs, more likely have impact on the risk of AD. In conclusion, we present evidence that a CNVR common to multiple races may influence a common biological pathway related to AD as a heritable disease.
Acknowledgments
This work was supported by National Institute of Health (R33DA027626) to JL.
Contributor Information
Alvaro Emilio Ulloa, Email: aulloa@mrn.org.
Jiayu Chen, Email: jchen@mrn.org.
Victor Manuel Vergara, Email: vvergara@mrn.org.
Jingyu Liu, Email: jliu@mrn.org.
References
- ABALKHAIL H, MITCHELL J, HABGOOD J, ORRELL R, DE BELLEROCHE J. A new familial amyotrophic lateral sclerosis locus on chromosome 16q12. 1–16q12. 2. The American Journal of Human Genetics. 2003;73:383–389. doi: 10.1086/377156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BAE JS, JUNG MH, LEE BC, CHEONG HS, PARK BL, KIM LH, KIM JH, PASAJE CFA, LEE JS, JUNG KH. The Genetic Effect of Copy Number Variations on the Risk of Alcoholism in a Korean Population. Alcoholism: Clinical and Experimental Research. 2011;36:35–42. doi: 10.1111/j.1530-0277.2011.01578.x. [DOI] [PubMed] [Google Scholar]
- BIERUT LJ, AGRAWAL A, BUCHOLZ KK, DOHENY KF, LAURIE C, PUGH E, FISHER S, FOX L, HOWELLS W, BERTELSEN S. A genome-wide association study of alcohol dependence. Proceedings of the National Academy of Sciences. 2010;107:5082–5087. doi: 10.1073/pnas.0911109107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BIERUT LJ, MADDEN PA, BRESLAU N, JOHNSON EO, HATSUKAMI D, POMERLEAU OF, SWAN GE, RUTTER J, BERTELSEN S, FOX L, FUGMAN D, GOATE AM, HINRICHS AL, KONVICKA K, MARTIN NG, MONTGOMERY GW, SACCONE NL, SACCONE SF, WANG JC, CHASE GA, RICE JP, BALLINGER DG. Novel genes identified in a high-density genome wide association study for nicotine dependence. Hum Mol Genet. 2007;16:24–35. doi: 10.1093/hmg/ddl441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BIERUT LJ, STRICKLAND JR, THOMPSON JR, AFFUL SE, COTTLER LB. Drug use and dependence in cocaine dependent subjects, community-based individuals, and their siblings. Drug Alcohol Depend. 2008;95:14–22. doi: 10.1016/j.drugalcdep.2007.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BOCCIA S, SAYED-TABATABAEI FA, PERSIANI R, GIANFAGNA F, RAUSEI S, ARZANI D, LA GRECA A, D’UGO D, LA TORRE G, VAN DUIJN CM. Polymorphisms in metabolic genes, their combination and interaction with tobacco smoke and alcohol consumption and risk of gastric cancer: a case-control study in an Italian population. BMC cancer. 2007;7:206. doi: 10.1186/1471-2407-7-206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BOUTTE D, CALHOUN VD, CHEN J, SABBINENI A, HUTCHISON K, LIU J. Association of genetic copy number variations at 11 q14. 2 with brain regional volume differences in an alcohol use disorder population. Alcohol. 2012 doi: 10.1016/j.alcohol.2012.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CAPPUZZO F, HIRSCH FR, ROSSI E, BARTOLINI S, CERESOLI GL, BEMIS L, HANEY J, WITTA S, DANENBERG K, DOMENICHINI I, LUDOVINI V, MAGRINI E, GREGORC V, DOGLIONI C, SIDONI A, TONATO M, FRANKLIN WA, CRINO L, BUNN PA, JR, VARELLA-GARCIA M. Epidermal growth factor receptor gene and protein and gefitinib sensitivity in non-small-cell lung cancer. J Natl Cancer Inst. 2005;97:643–55. doi: 10.1093/jnci/dji112. [DOI] [PubMed] [Google Scholar]
- CHEN J, LIU J, BOUTTE D, CALHOUN VD. A pipeline for copy number variation detection based on principal component analysis. Conf Proc IEEE Eng Med Biol Soc. 2011;2011:6975–8. doi: 10.1109/IEMBS.2011.6091763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CLAUS ED, EWING SWF, FILBEY FM, SABBINENI A, HUTCHISON KE. Identifying neurobiological phenotypes associated with alcohol use disorder severity. Neuropsychopharmacology. 2011;36:2086–2096. doi: 10.1038/npp.2011.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DICK DM, FOROUD T. Candidate genes for alcohol dependence: a review of genetic evidence from human studies. Alcoholism: Clinical and Experimental Research. 2003;27:868–879. doi: 10.1097/01.ALC.0000065436.24221.63. [DOI] [PubMed] [Google Scholar]
- FOROUD T, EDENBERG HJ, GOATE A, RICE J, FLURY L, KOLLER DL, BIERUT LJ, CONNEALLY PM, NURNBERGER JI, BUCHOLZ KK, LI TK, HESSELBROCK V, CROWE R, SCHUCKIT M, PORJESZ B, BEGLEITER H, REICH T. Alcoholism susceptibility loci: confirmation studies in a replicate sample and further mapping. Alcohol Clin Exp Res. 2000;24:933–45. [PubMed] [Google Scholar]
- FRIEDRICHSEN M, POULSEN P, WOJTASZEWSKI J, HANSEN PR, VAAG A, RASMUSSEN HB. Carboxylesterase 1 Gene Duplication and mRNA Expression in Adipose Tissue Are Linked to Obesity and Metabolic Function. PloS one. 2013;8:e56861. doi: 10.1371/journal.pone.0056861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GONZALEZ E, KULKARNI H, BOLIVAR H, MANGANO A, SANCHEZ R, CATANO G, NIBBS RJ, FREEDMAN BI, QUINONES MP, BAMSHAD MJ, MURTHY KK, ROVIN BH, BRADLEY W, CLARK RA, ANDERSON SA, O’CONNELL RJ, AGAN BK, AHUJA SS, BOLOGNA R, SEN L, DOLAN MJ, AHUJA SK. The influence of CCL3L1 gene-containing segmental duplications on HIV-1/AIDS susceptibility. Science. 2005;307:1434–40. doi: 10.1126/science.1101160. [DOI] [PubMed] [Google Scholar]
- GUILMATRE A, DUBOURG C, MOSCA AL, LEGALLIC S, GOLDENBERG A, DROUIN-GARRAUD V, LAYET V, ROSIER A, BRIAULT S, BONNET-BRILHAULT F, LAUMONNIER F, ODENT S, VACON GL, JOLY-HELAS G, DAVID V, BENDAVID C, PINOIT JM, HENRY C, IMPALLOMENI C, GERMANO E, TORTORELLA G, ROSA GD, BARTHELEMY C, ANDRES C, FAIVRE L, FRÉBOURG T, VEBER PS, CAMPION D. Recurrent Rearrangements in Synaptic and Neurodevelopmental Genes and Shared Biologic Pathways in Schizophrenia, Autism, and Mental RetardationSchizophrenia, Autism, and Mental Retardation Genes. Archives of General Psychiatry. 2012;66:947–956. doi: 10.1001/archgenpsychiatry.2009.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HASIN D. Classification of alcohol use disorders. Alcohol Research and Health. 2003;27:5–17. [PMC free article] [PubMed] [Google Scholar]
- HASIN DS, STINSON FS, OGBURN E, GRANT BF. Prevalence, correlates, disability, and comorbidity of DSM-IV alcohol abuse and dependence in the United States: results from the National Epidemiologic Survey on Alcohol and Related Conditions. Archives of General Psychiatry. 2007;64:830. doi: 10.1001/archpsyc.64.7.830. [DOI] [PubMed] [Google Scholar]
- HEATH AC, BUCHOLZ K, MADDEN P, DINWIDDIE S, SLUTSKE W, BIERUT L, STATHAM D, DUNNE M, WHITFIELD J, MARTIN N. Genetic and environmental contributions to alcohol dependence risk in a national twin sample: consistency of findings in women and men. Psychological medicine. 1997;27:1381–1396. doi: 10.1017/s0033291797005643. [DOI] [PubMed] [Google Scholar]
- HEMMINKI K, FÖRSTI A, BERMEJO JL. The ‘common disease-common variant’ hypothesis and familial risks. PLoS One. 2008;3:e2504. doi: 10.1371/journal.pone.0002504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HUANG RS, CHEN P, WISEL S, DUAN S, ZHANG W, COOK EH, DAS S, COX NJ, DOLAN ME. Population-specific GSTM1 copy number variation. Hum Mol Genet. 2009;18:366–72. doi: 10.1093/hmg/ddn345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- IAFRATE AJ, FEUK L, RIVERA MN, LISTEWNIK ML, DONAHOE PK, QI Y, SCHERER SW, LEE C. Detection of large-scale variation in the human genome. Nature genetics. 2004;36:949–951. doi: 10.1038/ng1416. [DOI] [PubMed] [Google Scholar]
- IMAI T, TAKETANI M, SHII M, HOSOKAWA M, CHIBA K. Substrate specificity of carboxylesterase isozymes and their contribution to hydrolase activity in human liver and small intestine. Drug metabolism and disposition. 2006;34:1734–1741. doi: 10.1124/dmd.106.009381. [DOI] [PubMed] [Google Scholar]
- JERNÅS M, OLSSON B, ARNER P, JACOBSON P, SJÖSTRÖM L, WALLEY A, FROGUEL P, MCTERNAN PG, HOFFSTEDT J, CARLSSON L. Regulation of carboxylesterase 1 (CES1) in human adipose tissue. Biochemical and biophysical research communications. 2009;383:63–67. doi: 10.1016/j.bbrc.2009.03.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JI B, LI M, WU WT, YICK LW, LEE X, SHAO Z, WANG J, SO KF, MCCOY JM, BLAKE PEPINSKY R. LINGO-1 antagonist promotes functional recovery and axonal sprouting after spinal cord injury. Molecular and Cellular Neuroscience. 2006;33:311–320. doi: 10.1016/j.mcn.2006.08.003. [DOI] [PubMed] [Google Scholar]
- LEE JA, LUPSKI JR. Genomic Rearrangements and Gene Copy-Number Alterations as a Cause of Nervous System Disorders. Neuron. 2006;52:103–121. doi: 10.1016/j.neuron.2006.09.027. [DOI] [PubMed] [Google Scholar]
- LERCHER MJ, URRUTIA AO, PAVLÍČEK A, HURST LD. A unification of mosaic structures in the human genome. Human molecular genetics. 2003;12:2411–2415. doi: 10.1093/hmg/ddg251. [DOI] [PubMed] [Google Scholar]
- LIN P, HARTZ SM, WANG JC, AGRAWAL A, ZHANG TX, MCKENNA N, BUCHOLZ K, BROOKS AI, TISCHFIELD JA, EDENBERG HJ. Copy Number Variations in 6q14. 1 and 5q13. 2 are Associated with Alcohol Dependence. Alcoholism: Clinical and Experimental Research. 2012 doi: 10.1111/j.1530-0277.2012.01758.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LIU J, CALHOUN VD, CHEN J, CLAUS ED, HUTCHISON KE. Effect of homozygous deletions at 22q13. 1 on alcohol dependence severity and cue-elicited BOLD response in the precuneus. Addiction Biology. 2011 doi: 10.1111/j.1369-1600.2011.00393.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LIU J, ULLOA A, PERRONE-BIZZOZERO N, YEO R, CHEN J, CALHOUN VD. A Pilot Study on Collective Effects of 22q13. 31 Deletions on Gray Matter Concentration in Schizophrenia. PloS one. 2012;7:e52865. doi: 10.1371/journal.pone.0052865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MANOLIO TA, COLLINS FS, COX NJ, GOLDSTEIN DB, HINDORFF LA, HUNTER DJ, MCCARTHY MI, RAMOS EM, CARDON LR, CHAKRAVARTI A. Finding the missing heritability of complex diseases. Nature. 2009;461:747–753. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OLSHEN AB, VENKATRAMAN E, LUCITO R, WIGLER M. Circular binary segmentation for the analysis of array-based DNA copy number data. Biostatistics. 2004;5:557–572. doi: 10.1093/biostatistics/kxh008. [DOI] [PubMed] [Google Scholar]
- PIAO X, BASEL-VANAGAITE L, STRAUSSBERG R, GRANT PE, PUGH EW, DOHENY K, DOAN B, HONG SE, SHUGART YY, WALSH CA. An autosomal recessive form of bilateral frontoparietal polymicrogyria maps to chromosome 16q12. 2–21. The American Journal of Human Genetics. 2002;70:1028–1033. doi: 10.1086/339552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PINTO D, MARSHALL C, FEUK L, SCHERER SW. Copy-number variation in control population cohorts. Human molecular genetics. 2007;16:R168–R173. doi: 10.1093/hmg/ddm241. [DOI] [PubMed] [Google Scholar]
- REDDY P, STAMATOYANNOPOULOS G, PAPAYANNOPOULOU T, SHEN C. Genomic footprinting and sequencing of human beta-globin locus. Tissue specificity and cell line artifact. Journal of Biological Chemistry. 1994;269:8287–8295. [PubMed] [Google Scholar]
- REDINBO M, BENCHARIT S, POTTER P. Human carboxylesterase 1: from drug metabolism to drug discovery. Biochemical Society Transactions. 2003;31:620–624. doi: 10.1042/bst0310620. [DOI] [PubMed] [Google Scholar]
- REICH T, EDENBERG HJ, GOATE A, WILLIAMS JT, RICE JP, VAN EERDEWEGH P, FOROUD T, HESSELBROCK V, SCHUCKIT MA, BUCHOLZ K. Genome-wide search for genes affecting the risk for alcohol dependence. American journal of medical genetics. 1998;81:207–215. [PubMed] [Google Scholar]
- SACCONE SF, HINRICHS AL, SACCONE NL, CHASE GA, KONVICKA K, MADDEN PA, BRESLAU N, JOHNSON EO, HATSUKAMI D, POMERLEAU O, SWAN GE, GOATE AM, RUTTER J, BERTELSEN S, FOX L, FUGMAN D, MARTIN NG, MONTGOMERY GW, WANG JC, BALLINGER DG, RICE JP, BIERUT LJ. Cholinergic nicotinic receptor genes implicated in a nicotine dependence association study targeting 348 candidate genes with 3713 SNPs. Hum Mol Genet. 2007;16:36–49. doi: 10.1093/hmg/ddl438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SAUNDERS JB, AASLAND OG, BABOR TF, GRANT M. Development of the alcohol use disorders identification test (AUDIT): WHO collaborative project on early detection of persons with harmful alcohol consumption-II. Addiction. 1993;88:791–804. doi: 10.1111/j.1360-0443.1993.tb02093.x. [DOI] [PubMed] [Google Scholar]
- SAXON AJ, KIVLAHAN DR, DOYLE S, DONOVAN DM. Further validation of the alcohol dependence scale as an index of severity. Journal of studies on alcohol and drugs. 2007;68:149. doi: 10.15288/jsad.2007.68.149. [DOI] [PubMed] [Google Scholar]
- SEBAT J, LAKSHMI B, MALHOTRA D, TROGE J, LESE-MARTIN C, WALSH T, YAMROM B, YOON S, KRASNITZ A, KENDALL J. Strong association of de novo copy number mutations with autism. Science. 2007;316:445–449. doi: 10.1126/science.1138659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SEBAT J, LAKSHMI B, TROGE J, ALEXANDER J, YOUNG J, LUNDIN P, MÅNÉR S, MASSA H, WALKER M, CHI M. Large-scale copy number polymorphism in the human genome. Science. 2004;305:525–528. doi: 10.1126/science.1098918. [DOI] [PubMed] [Google Scholar]
- SKINNER HA, ALLEN BA. Alcohol dependence syndrome: Measurement and validation. Journal of abnormal psychology. 1982;91:199–209. doi: 10.1037//0021-843x.91.3.199. [DOI] [PubMed] [Google Scholar]
- SONG J, KOLLER DL, FOROUD T, CARR K, ZHAO J, RICE J, NURNBERGER JI, BEGLEITER H, PORJESZ B, SMITH TL. Association of GABAA receptors and alcohol dependence and the effects of genetic imprinting. American Journal of Medical Genetics Part B: Neuropsychiatric Genetics. 2003;117:39–45. doi: 10.1002/ajmg.b.10022. [DOI] [PubMed] [Google Scholar]
- STEFANSSON H, RUJESCU D, CICHON S, PIETILÄINEN OP, INGASON A, STEINBERG S, FOSSDAL R, SIGURDSSON E, SIGMUNDSSON T, BUIZER-VOSKAMP JE. Large recurrent microdeletions associated with schizophrenia. Nature. 2008;455:232–236. doi: 10.1038/nature07229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- STONE JL, O’DONOVAN MC, GURLING H, KIROV GK, BLACKWOOD DH, CORVIN A, CRADDOCK NJ, GILL M, HULTMAN CM, LICHTENSTEIN P. Rare chromosomal deletions and duplications increase risk of schizophrenia. Nature. 2008;455:237–241. doi: 10.1038/nature07239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VIGILANT L, STONEKING M, HARPENDING H, HAWKES K, WILSON AC. African populations and the evolution of human mitochondrial DNA. Science. 1991;253:1503–7. doi: 10.1126/science.1840702. [DOI] [PubMed] [Google Scholar]
- VILARIÑO-GÜELL C, WIDER C, ROSS OA, JASINSKA-MYGA B, KACHERGUS J, COBB SA, SOTO-ORTOLAZA AI, BEHROUZ B, HECKMAN MG, DIEHL NN. LINGO1 and LINGO2 variants are associated with essential tremor and Parkinson disease. Neurogenetics. 2010;11:401–408. doi: 10.1007/s10048-010-0241-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WANG K, LI M, HADLEY D, LIU R, GLESSNER J, GRANT SF, HAKONARSON H, BUCAN M. PennCNV: an integrated hidden Markov model designed for high-resolution copy number variation detection in whole-genome SNP genotyping data. Genome research. 2007;17:1665–1674. doi: 10.1101/gr.6861907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WILSON GM, FLIBOTTE S, CHOPRA V, MELNYK BL, HONER WG, HOLT RA. DNA copy-number analysis in bipolar disorder and schizophrenia reveals aberrations in genes involved in glutamate signaling. Hum Mol Genet. 2006;15:743–9. doi: 10.1093/hmg/ddi489. [DOI] [PubMed] [Google Scholar]
- WU X, ZHU D, ZHANG J, ZHONG Y, XI Y, AN H, LIANG Y, YANG Y. The relationship between carboxylesterase 1 gene polymorphisms and susceptibility to antituberculosis drug-induced hepatotoxicity. Zhonghua nei ke za zhi [Chinese journal of internal medicine] 2012;51:524. [PubMed] [Google Scholar]
- WU YW, PRAKASH K, RONG TY, LI HH, XIAO Q, TAN LC, AU WL, DING JQ, TAN EK. Lingo2 variants associated with essential tremor and Parkinson’s disease. Human genetics. 2011;129:611–615. doi: 10.1007/s00439-011-0955-3. [DOI] [PubMed] [Google Scholar]
- YANG Y, CHUNG EK, WU YL, SAVELLI SL, NAGARAJA HN, ZHOU B, HEBERT M, JONES KN, SHU Y, KITZMILLER K, BLANCHONG CA, MCBRIDE KL, HIGGINS GC, RENNEBOHM RM, RICE RR, HACKSHAW KV, ROUBEY RA, GROSSMAN JM, TSAO BP, BIRMINGHAM DJ, ROVIN BH, HEBERT LA, YU CY. Gene copy-number variation and associated polymorphisms of complement component C4 in human systemic lupus erythematosus (SLE): low copy number is a risk factor for and high copy number is a protective factor against SLE susceptibility in European Americans. Am J Hum Genet. 2007;80:1037–54. doi: 10.1086/518257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YEO R. A., DEPARTMENT OF PSYCHOLOGY, U. O. N. M., ALBUQUERQUE, NEW MEXICO, UNITED STATES OF AMERICA, GANGESTAD, S. W., DEPARTMENT OF PSYCHOLOGY, U. O. N. M., ALBUQUERQUE, NEW MEXICO, UNITED STATES OF AMERICA, LIU, J., THE MIND RESEARCH NETWORK, A., NEW MEXICO, UNITED STATES OF AMERICA, DEPARTMENT OF ELECTRICAL AND COMPUTER ENGINEERING, U. O. N. M., ALBUQUERQUE, NEW MEXICO, UNITED STATES OF AMERICA, CALHOUN, V. D., THE MIND RESEARCH NETWORK, A., NEW MEXICO, UNITED STATES OF AMERICA, DEPARTMENT OF ELECTRICAL AND COMPUTER ENGINEERING, U. O. N. M., ALBUQUERQUE, NEW MEXICO, UNITED STATES OF AMERICA, HUTCHISON, K. E., DEPARTMENT OF PSYCHOLOGY, U. O. N. M., ALBUQUERQUE, NEW MEXICO, UNITED STATES OF AMERICA, THE MIND RESEARCH NETWORK, A., NEW MEXICO, UNITED STATES OF AMERICA & DEPARTMENT OF PSYCHOLOGY AND NEUROSCIENCE, U. O. C., BOULDER, COLORADO, UNITED STATES OF AMERICA 2012 Rare Copy Number Deletions Predict Individual Variation in Intelligence. PLoS ONE. :6. doi: 10.1371/journal.pone.0016339.
- ZHANG D, QIAN Y, AKULA N, ALLIEY-RODRIGUEZ N, TANG J, GERSHON ES, LIU C. Accuracy of CNV detection from GWAS data. PLoS One. 2011;6:e14511. doi: 10.1371/journal.pone.0014511. [DOI] [PMC free article] [PubMed] [Google Scholar]