

Abstract
Using a computationally driven approach, a class of inhibitors with picomolar potency known as the catechol diethers were developed targeting the non-nucleoside binding pocket (NNBP) of HIV-1 RT. Computational studies suggested that halogen bonding interactions between the C5 substituent of the inhibitor and backbone carbonyl of conserved residue Pro95 might be important. While the recently reported crystal structures of the RT complexes confirmed the interactions with the NNBP, they revealed the lack of a halogen bonding interaction with Pro95. In order to understand the effects of substituents at the C5 position, we determined additional crystal structures with 5-Br and 5-H derivatives. Using comparative structural analysis, we identified several conformations of the ethoxy uracil dependent on the strength of a van der Waals interaction with the Cγ of Pro95 and the C5 substitution. The 5-Cl and 5-F derivatives position the ethoxy uracil to make more hydrogen bonds, while the larger 5-Br and smaller 5-H position the ethoxy uracil to make fewer hydrogen bonds. EC50 values correlate with the trends observed in the crystal structures. The influence of C5 substitutions on the ethoxy uracil conformation may have strategic value, as future derivatives can possibly be modulated in order to gain additional hydrogen bonding interactions with resistant variants of RT.
Keywords: HIV-1 reverse transcriptase, non-nucleoside reverse transcriptase inhibitors (NNRTIs), structure based drug design, structure activity relationships (SAR), halogen bonds
Introduction
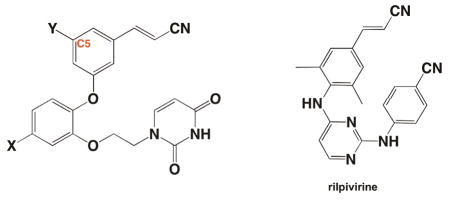
Highly active anti-retroviral therapy (HAART) is a major advancement in HIV-1 drug discovery research that improves the quality and duration of life for patients. The majority of antiretroviral therapies used in HAART target enzymes involved in infection and replication, which include HIV-1 protease, integrase (IN), and reverse transcriptase (RT) (1–3). Given the error prone nature of HIV-1 RT (4), antiretroviral drug discovery and development is an iterative process due to treatment failure from resistant strains and bioavailability issues (5, 6). In addition to ours, several other research programs have implemented a structure-based approach to develop new non-nucleoside reverse transcriptase inhibitors (NNRTIs) with improved pharmacological and resistance profiles (7–10). With the assistance of computational methods, a series of analogues known as the catechol diethers, have been optimized with guidance from free energy perturbation (FEP) calculations (11). Modifications suggested by the FEP calculations identified a lead scaffold with picomolar potency in HIV-1 MT2 cellular assays (11). In order to complement the computational studies, we previously determined two binary crystal structures in complex with our leading picomolar inhibitors (12). Analysis of the crystal structures identified key interactions made between the catechol diethers and the non-nucleoside binding pocket (NNBP) of HIV-1 RT. Several van der Waals interactions made with residues in the NNBP, in addition to multiple hydrogen bonds, all contribute to the picomolar potency of the catechol diether class of compounds (12). Given the success of these inhibitors, we continue to improve the catechol diether class of NNRTIs using structure-guided design.
In order to fully optimize the catechol diether series, additional considerations must be addressed using a rational approach. To address resistance, one goal has been to optimize our most potent catechol diethers to interact specifically with Pro95, an essential residue for RT activity that is not prone to mutation by the HIV-1 virus (13, 14). Earlier computational studies suggested that the carbonyl oxygen of Pro95 was capable of halogen bonding with a 5-Cl group of the cyanovinylphenyl substituent (11). However, analysis of the crystal structure revealed that the distance between the carbonyl oxygen and the chlorine was too far apart to make strong halogen bonding interactions (12). In order to probe the space near the carbonyl oxygen of Pro95, we have determined two additional RT crystal structures in complex with modified derivatives of the catechol diether series. These analogues include 5-H and 5-Br substituents on the cyanovinyl aryl ring. Structural analysis identifies the 5-Cl group as the most favorable substituent due to its optimal distance for van der Waals interactions with Pro95, whereas smaller or bulkier substituents are too far away or cause too much repulsion, respectively. In addition, comparison of these structures reveals a trend in which the ethoxy uracil substituent can adopt multiple conformations. In each conformation the hydrogen bonding interactions between the uracil and residues in the NNBP groove region are altered: compounds with picomolar potency maintain more hydrogen bonds than those with nanomolar potency. Interestingly, the strength of the van der Waals interaction between Pro95 and the C5 substituent seem to correlate with the observed phenomenon of the uracil hydrogen bond pattern. Thus, it appears that the substituent on the C5 position significantly affects the conformation of the uracil-containing side chain and thereby affects the interactions made between the compound and the binding pocket.
The detailed comparison of all of these structures suggests that the ethoxy uracil substituent is flexible—enabling the maintenance of potency against resistant strains—and that the compounds can possibly be modulated at the C5 position of the cyanovinylphenyl group to gain additional interactions. As observed in the FDA-approved NNRTI rilpivirine (TMC278), flexibility is presumably a key compound feature that may improve performance against resistant variants of RT (8). From this knowledge, further compound development targeting conserved residues such as Pro95 and promoting the optimal uracil side-chain conformation will assist in our efforts to optimize the catechol diethers against limitations such as resistance mutations.
Materials and Methods
The syntheses of compounds 1–4 have been reported previously (11, 12). Recombinant RT52A enzyme was expressed and purified to homogeneity using methods described previously (8, 12, 15). Crystals of RT52A in complex with 3 and 4 were prepared using similar methods as the catechol diether complexes (12). The final optimized condition for crystal growth consisted of 15% (w/v) PEG 8000, 100 mM ammonium sulfate, 15 mM magnesium sulfate, 5 mM spermine, and 50 mM citric acid pH 5.5. Crystals were transferred to a cryo-solution containing 27% (v/v) ethylene glycol and flash cooled with liquid nitrogen.
Diffraction data for the RT:3 and RT:4 crystals were collected at Brookhaven NSLS on beam line X29A. High-resolution data sets for the best diffracting crystals were scaled and merged in space group C2 using HKL2000 (16). In order to obtain phases, molecular replacement was performed with Phaser (17) using previously determined RT:1 (PDB code: 4H4M) as the search model (12). Alternatively, the structures could also be solved with Difference Fourier Methods using the former RT:1 model as Fc since the RT:1–4 crystals are isomorphous. Both solution methods yield identical structures for the RT:3 and RT:4 complex as suggested by low all atom rmsd (0.131 Å for RT:3, and 0.192 Å for RT:4) and small differences in Rfree and Rwork (Table S1) for the final refined models. The program Coot (18) was used for model building into the electron density. Maximum-likelihood restrained refinement in Phenix (19) was used to refine the structure after each cycle of model building until acceptable R-factors, geometry statistics (ideal rmsd for bonds and angles), and Ramachandran statistics were achieved (Table S1). PyMOL molecular viewer was used to visualize and analyze the structures (20). Iterative build omit σA-weighted 2mFo-Fc electron density maps were generated using Phenix Autobuild (21).
Results and Discussion
General Structure Details
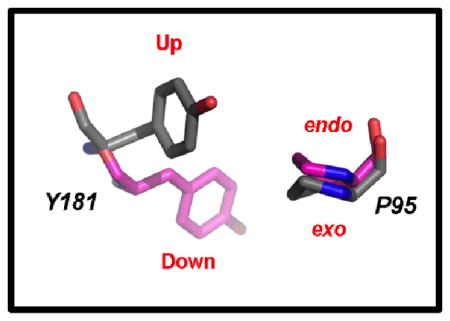
Similar to the earlier catechol diether structures, the electron density (Figure 1, Figure S3) reveals that RT is in the “open-cleft” conformation as observed in other NNRTI:RT crystal structures (8, 12, 22). Consistent with other NNRTIs, the primer grip (residues 227–235) shifts approximately 3–4 Å as a result of the catechol diether compounds binding near the “tunnel” region of the NNBP (23). Using a Cα backbone alignment for the p66 subunits and bound inhibitors, root mean square deviations (RMSD) were calculated comparing 4H4M with the RT:3 and RT:4 structures. The RMSD values for the 4H4M/RT:3 and 4H4M/RT:4 structures are 0.415 and 0.890 Å, respectively. Previous structures of RT bound to diarylpyrimidine inhibitors such as etravirine and rilpivirine reveal Tyr181 in the “up” conformation maintaining interactions with the aryl rings of such inhibitors (8, 24). For all of the catechol diether structures, Tyr181 is in the uncommon “down” conformation primarily to gain interactions with the catechol aryl ring (12). Besides the catechol diether complexes, the down conformation of Tyr181 is also found in other structures such as the RT:pyridinone derivative complex, PDB code 2BE2 (25). Previously, the 2BE2 crystal structure was used for computational modeling and the Tyr181 “down” conformation was accurately predicted upon catechol diether binding (11). The proximity of Tyr181 to the catechol aryl ring in the crystal structures suggests that π-π stacking may influence the “down” conformation observed in all of the catechol diether structures (Figure 2, Table 2). In addition to the unconventional conformation of Tyr181, Pro95 is also observed in the uncommon Cγ endo conformation, perhaps to avoid steric clash with the “down” conformation of Tyr181. This places the catechol diether class of compounds within a more optimal distance to interact with Pro95.
Figure 1.

Stereo view of omit, σA-weighted 2mFo-Fc electron density contoured to 1.0 σ for the RT:4 (A) and RT:3 (B) complexes. Both compounds 4 and 3 (green and orange, respectively) and Pro95 (yellow) were omitted from the model to generate iterative build omit maps using the original structure factors. Additional RT residues in the binding pocket are lavender.
Figure 2.

Stereo view of the catechol diether inhibitors (A) 4 (green) and (B) 3 (orange) bound to the NNBP of HIV-1 RT (lavender).
Table 2.
Comparison of Pro95 and Tyr181 conformations observed in the crystal structures for compound 1 and the other five FDA approved NNRTIs.
| |||
|---|---|---|---|
| RT:NNRTI Complex | PDB Code | P95 Rotamer | Y181 Conformation |
| Compound 1 | 4H4M | Cγ endo | “Down” |
| Rilpivirine | 2ZD1 | Cγ exo | “Up” |
| Etravirine | 3M8P | Cγ exo | “Up” |
| Efavirenz | 1IKW | Cγ exo | “Up” |
| Delaviridine | 1KLM | Cγ exo | “Up” |
| Nevirapine | 3HVT | Cγ exo | “Up” |
Analysis of the specific interactions made between the catechol diether compounds and the NNBP continually inform our multidisciplinary drug design efforts targeting HIV-1 RT. Comparison of all the catechol diether-RT crystal structures solved to date reveals a conserved binding mode between the diaryl core and the hydrophobic residues of the NNBP (Figure 2). Similar to the earlier structures, the diaryl core makes strong van der Waals interactions with Pro95, Leu100, Val106, Val108, Val179, Tyr181, Tyr188, Trp229, Leu234, and Tyr318, all residues within the p66 location of the NNBP. Most significantly, the cyanovinyl substituent appears to make favorable interactions with Trp229 (Figure 2) and is proximal to Pro95; these conserved residues are ideal for targeting in order to counteract the effects of resistance mutations in the NNBP (13, 14, 26).
It should be noted that the best RT:4 crystals diffracted to amplitudes extending to 2.69 Å, an improvement relative to the resolution of the other catechol diether structures (2.8–2.9 Å). The improvement in resolution for the RT:4 structure yields better overall electron density for water molecules, specifically, those interacting with residues in the NNBP. In the RT:4 structure, there is 2mFo-Fc electron density at 1.0 σ (Figure S3) for two coordinated water molecules near the halogen atom (X) at the 5-position in the B-ring, forming two water-mediated hydrogen bonds between the NH3 group of Lys103 and the hydroxyl group of Tyr181 (Figure 1A). While these water molecules are not apparent in the lower resolution structures, it seems likely that these water molecules are conserved and occupy the NNBP in a similar manner. Higher resolution structures for 1 and 2 are being pursued using crystal dehydration techniques and will provide a better solvent model to validate this hypothesis. Interestingly, there is one water molecule present in the RT:TMC278 structure (PDB code 2ZD1) which is analogous to the one near the chlorine atom in the RT:4 structure (Figure S1) (8). While these water molecules may be conserved in other NNRTI bound structures, additional water-mediated hydrogen bonds between the B-ring halogen and Lys103 and Tyr181 are unique to the catechol diether compounds and likely contribute to their low nanomolar and picomolar potency.
Effects of Halogen Substitutions at the C5 position
Despite the apparent benefit from the halogens, the crystal structure for 1 revealed that the chlorine did not participate in halogen bonding with the Pro95 carbonyl oxygen, a possibility that had been raised by computational modeling (11, 12). Although halogen bonding does not appear to occur, the EC50 values suggest that the 5-Cl and 5-F derivatives are optimal and replacement with heavier halogens such as Br does not improve potency (Table 1), possibly due to spatial penalties on complexation (12). In order to fully address this issue, we compared the crystal structure of RT:3 with earlier RT:catechol diether structures to analyze various sizes of halogen substitutions at the C5 position. In the RT:3 crystal structure, the bromine substituent has very large electron density relative to the chlorine and fluorine substituents at the C5 position in the RT:1 and RT:2 structures (Figure 1, Figure S3). The distance between the carbonyl oxygen of Pro95 and Br is 5.40 Å, even further than the distance observed for Cl and F (4.70 and 4.80 Å) in the structures with 1 and 2, respectively. In addition, the C-O-Br angle is 96°, again far from the linear geometry for an ideal halogen bond (12, 27). Thus, the bulkier Br atom may be too large to accommodate the limited space near Pro95, Tyr188 and Trp229.
Table 1.
EC50 Values in nanomolar (nM) for catechol diether derivatives.
| ||
|---|---|---|
| Compound | EC50 (nM) | |
| 1 | X=Y=Cl | 0.055 |
| 2 | X=Y=F | 0.320 |
| 3 | X=Cl, Y=Br | 5.2 |
| 4 | X=Cl, Y=H | 5.0 |
| Rilpivirine | 0.670 | |
To further study the effect of the C5 substituent on the molecular interactions in the NNBP and its effect on compound potency, we also analyzed the RT:4 crystal structure in detail. This compound replaces the C5 halogen with hydrogen yet has very similar potency to RT:3 (Table 1). Adding the hydrogen atom to the C5 position of 4 in PyMOL (20) allowed us to probe the space near Pro95. For better spatial interpretation, we viewed the C5 halogen/hydrogen atom of the compounds and Pro95 as van der Waals spheres. The Cγ atom of Pro95 is in close proximity to all of the C5 substituents (Figure 3). However, as the C5 substituent increases in size we see an increase in van der Waals contacts for the substituent and the Cγ atom of Pro95. This is apparent in Figure 3 (Top) in which the van der Waals spheres for these atoms get closer with larger C5 substituents and eventually maintain an optimal interaction distance (ca. 3.4 Å) with the 5-Cl of 1. Conversely, the larger Br atom in 3 seems to have a “repulsive effect” in which both the Cγ of Pro95 and the 5-Br atom slightly shift apart (interaction distance is 3.6 Å) from one another to avoid steric clash. The repulsive effect of the 5-Br with Pro95 results in a reorientation of the ethoxy uracil to accommodate the limited space in the pocket. In essence, the C5 substituents and their interaction with the Cγ atom of Pro95 follow a Lennard-Jones potential (28), in which the 5-Cl yields the optimal interaction distance for a van der Waals interaction. The smaller substituents also follow this trend in which the 5-H of 4 makes the weakest van der Waals contacts (interaction distance ca. 3.8 Å) and the 5-F of 2 gains more van der Waals contacts (interactions distance ca. 3.6 Å). This trend observed in the structure analysis is also reflected in the EC50 values of the compounds, in which the order of compound potency from least to greatest is 5-H=5-Br<5-F<5-Cl.
Figure 3.
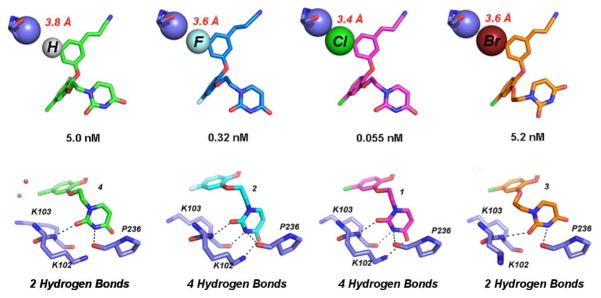
(Top) van der Waals sphere representation of the interaction between the Cγ atom of Pro 95 (lavender) and the C5 substituents of 4, 2, 1, and 3 from Left to Right. The 5-Cl (pink) and 5-F (blue) derivatives maintain the most hydrogen bonds and have potency in the picomolar range, while the interaction with 5-H (green) and 5-Br (orange) are not as strong. The repulsive effect causes the 5-Br to shift away from the Cγ causing a rearrangement of the uracil in order to avoid steric clash. (Bottom) Corresponding uracil conformation for the various C5 derivatives and the possible hydrogen bond interactions (black dashed lines) made with the donors and acceptors in the groove region of the NNBP. Hydrogen bonding potential for the C5 derivatives and strength of the C5-Cγ interaction correlate well with the EC50 values.
The RT complexes with the five FDA-approved NNRTIs, rilpivirine (TMC278), etravirine (TMC125), efavirenz, delavirdine, and nevirapine, were compared with the RT:1 structure (Table 2). In the RT:1 structure, as well as the other catechol diether structures, Pro95 specifically adopts the Cγ endo rotamer in which Cγ is pointed toward the 5-Cl group to maintain favorable van der Waals interactions as described above. Interactions between the 5-Cl containing ring and Tyr181 in the “down” conformation make space for the 5-Cl atom to move closer to the Cγ atom of Pro95 (Table 2). As observed in the approved RT:NNRTI complexes, interactions with Pro95 are generally weak. For example, distances between the closest NNRTI atom (which happens to be carbon for all the NNRTIs) and the Cγ of Pro95 range from 3.9–4.9 Å, which is greater than the sum of van der Waals radii for 2 carbon atoms. For all of the approved NNRTIs, the “up” conformation of Tyr181 restricts the space in the pocket and prevents the inhibitor from moving closer to Pro95 in order to gain favorable van der Waals interactions.
The rare conformation of Pro95 and Tyr181, EC50 value trends, and van der Waals analysis strongly suggest that the 5-Cl is ideally positioned on the catechol diether scaffold to interact with the Cγ of Pro95. While this interpretation suggests that the interactions with Cγ may compete with possible halogen bonding with the carbonyl, it is encouraging that our most potent inhibitor, compound 1, maintains favorable interactions with Pro95. In terms of the structure activity relationships, both the structures and the EC50 values strongly suggest that the chlorine atom is optimal at the C5 position of the cyanovinylphenyl ring. Perhaps the same approach can be used for Trp229 and other conserved residues in the NNBP, in which compounds are further modified to maintain optimal van der Waals interaction distances with target residues of interest.
Conformation of the uracil
In all RT crystal structures with the catechol diether compounds, the ethoxy uracil interacts with residues in the groove region of the NNBP, consisting of residues Lys102, Lys103, and Pro236. However, the ethoxy uracil is observed in multiple conformations in the different crystal structures. Structures of picomolar derivatives 1 and 2 reveal the ethoxy uracil in the syn-anti-gauche (sag) conformation (Figure 4) maintaining up to four hydrogen bonds with Lys102 and Lys103 (12). In the RT:3 crystal structure, we see the ethoxy linker in the reverse conformation, shifting the uracil slightly into the anti-anti-gauche (aag) conformation. Compound 4 is in a more intermediate conformation, in which the ethoxy linker is similar to 1 and 2 in the sag conformation, but the uracil is rotated slightly. Table 3 lists all of the possible hydrogen bonding interactions observed in the crystal structures for all of the catechol diether compounds. By examining all of these interactions, it is apparent that the picomolar inhibitors, compounds 1 and 2 in which the uracil is in the sag conformation, maintain the most hydrogen bonds with the donors and acceptors in the groove region, which include residues Lys102 and Lys103. There is even the possibility of making a fourth hydrogen bond with the backbone carbonyl of Pro236, in which the NH group of the uracil can participate in hydrogen bonds with either the carbonyl of Lys103 or Pro236. However, in the conformation observed in 3 and 4, only 2 hydrogen bonds can be formed between the uracil and groove residues Lys103 and Pro236.
Figure 4.
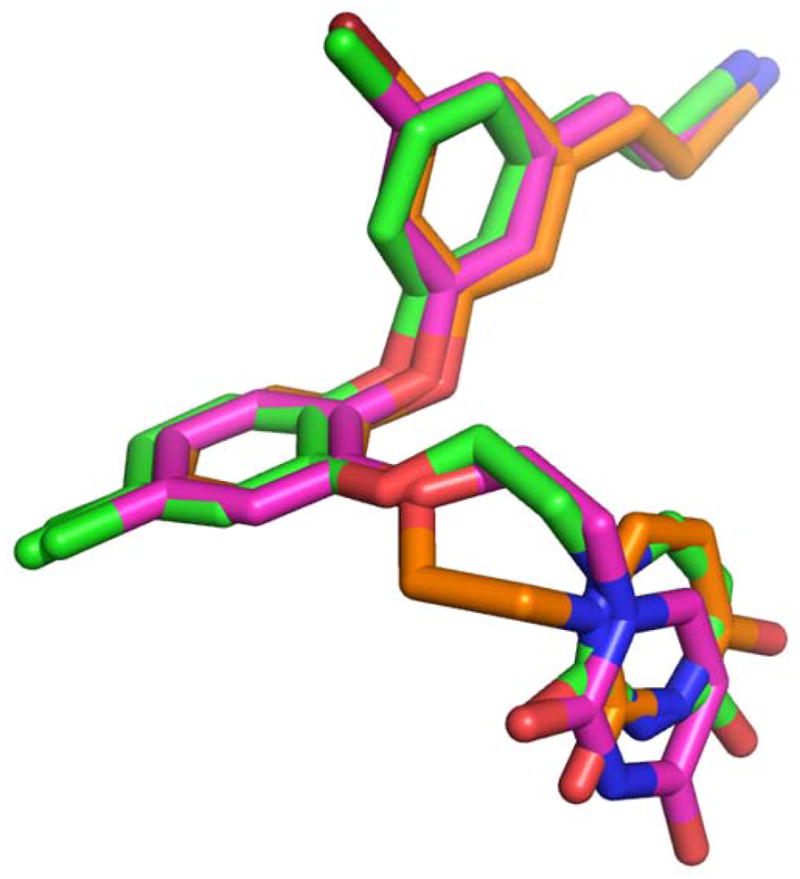
Overlay of the sag (pink), “Intermediate” (green), and aag (orange) conformations of the ethoxy uracil side chain as observed in the RT complexes bound to 1, 4, and 3, respectively
Table 3.
List of all possible hydrogen bonding interactions as observed in the crystal structures for compounds 1–4. The hydrogen bond distance cutoff for the analysis is 3.6 Å or less.
| NNRTI | Donor | Acceptor | Distance | Total Possible H bonds | Uracil Conformation |
|---|---|---|---|---|---|
| 4 | |||||
|
| |||||
| NH backbone K103 | CO uracil | 3.3 | 2 | Intermediate | |
| NH uracil | CO P236 backbone | 3.4 | |||
|
| |||||
| 3 | |||||
|
| |||||
| NH backbone K103 | CO uracil | 3.2 | 2 | aag | |
| NH uracil | CO P236 backbone | 3.6 | |||
|
| |||||
| 2 | |||||
|
| |||||
| NH backbone K103 | CO uracil | 2.9 | 4 | sag | |
| NH uracil | CO K103 backbone | 2.8 | |||
| NH uracil | CO P236 backbone | 3.2 | |||
| NH3 sidechain K102 | CO uracil | 3.2 | |||
|
| |||||
| 1 | |||||
|
| |||||
| NH backbone K103 | CO uraci | 3.2 | 4 | sag | |
| NH uracil | CO K103 backbone | 3.3 | |||
| NH uracil | CO P236 backbone | 3.4 | |||
| NH3 sidechain K102 | CO uracil | 3.5 | |||
Interestingly, the conformation and hydrogen bonding capacity of the uracil ring seems to correlate with the strength of van der Waals interaction between the Pro95 Cγ and the C5 substituent. Figure 3 shows the correlation between the strength of the van der Waals interaction and its effects on the hydrogen bonding potential of the uracil ring. Picomolar inhibitors 1 and 2 have stronger interactions with the Cγ atom of Pro95 and have uracil conformations that maintain the most hydrogen bonds. In the case of compound 4, the interaction between the Cγ and the hydrogen is not strong enough, causing the cyanovinylphenyl to shift slightly accompanied by a movement in the uracil ring. This movement in the uracil, considered as the “intermediate” conformation (Table 3,Figure 4), reduces the hydrogen bonding potential of compound 4. The interaction distances for 5-F of 2 and 5-Br of 3 with the Cγ atom are essentially the same; however, the larger Br atom causes the C5 aryl ring to also shift resulting in the rearrangement of the uracil in the aag conformation to prevent steric clash with residues in the groove. This conformation is more drastic than the “intermediate” conformation, in which the ethoxy linker rearranges and positions the uracil farther away from the hydrogen bond donors and acceptors within the groove region of the NNBP. The 15-fold loss in potency between the 5-F and 5-Br derivatives is most likely due to the loss of 2 hydrogen bonds observed with the uracil in the aag conformation for compound 3.
While it is difficult to quantify the direct relationship between molecular interactions observed in crystal structures and compound potency, we generally see a trend in which both the strength of van der Waals interactions with the Cγ of Pro95 and the positioning of the uracil ring contribute to picomolar potency. While the 5-F atom maintains the uracil conformation in the sag position to make four hydrogen bonds, its contacts with the Cγ of Pro95 are not as favorable, suggesting a possible explanation for the 6-fold increase in EC50 value compared to compound 1. Similarly, the weaker interactions observed between the 5-H and 5-Br derivatives, along with the fewer hydrogen bonds formed seem to contribute to their 100-fold difference in potency compared to 1 and 15-fold difference compared to 2. Previously, it has been reported that the contribution of a single hydrogen bond can vary significantly, ranging from 2- to 15-fold for weak, neutral-neutral hydrogen bonds, and up to 3000-fold for strong, charged hydrogen bonds (29). The energetic strength of van der Waals interactions has less variation but can contribute up to 4 kJ/mol (30). Thus, it seems possible that our structural observations somewhat correlate with the EC50 values for the different C5 analogues.
Unlike the hydrophobic core of other NNRTIs, the ethoxy uracil of our catechol diether compounds is flexible and may be a key advantage in adapting to changes in the NNBP such as mutations. As observed for rilpivirine (TMC278), torsional flexibility allows the compound to reorient and gain new interactions with the L100I/K103N RT variant (8, 31). Interestingly, we also observe interactions with Leu100, specifically with the Cδ1 and Cδ2 atoms of the side chain and the various C5 substituents. Distances between the C5 atoms and Cδ2 atom of Leu100 range from 3.4–4.2 Å, while distances between the C5 atoms and Cδ1 atom of Leu100 range from 3.2–4.1 Å. These values suggest that the C5 substituents are also optimally placed to form van der Waals interactions with Leu100. As examined in the four structures, the interaction with Leu100 is not enhanced by larger C5 substituents; however, binding of the four analogues may be impacted by the clinically relevant L100I mutation. Given the influence of the C5 substitution on compound orientation, future optimization can exploit the Pro95 Cγ van der Waals interaction to position the compound in the NNBP and anchor the uracil ring into the groove for maximal hydrogen bonding. Possible modulation of the ethoxy uracil at the C5 position to promote the sag conformation may be an effective strategy to maintain interactions with resistant variants of RT such as L100I/K103N while avoiding steric conflicts with residues in the binding pocket.
Conclusions and Future Directions
Although several anti-HIV therapeutics are currently available, there is still a compelling need to optimize new active compounds given the propensity of the HIV-1 virus to develop resistance against current treatments. Towards that goal, we recently reported an extraordinarily potent class of anti-HIV agents targeting RT that feature a catechol diether core scaffold. Additional crystal structures of RT in complex with modified derivatives of the catechol diether series assisted in the elucidation of key interactions and our structural observations correlate well with the reported EC50 values. The potential for halogen bonding between these compounds and Pro95 was explored extensively using comparative structural analysis to interpret the effects of different substitutions at the C5 position of the cyanovinylphenyl group. Upon analysis of the various crystal structures, it is apparent that Pro95 is not participating in halogen bonding and that, instead, a van der Waals interaction between the C5 halogen and Cγ of Pro95 is occurring with the 5-Cl of compound 1. In addition to these interactions, the ethoxy uracil substituent of the compounds adopts several different conformations of which the sag conformation observed in compounds 1 and 2 is optimal, allowing for the maintenance of the most hydrogen bonds. Conversely, the “intermediate” and aag conformations of the uracil allow for only two hydrogen bonds and clarify the differences in interactions between compounds with picomolar versus nanomolar potency. Interestingly, substitutions at the C5 position of the cyanovinylphenyl ring greatly influence the interaction with the Pro95 Cγ and thus the conformation of the ethoxy uracil in the groove pocket. As suggested by our structural observations, the flexibility of the ethoxy uracil, as well as the direct interaction of our compounds with conserved residue Pro95, is encouraging given the prevalence of mutations in the NNBP. Furthermore, these structural studies will aid future computational and structure-based efforts to optimize the catechol diether compound series against resistant variants of RT.
Supplementary Material
Conserved water molecules (spheres) in the NNBP of RT:TMC278 (gray) and RT:4 (green) as observed in the superimposed structures. The PDB code for RT:TMC278 structure is 2ZD1.
Stereo view of the omit mFo-Fc electron density for compounds (A) 4 and (B) 3 from the respective RT structures. Contour level is set to 3.0 σ.
Stereo view of 2mFo-Fc electron density for RT complexes bound to (A) compound 4 and (B) compound 3. Map contour level is set to 1.0 σ.
1 Phases determined from molecular replacement.
2 Phases determined from Difference Fourier Methods. The difference between Fc for 4H4M and Fo for the RT:4 complex is 0.39. The difference between Fc for 4H4M and Fo for the RT:3 complex is 0.42.
Acknowledgments
Gratitude is expressed to the National Institutes of Health (GM49551, AI44616, GM32136) for research support and (AI104334) for fellowship support to KMF. We also thank the National Synchrotron Light Source at Brookhaven National Laboratory for beam time on X29A.
References
- 1.Adamson CS, Freed EO. Anti-HIV-1 Therapeutics: From FDA-approved Drugs to Hypothetical Future Targets. Mol Interv. 2009;9:70–74. doi: 10.1124/mi.9.2.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Adamson CS, Freed EO. Novel approaches to inhibiting HIV-1 replication. Antivir Res. 2010;85 :119–141. doi: 10.1016/j.antiviral.2009.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pomerantz RJ, Horn DL. Twenty years of therapy for HIV-1 infection. Nat Med. 2003;9:867–873. doi: 10.1038/nm0703-867. [DOI] [PubMed] [Google Scholar]
- 4.Roberts JD, Bebenek K, Kunkel TA. The Accuracy of Reverse-Transcriptase from Hiv-1. Science. 1988;242:1171–1173. doi: 10.1126/science.2460925. [DOI] [PubMed] [Google Scholar]
- 5.Owen A. The impact of host pharmacogenetics on antiretroviral drug disposition. Curr Infect Dis Rep. 2006;8:401–408. doi: 10.1007/s11908-006-0052-2. [DOI] [PubMed] [Google Scholar]
- 6.Vandamme AM, Van Vaerenbergh K, De Clercq E. Anti-human immunodeficiency virus drug combination strategies. Antivir Chem Chemoth. 1998;9:187–203. doi: 10.1177/095632029800900301. [DOI] [PubMed] [Google Scholar]
- 7.Boyer J, Arnoult E, Medebielle M, Guillemont J, Unge J, Jochmans D. Difluoromethylbenzoxazole pyrimidine thioether derivatives: a novel class of potent non-nucleoside HIV-1 reverse transcriptase inhibitors. J Med Chem. 2011;54:7974–7985. doi: 10.1021/jm200766b. [DOI] [PubMed] [Google Scholar]
- 8.Das K, Bauman JD, Clark AD, Jr, Frenkel YV, Lewi PJ, Shatkin AJ, et al. High-resolution structures of HIV-1 reverse transcriptase/TMC278 complexes: strategic flexibility explains potency against resistance mutations. Proc Natl Acad Sci U S A. 2008;105:1466–1471. doi: 10.1073/pnas.0711209105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.De Clercq E. Anti-HIV drugs: 25 compounds approved within 25 years after the discovery of HIV. Int J Antimicrob Ag. 2009;33:307–320. doi: 10.1016/j.ijantimicag.2008.10.010. [DOI] [PubMed] [Google Scholar]
- 10.Lansdon EB, Brendza KM, Hung M, Wang R, Mukund S, Jin D, et al. Crystal structures of HIV-1 reverse transcriptase with etravirine (TMC125) and rilpivirine (TMC278): implications for drug design. J Med Chem. 2010;53:4295–4299. doi: 10.1021/jm1002233. [DOI] [PubMed] [Google Scholar]
- 11.Bollini M, Domaoal RA, Thakur VV, Gallardo-Macias R, Spasov KA, Anderson KS, et al. Computationally-guided optimization of a docking hit to yield catechol diethers as potent anti-HIV agents. J Med Chem. 2011;54:8582–8591. doi: 10.1021/jm201134m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Frey KM, Bollini M, Mislak AC, Cisneros JA, Gallardo-Macias R, Jorgensen WL, et al. Crystal structures of HIV-1 reverse transcriptase with picomolar inhibitors reveal key interactions for drug design. J Am Chem Soc. 2012;134:19501–19503. doi: 10.1021/ja3092642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Auwerx J, Van Nieuwenhove J, Rodriguez-Barrios F, de Castro S, Velazquez S, Ceccherini-Silberstein F, et al. The N137 and P140 amino acids in the p51 and the P95 amino acid in the p66 subunit of human immunodeficiency virus type 1 (HIV-1) reverse transcriptase are instrumental to maintain catalytic activity and to design new classes of anti-HIV-1 drugs. FEBS Lett. 2005;579:2294–2300. doi: 10.1016/j.febslet.2005.02.077. [DOI] [PubMed] [Google Scholar]
- 14.Ceccherini-Silberstein F, Gago F, Santoro M, Gori C, Svicher V, Rodriguez-Barrios F, et al. High sequence conservation of human immunodeficiency virus type 1 reverse transcriptase under drug pressure despite the continuous appearance of mutations. J Virol. 2005;79 :10718–10729. doi: 10.1128/JVI.79.16.10718-10729.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bauman JD, Das K, Ho WC, Baweja M, Himmel DM, Clark AD, Jr, et al. Crystal engineering of HIV-1 reverse transcriptase for structure-based drug design. Nucleic Acids Res. 2008;36:5083–5092. doi: 10.1093/nar/gkn464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Method Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 17.Mccoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, et al. PHENIX: A comprehensive python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.DeLano WL. PyMOL molecular viewer: Updates and refinements. Abstr Pap Am Chem S. 2009:238. [Google Scholar]
- 21.Terwilliger TC, Grosse-Kunstleve RW, Afonine PV, Moriarty NW, Adams PD, Read RJ, et al. Iterative-build OMIT maps: map improvement by iterative model building and refinement without model bias. Acta Crystallogr D. 2008;64:515–524. doi: 10.1107/S0907444908004319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Das K, Clark AD, Jr, Lewi PJ, Heeres J, De Jonge MR, Koymans LM, et al. Roles of conformational and positional adaptability in structure-based design of TMC125-R165335 (etravirine) and related non-nucleoside reverse transcriptase inhibitors that are highly potent and effective against wild-type and drug-resistant HIV-1 variants. J Med Chem. 2004;47:2550–2560. doi: 10.1021/jm030558s. [DOI] [PubMed] [Google Scholar]
- 23.Wisniewski M, Palaniappan C, Fu Z, Le Grice SF, Fay P, Bambara RA. Mutations in the primer grip region of HIV reverse transcriptase can increase replication fidelity. J Biol Chem. 1999;274:28175–28184. doi: 10.1074/jbc.274.40.28175. [DOI] [PubMed] [Google Scholar]
- 24.Kertesz DJ, Brotherton-Pleiss C, Yang M, Wang Z, Lin X, Qiu Z, et al. Discovery of piperidin-4-yl-aminopyrimidines as HIV-1 reverse transcriptase inhibitors. N-benzyl derivatives with broad potency against resistant mutant viruses. Bioorg Med Chem Lett. 2010;20:4215–4218. doi: 10.1016/j.bmcl.2010.05.040. [DOI] [PubMed] [Google Scholar]
- 25.Himmel DM, Das K, Clark AD, Jr, Hughes SH, Benjahad A, Oumouch S, et al. Crystal structures for HIV-1 reverse transcriptase in complexes with three pyridinone derivatives: a new class of non-nucleoside inhibitors effective against a broad range of drug-resistant strains. J Med Chem. 2005;48:7582–7591. doi: 10.1021/jm0500323. [DOI] [PubMed] [Google Scholar]
- 26.Pelemans H, Esnouf R, De Clercq E, Balzarini J. Mutational analysis of trp-229 of human immunodeficiency virus type 1 reverse transcriptase (RT) identifies this amino acid residue as a prime target for the rational design of new non-nucleoside RT inhibitors. Mol Pharmacol. 2000;57:954–960. [PubMed] [Google Scholar]
- 27.Wilcken R, Zimmermann MO, Lange A, Joerger AC, Boeckler FM. Principles and Applications of Halogen Bonding in Medicinal Chemistry and Chemical Biology. J Med Chem. 2013 doi: 10.1021/jm3012068. [DOI] [PubMed] [Google Scholar]
- 28.Boas FE, Harbury PB. Potential energy functions for protein design. Curr Opin Struc Biol. 2007;17:199–204. doi: 10.1016/j.sbi.2007.03.006. [DOI] [PubMed] [Google Scholar]
- 29.Davis AM, Teague SJ. Hydrogen bonding, hydrophobic interactions, and failure of the rigid receptor hypothesis. Angew Chem Int Edit. 1999;38:737–749. doi: 10.1002/(SICI)1521-3773(19990315)38:6<736::AID-ANIE736>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 30.Atkins PaJdP. Physical Chemistry for the Life Sciences. Oxford, UK: Oxford University Press; 2006. [Google Scholar]
- 31.La Regina G, Coluccia A, Silvestri R. Looking for an active conformation of the future HIV type-1 non-nucleoside reverse transcriptase inhibitors. Antivir Chem Chemother. 2010;20:213–237. doi: 10.3851/IMP1607. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Conserved water molecules (spheres) in the NNBP of RT:TMC278 (gray) and RT:4 (green) as observed in the superimposed structures. The PDB code for RT:TMC278 structure is 2ZD1.
Stereo view of the omit mFo-Fc electron density for compounds (A) 4 and (B) 3 from the respective RT structures. Contour level is set to 3.0 σ.
Stereo view of 2mFo-Fc electron density for RT complexes bound to (A) compound 4 and (B) compound 3. Map contour level is set to 1.0 σ.
1 Phases determined from molecular replacement.
2 Phases determined from Difference Fourier Methods. The difference between Fc for 4H4M and Fo for the RT:4 complex is 0.39. The difference between Fc for 4H4M and Fo for the RT:3 complex is 0.42.