

1 Introduction
Protein and nucleic acid modification processes play a central role in cellular viability. These range from the regulation of transcription via the modification of DNA,1 the splicing of RNA prior to translation2 and the control of cell signaling at a multitude of levels via covalent modifications of proteins, e.g. refs. 3 and 4. Peptides are also known to undergo extensive modifications, in particular, during the bacterial generation of cellular defense molecules that are increasingly being studied as possible mammalian antibiotics.5
This review is focused on a unique set of posttranslational modifications that convert canonical amino acid side chains within either a peptide or folded protein into quinone-containing redox cofactors.6,7 The peptide-derived quinocofactor, pyrroloquinoline quinone (PQQ), was the first to be detected in 1964, in association with the bacterial enzyme, glucose dehydrogenase.8 This was followed by X-ray characterizations of PQQ, either alone9 or in a non-covalent complex with several dehydrogenases.10-12 The property of PQQ as a reversibly-bound cofactor that can be shared among many redox proteins contrasts with the remaining quinocofactors presented in Scheme 1, each of which is found to be covalently associated with its cognate protein. There was a considerable lag between the characterization of PQQ and the remainder of the quinocofactors, with identification of trihydroxyphenylalanine quinone (TPQ)13 and tryptophan tryptophylquinone (TTQ)14 occurring in the early 1990s, followed by lysyl tyrosine quinone (LTQ)15 in 1996 and cysteine tryptophylquinone (CTQ)16 in 2001. These quinocofactors are found to function either in prokaryotes or eukaryotes, with TPQ being the exception that spans these two biological domains. The distinguishing features of each cofactor are highlighted in Table 1, with the underlying commonality being that each is constructed around an aromatic side chain (tryptophan or tyrosine). This review brings a primary focus to the variety of biosynthetic pathways for the production of quinocofactors [cf. refs. 7,17,18] and the reader is referred to a number of treatises that are centered primarily on the enzymatic mechanisms surrounding the mature cofactors.19-21
Scheme 1.
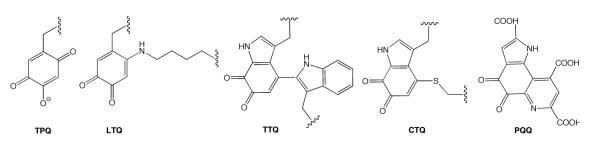
Structures of the established quinocofactors, TPQ, LTQ, TTQ, CTQ, and PQQ.
Table 1.
Properties of the Quinocofactors Illustrated in Scheme 1.
| PQQ | TTQ | CTQ | TPQ | LTQ | |
|---|---|---|---|---|---|
| Biological niche |
Solely prokaryotic |
Solely prokaryotic |
Solely prokaryotic |
Eukaryotic/ prokaryotic |
Solely eukaryotic |
|
| |||||
| Type of enzyme activity |
Dehydrogenase | Dehydrogenase | Dehydrogenase | Oxidase | Oxidase |
|
| |||||
| Aromatic amino acid scaffold |
Tyr a | Trp b | Trp c | Tyr d | Tyr e |
|
| |||||
| Second amino acid in cofactor |
Glu f | Trp b | Cys c | None | Lys e |
|
| |||||
| Requires new C–C bond in cofactor |
Yes f | Yes b | No c | No d | No e |
|
| |||||
| X-ray structures |
Yes g | Yes h | Yes c | Yes i | No |
|
| |||||
| Number of gene products required for biosynthesis j |
5(6) k | 11 l (12) m | 5(6) n | 1 o | 1 p |
|
| |||||
| Metal ions implicated in the pathway for either biogenesis and/or catalysis |
Fe, Zn2+, Ca2+ j |
Fe, Cu, q
Ca2+ r |
Fe, Cu s | Cu, o,t
(Ca2+) u |
Cu p |
Ref. (31);
Ref. (14);
Ref. (16);
Ref. (13);
Ref. (15);
Ref. (30);
Ref. (9);
Ref. (63);
Refs. (94−97, 99, 114);
In the cases where ambiguity exists as to the number of essential genes, the greater number is included in parentheses.
Ref. (23);
Ref. (64);
Ref. (65);
Ref. (86);
Refs. (101, 104);
Ref. (137);
Ref. (85);
Ref. (157);
Refs. (87, 89);
Ref. (113);
Ref. (158).
2 PQQ (Pyrroloquinoline Quinone)
2.1 General Background
The biological role for PQQ has traditionally been described as a prokaryotic “vitamin”, with the observation that non-PQQ-producing bacterial species such as Escherichia coli (E. coli) show enhanced growth rate in the presence of PQQ.22 Recently completed bioinformatics analyses present a method for the identification of intact operons for the production of PQQ, allowing us to estimate ≥ 125 bacterial species that are capable of PQQ production.23 The best-described enzymes that utilize PQQ as catalytic cofactor are methanol and glucose dehydrogenases24-26 (cf. Figure 1). These enzymes function in the periplasmic space of Gram-negative bacteria to oxidize their respective substrate to aldehyde, concomitant with the production of reduced PQQ (PQQH2). The reoxidation of PQQH2 by one-electron acceptors (such as ubiquinone) is linked to a plasma membrane electron transfer chain, providing a venue for the direct utilization of the energy derived from the oxidation of one-carbon units and glucose in maintaining cell viability. Within a community of multiple species of bacteria, this provides the PQQ-producing species with a competitive growth advantage. The role of PQQ in mammals has been much more controversial, with careful exclusion of PQQ from the diets of laboratory mice leading, after several generations, to infertility,27 with more recent studies point toward a role for PQQ in mitochondrial biogenesis.28 While these studies indicate a requirement for low levels of PQQ in a mammalian diet, the full range of mammalian targets and precise mechanism of action remain to be elucidated.
Figure 1.

Ribbon diagram of the overall structure of the dimer of glucose dehydrogenase from Acetinobacter calcoaceticus with PQQ (red). The spheres in yellow are Ca2+. Modified from ref. (11).
2.2 Biosynthetic Process
Although the chemical structure of PQQ has been determined and the mechanism of PQQ-utilizing enzymes is well studied,19 the biosynthetic pathway leading to PQQ has not yet been solved. The first step in the elucidation of PQQ biosynthesis was the identification of the amino acid precursors by 13C-labeling and nuclear magnetic resonance (NMR). Methylobacterium extorquens (M. extorquens) AM1 was grown on 1-13C- or 2-13C ethanol or 13C methanol and the resulting 13C enrichments in PQQ were compared to the labeling patterns in amino acids.29-31 From such studies, PQQ is concluded to be formed from the crosslinking of a glutamate and tyrosine side chain (Figure 2).
Figure 2.
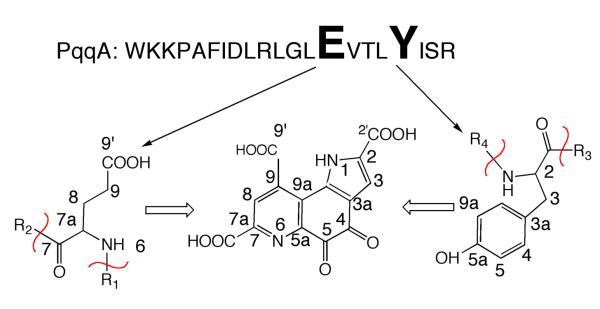
PQQ is derived from the conserved Glu and Tyr within PqqA. The sequence shown for PqqA is from K. pneumoniae. From ref. (33).
The biosynthesis of PQQ is accomplished by the gene products of a specific pqq operon. Genes involved in PQQ synthesis have been cloned from Acinetobacter calcoaceticus,32Klebsiella pneumoniae (K. pneumoniae),33Pseudomonas fluorescens CHAO,34Methylobacterium organophilum DSM 760,35 and M. extorquens AM1.36 In K. pneumoniae, the pqq operon comprises six genes (designated pqqA-F) (Figure 3).33 These genes have been expressed in E. coli, a non-PQQ producer and lead to the production of PQQ.37 Genetic knockout studies of each of these genes show that four of the six gene products (PqqA, PqqC, PqqD, and PqqE) are absolutely required for PQQ production.38 Although genetic knockout studies of pqqB had provided ambiguous results regarding the absolute requirement for PqqB in PQQ biosynthesis,38 bioinformatic analyses indicate that this gene likely performs an essential function in PQQ production.23
Figure 4.

Structure of PqqD in K. pneumoniae from X. campestris. While this structure is illustrated as a single subunit, it is a dimer in the crystal. From ref. (23).
The formation of PQQ from PqqA necessitates the formation of a new carbon–carbon bond between atoms C9 and C9a (Figure 2). Such reactions are chemically challenging and generally require specialized enzymes capable of free radical generation. The only candidate for this type of reaction within the pqq operon is the gene encoding PqqE. PqqE contains a highly conserved cysteine motif unique to the radical S-adenosyl-l-methionine (SAM) family (CX3CX2C), has been demonstrated to contain a second [4Fe-4S]2+ cluster and to be capable of cleaving SAM to methionine and 5'-deoxyadenosine in an uncoupled reaction.39 The N-terminal 4Fe-4S center of PqqE is the site of the canonical motif, and it is assigned to the role of SAM cleavage and subsequent hydrogen atom abstraction from substrate. The second 4Fe-4S center (found at the protein’s C-terminus) is much more divergent among identified proteins with two (4Fe- 4S) centers,40 and may function either as a recognition site for the peptide substrate (PqqA) by analogy to MoaA, an ortholog with 30% identity,41 or as an electron sink during the catalytic mechanism.40 Thus far, it has not been possible to demonstrate that PqqE acts upon PqqA, implying that PqqA may need to be modified by other proteins in the pathway, before it can behave as a substrate for PqqE (cf. Scheme 2).
Scheme 2.
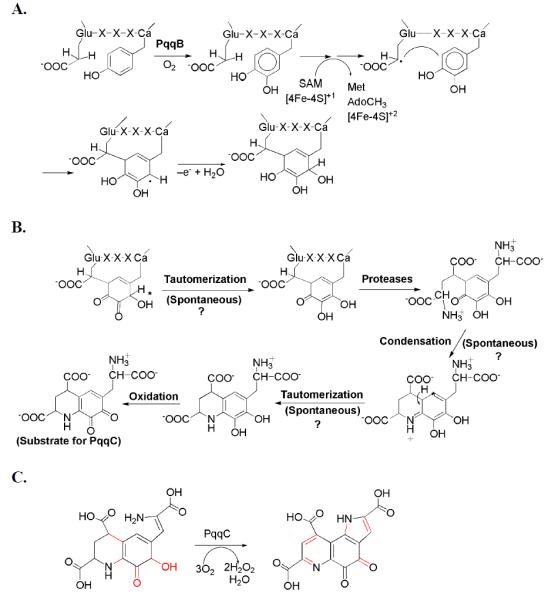
A speculative pathway for the production of PQQ, illustrating the large number of unknown features. A. A working mechanism invokes hydroxylation of the conserved Tyr within PqqA by PqqB, prior to the action of PqqE. B. The limited number of conserved components of the PQQ operon suggests the presence of a number of spontaneous (non-catalyzed) steps. C. The last step, in which AHQQ is converted to PQQ, is established.
Velterop et al. carried out a series of experiments in which cell extracts of E. coli containing all but one of the Pqq proteins were combined with those containing the missing Pqq protein.38 PQQ was produced in only one of these sets involving the back addition of PqqC. E. coli cells containing a clone encoding all but the PqqC protein produced an intermediate of PQQ that, while unstable, was shown to accumulate in both the culture medium and inside of cells. Although the amount of the intermediate was low, it was purified42 and identified as 3a-(2-amino-2-carboxyethyl)-4,5-dioxo-4,5,6,7,8,9-hexahydroquinoline-7,9-dicarboxylic acid (AHQQ).43 Conversion of AHQQ to PQQ, catalyzed by PqqC was, thus, attributed to the final step in the overall pathway.44 This has been confirmed by in vitro assays of PqqC, showing an eight-electron oxidation and ring cyclization of AHQQ to PQQ. Three equivalents of O2 are required for a single turnover producing two equivalents of H2O2, indicating that one equivalent of H2O2 produced by the enzyme is used as an electron acceptor during PQQ synthesis45 (Scheme 3). The activation of O2 in the absence of a metal or cofactor is of particular interest (see below).
Scheme 3.
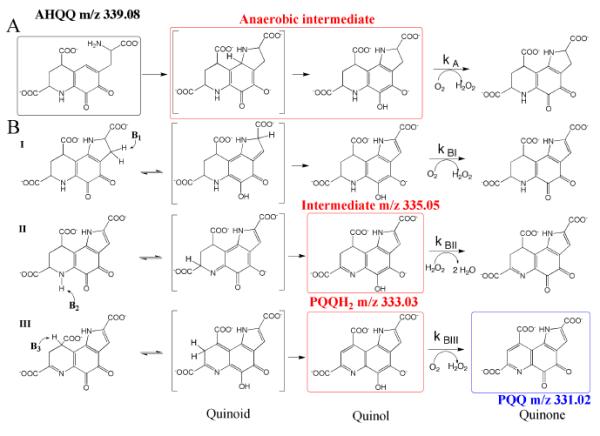
The four partial reactions catalyzed by PqqC. The reaction intermediates that have been detected are shown in the red boxes and the oxidized product is in blue. The final species formed in vivo is likely to be PQQH2, which is proposed to dissociate from PqqC before its final oxidation to PQQ. Adapted from ref. (53).
The small, 10-KDa protein PqqD has engendered considerable interest, as it has no homology to any characterized protein. The Xanthomonas campestris (X. campestris) PqqD crystal structure was solved46 (PDB ID: 3G2B) (Figure 4) and, surprisingly, the conserved side chains are in a region that acts as a linker between the helices and β turn. The paucity of secondary structure within PqqD suggests a requirement for a binding partner, i.e., its participation in protein/protein interactions. It will be important to determine if the functional form of PqqD is a dimer, since this is the form characterized by X-ray crystallography. Of interest in this context, gene fusions have been identified between pqqD and either pqqC47 or pqqE.23 The addition of PqqD to anaerobic PqqE in the presence of SAM has been found to lead to an increase in α-helical content compared to the individual proteins (by circular dichroism (CD)), and to changes in the electronic environment of the [4Fe-4S]1+ clusters of PqqE (by electron paramagnetic resonance (EPR)), indicating a specific interaction between the two proteins.48 The precise role of this interaction, as well as the possible interaction of PqqD with other proteins within the pathway, remains to be elucidated.
The role of PqqB is still unknown. PqqB shares homology with the metallo-beta-lactamase family and a crystal structure is available (PDB ID: 3JXP) (Figure 5). The comparison of the X-ray structure for PqqB to its closest homolog within the metallo-beta-lactamase superfamily, PhnP, a metallo-protein with two Mn2+ in the active site, is extremely revealing regarding putative function. Figure 5 shows the overall structural similarity between PhnP and PqqB, together with the loss of the ligands for one of the two metals observed to be bound to the PhnP active site from PqqB. The metal ligand residues that are retained in PqqB represent a 2-His/1-carboxylate facial triad configuration, characteristic of the non-heme metal-binding oxygenase family.49 In the X-ray structure of PqqB, a Zn2+ has been identified at a second structural site, while the presumed iron atom at the active site is absent. Nonetheless, it appears likely that the gene product, PqqB, functions as a non-heme metallo-oxygenase in the course of the conversion of the peptide PqqA to PQQ (cf. Scheme 2).
Figure 5.

Structure of PqqB from Pseudomonas putida (A) in relation to its closest homolog; PhnP from E. coli (B). The active site of PqqB shows a 2-His/1-carboxylate facial triad configuration, characteristic of the non-heme metal-binding oxygenase family. As shown, PqqB contains Zn2+ (green) at the structural site but no metal at the active site. The Mn in PhnP is colored purple. From ref. (23).
PqqF is homologous to zinc-dependent proteases, initially suggesting its role in excising a crosslinked Tyr-Glu intermediate from the PqqA peptide. However, PqqF has not been shown to be essential for PQQ biosynthesis, and is absent from 53 of the identified PQQ operons.23 Thus, either many or all of the proteolytic steps necessary for the excision of the cofactor from its peptide are likely to be catalyzed by non-specific, cell-associated proteases.
Although PQQ was the first quinone cofactor discovered, the pathway for its production remains the least understood. Nonetheless, a number of distinctive and interesting features have emerged that form the basis for future investigations. First, PqqC is able to activate O2 in the absence of a metal or cofactor. The combination of kinetic studies, site-directed mutagenesis, and protein crystal structures has begun to define the roles of individual amino acids in O2 activation.50,51 From X-ray crystallography, a molecule of O2 or H2O2 is observed52 at a distance of 3.4 Å from a carbon of bound PQQ.50 Moreover, a progressive closure from an observed open to a closed conformation plays a primordial role in the catalysis (Figure 6).53 Second, PqqD, a small 10-kDa protein with no homology to any characterized protein, has an indispensable role in the biosynthesis of PQQ that is not yet elucidated. Third, a strictly anaerobic PqqE must function with an O2-dependent PqqC and likely, an O2-dependent non-heme iron oxygenase (PqqB). PqqE appears highly sensitive to O2, with catalytic SAM cleavage to generate the 5'-deoxyadenosine radical occurring only with PqqE that has been expressed and purified under anaerobic conditions.39 PQQ is not produced in the absence of O2, even when all the pqq gene products are present.38
Figure 6.

Structure of PqqC from K. pneumoniae. [(From ref. (53)] The structure in green is the open configuration and the black structure is the closed configuration containing bound PQQ. The red diatomic is labeled as O2, but could be H2O2. The R157 in the closed structure is to the left of R179.
A possible solution to this necessary combination of aerobic and anaerobic conditions may be via the discrete formation of protein-protein complexes. A number of findings strongly suggest a formation of such a complex during PQQ biosynthesis. These include the conservation of the ordering of ORFs within the operons for PQQ producers (strictly conserved in the order, pqqA–E within 98% of the sequenced genomes), the observation of gene fusions (D/C and D/E) and the demonstrated interaction of PqqD with PqqE. Conservation of gene order within an operon is not generally characteristic of prokaryotic organisms,54,55 and when such ordering is observed the corresponding gene products have been shown to physically interact.56,57 Protein–protein interactions are common in metabolic pathways, and these interactions can serve to increase the efficiency of the overall process by positioning consecutively acting enzymes in proximity to one another and by protecting sensitive intermediates from degradation via direct channeling between enzymes. In the PQQ biosynthetic pathway, protein complex formation may both shield PqqA from proteolytic degradation before completion of the necessary modifications and protect PqqE from extensive exposure to oxygen during turnover.
3 TTQ (Tryptophan Tryptophylquinone)
3.1 General Background
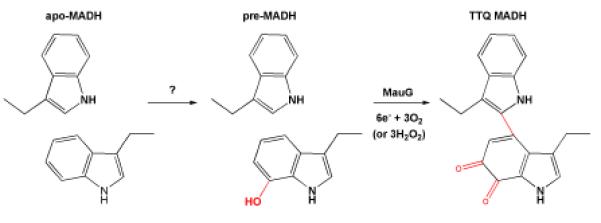
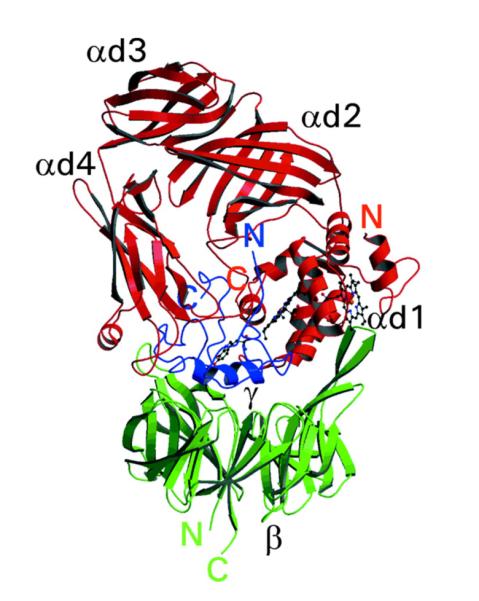
TTQ has been identified in the active site of the prokaryotic enzymes, methylamine dehydrogenase (MADH)14 and aromatic amine dehydrogenase (AADH).58 These enzymes allow the host bacterium to grow with amines as a sole source of carbon, nitrogen and energy. TTQ is an in situ cofactor formed by posttranslational modification of two tryptophan residues of the polypeptide chain (e.g. the ßTrp57 and ßTrp108 within the α2ß2 MADH tetramer from Paracoccus denitrificans (P. denitrificans) MADH, (cf. Figure 7)). In the course of cofactor production, two atoms of oxygen are incorporated into the indole ring of one tryptophan, and a covalent bond forms between the indole rings to crosslink the tryptophans (Scheme 4). The enzymes that function with TTQ are, as for the PQQ enzymes described above, dehydrogenases (Table 1) that catalyze the conversion of a primary amine to its corresponding aldehyde concomitant with formation of a reduced aminoquinol, TTQH2; the latter is recycled back to TTQ via two one-electron transfers to an external, copper-containing electron acceptor protein (amicyanin59 for MADH and azurin60 for AADH). The reductive portion of the reaction begins with covalent adduct formation between the amine of substrate and a carbonyl TTQ, while the oxidative portion of the catalytic reaction produces an aminosemiquinone intermediate61 that subsequently proceeds to the aminoquinone.62 Both the dehydrogenase and electron acceptor are localized in the periplasmic space of the host bacterium. As illustrated in Figure 8, the mature form of MADH is a heterodimer of two 45-kDa α subunits and two 14-kDa β subunits, the latter of which possess the mature TTQ cofactor.
Figure 7.

Three-dimensional structure of the preMADH-MauG complex. Color scheme is MauG (pink); preMADH α (blue), and β (green). A. Three-dimensional structure of the preMADH-MauG complex (PDB code 3L4M). B. Spatial layout of potential redox groups. A portion of the crystal structure. Residues Trp57 and Trp108 at the β subunit of MADH, Trp199 of the α subunit of MADH and the two hemes of MauG are displayed as sticks. From ref. (73).
Scheme 4.
Schematic for the production of TTQ.
Figure 8.

Structure of the mature form of MADH (PDB code: 2bbk). The MADH α subunit is blue and the β subunit is green. From ref. (85).
3.2 Biosynthetic Process
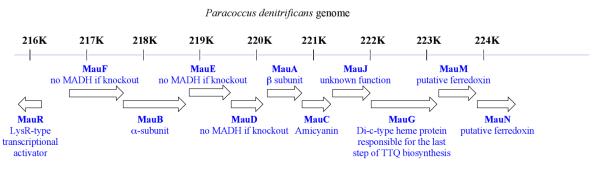
The biogenesis of MADH requires the posttranslational modifications that involve the generation of TTQ, the formation of six disulfide bonds in the β subunit, the export of α and β subunits to the periplasm, and their assembly into the α2β2 quarternary structure.63 The mau cluster of P. denitrificans contains 11 genes with a gene order of mauRFBEDACJGMN (Figure 9).64 In M. extorquens AM1, mauL is present between mauG and mauM.65 The α and β subunits of MADH are encoded by mauB and mauA, respectively,66 and mauC67 encodes amicyanin. Amicyanin is a type I blue copper protein that transfers electrons received from TTQH2 to the heme group of cytochrome c-551i. Complex formation between amicyanin, MADH, and cytochrome c- 551i has been demonstrated in the periplasm and a crystal structure of the complex is solved.68 Four other genes are essential for MADH biogenesis in P. denitrificans, with deletions of mauF,65mauD,69mauE69 or mauG64 resulting in loss of MADH activity, as well as the ability of the bacterium to grow on methylamine. In the single deletions of mauD, mauE or mauF, no MADH β subunit could be detected in cell extracts, and the levels of the MADH α subunit were low. MauE and MauD are specifically involved in the processing, transport, and/or maturation of the β subunit and the absence of each of these proteins leads to production of a non-functional β subunit, which becomes rapidly degraded. The mauG deletion results in loss of MADH activity and the inability to grow on methylamine; however, the levels of the α and β subunits expressed are similar to the wild-type (WT).64 Importantly, with regard to our understanding of the TTQ biogenesis pathway, the deletion of mauG leads to the accumulation of preMADH70 that contains a partially assembled cofactor (preTTQ) consisting of a monohydroxylated βTrp57 at C7 of the indole ring; the latter is the MADH substrate for MauG. The in vitro incubation of preMADH with purified MauG and oxidizing equivalents provided either by O2 plus an electron donor, or by H2O2,71 results in completion of TTQ biosynthesis and formation of active MADH. The enzyme responsible for the addition of this initial –OH group in pre- MADH is currently unknown. MauG was shown to be a 42.3-kDa monomer that possesses two covalently bound c-type hemes, as predicted from the gene sequence72 and catalyzes the final six-electron oxidation in three two-electron reactions using either O2 or H2O2 to complete TTQ formation. The mechanism of MauG is discussed below.
Figure 9.
The operon structure of mau gene cluster of P. denitrificans. From ref. (85).
3.3 Mechanism of MauG
The complex structure of MauG with pre-MADH (PDB ID: 3L4M) (Figure 7) is fully active in the crystal.73 A major mechanistic surprise was the realization that the activated oxygen species responsible for the six-electron oxidation at the preTTQ resides 40 Å from the site of TTQ formation. Further, the posttranslational modifications on Trp occur at ca. 15 Å from the di-heme unit. The oxidation of TTQ has been concluded to occur via a radical hopping mechanism, to overcome the large distance between the MauG bis-Fe(IV) oxidant and the TTQ synthetic site, contrasting with single-step electron tunneling processes that operate more usually over shorter distances.74 The electron transfer from preTTQ to the bis-Fe(IV) requires that Trp199 of MauG be transiently and reversibly oxidized to a free radical.74,75
The net reaction consists of three MauG-dependent two-electron events in the following order: [1] crosslink formation between βTrp57 and βTrp108;76 [2] hydroxylation of βTrp57 C6 to form a quinol; and [3] oxidation of quinol to quinone.77 Remarkably, it has been possible to monitor this reaction in the crystal (Figure 10).73,78 As noted above, the MauG redox form responsible for the catalytic activity is an unprecedented bis-Fe(IV) species,79,80 and can be formed either by addition of O2 to diferrous MauG or addition of H2O2 to diferric MauG.81 Whether O2 or H2O2 is the physiological oxidant is unknown. One MauG heme is high-spin and five-coordinate, and th e other heme, closer to preMADH, is low-spin with an unusual six-coordinate, His/Tyr axial ligation.82 This is the first observation of a His/Tyr axial ligation in a c-type heme. The intrinsic reduction potentials of the two hemes are similar, and facile electron transfer between them occurs via a conserved Trp93,79 with the implication that the di-heme unit acts as a single redox cofactor rather than as two independent hemes.83 This unprecedented bis-Fe(IV) redox state is quite long-lived in comparison to other Fe(V)-equivalent species.81 A charge-resonance-transition phenomenon has been put forth to explain why the bis-Fe(IV) intermediate is stabilized in MauG and does not permanently oxidize its own aromatic residues.84 The axial Tyr294 ligand to the low-spin heme appears key to attaining the bis-Fe(IV) state; a His variant at this position could not catalyze TTQ formation.82 A recent review addresses the highly unusual aspects of the long-range catalytic reaction mediated by MauG.85 A proposed mechanism for MauG showing a two-radical intermediate is given in Scheme 5.
Figure 10.

A. X-ray detected intermediates during TTQ biosynthesis. B. The observed rotation in β Trp57–OH during cross-link formation. From ref. (78).
Scheme 5.
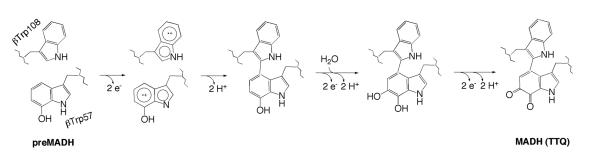
Proposed mechanism of MauG. From ref. (78).
4 CTQ (Cysteine Tryptophyl Quinone)
4.1 General Background
Among the known prokaryotic quinocofactors, CTQ was the last to be discovered. A seminal paper appeared in 2001, representing an elegant collaboration between the X-ray crystallographic and biochemical laboratories of Mathews and Tanizawa.16 The resulting structure of a quinohemoprotein amine dehydrogenase (QHNDH) from P. denitrificans indicated an active site tryptophan- derived quinone that had undergone crosslinking to a cysteine side chain. This was located in one of the three subunits (γ subunit) of a heterotrimeric protein. The arrangements of the three subunits in QHNDH is shown in Figure 11, and reveals many distinctive features. The γ subunit (dark blue) contains, in addition to a crosslinked tryptophylquinone, three additional crosslinks between the sulfur atoms of cysteine side chains and the γ carbon of Glu or β carbon of Asp side chains. Within the α subunit, there are two heme-iron groups organized in a manner reminiscent of mauG: one of the heme groups resides near the protein surface and the second is buried near the interior of the protein. The β subunit, making contact with both the α and γ subunits, has the distinctive β propeller domain structure seen in the family of alcohol dehydrogenases that utilize PQQ as cofactor.
Figure 11.
Three-dimensional structure of QHNDH from P. denitrificans. From ref. (16).
The type of protein shown in Figure 11 has been predicted to be present in at least 17 Gram-negative bacterial species86 and, analogous to PQQ- and TTQ-dependent enzymes, resides in its mature form within the periplasmic space. Analogous to TTQ enzymes, the net reaction catalyzed by QHNDH involves oxidation of amines to form aldehydes and the aminoquinol of CTQ (CTQH2), which undergoes reoxidation via two one-electron transfer steps to either azurin or cytochrome c as the electron acceptor.87 The latter is presumed to take place via the two heme-iron centers in the α subunit, providing a pathway for long-range electron transfer from the buried active site quinol to a solvent-accessible surface. These electron carriers subsequently donate their reducing equivalents to a membrane-associated electron transfer chain active in adenosine triphosphate (ATP) formation.
Consistent with the reaction mechanisms demonstrated for other amine-utilizing quinocofactors, covalent adduct formation between substrate and cofactor is anticipated during the catalytic cycle. The position of nitrogen attack on CTQ can be inferred from an X-ray structure of inhibitory complex of QHNDH and phenylhydrazine,88 Figure 12. The active site base, catalyzing proton loss from the α carbon of substrate in complex with CTQ, is ascribed to Asp33, which is constrained via a loop formed between Cys27 and the γ carbon of Asp33 to a position close to the reactive carbonyl of CTQ. A second structural loop, derived from the crosslink between Cys41 and Asp49, also appears to play an important role in the overall positioning of the CTQ for optimal catalytic reactivity.87
Figure 12.

Structure of the phenylhydrazine complex (Phz) of QHNDH and its relationship to the active site base, Asp33. Trq is tryptophylquinone. The gray netting represents electron density. From ref. (88).
4.2 Biosynthetic Process
The operon encoding CTQ production was originally described as containing four ORFs (cf. Figure 13), with ORFs 1, 3 and 4 encoding the enzymatic α, γ and β subunits, respectively, and ORF 2 encoding a radical SAM enzyme.16 N- terminal extensions were identified in the α and β subunits and concluded to be essential for transport of these gene products to the periplasmic space.89 An additional ORF that is located 3' to ORF 4 has been recently identified, ORF 5.86 Of further potential consequence, ORF 5 is found to be located immediately upstream from the ORF for an ABC-type transporter. It is postulated that the latter may play a role in the export of QHNDH or its subunits from the cytoplasm to periplasm.86
Figure 13.
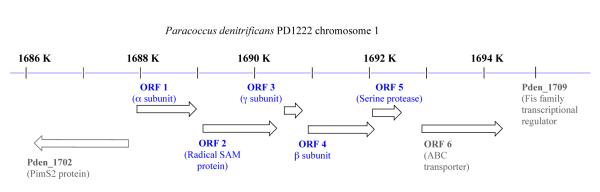
The operon structure of QHNDH and associated genes. From ref (89).
The initial implication of a radical SAM enzyme in CTQ maturation was tested by a series of ORF 2 knockouts, rescue experiments and mutagenesis studies.89 These have shown that the ORF 2 protein is essential for the formation of the three Cys to Glu/Asp crosslinks, together with the redistribution of the γ subunit from the cytoplasm to the periplasm. The inability to detect a quinone in the γ subunit of QNHDH in the absence of a functional ORF 2 protein further suggested that the formation of the Cys to Glu crosslinks was a prerequisite to CTQ formation. An unexpected finding was a mass for the γ subunit that was considerably larger than expected from the initially assigned methionine at its N-terminus.89 After re-examination of the sequence of the ORF operon, a second start site for translation of the γ subunit was found at -28 amino acids. This precursor form of the γ subunit has been concluded to contain a propeptide at its N-terminus, in analogy to the presence of N-terminal extensions identified in the ORFs encoding the α and β subunits of QNHDH.
The fifth gene product (ORF 5) is produced when cells are grown on n-butylamine, conditions that also induce the production of the gene products from ORF 1 to 4.86 ORF 5 encodes a peptidase that belongs to the subtilisin family, with the presence of a conserved catalytic triad comprised of His, Asp, and Ser. While disruption of ORF 5 leads to production of the individual subunits of QNHDH (under conditions of growth in n-butylamine), the absence of a functional ORF 5 fails to yield the final quinone-containing QNHDH in the periplasm.86 Under this condition, the α and β subunits make their way into the periplasm, and the majority of the γ subunit remains in the cytoplasm. This form of the γ subunit has retained the propeptide first identified in the ORF 2 knockout, contains the three Cys-Glu/Asp crosslinks of the mature QHNDH but lacks CTQ. A sequence of events can be envisaged in which the ORF 2 gene product catalyzes crosslinking of the full-length γ subunit, which subsequently undergoes translocation to the periplasm concomitant with cleavage of its propeptide via the ORF 5-encoded protease. The subsequent assembly of the heterotrimeric QNHDH may, in fact, be a prerequisite for the formation of the CTQ cofactor, though there are no data yet to bear on this point. It is tantalizing to speculate that the oxidation of the Trp precursor may be assisted by the two hemes contained within the α subunit, analogous to the MauG action outlined under TTQ (see Relationship to Biosynthesis of TTQ below). The activity of the protease toward short peptides has shown only limited or single turnover kinetics, and this has allowed the identification of a covalent adduct between the ORF 5 protein and the N-terminal of the cleaved peptide, further confirming a function for the CTQ-linked protease that falls squarely within the category of serine proteases.86 Based on the available data, a tentative pathway for the sequence of events in CTQ production is outlined in Scheme 6.
Scheme 6.
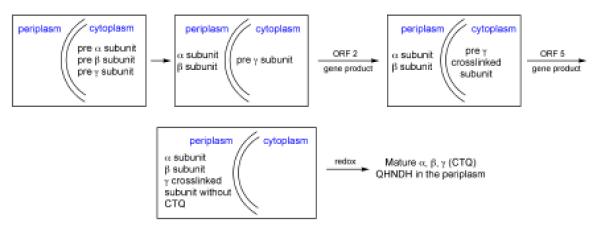
Summary of likely sequence for the processing steps in CTQ production.
4.3 Relationship to Biosynthesis of TTQ
CTQ is one of only two identified quinocofactors that is derived from Trp (Table 1). In this context, it might have been expected that the biosynthetic pathways for TTQ and CTQ would indicate similarities to one another. At first appearances, the differences between the two pathways appear greater than the similarities. To begin with, the crosslink between the two tryptophans in TTQ requires the generation of a new carbon-carbon bond, whereas the crosslink in CTQ involves the addition of a sulfur atom to the parent Trp ring. Formation of a new C–C bond is intrinsically more difficult than the S–C bond, which could form subsequent to quinone formation on the parent Trp ring, via a thiolate attack on a tryptophanyl quinone (see the discussion of TPQ and LTQ below).
The modifying enzymes identified within the respective operons for TTQ and CTQ are also quite different, with a radical SAM enzyme seen solely in the CTQ pathway. Very unexpectedly, this radical SAM activity is necessary for formation of the sulfur-carbon crosslinks that define the structure of QHNDH, in particular the relationship of the CTQ cofactor to the active site base required for catalysis, and not for the generation of the CTQ itself. In the case of TTQ, overall protein structure and active site geometry is maintained via a series of disulfide bonds instead of thioether bonds, raising the question of why the CTQ pathway would have resorted to radical SAM chemistry to sculpt its active site. The fact that a fourth Cys must be “reserved” in QHNDH for the final crosslink in CTQ production may explain this peculiarity. The formation of three disulfide bonds in the presence of an active site free thiol in QHNDH would have introduced the possibility of disulfide interchange reactions, possibly leading to incorrectly folded protein and the resulting unavailability of Cys37 for crosslinking to Trp43 in the generation of CTQ.
In this context, it is interesting that the parent Trp in the protein precursor to TTQ may be able to undergo crosslinking to Cys, when the second active site Trp(β108) is replaced by Cys via site-specific mutagenesis.90 However, the generation of a catalytically active CTQ-like cofactor in MADH is quite limited, if it occurs at all. Mutation of Trp(β108) to either cysteine or histidine produces in the main a partially processed tryptophyl cofactor containing a single oxygen, together with the property of an increased dissociation of the α2β2 heterodimer into its subunits.90 The most prominent similarity between TTQ and CTQ production is the presence of gene products that contain two distal heme iron prosthetic groups, with one heme located near the solvent and the second heme near the active site of the final dehydrogenase. In the case of TTQ production, the protein bearing these two heme groups is MauG, which performs a processing role but is absent from the final dehydrogenase. In CTQ production, the two heme groups are located in the α subunit of the dehydrogenase QHNDH. This raises the question of whether the α subunit of QHNDH performs dual roles: the first would involve the oxidation of Trp43 in CTQ formation in analogy to the role played by MauG in TTQ formation, while the second role would be to provide a pathway for electron transfer during catalytic turnover from the substrate-reduced CTQ to the exogenous electron acceptor.
5 TPQ (Trihydroxyphenylalanine Quinone)
5.1 General Background
The discovery of TPQ in a copper amine oxidase (CAO) from bovine serum (BSAO)13 represented a turning away of researchers from a primary focus on PQQ. While it had been expected that the family of CAOs would contain a covalently- bound cofactor, this was initially thought to involve the posttranslational insertion of a normally dissociable cofactor such as the carbonyl cofactor pyridoxal phosphate91 or PQQ itself.92 When a structural proof for TPQ was put forth in 1990, it confirmed the presence of a covalently bound, active site quinone, but one that was quite different from PQQ and derived from a single tyrosine side chain of the protein backbone.13
Among the cofactors in Figure 1, TPQ is unique in that it has not undergone crosslinking to a second amino acid. This feature simplifies the biosynthetic process but actually complicates the subsequent mechanistic involvement of TPQ in redox chemistry. All of the quinocofactor-containing enzymes are inferred to function by a similar mechanism. This involves covalent attachment of the substrate to one of the carbonyls of the quinone moiety, followed by proton loss from substrate that is catalyzed by a highly conserved active site aspartate93 (cf. boxed region of Scheme 7 for an illustration of the reaction mechanism with TPQ). While a possible non-covalent mechanism has been proposed in the case of reaction of PQQ enzymes and their substrates,19 and an X-ray structure is not yet available to identify the active site base in the LTQ-containing lysyl oxidase, the mechanistic features illustrated for TPQ provide a reasonable starting point for understanding the reactivity of all the quinocofactors. Key features are: [i] the creation of an electrophilic sink to facilitate proton abstraction from substrate by an active site base, [ii] the formation of reduced cofactor concomitant with substrate oxidation (as a direct result of substrate deprotonation and electron delocalization) and [iii] reoxidation of the reduced, quinol form of cofactor back to the starting quinone, either with molecular oxygen or an exogenous electron acceptor.21
Scheme 7.

Impact of mutation of the consensus sequence within the TPQ-containing H. polymorpha amine oxidase on catalytic intermediates. The catalytically productive intermediates are shown in the box. Adapted from ref. (102).
In the context of the two tryptophan-derived quinocofactors discussed above, TTQ and CTQ, the crosslinking of the quinone bearing Trp to a second amino acid might have been expected to provide the principal role in the positioning of the carbonyls of cofactor in relation to the incoming substrate and the active site base. Yet, as described above, the active site of the TTQ-containing enzyme contains six disulfide bonds,63 while the active site of the CTQ-containing QNHDH has a series of unusual thioether linkages inserted posttranslationally.89 In both instances, these structural modifications to the respective active sites have been concluded to play additional roles in defining the distances and orientations among the cofactor, substrate and catalytic base.
As X-ray structures began to emerge for the mature, TPQ-containing enzymes, it became clear that the positioning of the TPQ could be variable and protein-dependent94-99 (Figure 14). This ability to visualize TPQ within different positions of the active site now appears directly related to the mechanism of biogenesis (see below). However, the mobility of TPQ also raised a question regarding the mechanism whereby active site interactions could be controlled during catalytic turnover of substrate. A possible answer to this question arose from the early efforts to identify the amino acid precursor to the mature TPQ. Alignment of DNA and protein sequences for individual TPQ-containing proteins both confirmed that tyrosine was the precursor to TPQ and identified a conserved consensus sequence that flanks the Tyr precursor to TPQ: Asn-Tyr(TPQ)- Asp/Glu, where the C-terminal Asp/Glu is distinct from the Asp involved in proton removal from substrate during catalysis.100
Figure 14.

Active site in (left) apo-A. globiformis CAO and in (right) a superposition of the active form of holo-AGAO (red) with the inactive form of holo-AGAO (green) and the apo-AGAO (black). His431, 433, and 592 act as ligands to the active site copper in the holo-forms. Adapted from ref. (99).
It was initially thought that this consensus sequence might function as a recognition site for the binding and action of ancillary proteins during cofactor maturation. However, when mutations were inserted, at either the N-terminal Asn or C- terminal Asp/Glu of the consensus sequence, biosynthesis continued to occur (albeit at variable and somewhat reduced rates).101 As now known, TPQ arises via an autocatalytic process (see below) with no requirement for additional gene products, and a major consequence of mutagenesis with the consensus sequence is to introduce a rotation of TPQ-linked intermediates into inactive conformers during catalytic turnover.93,102,103 An illustration of this type of behavior for several mutants within a CAO from Hansenula polymorpha (H. polymorpha) (HPAO) is shown in Scheme 7, which highlights the production of non-productive, ring-flipped complexes during catalytic turnover. Careful inspection of X-ray structures for TPQ-containing CAOs reveals a structural motif that involves a number of interactions among active site side chains that link the Asn and carboxylate of the consensus sequence. As illustrated in Figure 15 for HPAO, a network of hydrogen bonds from Glu406 to Tyr186 is seen to position Asn404 behind the TPQ.102 The major role of the consensus sequence has, thus, been concluded to be that of providing an anchor for the positioning of TPQ during catalysis. The use of a protein motif, as opposed to active site disulfide bonds or thioether linkages, to immobilize this quinocofactor appears to satisfy the dual needs of cofactor immobilization during catalysis and mobility of the Tyr precursor as it undergoes the multiple steps of biogenesis.
Figure 15.

Proposed loop structure provided by Asn404 and Glu406 within the active site of HPAO. Residues from the A subunit are gray and those from the B subunit yellow. Oxygen atoms are red, and nitrogen atoms are blue. Connectivity is shown by solid lines between residues, and hydrogen bonding distances are given for these interactions (from PDB file 1A2V). All CAOs are homodimers. From ref. (102).
5.2 Biosynthetic Process
The study of the posttranslational generation of TPQ began with a key experiment by Tanizawa and co-workers, which showed that expression of the gene for a CAO from Arthrobacter globiformis (A. globiformis) in E. coli under limiting copper conditions produced the precursor form of enzyme. Subsequent addition of Cu(II) in the presence of air led to TPQ, whereas anaerobic conditions failed to give any reaction, implicating the dual requirements for copper ion and molecular oxygen.104 Although E. coli is capable of producing its own TPQ-containing CAO, this did not appear to compromise interpretations from the heterologous over-expression of the A. globiformis amine oxidase (AGAO). Important follow-up studies established a number of key features of this process. Using resonance Raman spectroscopy, it was shown that the oxygen at carbon-2 of the mature cofactor is derived from water. Further, it could be concluded that at neutral pH, electron density at the oxygen of carbon-4 undergoes delocalization into the oxygen at carbon-2, in contrast to the retention of full carbonyl character at carbon-5,104:
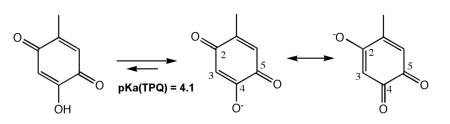
While the oxygen of the carbonyl at carbon-5 is believed to arise from molecular oxygen during biogenesis, detection of O-18 transfer from labeled dioxygen is precluded by a rapid exchange reaction with water at this position.105 Finally, Ruggiero and Dooley established a stoichiometry of two moles of O2 consumed and one mole of H2O2 produced per TPQ produced in the AGAO.106
Cai and Klinman pursued a different approach, using Saccharomyces cerevisiae (S. cerevisiae) as the host for expression of a gene for CAO from a second yeast strain, H. polymorpha (HPAO).106 The laboratory strain of S. cerevisiae is unable to produce its own CAO, eliminating a potential complication of TPQ production within a heterologously expressed CAO that could involve enzymes native to the host. The fact that active HPAO arises in an S. cerevisiae expression system provided early evidence for a self-processing mechanism.107 However, efforts to isolate a precursor form of HPAO, by carrying out the growth of S. cerevisiae in copper ion-depleted media, were unsuccessful. It was ultimately found that depletion of cupric ion in the yeast growth medium led to metalation of HPAO by zinc ion (and as a result inactive enzyme).108 Many years later, multi-energy X-ray data analysis of a second eukaryotic CAO (human diamine oxidase) that contained a mixture of copper and zinc ions at its active sites showed that only the subunits with copper occupancy were able to produce TPQ.109 While it was not possible to find conditions for removal of the tightly bound Zn+2 from recombinant HPAO and its replacement by copper ion, site-specific mutagenesis proved very useful in showing an essential role for copper. These studies involved mutagenizing either one of the inferred ligands to copper (H456D) or the glutamate at the C-terminal end of the consensus sequence (E406N). Whereas E406N produced active enzyme, the H456D variant contained only a trace amount of copper (ca. 4%) and very little TPQ, implicating an essential role for copper-binding in the formation of TPQ within HPAO.101 Ultimately, the E. coli expression system of Tanizawa was found to adapt quite well to HPAO, opening up the door for the series of detailed mechanistic studies using the metal- free precursor forms of enzyme from H. polymorpha and its comparison to AGAO.
As noted, the basic reaction leading to TPQ requires the insertion of two oxygen atoms into the phenolic ring of Tyr to form a 2,4,5-trihydroxyphenethylalanine residue, which then undergoes a two-electron oxidation to TPQ; the latter reaction is the source of the one mole of H2O2 produced during biogenesis. A major goal from the beginning was the direct detection of each reaction intermediate, in particular to understand the stepwise oxygen insertion steps. Three dominant methods were pursued: EPR (to search for free radical intermediates), UV/Vis (to detect intermediate absorbing species with signatures distinct from the initial Tyr and final TPQ (λmax = 480 nm at pH 7, where TPQ has ionized to its anionic state) and X-ray crystallography.
EPR studies proved largely uninformative using either the AGAO or HPAO enzymes, despite the possibility of an intramolecular one-electron transfer between the active site Tyr and Cu(II) to form a Tyr-derived free radical and the reduced Cu(I):
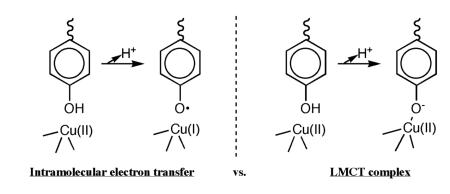
The virtue of an electron redistribution via intramolecular transfer would have been the generation of a Cu(I) metal center that could bind and reductively activate molecular oxygen (ground state triplet) for combination with an adjacent Tyr radical. Given the large difference in redox potentials between the copper half reaction (ε° ~ 0.1 to 0.3 V vs. NHE)110 and tyrosinate (ε ~ 0.7 V vs. NHE),111 the failure to see any detectable accumulation of a Tyr radical may not be surprising. As an alternative to a discrete tyrosine to copper electron transfer step, the generation of a ligand-to-metal charge transfer (LMCT) band between copper and tyrosinate emerged as the key initial step of the reaction mechanism.
The first intimation of a possible LMCT intermediate came from the X-ray structure of the metal-free, precursor form of the AGAO, where the unprocessed Tyr could be seen pointing directly into the empty metal site.99 This contrasted directly with the mature TPQ in which the modified aromatic ring had rotated off of the metal center into the catalytically active conformation94 (Figure 14). Subsequently, UV/Vis spectroscopy provided more direct evidence for a LMCT species, using a specific experimental protocol in which HPAO was preloaded with cupric ion under anaerobic conditions and then exposed to molecular oxygen.112 Under such conditions, a new species at 350 nm appears, displaying a classic precursor-product relationship with the appearance of TPQ, Figure 16. It was noted at the outset that a 350 nm absorption band could have represented the accumulation of a peroxo-intermediate,112 expected to arise from the addition of O2 to the tyrosine ring - in the event that this intermediate were to undergo a rate-determining breakdown via proton abstraction from the aromatic ring to form dopaquinone (cf. intermediates (III) and (IV) in Scheme 8). This possibility was, however, ruled out by the observation of an unchanged rate for cofactor biogenesis using an engineered precursor form of HPAO in which all of the protein-bound tyrosines had been deuterated at the carbon 3 and 5 positions of the aromatic ring.113 Experimental support for the 350 nm species as a LMCT band comes from the similarity of this absorbance band to authentic tyrosinate-copper model complexes, together with the red shift of λmax to 390 nm (accompanied by a ca. ten-fold slower rate of biogenesis) when one of the His ligands to the active site copper is changed to Cys (H624C).112 Additionally, X-ray crystallographic studies of the redox inactive, zinc ion substituted form of HPAO complex show a direct coordination of the 4-hydroxyl of the precursor Tyr to the metal center.114 In general, complexes between tyrosine and zinc ion are rare in proteins, implicating specific active site features/interactions within HPAO that act to “drive” the precursor Tyr onto the copper.
Figure 16.

Precursor-product relationship for the active 350 nm species in TPQ formation within HPAO. These graphs represent spectroscopic changes following the oxygenation of enzyme where Cu2+ has been pre-bound anaerobically. A. Absorbance changes following exposure to O2. Spectra are at 1, 2, 3, 4, and 5 min following exposure to oxygen; the spectrum before oxygenation has been subtracted. Arrows indicate the direction of change. B. Absorbance changes during the decay of the 350 nm species. Spectra are 8, 10, 14, 20, and 30 min following the introduction of O2; the spectrum at 5 min after aeration has been subtracted. Arrows indicate the direction of change. C. Kinetics of the reaction with O2 at 350 nm (○) and 480 nm (●). From ref. (112).
Scheme 8.
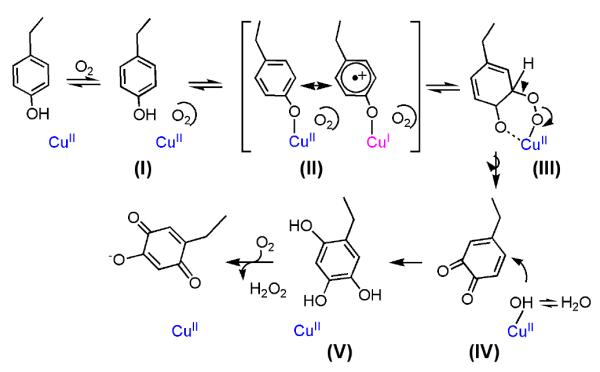
Mechanism of TPQ production. [Adapted fron ref. (17)].
The protocol used for copper insertion was very important in assigning the 350 nm species to a catalytic intermediate in biogenesis. When UV/Vis is used to monitor the status of protein after addition of copper ion in the absence of O2, another 350 nm band is observed that serendipitously decays with a similar rate constant to the appearance of TPQ in the presence of O2 for WT enzyme. This was initially very confusing, until the aerobic reaction of metal with two consensus site mutants (E406Q and N404D) was shown to produce a much faster decay of the 350 nm band than TPQ formation,112 (Table 2):
Table 2.
Kinetics of Copper-Dependent Spectroscopic Changes during Biogenesisa. From ref. (112).
|
|
||
|---|---|---|
| Decrease, k380 (min−1) | increase, k480 (min−1) | |
| WT HPAO | (6 ± 1) × 10−2 | (8 ± 3) × 10−2 |
| E406Q | (8 ± 1) × 10−2 | (1.3 ± 0.5) 10−2 |
| N404D | (3.4 ± 0.6) × 10−2 | (6 ± 2) × 10−4 |
Assays were conducted at 25°C and pH 7.0. Reactions were begun by the addition of substoichiometric Cu(II) (0.7–0.8 equiv) to an air-saturated enzyme solution.
Overall, the data have shown that copper binding is a multi-step process, involving a series of LMCT interactions that can, under certain conditions, produce a signature that is similar to the catalytically productive intermediate that ultimately governs TPQ formation. For this reason, successful monitoring of biogenesis intermediates requires a preloading of protein with copper in the absence of O2 and then exposure to O2 to initiate the reaction. Under these conditions, both WT and consensus site mutants show a 1:1 correspondence between disappearance of the 350 nm species and TPQ production,112 (Figure 16).
Understanding the nature of the dependence of the 350 nm band on the presence of O2 was initially challenging. Consistent with an essential role for O2 in LMCT formation, time-resolved X-ray crystallography of biogenesis within the Arthrobacter enzyme showed two new intermediates, one of which forms in the absence of O2 and shows the precursor Tyr pointing toward the active site copper but too far away (2.5 Å) for direct coordination.115 (The second intermediate, seen after addition of O2, is dopaquinone, and will be discussed below.) The impact of changes in O2 concentration on the properties of TPQ formation was subjected to some scrutiny for HPAO, using either UV/Vis to follow spectroscopic intermediates or an oxygen electrode to follow O2 uptake.113,116 Under conditions of air, identical rate constants are observed independent of the assay method. Elevation of the O2 concentration toward 100% initially showed a linear (non-saturating) dependence on O2 concentration when measuring oxygen uptake, whereas the formation of TPQ indicated a Km for O2 of ca. 180 μM. Interpretation of this difference in kinetic order could have been complicated by the fact that biogenesis involves two O2-dependent steps. However, the second O2-dependent reaction of biogenesis involving conversion of reduced quinol to the final quinone is germane to catalysis, can be studied independently and is known to occur much more rapidly than the rate of TPQ formation.117 In fact, a large fraction (if not all) of the divergence between the dependence of the measured rates on O2 concentration was later shown to be due to the extreme difficulty of following the slow reaction of biogenesis at elevated O2 levels in an oxygen electrode chamber.113 Once extreme efforts were made to reduce a time-dependent exchange between the high O2 level in the reaction chamber and ambient air, both the uptake of O2 and production of TPQ displayed similar Km values.113 The observation of saturation kinetics with regard to O2 thus provides support for an O2 binding site on the precursor protein. The final model for the initiation of the O2-dependent portion of the TPQ biogenesis reaction is shown in Scheme 8, and involves a binding of O2 into a protein-derived pocket (species (I)) as a prerequisite for movement of the precursor Tyr onto the active site metal ion (species (II)). The ability to detect these first O2-dependent steps in TPQ formation is a direct result of the accumulation of the O2-dependent LMCT that precedes a rate-limiting attack of O2 onto the metal-activated precursor tyrosine ring.113
The idea of a protein-derived O2 binding pocket in HPAO has been similarly proposed during catalytic turnover and has met some resistance in the inorganic community.118 The arguments in favor of an off-metal site for O2 binding site and reactivity have been published elsewhere and will not be repeated here. [cf. refs. 119-122] Quite importantly, a series of mutagenesis studies provide strong evidence for a similar O2 binding pocket during biogenesis and catalysis. In particular, a methionine residue, identified in HPAO to lie near the copper-binding site (Figure 17), was suggested to be threonine in a related eukaryotic CAO from bovine source (BSAO).122 When the Met634 in HPAO was mutated to the Thr of BSAO, the second order rate constant for reaction of HPAO with O2 was found to be reduced to the value seen in BSAO.119 A series of hydrophobic amino acids were subsequently inserted into the Met position, showing a trend in reactivity toward O2 that correlated with the volume of the amino acid side chain.122 Remarkably, when the same series of mutants were analyzed for their rate of biogenesis, a similar dependence on side chain volume was observed.113 These findings, together with the observations outlined above, provide strong support for a similar protein pocket that functions to accommodate molecular O2 during both catalytic turnover and TPQ biogenesis.
Figure 17.

Active conformation of TPQ within the active site of copper amine oxidase from H. polymorpha. Hydrogen bonds are indicated by dashed lines and distances are in angstroms. A water molecule is represented by a red sphere. The Met634 that is implicated in O2 binding is seen adjacent to the active site Cu2+. From ref. (94).
The absolute requirement of Cu(II) for the autocatalytic formation of TPQ has been examined in a number of ways. Using precursor forms of both the AGAO and HPAO, biogenesis has been initiated by the insertion of alternate metals into the empty metal site, with the results shown in Tables 3 and 4, and Figure 18, respectively.
Table 3.
Rates of TPQ Biogenesis in AGAO Assisted by Cu(II), Co(II), and Ni(II) Ions. From ref. (123).
| metalionb | biogenesis rate constantsa (min−1) |
|---|---|
| Cu(II) = 0.5 mM | 1.50 ± 0.2 |
| Co(II) = 0.5 mM | (1.32 ± 0.04) × 10−3 |
| Co(II) = 0.5 mM, Cu(II) = 0.005 mM | (1.36 ± 0.05) × 10−3 |
| Ni(II) = 0.5 mM | (1.25 ± 0.02) × 10−3 |
| Ni(II) = 0.5 mM, Cu(II) = 0.005 mM | (1.18 ± 0.02) × 10−3 |
| Ni(II) = 0.5 mM, Cu(II) = 0.05 mM | (1.41 ± 0.03) × 10−3 |
Determined from the increase in absorbance at 480 nm at 30°C and pH 6.8.
Assays with the alternate metals, Co(II) or Ni(II), show similar rate constants, either in the absence or presence of trace Cu(II), ruling out a small amount of copper as the origin of the observed biogenesis rate.
Table 4.
Kinetic Properties of HPAO, Reconstituted with Cu(II) vs. Ni(II). From ref. (124).
| kTPQa,b with air (h−1) kTPQa,c with O2 (h−1) 2H2kTPQa,b,d with air (h−1) kcatb,e with air (s−1) | |||
|---|---|---|---|
| Cu(II) 3.3 (0.6) | – | – | 3.80 (0.06) |
| Ni(II) 0.028 (0.006) | 0.044 (0.009) | 0.031 (0.006) | 0.09 (0.01) |
Measured at 480 nm with 40 μM HPAO and 40 μM metal ion.
Reactions performed in air-saturated buffer.
Reactions performed in oxygen-saturated buffer.
Reactions assessed for HPAO containing 3,5-[2H2]tyrosine.
Methylamine oxidation measured by the oxygen electrode assay with protein concentrations of < 0.5 μM.
Figure 18.
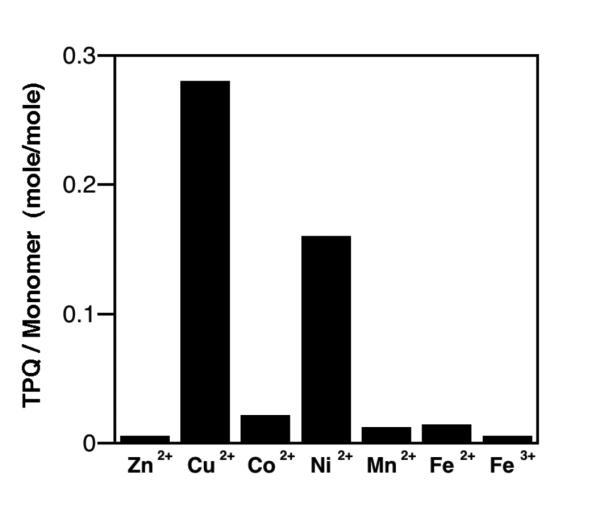
Bar graph for impact of metal replacement in HPAO biogenesis. From ref. (124).
Both Ni(II) and Co(II) lead to a very slow TPQ production in AGAO.123 The rate of biogenesis with Ni(II) is ca. 7-fold faster in HPAO,124 Table 4, analogous to findings during catalytic turnover where Co(II) has been found to substitute for copper in HPAO to yield an almost identical kcat.120,121
The very low potential for the reduction of Ni(II) to Ni(I) (ε° = -1.16 V vs. NHE),125 together with the failure to detect a UV/Vis intermediate with a precursor/product relationship to TPQ formation, has led to the suggestion of a rate- limiting outer sphere electron transfer from the Tyr precursor to pre-bound O2 that is followed by a series of rapid kinetic steps.124 In an important control study, Samuels and Klinman tested the hypothesis that Cu(II), and not Cu(I), was the catalyst for TPQ formation. Inserting Cu(I) under anaerobic conditions and then initiating biogenesis with air, showed that the Cu(I) was required to undergo an oxidation to Cu(II) before the chemical steps converting Tyr to TPQ were initiated.126 Schwartz et al. used a Cu(II)- specific ligand azide to test the impact of blocking ligation to copper on the biogenesis rate. Fully consistent with the model in Scheme 8, the 400-nm band characteristic of a copper-azide complex was found to be lost concomitant with TPQ formation, implicating dissociation of azide as the rate-limiting step under these conditions.127
The last component to be considered in depth for its impact on biogenesis is the role of a second, absolutely conserved Tyr within the active site of all characterized CAOs (Tyr407 in HPAO, Figure 17). A very surprising result was that the impact of a Tyr to Phe vs. Ala conversion in HPAO leads to very different results for catalysis,128 Table 5, than biogenesis,129 Table 6.
Table 5.
Rate Constants for Ethylamine Oxidation by WT HPAO and Y305 Mutants (37°C, pH 7). From (128).
| Enzyme | kcat (s−1) | kcat/Km (M−1 s−1) |
|---|---|---|
| WT | 20 | 5.2 × 104 |
| Y305A | 7.5 (0.4) | 1.4 × 104 (0.3) |
| Y305F | 0.16 (0.01) | 0.010 × 104 (0.002) |
Table 6.
Rate Constants (min−1) for Peak Formation during Cofactor Biogenesis in WT HPAO and Y305 Mutants (25°C). From (129).
| Y305A | Y305F | WT | |||
|---|---|---|---|---|---|
| k 480 | k 400 | k 480 | k 420 | k 480 | |
|
|
|||||
| pH 6.5 | 0.0015 | NDa | 0.027 | NAb | 0.067 |
| pH 7c | 0.0025 | 0.0022 | 0.024 | 0.084 | 0.08 |
| pH 8 | 0.0035 | 0.002 | 0.041 | 0.030 | 0.11 |
Not determined; a significant amount of the species was not observed.
A formation rate constant could not be determined due to significant overlap between the 420 and 480 nm bands.
Note the greater impact of Y305F on the rate of formation of the alternate product(s) detected at 420 nm (cf. Figure 19).
The role for the alternate Tyr during catalytic turnover has been ascribed to its hydrogen bonding to the O-4 of TPQ. The fact that an Ala substitution is so much better tolerated at this position during catalysis was suggested to be due to an ability of water to access the active site and provide an alternate hydrogen bonding capability.129 A similar type of behavior has also been reported by Herschlag and co-workers for the enzyme ketosteroid isomerase.130 In the biosynthetic portion of the reaction, the size of the side chain appears to be of greater importance, most likely in guiding the movement of the Tyr side chain as it moves through its various catalytic intermediates on its way to the mature TPQ. Another unexpected and extremely important property of the Tyr to Phe mutant came from the UV/Vis features of its biogenesis that indicated a pathway parallel to TPQ formation (producing a species with λmax = 420 nm).131 After considerable effort, the structure of the product from the alternate pathway was solved by X-ray crystallography and the result is presented in Figure 19. As illustrated, extra electron density appears at several position within the ring of Tyr and is the same, independent of whether gene expression is carried out in S. cerevisiae (with sufficient copper ion present in the growth medium to reconstitute the HPAO during cell growth) or in E. coli (where protein is reconstituted with Cu(II) after isolation of the purified apo-protein). In both instances, the observed electron density is consistent with the generation of hydroperoxo-derivatives on the aromatic ring. It appears that the ring hydroxyl of Tyr305 performs an important function in acid/base catalysis during TPQ production, presumably aiding in the non-rate- determining cleavage of the O-O bond within the peroxy- intermediate formed from addition of O2 to the precursor Tyr ring ((III) in Scheme 8). In the absence of Tyr305, an abortive reaction path becomes activated, leading instead, to proton loss and re-aromatization concomitant with retention of the peroxide adduct(s) within the ring structure.131
Figure 19.

Final electron density maps of the Y305F mutants of HPAO computed using coefficients (2Fo – Fc) and (Fo – Fc) where Fc was calculated from the models refined with TPO and the modified Met634 present. The (2Fo – Fc) maps are contoured at the 0.8 (cyan), 1.0 (crimson), and 8.0 (dark blue) σ levels, and the (F° – Fc) maps are contoured at the –3.0σ level (gold); no contours above the +3.0σ level are present. Hydrogen and coordination bonds are indicated by black dashed lines. A. The E. coli-expressed structure is shown. B. The yeast-expressed structure is shown. TPO is the abbreviation for the hyper-oxidized derivative of Y405. From ref. (131).
This final study on HPAO, together with the spectroscopic and structural studies outlined above for AGAO and HPAO, satisfied the initial goal of detecting each of the proposed reaction intermediates in TPQ biosynthesis. The postulated mechanism of Scheme 8 proceeds as follows. First, O2 binds to a non-metal site initiating the movement of the Tyr precursor ring onto the Cu(II). This intermediate undergoes sufficient charge transfer to activate the Tyr ring for a rate-determining O2 attack. A resulting metal coordinated peroxy- intermediate undergoes acid assisted cleavage to yield dopaquinone. In a last step, the electrophilic ring of dopaquinone undergoes nucleophilic attack by a Cu(II)–OH according to the mechanistic conclusions from model studies of dopaquinone reactivity.132 This “mechanistic dance” is quite remarkable, given the need for a series of precise active site configurations that starts with the off metal Tyr precursor and changes during each successive chemical transformation. This includes the final rotation of the product TPQ off of the metal to a position that is close to the active site base and can initiate catalysis.
5.3 Relationship of the Pathway for TPQ Biosynthesis to that for PQQ, TTQ and CTQ
Ostensibly, the formation of TPQ is completely different from the processes that lead to PQQ, TTQ and CTQ, given the requirement for multiple and distinctive gene products to produce the latter three cofactors. There is also variability regarding the depth of our understanding for the production of the individual quinocofactors (Schemes 2, 4, 6, 8). Nonetheless, some interesting and potentially important similarities emerge. All of the above cofactors are produced by highly oxidative processes that are initiated by the addition of molecular oxygen to a Tyr or Trp side chain. Further, the subsequent oxidation of the respective aromatic ring leads to new electrophilic species that play a role in downstream chemical modifications. It would appear that these pathways represent a remarkable example of chemical convergence that has occurred via the recruitment of remarkably different biological tool boxes.
6. LTQ (Lysyl Tyrosine Quinone)
6.1 General Background
The sole enzyme reported to contain LTQ, lysyl oxidase, is yet again, quite distinct from the other demonstrated quinone-containing oxidases. This monomeric, extracellular mammalian enzyme plays an essential role in the oxidative crosslinking of collagen and elastin, with defects in enzyme expression correlating with a plethora of disease states.133 The enzyme is expressed as a pre-propeptide, with the release of a 21 amino acid signal peptide occurring concomitant with the localization of the enzyme to the extracellular matrix.134 The resulting 50 kDa product undergoes further proteolytic processing to yield a catalytically active 32 kDa oxidase.135 Though much smaller in size than the dimeric TPQ-containing enzymes, lysyl oxidases share the property of a Cu(II)-binding site, presumed to lie in close proximity to the precursor tyrosine and mature LTQ. It might, therefore, be anticipated that the mechanism of cofactor biosynthesis in lysyl oxidase would show strong similarities to TPQ formation.
A severe limitation in characterizing the properties and mechanistic origins of LTQ has been the lack of a viable, high-level expression system for lysyl oxidase, together with the absence of an X-ray structure for either a precursor or mature form of lysyl oxidase. The initial demonstration of LTQ depended on isolation of phenylhydrazine-derivatized active site peptides, their subsequent sequencing and alignment against the corresponding cDNA.15 Repeatedly, two amino acids were found to be released at each round of peptide sequencing which, after subtraction of the masses for both the N- and C-terminal flanking amino acids, led to identification of lysine as the crosslinking moiety to a tyrosine-derived quinone. In light of the possibility of a side reaction involving, for example, a spurious crosslinking of a non-active site lysine to a derivatized dopaquinone cofactor during peptide isolation and analysis, a number of key controls were carried out. These involved chemical synthesis of an authentic model for the phenylhydrazine adduct of LTQ and the direct comparison of its UV/Vis and resonance Raman (RR) properties to the phenylhydrazone of lysyl oxidase and peptides derived thereof.15 Additionally, mutagenesis studies indicated the essentiality of both Tyr349 and Lys314 to LTQ formation.15 A subsequent comparison of RR data obtained with native lysyl oxidase to the underivatized model compound of LTQ, Figure 20, provided strong confirmatory evidence for the proposed enzymatic crosslinked cofactor structure.136
Figure 20.

RR spectra of native lysyl oxidase in relation to a representative model compound. From ref. (136).
6.2 Biosynthetic Process
A possible mechanism for LTQ production in lysyl oxidase is illustrated in Scheme 9. Of particular note, and by analogy to TPQ formation (Scheme 8), are the roles of cupric ion in activation of the Tyr349 precursor and the intermediacy of dopaquinone as an electrophilic site for nucleophilic attack by an active site functional group (hydroxide ion in the case of TPQ formation vs. the ε-amino group of Lys314 for LTQ). Despite the difficulties in obtaining a high-yield expression system for lysyl oxidase, Bollinger et al. were able to isolate a copper-depleted form of lysyl oxidase and to show that enzyme activity was dependent on the addition of exogenous Cu(II) in the presence of molecular oxygen.137
Scheme 9.

Postulated pathway for the production of LTQ. [Adapted from ref. (139)]. DPQ is dopaquinone.
While small molecule, model studies could, in principle, provide insight into the pathway(s) for LTQ biosynthesis, primary amines were found to interact dominantly with the carbonyls of dopaquinone.138 However, using secondary amines, a 1,4- addition reaction between dopaquinone and amines was observed, with the product of this reaction providing the reference compounds used for assignment of the spectroscopic properties of lysyl oxidase.138 Ultimately, significant mechanistic insight into LTQ formation has come from studies of a mutant form of AGAO, in which the active site base, Asp298, had been converted to Lys. Using the copper-depleted E. coli expression system developed by Tanizawa for studies of cofactor maturation in native Arthrobacter enzyme,104 Mure and co-workers obtained both the precursor and mature forms of the D298K variant of AGAO.139 Initiation of biogenesis with the former, by addition of Cu(II) in the presence of O2, showed time-dependent, biphasic UV/Vis traces. A rapid initial phase leads to a ca. 500 nm species, whose formation is dependent on the level of O2; this is converted in a slower, O2-independent process to a blue-shifted species at 454 nm. Characterization of both species by RR, led to the proposal of an 1,4-addition product between the Lys at position 298 and a dopaquinone intermediate, that is slowly tautomerized to a more stable iminoquinone:139
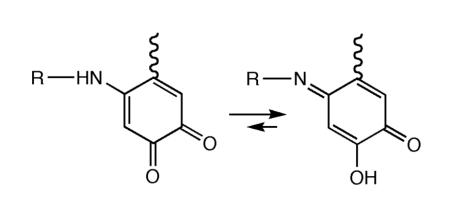
An X-ray structure of the D298K variant of AGAO clearly shows an LTQ-like structure, in contrast to the canonical TPQ generated in the native enzyme. These structural studies provide strong confirmatory evidence for the generation of a lysine-crosslinked dopaquinone in the Lys-containing variant, Figure 21. Consistent with spectroscopic work, the product visualized by X-ray crystallography is an iminoquinone. The propensity for iminoquinone formation has been attributed to the presence of the second conserved tyrosine (Y284) in the active site of the AGAO that normally forms a short hydrogen bond to the oxyanion at carbon 4 in TPQ. In contrast to TPQ, which ionizes with a pKa of ca. 4,140 LTQ appears unionized under all conditions studied, both in the modified AGAO and in lysyl oxidase. The tautomerization of an initially formed LTQ to a more stable iminoquinone, which provides a more suitable hydrogen-bonding partner for Tyr284, appears to dictate the final structure of the crosslinked structure.
Figure 21.

X-ray structures for AGAO. A. The product formed in the D298Kmutant of AGAO. B. The putative dopaquinone intermediate. From ref. (159). The blue spheres are water molecules, the red lines show ligands to the Cu2+ and the blue lines show hydrogen-bonded networks. From ref. (139).
While the above-described study of LTQ biogenesis in the mutant AGAO, provides considerable weight for the postulated pathway of Scheme 9, there are a number of important differences between this study and lysyl oxidase: First, there is the driving force for tautomerization of the LTQ-like cofactor to its iminoquinone product, that is provided by Tyr284 in AGAO. Second, there is a lack of reactivity of the final iminoquinone toward phenylhydrazine reagents and amine substrates. The latter is attributed, in large part, to a final movement of the crosslinked structure into a site that buries and protects the quinone from further chemical reactivity.139 Future detailed studies on the mechanism of LTQ formation in the lysyl oxidases are clearly dependent on the availability of a high-level expression system for the generation of the mature 32-kDa form of protein, together with its copper-free, precursor form. Given the level of posttranslational processing of lysyl oxidases that occurs in vivo, and the uncertainty as to which form of lysyl oxidase undergoes metal insertion, this may continue to be a challenging experimental problem.
7 Overview
Within the context of bioinorganic chemistry, the role of metal ions can be seen to be pervasive in quinone cofactor biosynthesis. For example, the iron-sulfur containing radical SAM enzymes play important roles in PQQ and CTQ biosynthesis. Proteins containing a unique pair of heme iron enzymes are present in the pathways for the production of TTQ and CTQ. A non-heme iron monooxygenase is strongly implicated in PQQ production and copper ions are essential for the production of TPQ and LTQ. The variability with regard to metal ion usage parallels the diversity of the enzymes and pathways utilized for the production of each of the quinone cofactors.
In searching for common features among the pathways, in all cases the initial hydroxylation of either a Tyr or Trp appears necessary. By nature of the structure of Trp, a second ring hydroxylation must occur in some manner during the production of TTQ and CTQ. These hydroxylations are followed by the addition of either a hydroxide ion (TPQ) or a second amino acid side chain (PQQ, TTQ, CTQ and LTQ), to initiate the final segment of cofactor production. The latter occurs with varying degrees of complexity (cf. Schemes 2, 4, 6, 8 and 9). In the examples where formation of a new carbon-carbon bond is not part of the pathway (Schemes 6, 8 and 9 and Table 1) the creation of an electrophilic center on the parent Tyr or Trp ring appears sufficient for subsequent nucleophilic attack by hydroxide ion, Cys or Lys. This property has raised the question of whether new quinone cofactors have yet to be discovered, originating via the addition of additional amino acid side chains to an electrophilic tryptophanyl quinone or dopaquinone (illustrated below):
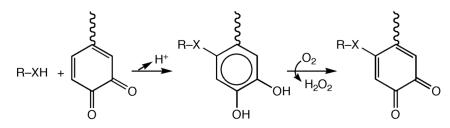
where R–XH is a side chain distinct from Lys or Cys.
It had originally been proposed that the CAO from Aspergillus niger contained a Glu crosslinked dopaquinone.141 This proposal was tested by the synthesis of suitable carboxylate-crosslinked model compounds and the comparison of their spectroscopic properties to those of the AGAO.142 This exercise demonstrated that the previously established TPQ was, in fact, the cofactor in the AGAO. In search of a deeper understanding of why the carboxylate-crosslinked cofactor might be inappropriate as a catalyst within the family of quinone-containing enzymes, the redox potential of a model compound was tested and shown to be +133 mV vs. SCE. This value is in marked contrast to all of the other quinocofactors that show redox potentials in the range of −150 to −180 mV vs. SCE.140 The evolutionary selection of viable quinone-based redox cofactors appears strongly linked to the catalytic behavior of the quinone-dependent enzymes, in particular the ability of the catalytically produced quinols to undergo facile reoxidation back to their starting quinones.
It is possible that we have to look in a completely different direction to understand the factors that led to the appearance of this unique family of enzymatic cofactors. Nature is often redundant with regard to the generation of different protein families that catalyze identical chemical reaction and flavin-dependent enzymes are known to bring about net amine and alcohol oxidations that are very similar to the reactions catalyzed by the quinocofactor-dependent enzymes discussed herein.143,144 The quinone-dependent enzymes are, in fact, more restrictive than flavin-dependent systems, either acting exclusively on primary amine substrates (TTQ, CTQ, TPQ, and LTQ) or a select number of primary alcohols (in the case of PQQ). One very intriguing feature of this class of redox cofactors is their predominant function outside of the cell: either within the periplasmic space of Gram-negative bacteria (for the PQQ-, TTQ- and CTQ-dependent enzymes)145-147 or within the extracellular matrix of mammals (the LTQ-dependent reaction of lysyl oxidases).148,149 Even in the case of the mammalian TPQ-dependent enzymes (where several intracellular enzymes are documented),150,151 important ectopic enzymatic variants have been identified on the outer plasma membrane of the endothelium of the vasculature and of adipocytes.152-154 Soluble forms of TPQ-containing CAOs have also been identified in serum.155,156 These considerations raise a very intriguing possibility: that the elusive evolutionary link among the structurally divergent quinone cofactors lies with the subcellular location in which their respective enzymes function. For the future, the rich chemistry and biology of these cofactors and enzymes is expected to drive many new inquiries and discoveries.
Figure 3.
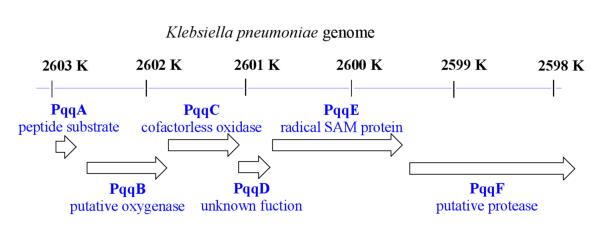
The PQQ biosynthetic operon in K. pneumoniae. From ref. (33).
8 Acknowledgements
We thank Mae Tulfo for invaluable assistance in the preparation of the manuscript. This work is supported by a grant from the NIH (GM039296) to J.P.K.
Biographies

Bio for Judith P. Klinman
Judith Klinman received her A.B. and Ph. D. from the University of Pennsylvania in l962 and l966 and then carried out postdoctoral research with Dr. David Samuel at the Weizmann Institute of Science, Israel, and Dr. Irwin Rose at the Institute for Cancer Research, Philadelphia. She was an independent researcher at the Institute for Cancer Research for many years, before moving to the University of California at Berkeley in l978, where she is now a Professor of the Graduate School in the Departments of Chemistry and of Molecular and Cell Biology. Her research is focused on four areas: (i) nuclear tunneling in enzyme-catalyzed reactions and the relationship of this phenomenon to protein dynamics and the origin of enzymatic rate accelerations; (ii) enzymatic methyl transferases and the role of active site compaction in catalysis; (iii) the mechanism of dioxygen activation by enzymes; and (iv) the biogenesis and catalytic mechanism of quinoproteins and cofactors. In addition to her lifelong fascination with enzymes, Dr. Klinman enjoys adventure travel, a stack of good books, gardening, experimenting with art, and not least, the amazement of four children and their eight children.

Bio for Florence Bonnot
Florence Bonnot received her undergraduate degree in biochemistry from the Institut National des Sciences Appliqués, Lyon, France in 2005. During her PhD at the Laboratoire de Chimie et Biologie des Métaux under the supervision of Dr. Vincent Nivière in the team of Pr. Marc Fontecave, Florence worked on the superoxide reductase, and more specifically, on the mechanism of this enzyme implied in the detoxification of superoxide, and has demonstrated the importance of the second sphere of coordination on the control of the evolution of iron peroxide intermediate. She spent her postdoctoral years in the Klinman lab at UC Berkeley from 2010-2013, working on PQQ biogenesis. She is now a researcher at the Laboratoire d’Ingénierie des Systémes Biologiques et des Procedés in Toulouse, France.
Footnotes
9 Note added in proof:
Since the completion of this review, Okazaki et al. have reported the first example of the presence of CTQ within the active site of an amine oxidase (Okazaki, S.; Nakano, S.; Matsui, D.; Akaji, S.; Inagaki, K.; Asano, Y. J. Biochemistry (2013) 154, 233). It has also been reported that a flavoprotein LodB is required for maturation of lysine-ε-oxidase (LodA) (Chacón-Verdú, M.D.; Gomez, D.; Solano, F.; Lucas-Eio, P.; Sánchez-Amat, A. Appl. Microbiol. Biotechnol. DOI 10.1007/S00253-013-5168-3).
10 References
- 1.Bergman Y, Cedar H. Nat. Struct. Mol. Biol. 2013;20:274. doi: 10.1038/nsmb.2518. [DOI] [PubMed] [Google Scholar]
- 2.Adeli K. Am. J. Physiol.-Endoc. M. 2011;301:E1051. doi: 10.1152/ajpendo.00399.2011. [DOI] [PubMed] [Google Scholar]
- 3.Tarrant MK, Cole PA. Annu. Rev. Biochem. 2009;78:797. doi: 10.1146/annurev.biochem.78.070907.103047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Glozak MA, Sengupta N, Zhang XH, Seto E. Gene. 2005;363:15. doi: 10.1016/j.gene.2005.09.010. [DOI] [PubMed] [Google Scholar]
- 5.Mukherjee S, Vaishnava S, Hooper LV. Cell Mol. Life Sci. 2008;65:3019. doi: 10.1007/s00018-008-8182-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yukl ET, Wilmot CM. Curr. Opin. Chem. Biol. 2012;16:54. doi: 10.1016/j.cbpa.2012.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Davidson VL. Mol. Biosyst. 2011;7:29. doi: 10.1039/c005311b. [DOI] [PubMed] [Google Scholar]
- 8.Hauge JG. J. Biol. Chem. 1964;239:3630. [PubMed] [Google Scholar]
- 9.Salisbury SA, Forrest HS, Cruse WBT, Kennard O. Nature. 1979;280:843. doi: 10.1038/280843a0. [DOI] [PubMed] [Google Scholar]
- 10.Blake CCF, Ghosh M, Harlos K, Avezoux A, Anthony C. Nat. Struct. Mol. Biol. 1994;1:102. doi: 10.1038/nsb0294-102. [DOI] [PubMed] [Google Scholar]
- 11.Oubrie A, Rozeboom HJ, Kalk KH, Olsthoorn AJJ, Duine JA, Dijkstra BW. Embo J. 1999;18:5187. doi: 10.1093/emboj/18.19.5187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Oubrie A, Rozeboom HJ, Dijkstra BW. Proc. Natl. Acad. Sci. U.S.A. 1999;96:11787. doi: 10.1073/pnas.96.21.11787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Janes SM, Mu D, Wemmer D, Smith AJ, Kaur S, Maltby D, Burlingame AL, Klinman JP. Science. 1990;248:981. doi: 10.1126/science.2111581. [DOI] [PubMed] [Google Scholar]
- 14.McIntire WS, Wemmer DE, Chistoserdov A, Lidstrom ME. Science. 1991;252:817. doi: 10.1126/science.2028257. [DOI] [PubMed] [Google Scholar]
- 15.Wang SX, Mure M, Medzihradszky KF, Burlingame AL, Brown DE, Dooley DM, Smith AJ, Kagan HM, Klinman JP. Science. 1996;273:1078. doi: 10.1126/science.273.5278.1078. [DOI] [PubMed] [Google Scholar]
- 16.Datta S, Mori Y, Takagi K, Kawaguchi K, Chen ZW, Okajima T, Kuroda S, Ikeda T, Kano K, Tanizawa K, Mathews FS. Proc. Natl. Acad. Sci. U.S.A. 2001;98:14268. doi: 10.1073/pnas.241429098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.DuBois JL, Klinman JP. Arch. Biochem. Biophys. 2005;433:255. doi: 10.1016/j.abb.2004.08.036. [DOI] [PubMed] [Google Scholar]
- 18.Schwartz B, Klinman JP. Vitam. Horm. 2001;61:219. doi: 10.1016/s0083-6729(01)61007-0. [DOI] [PubMed] [Google Scholar]
- 19.Anthony C. Arch. Biochem. Biophys. 2004;428:2. doi: 10.1016/j.abb.2004.03.038. [DOI] [PubMed] [Google Scholar]
- 20.Klinman JP, Mu D. Annu. Rev. Biochem. 1994;63:299. doi: 10.1146/annurev.bi.63.070194.001503. [DOI] [PubMed] [Google Scholar]
- 21.Mure M, Mills SA, Klinman JP. Biochemistry. 2002;41:9269. doi: 10.1021/bi020246b. [DOI] [PubMed] [Google Scholar]
- 22.Ameyama M, Matsushita K, Shinagawa E, Hayashi M, Adachi O. Biofactors. 1988;1:51. [PubMed] [Google Scholar]
- 23.Shen YQ, Bonnot F, Imsand EM, RoseFigura JM, Sjolander K, Klinman JP. Biochemistry. 2012;51:2265. doi: 10.1021/bi201763d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Anthony C, Zatman LJ. Biochem. J. 1967;104:953. doi: 10.1042/bj1040953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Duine JA, Frank J, van Zeeland JK. FEBS Lett. 1979;108:443. doi: 10.1016/0014-5793(79)80584-0. [DOI] [PubMed] [Google Scholar]
- 26.Anthony C. Antioxid. Redox. Sign. 2001;3:757. doi: 10.1089/15230860152664966. [DOI] [PubMed] [Google Scholar]
- 27.Steinberg F, Stites TE, Anderson P, Storms D, Chan I, Eghbali S, Rucker R. Exp. Biol. Med. 2003;228:160. doi: 10.1177/153537020322800205. [DOI] [PubMed] [Google Scholar]
- 28.Chowanadisai W, Bauerly KA, Tchaparian E, Wong A, Cortopassi GA, Rucker RB. J. Biol. Chem. 2010;285:142. doi: 10.1074/jbc.M109.030130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Houck DR, Hanners JL, Unkefer CJ. J. Am. Chem. Soc. 1988;110:6920. [Google Scholar]
- 30.Houck DR, Hanners JL, Unkefer CJ. J. Am. Chem. Soc. 1991;113:3162. [Google Scholar]
- 31.Vankleef MAG, Duine JA. FEBS Lett. 1988;237:91. doi: 10.1016/0014-5793(88)80178-9. [DOI] [PubMed] [Google Scholar]
- 32.Goosen N, Horsman HPA, Huinen RGM, Vandeputte PJ. Bacteriol. 1989;171:447. doi: 10.1128/jb.171.1.447-455.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Meulenberg JJM, Sellink E, Riegman NH, Postma WH. Mol. Gen. Genet. 1992;232:284. doi: 10.1007/BF00280008. [DOI] [PubMed] [Google Scholar]
- 34.Schnider U, Keel C, Voisard C, Defago G, Haas D. Appl. Environ. Microb. 1995;61:3856. doi: 10.1128/aem.61.11.3856-3864.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Biville F, Mazodier P, Turlin E, Gasser F. A Van Leeuw J Microb. 1989;56:103. doi: 10.1007/BF00822590. [DOI] [PubMed] [Google Scholar]
- 36.Morris CJ, Biville F, Turlin E, Lee E, Ellermann K, Fan WH, Ramamoorthi R, Springer AL, Lidstrom ME. J. Bacteriol. 1994;176:1746. doi: 10.1128/jb.176.6.1746-1755.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Meulenberg JJ, Sellink E, Loenen WA, Riegman NH, van Kleef M, Postma PW. FEMS Microbiol. Lett. 1990;59:337. doi: 10.1016/0378-1097(90)90244-k. [DOI] [PubMed] [Google Scholar]
- 38.Velterop JS, Sellink E, Meulenberg JJM, David S, Bulder I, Postma PW. J. Bacteriol. 1995;177:5088. doi: 10.1128/jb.177.17.5088-5098.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wecksler SR, Stoll S, Tran H, Magnusson OT, Wu SP, King D, Britt RD, Klinman JP. Biochemistry. 2009;48:10151. doi: 10.1021/bi900918b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lanz ND, Booker SJ. BBA-Proteins Proteom. 2012;1824:1196. doi: 10.1016/j.bbapap.2012.07.009. [DOI] [PubMed] [Google Scholar]
- 41.Hanzelmann P, Schindelin H. Proc. Natl. Acad. Sci. U.S.A. 2006;103:6829. doi: 10.1073/pnas.0510711103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Toyama H, Chistoserdova L, Lidstrom ME. Microbiol.-UK. 1997;143:595. doi: 10.1099/00221287-143-2-595. [DOI] [PubMed] [Google Scholar]
- 43.Magnusson OT, Toyama H, Saeki M, Schwarzenbacher R, Klinman JP. J. Am. Chem. Soc. 2004;126:5342. doi: 10.1021/ja0493852. [DOI] [PubMed] [Google Scholar]
- 44.Toyama H, Fukumoto H, Saeki M, Matsushita K, Adachi O, Lidstrom ME. Biochem. Bioph. Res. Co. 2002;299:268. doi: 10.1016/s0006-291x(02)02603-7. [DOI] [PubMed] [Google Scholar]
- 45.Magnusson OT, Toyama H, Saeki M, Rojas A, Reed JC, Liddington RC, Klinman JP, Schwarzenbacher R. Proc. Natl. Acad. Sci. U.S.A. 2004;101:7913. doi: 10.1073/pnas.0402640101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tsai TY, Yang CY, Shih HL, Wang AH, Chou SH. Proteins. 2009;76:1042. doi: 10.1002/prot.22461. [DOI] [PubMed] [Google Scholar]
- 47.Toyama H, Nishibayashi E, Saeki M, Adachi O, Matsushita K. Biochem. Bioph. Res. Co. 2007;354:290. doi: 10.1016/j.bbrc.2007.01.001. [DOI] [PubMed] [Google Scholar]
- 48.Wecksler SR, Stoll S, Iavarone AT, Imsand EM, Tran H, Britt RD, Klinman JP. Chem. Commun. (Camb) 2010;46:7031. doi: 10.1039/c0cc00968g. [DOI] [PubMed] [Google Scholar]
- 49.Koehntop KD, Emerson JP, Que LJ. Biol. Inorg. Chem. 2005;10:87. doi: 10.1007/s00775-005-0624-x. [DOI] [PubMed] [Google Scholar]
- 50.Puehringer S, RoseFigura J, Metlitzky M, Toyama H, Klinman JP, Schwarzenbacher R. Proteins-Structure Function and Bioinformatics. 2010;78:2554. doi: 10.1002/prot.22769. [DOI] [PubMed] [Google Scholar]
- 51.Magnusson OT, RoseFigura JM, Toyama H, Schwarzenbacher R, Klinman JP. Biochemistry. 2007;46:7174. doi: 10.1021/bi700162n. [DOI] [PubMed] [Google Scholar]
- 52.RoseFigura JM, Puehringer S, Schwarzenbacher R, Toyama H, Klinman JP. Biochemistry. 2011;50:1556. doi: 10.1021/bi1015474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bonnot F, Iavarone AT, Klinman JP. Biochemistry. 2013;52:4667. doi: 10.1021/bi4003315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mushegian AR, Koonin EV. Trends Genet. 1996;12:289. doi: 10.1016/0168-9525(96)20006-x. [DOI] [PubMed] [Google Scholar]
- 55.Omelchenko MV, Makarova KS, Wolf YI, Rogozin IB, Koonin EV. Genome Biol. 2003:4. doi: 10.1186/gb-2003-4-9-r55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dandekar T, Snel B, Huynen M, Bork P. Trends Biochem. Sci. 1998;23:324. doi: 10.1016/s0968-0004(98)01274-2. [DOI] [PubMed] [Google Scholar]
- 57.Fondi M, Emiliani G, Fani R. Res. Microbiol. 2009;160:502. doi: 10.1016/j.resmic.2009.05.001. [DOI] [PubMed] [Google Scholar]
- 58.Govindaraj S, Eisenstein E, Jones LH, Sandersloehr J, Chistoserdov AY, Davidson VL, Edwards SL. J. Bacteriol. 1994;176:2922. doi: 10.1128/jb.176.10.2922-2929.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Husain M, Davidson VL. J. Biol. Chem. 1985;260:4626. [PubMed] [Google Scholar]
- 60.Hyun YL, Davidson VL. Biochemistry. 1995;34:12249. doi: 10.1021/bi00038a020. [DOI] [PubMed] [Google Scholar]
- 61.Bishop GR, Brooks HB, Davidson VL. Biochemistry. 1996;35:8948. doi: 10.1021/bi960404x. [DOI] [PubMed] [Google Scholar]
- 62.Zhu ZY, Davidson VL. Biochemistry. 1999;38:4862. doi: 10.1021/bi982939r. [DOI] [PubMed] [Google Scholar]
- 63.Chen LY, Doi N, Durley RCE, Chistoserdov AY, Lidstrom ME, Davidson VL, Mathews FS. J. Mol. Biol. 1998;276:131. doi: 10.1006/jmbi.1997.1511. [DOI] [PubMed] [Google Scholar]
- 64.Vanderpalen CJNM, Slotboom DJ, Jongejan L, Reijnders WNM, Harms N, Duine JA, Vanspanning RJ. M. Eur. J. Biochem. 1995;230:860. doi: 10.1111/j.1432-1033.1995.tb20629.x. [DOI] [PubMed] [Google Scholar]
- 65.Chistoserdov AY, Chistoserdova LV, Mcintire WS, Lidstrom ME. J. Bacteriol. 1994;176:4052. doi: 10.1128/jb.176.13.4052-4065.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chistoserdov AY, Boyd J, Mathews FS, Lidstrom ME. Biochem. Bioph. Res. Co. 1992;184:1181. doi: 10.1016/s0006-291x(05)80007-5. [DOI] [PubMed] [Google Scholar]
- 67.Vanspanning RJM, Wansell CW, Reijnders WNM, Oltmann LF, Stouthamer AH. FEBS Lett. 1990;275:217. doi: 10.1016/0014-5793(90)81475-4. [DOI] [PubMed] [Google Scholar]
- 68.Chen LY, Durley RCE, Mathews FS, Davidson VL. Science. 1994;264:86. doi: 10.1126/science.8140419. [DOI] [PubMed] [Google Scholar]
- 69.van der Palen CJ, Reijnders WNM, de Vries S, Duine JA, van Spanning RJ. A. van Leeuw. 1997;72:219. doi: 10.1023/a:1000441925796. [DOI] [PubMed] [Google Scholar]
- 70.Pearson AR, De la Mora-Rey T, Graichen ME, Wang YT, Jones LH, Marimanikkupam S, Agger SA, Grimsrud PA, Davidson VL, Wilmot CM. Biochemistry. 2004;43:5494. doi: 10.1021/bi049863l. [DOI] [PubMed] [Google Scholar]
- 71.Wang YT, Li XH, Jones LH, Pearson AR, Wilmot CM, Davidson VL. J. Am. Chem. Soc. 2005;127:8258. doi: 10.1021/ja051734k. [DOI] [PubMed] [Google Scholar]
- 72.Wang YT, Graichen ME, Liu AM, Pearson AR, Wilmot CM, Davidson VL. Biochemistry. 2003;42:7318. doi: 10.1021/bi034243q. [DOI] [PubMed] [Google Scholar]
- 73.Jensen LMR, Sanishvili R, Davidson VL, Wilmot CM. Science. 2010;327:1392. doi: 10.1126/science.1182492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Abu Tarboush N, Jensen LMR, Yukl ET, Geng JF, Liu AM, Wilmot CM, Davidson VL. Proc. Natl. Acad. Sci. U.S.A. 2011;108:16956. doi: 10.1073/pnas.1109423108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Abu Tarboush N, Jensen LMR, Wilmot CM, Davidson DL. FEBS Lett. 2013;587:1736. doi: 10.1016/j.febslet.2013.04.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lee S, Shin S, Li X, Davidson VL. Biochemistry. 2009;48:2442. doi: 10.1021/bi802166c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Shin S, Abu Tarboush N, Davidson VL. Biochemistry. 2010;49:5810. doi: 10.1021/bi1004969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yukl ET, Liu F, Krzystek J, Shin S, Jensen LMR, Davidson VL, Wilmot CM. Proc. Natl. Acad. Sci. U.S.A. 2013;110:4569. doi: 10.1073/pnas.1215011110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Li X, Fu R, Lee S, Krebs C, Davidson VL, Liu A. Proc. Natl. Acad. Sci. U.S.A. 2008;105:8597. doi: 10.1073/pnas.0801643105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Li XH, Feng ML, Wang YT, Tachikawa H, Davidson VL. Biochemistry. 2006;45:821. doi: 10.1021/bi052000n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lee SY, Shin S, Li XH, Davidson VL. Biochemistry. 2009;48:2442. doi: 10.1021/bi802166c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Abu Tarboush N, Jensen LMR, Feng ML, Tachikawa H, Wilmot CM, Davidson VL. Biochemistry. 2010;49:9783. doi: 10.1021/bi101254p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Fu R, Liu FG, Davidson VL, Liu AM. Biochemistry. 2009;48:11603. doi: 10.1021/bi9017544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Geng JF, Dornevil K, Davidson VL, Liu AM. Proc. Natl. Acad. Sci. U.S.A. 2013;110:9639. doi: 10.1073/pnas.1301544110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Davidson VL, Wilmot CM. Annu. Rev. Biochem. 2013;82:531. doi: 10.1146/annurev-biochem-051110-133601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Nakai T, Ono K, Kuroda S, Tanizawa K, Okajima TJ. Biol. Chem. 2012;287:6530. doi: 10.1074/jbc.M111.324756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sun DP, Ono K, Okajima T, Tanizawa K, Uchida M, Yamamoto Y, Mathews FS, Davidson VL. Biochemistry. 2003;42:10896. doi: 10.1021/bi035062r. [DOI] [PubMed] [Google Scholar]
- 88.Datta S, Ikeda T, Kano K, Mathews FS. Acta Crystallogr. D. 2003;59:1551. doi: 10.1107/s090744490301429x. [DOI] [PubMed] [Google Scholar]
- 89.Ono K, Okajima T, Tani M, Kuroda S, Sun D, Davidson VL, Tanizawa K. J. Biol.Chem. 2006;281:13672. doi: 10.1074/jbc.M600029200. [DOI] [PubMed] [Google Scholar]
- 90.Pearson AR, Jones LH, Higgins LA, Ashcroft AE, Wilmot CM, Davidson VL. Biochemistry. 2003;42:3224. doi: 10.1021/bi027073a. [DOI] [PubMed] [Google Scholar]
- 91.Blaschko H, Buffoni FP. Roy. Soc. Lond. B-Bio. 1965;163:45. doi: 10.1098/rspb.1965.0059. [DOI] [PubMed] [Google Scholar]
- 92.Knowles PF, Pandeya KB, Rius FX, Spencer CM, Moog RS, Mcguirl MA, Dooley DM. Biochem. J. 1987;241:603. doi: 10.1042/bj2410603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Plastino J, Green EL, Sanders-Loehr J, Klinman JP. Biochemistry. 1999;38:8204. doi: 10.1021/bi9826660. [DOI] [PubMed] [Google Scholar]
- 94.Li R, Klinman JP, Mathews FS. Structure. 1998;6:293. doi: 10.1016/s0969-2126(98)00033-1. [DOI] [PubMed] [Google Scholar]
- 95.Parsons MR, Convery MA, Wilmot CM, Yadav KDS, Blakely V, Corner AS, Phillips SEV, Mcpherson MJ, Knowles PF. Structure. 1995;3:1171. doi: 10.1016/s0969-2126(01)00253-2. [DOI] [PubMed] [Google Scholar]
- 96.Freeman HC, Guss JM, Kumar V, McIntire WS, Zubak VM. Acta Crystallogr. D. 1996;52:197. doi: 10.1107/S0907444995007529. [DOI] [PubMed] [Google Scholar]
- 97.Kumar V, Dooley DM, Freeman HC, Guss JM, Harvey I, McGuirl MA, Wilce MCJ, Zubak VM. Structure. 1996;4:943. doi: 10.1016/s0969-2126(96)00101-3. [DOI] [PubMed] [Google Scholar]
- 98.Nakamura N, Moenne-Loccoz P, Tanizawa K, Mure M, Suzuki S, Klinman JP, Sanders-Loehr J. Biochemistry. 1997;36:11479. doi: 10.1021/bi9708139. [DOI] [PubMed] [Google Scholar]
- 99.Wilce MCJ, Dooley DM, Freeman HC, Guss JM, Matsunami H, McIntire WS, Ruggiero CE, Tanizawa K, Yamaguchi H. Biochemistry. 1997;36:16116. doi: 10.1021/bi971797i. [DOI] [PubMed] [Google Scholar]
- 100.Janes SM, Palcic MM, Scaman CH, Smith AJ, Brown DE, Dooley DM, Mure M, Klinman JP. Biochemistry. 1992;31:12147. doi: 10.1021/bi00163a025. [DOI] [PubMed] [Google Scholar]
- 101.Cai D, Klinman JP. J. Biol. Chem. 1994;269:32039. [PubMed] [Google Scholar]
- 102.Schwartz B, Green EI, Sanders-Loehr J, Klinman JP. Biochemistry. 1998;37:16591. doi: 10.1021/bi981541s. [DOI] [PubMed] [Google Scholar]
- 103.Cai D, Dove J, Nakamura N, Sanders-Loehr J, Klinman JP. Biochemistry. 1997;36:11472. doi: 10.1021/bi970812g. [DOI] [PubMed] [Google Scholar]
- 104.Matsuzaki R, Fukui T, Sato H, Ozaki Y, Tanizawa K. FEBS Lett. 1994;351:360. doi: 10.1016/0014-5793(94)00884-1. [DOI] [PubMed] [Google Scholar]
- 105.Nakamura N, Matsuzaki R, Choi YH, Tanizawa K, SandersLoehr JJ. Biol. Chem. 1996;271:4718. doi: 10.1074/jbc.271.9.4718. [DOI] [PubMed] [Google Scholar]
- 106.Ruggiero CE, Dooley DM. Biochemistry. 1999;38:2892. doi: 10.1021/bi9824994. [DOI] [PubMed] [Google Scholar]
- 107.Cai DY, Klinman JP. Biochemistry. 1994;33:7647. doi: 10.1021/bi00190a019. [DOI] [PubMed] [Google Scholar]
- 108.Cai D, Williams NK, Klinman JP. J. Biol. Chem. 1997;272:19277. doi: 10.1074/jbc.272.31.19277. [DOI] [PubMed] [Google Scholar]
- 109.McGrath AP, Caradoc-Davies T, Collyer CA, Guss JM. Biochemistry. 2010;49:8316. doi: 10.1021/bi1010915. [DOI] [PubMed] [Google Scholar]
- 110.Verhagen MFJM, Meussen ETM, Hagen WR. BBA-Gen. Subjects. 1995;1244:99. doi: 10.1016/0304-4165(94)00201-8. [DOI] [PubMed] [Google Scholar]
- 111.Stubbe J, van der Donk WA. Chem. Rev. 1998;98:705. doi: 10.1021/cr980059c. [DOI] [PubMed] [Google Scholar]
- 112.Dove JE, Schwartz B, Williams NK, Klinman JP. Biochemistry. 2000;39:3690. doi: 10.1021/bi992225w. [DOI] [PubMed] [Google Scholar]
- 113.DuBois JL, Klinman JP. Biochemistry. 2005;44:11381. doi: 10.1021/bi0504759. [DOI] [PubMed] [Google Scholar]
- 114.Chen Z, Schwartz B, Williams NK, Li R, Klinman JP, Mathews FS. Biochemistry. 2000;39:9709. doi: 10.1021/bi000639f. [DOI] [PubMed] [Google Scholar]
- 115.Kim M, Okajima T, Kishishita S, Yoshimura M, Kawamori A, Tanizawa K, Yamaguchi H. Nat. Struct. Biol. 2002;9:591. doi: 10.1038/nsb824. [DOI] [PubMed] [Google Scholar]
- 116.Schwartz B, Dove JE, Klinman JP. Biochemistry. 2000;39:3699. doi: 10.1021/bi9922244. [DOI] [PubMed] [Google Scholar]
- 117.Takahashi K, Klinman JP. Biochemistry. 2006;45:4683. doi: 10.1021/bi0521893. [DOI] [PubMed] [Google Scholar]
- 118.Mukherjee A, Smirnov VV, Lanci MP, Brown DE, Shepard EM, Dooley DM, Roth JP. J. Am. Chem. Soc. 2008;130:9459. doi: 10.1021/ja801378f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Su Q, Klinman JP. Biochemistry. 1998;37:12513. doi: 10.1021/bi981103l. [DOI] [PubMed] [Google Scholar]
- 120.Mills SA, Klinman JP. J. Am. Chem. Soc. 2000;122:9897. [Google Scholar]
- 121.Mills SA, Goto Y, Su QJ, Plastino J, Klinman JP. Biochemistry. 2002;41:10577. doi: 10.1021/bi0200864. [DOI] [PubMed] [Google Scholar]
- 122.Goto Y, Klinman JP. Biochemistry. 2002;41:13637. doi: 10.1021/bi0204591. [DOI] [PubMed] [Google Scholar]
- 123.Okajima T, Kishishita S, Chiu YC, Murakawa T, Kim M, Yamaguchi H, Hirota S, Kuroda S, Tanizawa K. Biochemistry. 2005;44:12041. doi: 10.1021/bi051070r. [DOI] [PubMed] [Google Scholar]
- 124.Samuels NM, Klinman JP. Biochemistry. 2005;44:14308. doi: 10.1021/bi051176m. [DOI] [PubMed] [Google Scholar]
- 125.Gomes L, Pereira E, de Castro B. J. Chem. Soc. Dalton. 2000;1373 [Google Scholar]
- 126.Samuels NM, Klinman JP. J. Biol. Chem. 2006;281:21114. doi: 10.1074/jbc.M601501200. [DOI] [PubMed] [Google Scholar]
- 127.Schwartz B, Olgin AK, Klinman JP. Biochemistry. 2001;40:2954. doi: 10.1021/bi0021378. [DOI] [PubMed] [Google Scholar]
- 128.Hevel JM, Mills SA, Klinman JP. Biochemistry. 1999;38:3683. doi: 10.1021/bi982199m. [DOI] [PubMed] [Google Scholar]
- 129.DuBois JL, Klinman JP. Biochemistry. 2006;45:3178. doi: 10.1021/bi052025m. [DOI] [PubMed] [Google Scholar]
- 130.Kraut DA, Sigala PA, Fenn TD, Herschlag D. Proc. Natl. Acad. Sci. U.S.A. 2010;107:1960. doi: 10.1073/pnas.0911168107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Chen ZW, Datta S, DuBois JL, Klinman JP, Mathews FS. Biochemistry. 2010;49:7393. doi: 10.1021/bi100643y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Mandal S, Lee Y, Purdy MM, Sayre LM. J. Am. Chem. Soc. 2000;122:3574. [Google Scholar]
- 133.Kagan HM, Li WD. J. Cell Biochem. 2003;88:660. doi: 10.1002/jcb.10413. [DOI] [PubMed] [Google Scholar]
- 134.Hamalainen ER, Jones TA, Sheer D, Taskinen K, Pihlajaniemi T, Kivirikko KI. Genomics. 1991;11:508. doi: 10.1016/0888-7543(91)90057-l. [DOI] [PubMed] [Google Scholar]
- 135.Gacheru SN, Trackman PC, Shah MA, Ogara CY, Spacciapoli P, Greenaway FT, Kagan HM. J. Biol. Chem. 1990;265:19022. [PubMed] [Google Scholar]
- 136.Wang S-X, Nakamura N, Mure M, Klinman JP, Sanders-Loehr J. J. Biol. Chem. 1997;272:28841. doi: 10.1074/jbc.272.46.28841. [DOI] [PubMed] [Google Scholar]
- 137.Bollinger JA, Brown DE, Dooley DM. Biochemistry. 2005;44:11708. doi: 10.1021/bi0504310. [DOI] [PubMed] [Google Scholar]
- 138.Mure M, Wang SX, Klinman JP. J. Am. Chem. Soc. 2003;125:6113. doi: 10.1021/ja0214274. [DOI] [PubMed] [Google Scholar]
- 139.Moore RH, Spies MA, Culpepper MB, Murakawa T, Hirota S, Okajima T, Tanizawa K, Mure M. J. Am. Chem. Soc. 2007;129:11524. doi: 10.1021/ja0731165. [DOI] [PubMed] [Google Scholar]
- 140.Mure M, Klinman JP. J. Am. Chem. Soc. 1993;115:7117. [Google Scholar]
- 141.Frebort I, Pec P, Luhova L, Toyama H, Matsushita K, Hirota S, Kitagawa T, Ueno T, Asano Y, Kato Y, Adachi O. BBA-Protein Struct. M. 1996;1295:59. doi: 10.1016/0167-4838(96)00014-3. [DOI] [PubMed] [Google Scholar]
- 142.Melville CR, Green EI, Sanders-Loehr J, Klinman JP. Biochemistry. 2000;39:7589. doi: 10.1021/bi000507m. [DOI] [PubMed] [Google Scholar]
- 143.Rigby SE, Basran J, Combe JP, Mohsen AW, Toogood H, van Thiel A, Suitcliffe MJ, Leys D, Munro AW, Scrutton NS. Biochem. Soc. T. 2005;33:754. doi: 10.1042/BST0330754. [DOI] [PubMed] [Google Scholar]
- 144.Edmondson DE, Mattevi A, Binda C, Li M, Hubalek F. Curr. Med. Chem. 2004;11:1983. doi: 10.2174/0929867043364784. [DOI] [PubMed] [Google Scholar]
- 145.Goodwin PM, Anthony C. Adv. Microb. Physiol. 1998;40:1. doi: 10.1016/s0065-2911(08)60129-0. [DOI] [PubMed] [Google Scholar]
- 146.Husain M, Davidson VL. J. Bacteriol. 1987;169:1712. doi: 10.1128/jb.169.4.1712-1717.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Adachi O, Kubota T, Hacisalihoglu A, Toyama H, Shinagawa E, Duine JA, Matsushita K. Biosci. Biotech. Bioch. 1998;62:469. doi: 10.1271/bbb.62.469. [DOI] [PubMed] [Google Scholar]
- 148.Kim MS, Kim SS, Jung ST, Park JY, Yoo HW, Ko J, Csiszar K, Choi SY, Kim Y. J. Biol. Chem. 2003;278:52071. doi: 10.1074/jbc.M308856200. [DOI] [PubMed] [Google Scholar]
- 149.Csiszar K. Prog. Nucleic Acid. Re. 2001;70:1. doi: 10.1016/s0079-6603(01)70012-8. [DOI] [PubMed] [Google Scholar]
- 150.Crabbe JC, Waight RD, Bardsley WG, Barker RW, Kelly ID, Knowles PF. Biochem. J. 1976;155:679. doi: 10.1042/bj1550679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Mondovi B, Finazzi A, Scioscia A, Rotilio G. Biochem. J. 1964;91:408. doi: 10.1042/bj0910408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Smith DJ, Salmi M, Bono P, Hellman J, Leu T, Jalkanen S. J. Exp. Med. 1998;188:17. doi: 10.1084/jem.188.1.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Salmi M, Jalkanen S. Nat. Rev. Immunol. 2005;5:760. doi: 10.1038/nri1705. [DOI] [PubMed] [Google Scholar]
- 154.Shen SH, Wertz DL, Klinman JP. PLoS One. 2012 doi: 10.1371/journal.pone.0029270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Mu D, Medzihradszky KF, Adams GW, Mayer P, Hines WM, Burlingame AL, Smith AJ, Cai D, Klinman JP. J. Biol. Chem. 1994;269:9926. [PubMed] [Google Scholar]
- 156.Meszaros Z, Karadi I, Csanyi A, Szombathy T, Romics L, Magyar K. Eur. J. Drug Metab. Ph. 1999;24:299. doi: 10.1007/BF03190036. [DOI] [PubMed] [Google Scholar]
- 157.Shin S, Feng ML, Chen Y, Jensen LMR, Tachikawa H, Wilmot CM, Liu AM, Davidson VL. Biochemistry. 2011;50:144. doi: 10.1021/bi101819m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Smith MA, Pirrat P, Pearson AR, Kurtis CRP, Trinh CH, Gaule TG, Knowles PF, Phillips SEV, McPherson MJ. Biochemistry. 2010;49:1268. doi: 10.1021/bi901738k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Kim M, Okajima T, Kishishita S, Yoshimura M, Kawamori A, Tanizawa K, Yamaguchi H. Nat. Struct. Biol. 2002;9:591. doi: 10.1038/nsb824. [DOI] [PubMed] [Google Scholar]