

Abstract
Aims
Mice unable to locally regenerate corticosterone due to deficiency of 11β-hydroxysteroid dehydrogenase type 1 (11βHSD1) have enhanced angiogenesis during acute myocardial infarct healing. The present study investigates the hypotheses that in these mice (i) inflammation and angiogenic signalling are promoted and (ii) longer-term remodelling and function are improved.
Methods and results
Myocardial infarction (MI) was induced by coronary artery ligation in 11βHSD1−/− and wild-type (C57BL/6) mice. Studies were terminated 2, 4, 7, and 28 days post-surgery. Increased vessel density (CD31 immunoreactivity) on the infarct border was confirmed 7 days after MI in 11βHSD1−/− hearts (P < 0.05) and was accompanied by improved ejection fraction (ultrasound) compared with C57BL/6. During wound healing, recruitment of neutrophils (at 2 days after MI) and macrophages (from 4 days after MI) and expression of monocyte-chemoattractant protein-1 was increased in 11βHSD1−/− compared with C57BL/6 hearts (P < 0.05). Recruitment of alternatively activated YM1-positive macrophages was particularly enhanced in the period preceding increased vessel density and was accompanied by increased expression of pro-angiogenic IL-8. By 28 days post-MI, when the infarct scar had matured, higher vessel density was maintained in 11βHSD1−/− hearts and vessels were smooth-muscle coated. Infarct scars were thicker (P < 0.001) in 11βHSD1−/− compared with C57BL/6 hearts and ejection fraction was higher (P < 0.05).
Conclusion
Increased vessel density in healing infarcts of mice deficient in 11−/−HSD1 follows recruitment of pro-reparative macrophages and increased pro-angiogenic signalling. Mature infarcts show less thinning and cardiac function is improved relative to wild-type mice, suggesting that 11βHSD1 may be a novel therapeutic target after MI.
Keywords: Angiogenesis, Inflammation, Alternatively activated macrophages, YM1, IL-8
1. Introduction
Infarct expansion is an important determinant of long-term outcome following myocardial infarction (MI). Therapeutic strategies aimed at limiting infarct size, such as reperfusion, have led to reduction of acute mortality post-MI. Despite this intervention, patients still develop heart failure.1 Enhancing blood supply to the infarct border through stimulation of angiogenesis, e.g. by injection of putative cell progenitors or pro-angiogenic factors into the myocardium, reduces infarct expansion and remodelling, and improves heart function in experimental MI.1–3 However, translation of this strategy to the clinic has had limited success to date.4,5 An alternative approach is to manipulate endogenous mechanisms involved in infarct healing so that the associated angiogenic response is promoted.
Activation of corticosteroid receptors is regulated in target tissues by pre-receptor metabolism of glucocorticoids by the isozymes of 11β-hydroxysteroid dehydrogenase (11βHSD).6 Following secretion of active glucocorticoids (principally cortisol in humans and corticosterone in rodents) from the adrenal cortex under the control of ACTH, these steroids are inactivated by 11βHSD2 (to cortisone and 11-dehydrocorticosterone, respectively) in the kidney and a few other tissues. These inert metabolites are then regenerated into active glucocorticoid by 11βHSD1, which is expressed primarily in glucocorticoid target tissues and has been implicated in the regulation of cognitive, metabolic, and cardiovascular function.7,8 Importantly, 11βHSDs influence the intracellular concentrations of active glucocorticoids independently of any change in circulating plasma glucocorticoids levels; changes in 11βHSD activity do alter metabolic clearance rate of glucocorticoids, but the circulating level of cortisol (or corticosterone) is maintained by compensatory changes in ACTH and adrenal secretion. In the cardiovascular system, 11βHSD1 is expressed in the heart9,10 and in the vascular wall.11,12 In a previous study, we showed that mice deficient in 11βHSD1 have enhanced capacity for angiogenesis, and that vessel density is increased during infarct healing after MI.13 However, the mechanism of increased neovascularization in the hearts of 11βHSD1−/− mice is unknown, as are the functional consequences for the heart. Inflammatory cells, particularly monocyte/macrophages, are an important source of pro-angiogenic cytokines, such as vascular endothelial growth factor (VEGF) and interleukin-8 (IL-8).14 Depletion of monocytes/macrophages after experimental MI results in impaired angiogenesis on the infarct border.15,16 In contrast, enhancement of monocyte recruitment by overexpression of monocyte chemoattractant protein-1 (MCP-1),17 or direct injection of activated macrophages into the heart,18 increases vessel density on the infarct border and improves heart function.
Glucocorticoids are released into the systemic circulation acutely after MI and appear to be initially cardioprotective, as blockade of the glucocorticoid receptor (GR) increases infarct size.19,20 However, experimental and clinical studies have shown that prolonged exposure to high systemic levels of glucocorticoid, achieved by administration of synthetic glucocorticoid, is detrimental following MI,21,22 consistent with the ability of glucocorticoid to suppress inflammatory processes essential for infarct healing.13,23,24 Deficiency of 11βHSD1 exacerbates inflammation in murine models of arthritis and sterile peritonitis,25 suggesting that locally regenerated glucocorticoid also suppresses inflammation.
The present study was designed to investigate the hypothesis that the lack of local glucocorticoid regeneration in 11βHSD1−/− mice results in modification of the inflammatory response after MI resulting in promotion of pro-angiogenic signalling in the healing myocardial infarct. We also aimed to investigate whether the vessels formed early after MI are retained after infarct healing is complete and importantly, whether there are beneficial consequences for cardiac remodelling and function in 11βHSD1−/− mice.
2. Methods
2.1. Animals
Ten- to 12-week-old male 11βHSD1 knock out (11βHSD1−/− congenic with C57BL/6) mice were bred from an in-house colony.26 Controls were commercial C57BL/6 mice (Harlan). The investigation conforms with the Guide for the Care and Use of Laboratory Animals published by the US National Institute of Health (NIH Publication No 82-23 revised 1996) and was approved by the University of Edinburgh ethics committee.
2.2. Coronary artery ligation
Mice (n = 128) were anaesthetized (1 mg/kg medetomidine, 75 mg/kg ketamine, and 600 μg/kg atropine) and received buprenorphine (0.05 mg/kg) for analgesia. The trachea was intubated for mechanical ventilation (120 strokes/min, 200 μL stroke volume, Hugo Sachs Elektronik Minivent). The left thorax was opened at the fourth intercostal space and the left main descending coronary artery was ligated with a 6.0 prolene suture. Sham animals did not have the ligature tied. Following surgery mice received the reversal agent atipamezole (5 mg/kg) and 1.5 mL sterile saline intraperitoneally, and oxygen-enriched air until fully recovered.
2.3. Corticosterone radioimmunoassay
Plasma corticosterone levels 24 h and every 7 days after surgery at the diurnal nadir were measured by radioimmunoassay as described previously.26
2.4. Ultrasound and tissue collection
At 2, 4, 7, or 28 days after surgery, heart function was assessed by ultrasound (Diasus 10–22 MHz probe, Dynamic Imaging, Livingstone, UK). Left ventricular ejection fraction (%EF) was calculated as detailed in the Supplementary Methods. The observer was blinded for ultrasound measurements and all other analyses.
2.5 mg BrdU (Sigma) was administered i.p. 1 h prior to sacrifice to label proliferating cells.27 The heart was removed and bisected down the longitudinal axis through the infarct. One half was fixed in 10% neutral buffered formalin for histology and immunohistochemistry. The other half was frozen immediately and stored at −80°C.
2.5. Infarct size measurement
Infarct size at 24 h after MI was measured by triphenyltetrazolium chloride (TTC, Sigma) staining as described previously.28
2.6. Histology and immunohistochemistry
Haematoxylin and eosin, Picrosirius Red, and Masson's Trichrome stains were used to identify neutrophils, collagen, and the infarct scar, respectively. Immunohistochemistry was used to identify endothelial cells (anti-CD31, BD Pharmingen), proliferating cells (anti-BrdU, Sigma), macrophages (anti-mac 2, Cedarlane), alternatively activated macrophages (anti-YM1, Stem cell Technologies), and activated myofibroblasts [anti-α smooth muscle actin (SMA), Sigma]. Biotinylated secondary antibodies (rabbit anti-rat, goat anti-mouse, and goat anti-rabbit, 1:200, Vector) were subsequently added prior to extravidin peroxidase (Sigma). Detection of peroxidase activity was with the 3,3-diaminobenzidine kit (Vector). Sections were counterstained with haematoxylin, dehydrated, and mounted in DPX resin (Fluka). See Supplementary Methods for further details.
For quantification, sections were tiled at ×25 or ×100 magnification (Image Pro6.2, Stereologer Analyser 6 MediaCybernetics). Neutrophils (identified morphologically) and counted in 10 randomly assigned 40 μm2 areas of left ventricle (LV); CD31 and αSMA positive vessels (less than 200 μm in diameter) were counted in 10 randomly assigned 400 μm2 areas of LV, and an average per LV calculated.29 BrdU positive cells were counted per mm2. Macrophage (Mac 2 and YM1) staining was calculated based on percentage staining of the infarct border. The area of collagen deposition (Picrosirius Red) and scar area (Masson's Trichrome) were quantified as a percentage of the total LV. Scar thickness was calculated from the thickness of three points on the scar and averaged. Epicardial infarct length was calculated as epicardial infarct length/epicardial length × 100.28
2.7. RNA isolation and quantitative real-time reverse-transcription polymerase chain reaction
One microgram RNA extracted from the frozen half heart using Trizol (Invitrogen, Paisley, UK) was reverse transcribed to cDNA (Applied Biosystems high capacity cDNA reverse transcription kit). TAQman® gene expression assays were used to detect interleukin 6 (IL-6 Mm99999064_m1), monocyte chemotactic protein 1 (MCP-1 Mm00441242_m1), and KC (mouse homologue of interleukin 8 which will be referred to as IL-8, Mm00433859_m1). mRNA expression levels were normalized for GAPDH expression and presented as fold increases over sham control analysed in parallel.
2.8. Statistical analysis
All values are expressed as mean ± SEM. Comparisons of day 2–7 data are by two-way ANOVA with Bonferroni post hoc tests, with the exception of qRT–PCR data which are by Kruskal–Wallis testing. Two-tailed unpaired Student's t-tests were used to compare infarct size and 28 day data. P-values < 0.05 denote statistical significance.
3. Results
3.1. Survival, infarct size, and plasma corticosterone after MI
Survival rate was 76% (63/83 total) in C57BL/6 and 78% (35/45 total) in 11βHSD1−/− mice, and in both groups, death was due to acute heart failure or rupture during early infarct healing. TTC staining showed that the extent of LV damage 24 h after MI was comparable in C57Bl6 (35.1 ± 1.0% of LV, n = 6) and 11βHSD1−/− (37.8 ± 2.4%, n = 6) mice. At this time, the plasma corticosterone concentration was 10-fold higher than basal levels.26 Plasma corticosterone declined by day 7 to approximately 75 nmol/L (Supplementary material online, Figure S1) in both C57BL/6 and 11βHSD1−/− mice.
3.2. Inflammatory cell recruitment during infarct healing
At 2 days after MI, there was significant neutrophil recruitment to the LV relative to sham operation (P < 0.01, Figure 1). In 11βHSD1−/− mice, the number of neutrophils in the LV at this time was significantly enhanced compared with C57BL/6 (P < 0.01). Thereafter, neutrophil content decreased to similar levels as the sham-operated controls in both groups (Figure 1). Early expression of the neutrophil chemo-attractants IL-6 and IL-8 did not increase significantly in 11βHSD1−/− compared with C57BL/6 mice (day 2 IL-6 expression 1.14 ± 0.01 relative to sham in C57BL/6 and 1.22 ± 0.02 relative to sham in 11βHSD1−/−; day 2 IL-8 expression 1.14 ± 0.01 relative to sham in C57BL/6 and 1.17 ± 0.02 relative to sham in 11βHSD1−/−, n = 6 per group).
Figure 1.
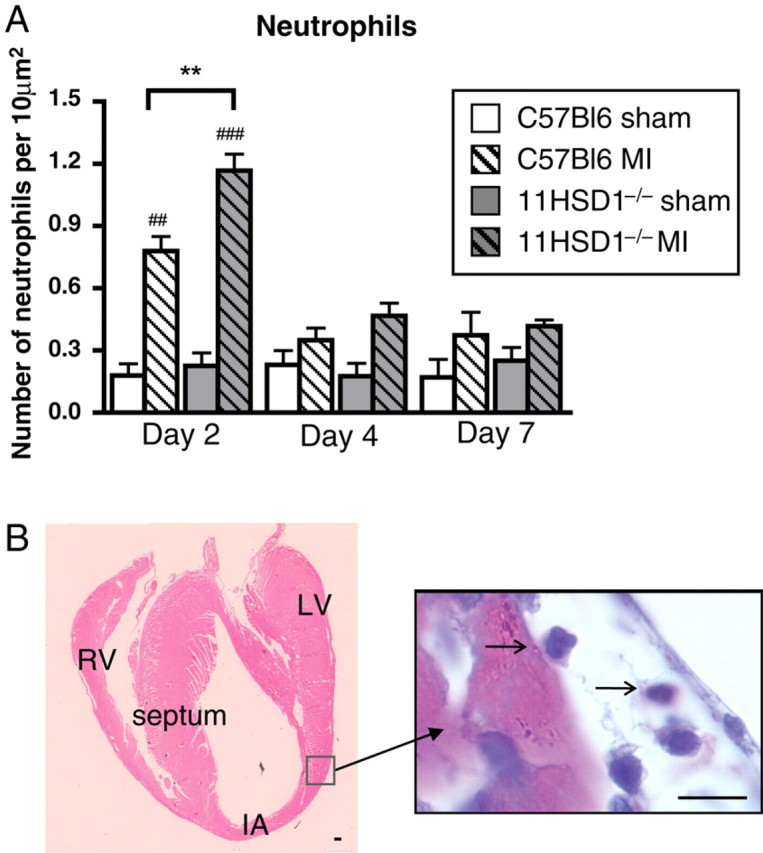
Neutrophil infiltration during infarct healing. (A and B) Neutrophils were identified in the left ventricular myocardium (LV) by their distinctive multi-lobed nuclei in haematoxylin and eosin-stained sections, counted and expressed as number per 10 μm2. (B) Tiled heart 2 days post-infarction showing neutrophil invading infarct border. IA, infarct area; RV, right ventricle. Arrows point to neutrophils. n = 8, C57BL/6 sham; n = 12, C57BL/6 MI; n = 4, 11βHSD1−/− sham; n = 6, 11βHSD1−/− MI. ##P < 0.01, ###P < 0.001 (versus matched sham), **P < 0.01 (C57BL/6 versus 11βHSD1−/−). Scale bar, 10 μm.
Mac 2 immunoreactive macrophages were identified predominantly in the infarct and border zone, becoming evident from 2 days after MI (Figure 2D and E). The extent of mac 2 immunoreactivity was further increased at 4 and 7 days post-MI relative to sham operation (P < 0.001, Figure 2A and E). In hearts from 11βHSD1−/− mice, macrophage accumulation tended to be increased relative to controls and this was significant by 7 days after MI (P < 0.01, Figure 2A). Flow cytometric analysis of LV homogenates also demonstrated increased content of CD11b+ve monocyte/macrophages in 11βHSD1−/− hearts at this time (data not shown). Expression of the macrophage chemoattractant MCP-1 mRNA was also greater in the LV of 11βHSD1−/− compared with control mice 7 days after MI (P < 0.01, Figure 2B). Specific immunostaining for the subset of pro-angiogenic and reparative ‘alternatively-activated’ macrophages, identified by YM1, revealed increased accumulation at 4 and 7 days post-MI relative to sham-operated mice, in which staining was negligible (Figure 2C and F). In 11βHSD1−/− mice, accumulation of YM1-positive macrophages was significantly greater than in C57BL/6 controls (P < 0.05) by 4 days after MI (Figure 2C). Detection of activated myofibroblasts using α-smooth muscle actin (αSMA) showed significant accumulation of immunopositive cells from 4 days after MI (Supplementary material online, Figure S2), consistent with scar maturation. However, there was no significant influence of genotype on αSMA immunoreactivity at either time point.
Figure 2.

Macrophage accumulation during infarct healing. (A, D, E) mac 2 immunohistochemistry was used to detect macrophage infiltration and was quantified as percent staining of the infarct border (IB). (B) Monocyte chemoattractant protein-1 (MCP-1) mRNA levels in heart tissue normalized to GAPDH housekeeping gene. (C, F) YM1-positive staining in heart tissue showing alternatively activated macrophages and quantified as percent staining of the infarct border. YM1 staining was absent from the hearts of mice that underwent sham operation (C). n = 8, C57BL/6 sham; n = 12, C57BL/6 MI; n = 4, 11βHSD1−/− sham; n = 6, 11βHSD1−/− MI; for RT–PCR n = 6 per group. ##P < 0.01, ###P < 0.001 (sham versus MI). *P < 0.05, **P < 0.01, ***P < 0.001 (C57BL/6 versus 11βHSD1−/−). Scale bar, 10 μm.
3.3. Post-infarct neovascularization and pro-angiogenic signalling
The density of small (<200 μm in diameter) CD31-positive blood vessels increased after MI, particularly on the infarct border (Figure 3A and C). Consistent with increased vessel formation, cell proliferation, identified by BrdU incorporation, was also increased at the same site (Figure 3B and C). By 7 days after MI, both vessel density (P < 0.05, Figure 3A) and cell proliferation (P < 0.01, Figure 3B and C) were significantly higher in 11βHSD1−/− than in C57BL/6 control hearts. This was accompanied by increased abundance of the pro-angiogenic signalling molecule IL-8 mRNA in the LV of 11βHSD1−/− hearts (P < 0.01, Figure 3D). Expression of VEGFα mRNA was not increased after MI in either C57BL/6 or 11βHSD1−/− hearts at this time (Figure 3D) or earlier during infarct healing (e.g. VEGF expression was 1.02 ± 0.09 and 1.00 ± 0.05 compared with sham in C57BL/6 at 2 and 4 days after MI; 1.02 ± 0.03 and 0.98 ± 0.02 compared with sham in 11βHSD1−/− hearts at 2 and 4 days after MI, n = 6 per group).
Figure 3.

Vessel density and cell proliferation during infarct healing. (A) CD31-positive vessels <200 μm in diameter in the LV, expressed per 400 μm2. (B) Nuclei positive for BrdU incorporation in the LV, expressed per mm2. (C) Representative sections of infarct border at 7 days after infarction, arrows point to CD31-positive vessels. (D) Interleukin 8 (IL-8) and vascular endothelial growth factor α (VEGFα) mRNA expression levels in heart tissue normalized to GAPDH housekeeping gene at 7 days after MI. n = 8, C57BL/6 sham; n = 12, C57BL/6 MI; n = 4, 11βHSD1−/− sham; n = 6, 11βHSD1−/− MI; for RT–PCR n = 6 per group. #P < 0.05, ###P < 0.001 (sham versus MI). *P < 0.05, **P < 0.01 (C57BL/6 versus 11βHSD1−/−). Scale bar, 10 μm.
3.4. Cardiac function during early infarct healing
There were no differences in basal cardiac function between un-operated 11βHSD1−/− and C57BL/6 mice (Supplementary material online, Table S1). After sham operation, ejection fraction (EF) was consistently above 60% in both C57BL/6 and 11βHSD1−/− mice, and was not different between these two groups (see Supplementary material online, Table S1). After MI, EF was significantly reduced in all mice, compared with sham operation (e.g. at 7 days after surgery EF was 32 ± 4% in MI compared with 64 ± 5% in sham-operated C57BL/6 mice, P < 0.01). EF was similarly depressed in both C57BL/6 and 11βHSD1−/− at 2 and 4 days after MI (Figure 4). However, by 7 days after MI, EF was significantly improved in 11βHSD1−/− compared with control mice (P < 0.05, Figure 4).
Figure 4.

Heart function during and after infarct healing. Ejection fraction was calculated from the LV end-diastolic area (LVEDA) and LV end-systolic area (LVESA) and expressed as a percentage. Data are presented as mean ± SEM, lighter columns C57BL/6, darker columns 11βHSD1−/−. (for C57BL/6, n = 12 MI during early healing at 2, 4, and 7 days after MI, n = 10 for MI at day 28; for 11βHSD1−/− n = 6 MI day 2, 4, and 7, and n = 9 for MI day 28). *P < 0.05, **P < 0.01 C57BL/6 versus 11βHSD1−/−.
3.5. Post-infarct healing characterization
Investigation of cardiac function at 28 days after MI, when infarct healing was largely complete, revealed that the improvement in function in 11βHSD1−/− mice apparent at 7 days after MI was retained (P < 0.01, Figure 4). Dysfunction post-MI was not associated with chamber dilatation at this point, as LV end-diastolic area remained consistent throughout the post-operative period in all mice (data not shown).
In hearts collected 28 days after MI, the density of small CD31-positive vessels also remained higher in 11βHSD1−/− relative to C57BL/6 hearts (P < 0.05, Figure 5A). Furthermore, the number of αSMA immunopositive vessels was also increased in 11βHSD1−/− hearts (P < 0.001, Figure 5B), suggesting that some of the new vessels had matured and become pericyte coated (Figure 5C). The extent of fibrosis and scar area, evaluated by Picrosirius Red and Masson's Trichrome staining, were comparable between control and 11βHSD1−/− mice 28 days post-MI (Figure 6A and B). However, 11βHSD1−/− mice had significantly thicker infarcts than C57BL/6 mice (P < 0.001, Figure 6C and E) and the scars had a tendency to be shorter (Figure 6D).
Figure 5.
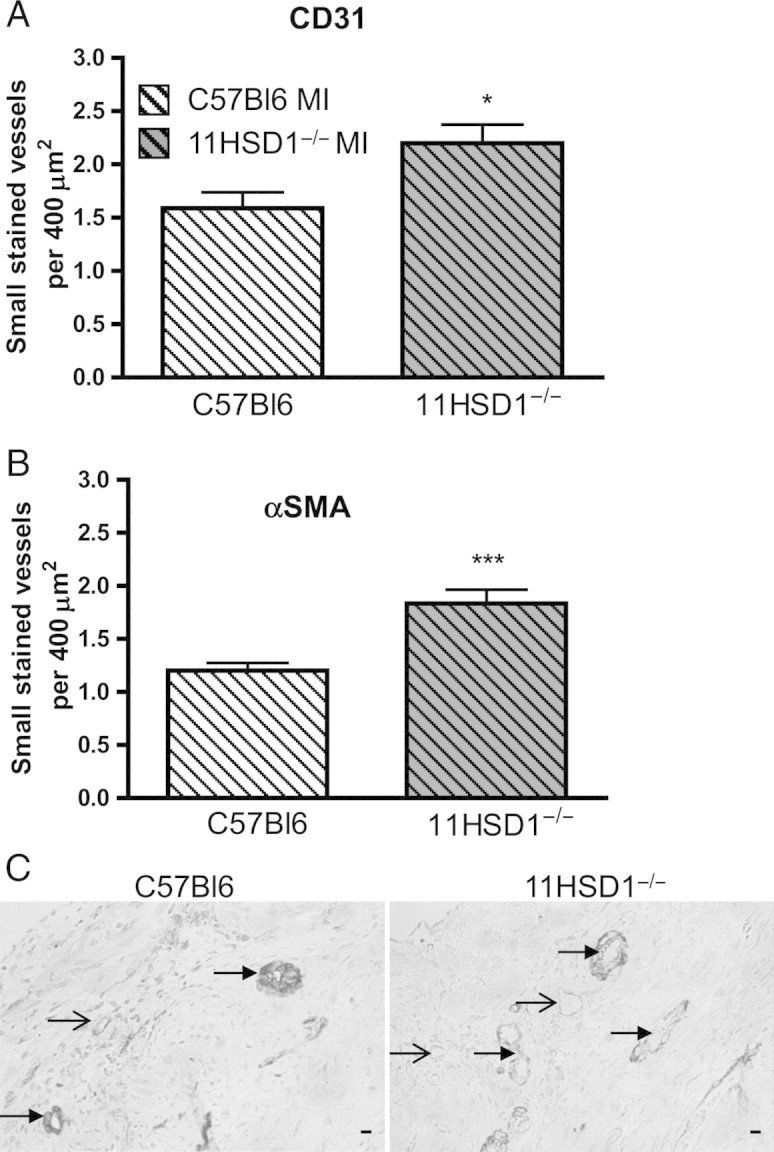
Blood vessel density and pericyte coverage 28 days after MI. (A) CD31 and (B) α smooth muscle actin (αSMA smooth muscle cells) positive vessels <200 μm in diameter counted in sequential sections from the LV, expressed per 400 μm2. (C) Representative sections showing typical double immuno-staining for CD31 (brown) and αSMA (blue) on the infarct border of C57BL/6 (C57BL6) and 11βHSD1−/− (11HSD1−/−) hearts . Filled arrows point to pericyte, smooth muscle coated vessels, open arrows point to pericyte poor, smooth muscle negative vessels. n = 10, C57BL/6 MI; n = 9, 11βHSD1−/− MI. *P < 0.05, ***P < 0.001. Scale bar, 10 μm.
Figure 6.

Fibrosis and scar formation 28 days after MI. (A) Collagen deposition measured from Picrosirius Red stained sections and expressed as percent staining of the LV in C57BL/6 (C57BL6, light columns) and 11βHSD1−/− (11HSD1−/−, dark columns) hearts . (B–E) Scar dimensions and infarct lengths were assessed from Masson's Trichrome stained sections (B). (C) Scar area expressed as the percentage of the LV. (D) Scar thickness was averaged from three points taken across the scar. (E) Epicardial infarct length expressed as a percentage of the epicardial LV length. n = 10, C57BL/6 MI; n = 9, 11βHSD1−/− MI for Picrosirius Red and Masson's Trichrome staining. ***P < 0.001 (C57BL/6 versus 11βHSD1−/−). Scale bar, 10 μm.
4. Discussion
We have previously shown that deficiency of 11βHSD1 is associated with increased vessel formation in experimental models of angiogenesis in vitro and in vivo, including in the healing myocardium of mice that have undergone MI.13 In the current study, we aimed to extend these observations by investigating whether modification of inflammation during infarct healing might provide a stimulus for increased vascularization in 11βHSD1−/− mice. We additionally aimed to investigate whether these acute vascular changes translated into sustained functional improvement. The data demonstrate that increased neovascularization during infarct healing in 11βHSD1−/− mice follows increased accumulation of neutrophils and of alternatively activated macrophages, and occurs in parallel with increased expression of the pro-angiogenic chemokine, IL-8. Furthermore, enhanced blood vessel density is retained at 28 days post-MI in 11βHSD1−/− mice, by which time vessels on the infarct border have matured, scar thinning is reduced, and cardiac function is improved.
Neovascularization on the infarct border typically begins during the reparative phase of healing.30,31 In the present study, the number of CD31-positive blood vessels in the LV increases during this period between 4 and 7 days after MI. Increased incorporation of BrdU into cells on the infarct border over the same period is consistent with the endothelial cell proliferation that is known to contribute to new blood vessel formation.27 In mice deficient in 11βHSD1, vessel density is higher during infarct healing, in agreement with our previous observations,13 and cellular proliferation is also significantly increased. Glucocorticoids are known to suppress endothelial cell proliferation.32 Promotion of angiogenesis in the hearts of 11βHSD1−/− mice is consistent with lifting of this suppression in mice unable to regenerate glucocorticoid, permitting enhanced proliferation. The primary stimulus for neovascularization after MI is from pro-angiogenic cytokines and chemokines released by neighbouring cells in the infarct. IL-8 is secreted by macrophages and endothelial cells in the healing infarcts, acts as a chemoattractant for bone marrow-derived endothelial progenitor cells, and can promote endothelial cell proliferation.33,34 In the present study, we find that expression of IL-8 is significantly increased in the LV of 11βHSD1−/− mice, suggesting that mechanisms other than direct regulation of cellular proliferation may contribute to increased neovascularization in the absence of 11βHSD1 activity.
Monocyte/macrophages have a key role in regulation of injury-associated angiogenesis.15,35 When macrophage accumulation in the infarct is prevented, following depletion by liposome-encapsulated clodronate, angiogenesis is abolished.15,16,36 Glucocorticoids downregulate inflammatory cytokines, upregulate anti-inflammatory cytokines, and modulate phagocytosis by macrophages.23,24,37 Macrophages express 11βHSD1 and its expression is upregulated after activation,37,38 which may serve to curtail the inflammatory response in the healing myocardial infarct. In support of this hypothesis, investigation of hearts from 11βHSD1−/− mice after MI reveals that accumulation of both neutrophils and macrophages is magnified in comparison to hearts from C57BL/6 mice. Infarct size is a key stimulus for inflammatory cell infiltration after MI,39,40 but deficiency of 11βHSD1 has no effect on ischaemia-associated damage to the myocardium. Systemic corticosterone, released from the adrenal gland in response to surgical stress and MI, is another potential modulator of myocardial inflammation. However, the 11βHSD1−/− mice have comparable basal systemic corticosterone and response to MI as the wild-type mice; this is consistent with control of circulating corticosterone by ACTH being independent of peripheral 11βHSD activity, and with previous reports of normal plasma corticosterone in 11βHSD1−/− mice on a C57BL/6 background.41 If anything, plasma corticosterone levels tended to be a little higher in 11βHSD1−/− mice, which also occurs with 11βHSD1 deletion on a mixed genetic background42 and would be anticipated to oppose the beneficial effects on outcome from MI observed here. This emphasizes that local intracellular changes in glucocorticoid generation in the heart, or elsewhere, are more likely to be regulating the inflammatory response post-MI.
Neutrophils are attracted to the heart very soon after infarction and contribute to cell removal during the early inflammatory phase of infarct healing as well as releasing cytokines e.g. IL-4 that regulate the later reparative phase. Enhanced neutrophil accumulation in 11βHSD1 hearts may therefore impact on both of these phases of infarct repair. The mechanism of increased neutrophil accumulation in 11βHSD1−/− hearts is not clear. Recruitment is regulated by IL-6 and IL-8,40 but there is no difference in expression of these cytokines in 11βHSD1−/− compared with C57BL/6 hearts, at least at 2-day post-MI mice. This may indicate that early cytokine expression is not a mechanism for enhanced neutrophil recruitment in 11βHSD1−/− mice. However, we cannot rule out the possibility that acute differences in expression may have been masked by the changes in cytokine expression that are associated with acute surgical trauma, as previously described.43 Mice deficient in glucocorticoids following adrenalectomy have increased expression of the adhesion molecules that have a role in tethering neutrophils to the endothelium prior to translocation into inflamed tissue.44 Reduced availability of glucocorticoids in the 11βHSD1−/− mice may therefore provide a similar stimulus for enhanced retention of neutrophils post-MI. Once in the infarct, neutrophils undergo apoptosis and are removed by the phagocytic activity of macrophages.45 An alternative explanation for our findings is that neutrophils undergo less or delayed apoptosis in 11βHSD1−/− mice. There is, however, no evidence for delayed neutrophil removal in 11βHSD1−/− compared with C57Bl/6 mice. Furthermore, this mechanism appears unlikely as glucocorticoids tend to inhibit neutrophil apoptosis46 and reduced local glucocorticoid availability would therefore be expected to enhance, rather than reduce neutrophil apoptosis.
Monocytes are attracted to the infarct by monocyte chemotactic protein-1 (MCP-1), also secreted by macrophages.17,47 In the present study, expression of MCP-1 mRNA is increased in 11βHSD1−/− hearts in parallel with increased macrophage accumulation. Nahrendorf et al.15 have suggested that alternatively activated, pro-angiogenic monocytes present during the reparative phase of healing play a vital role in infarct healing. Unlike classically activated macrophages that secrete pro-inflammatory mediators and display phagocytic behaviour, alternatively activated macrophages secrete anti-inflammatory and angiogenic cytokines such as TGFβ, IL-4, and IL-8 and can be identified by secretion of the chitinase-like molecule YM1.48,49 Data presented here shows for the first time that YM1-positive macrophages are indeed present in healing myocardial infarcts. Furthermore, immunostaining revealed the presence of a greater proportion of YM1-positive, alternatively activated macrophages in the infarct border of 11βHSD1−/− mice during the reparative phase of healing. These macrophages are the likely source of IL-8, expression of which is increased at the time of angiogenesis in the 11βHSD1−/− mice. Further studies are required to elucidate the mechanism for preferential assumption of the pro-reparative phenotype in 11βHSD1−/− mice and to investigate its importance in determining the increased vessel density in these mice.
It is clear from many studies that enhancement of angiogenesis on the infarct border post-MI improves heart function.1–3,28,34,50 In the present study, we show that cardiac function in 11βHSD1−/− mice is similar to that in C57BL/6 mice early after infarction, but by 7 days post-MI, at the time when increased vessel density is clear, ejection fraction is also enhanced. Increased vessel density early after infarction therefore appears to be of benefit, potentially by increasing blood supply to cardiomyocytes on the infarct border, but if this is to remain it is important that the early capillaries mature so that blood supply is maintained. As the scar heals, the early vessels are either pruned or they mature by gaining pericyte coverage.30,31 In hearts of 11βHSD1−/− mice, we show that increased vessel density is retained at 28 days post-MI, by which time many of the vessels have become smooth muscle coated. Correspondingly, ejection fraction also remained higher in 11βHSD1−/− compared with control mice at this time. By 28 days after MI, the infarcted myocardium was replaced by a collagen rich scar. Assessment of the scar structure revealed that although the overall scar area was similar, scars from 11βHSD1−/− mice were thicker and tended to be shorter than those from control mice. Failure to show a significant reduction in scar length is a limitation of the study, likely because of insufficient mice to account for the variability in this parameter. It is possible that enhancement of blood supply to the infarct border resulted in salvage of cardiomyocytes, as has previously been reported,50 reducing infarct expansion and contributing to increased cardiac contractility. The chitinase-like activity of YM1, present to a greater extent in 11βHSD1−/− hearts, can aid in matrix reorganization and wound healing,48,51 and this may also have contributed to the reduction in infarct thinning observed in these mice. Macrophages have an important role in scar formation by enhancing fibrosis.16 Macrophage secretion of transforming growth factor-β can activate myofibroblasts subsequently leading to collagen production.52,53 However, the data presented here indicate that despite greater macrophage accumulation in 11βHSD1−/− mice, myofibroblast activation and fibrosis were not increased relative to controls.
In summary, the present results support the hypothesis that inflammatory cell recruitment after MI is modified in mice deficient in 11βHSD1 and that this provides an enhanced stimulus for angiogenesis in the healing infarcts of these mice. Furthermore, increased vessel density is associated with reduction of infarct thinning and sustained functional improvement after MI. Glucocorticoids can activate both GR and mineralocorticoid receptor (MR). Blockade of MR soon after MI resulted in improvement in cardiac function in the EPHESUS clinical trial 54 and in an experimental model of MI.36 It may therefore be prevention of MR activation by locally generated corticosterone that accounts for the present observations in 11βHSD1−/− mice, and this requires confirmation in further studies. While the MR antagonist Eplerenone is currently being used clinically post-MI, it can lead to hyperkalaemia.54 Inhibitors of 11βHSD1 are in phase II trials for treatment of diabetes and other data suggest that they will prove beneficial in obesity, and atherosclerosis.55 The present data suggest that they may also provide an alternative approach for regulation of corticosteroid activity after MI.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Funding
This work was supported by The Hypertension Trust.
Supplementary Material
Acknowledgements
We thank Jonathan R. Seckl and John J. Mullins for the provision of the 11βHSD1−/− mice and Hector Scott and Susan Harvey for their assistance with histology and immunohistochemistry. We acknowledge the support of the British Heart Foundation Centre of Research Excellence.
Conflicts of interest: none declared.
References
- 1.Engel FB, Hsieh PC, Lee RT, Keating MT. FGF1/p38 MAP kinase inhibitor therapy induces cardiomyocyte mitosis, reduces scarring, and rescues function after myocardial infarction. Proc Natl Acad Sci USA. 2006;103:15546–15551. doi: 10.1073/pnas.0607382103. doi:10.1073/pnas.0607382103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sasaki T, Fukazawa R, Ogawa S, Kanno S, Nitta T, Ochi M, et al. Stromal cell-derived factor-1alpha improves infarcted heart function through angiogenesis in mice. Pediatr Int. 2007;49:966–971. doi: 10.1111/j.1442-200X.2007.02491.x. doi:10.1111/j.1442-200X.2007.02491.x. [DOI] [PubMed] [Google Scholar]
- 3.Orlic D, Kajstura J, Chimenti S, Limana F, Jakoniuk I, Quaini F, et al. Mobilized bone marrow cells repair the infarcted heart, improving function and survival. Proc Natl Acad Sci USA. 2001;98:10344–10349. doi: 10.1073/pnas.181177898. doi:10.1073/pnas.181177898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lasala GP, Minguell JJ. Bone marrow-derived stem/progenitor cells: their use in clinical studies for the treatment of myocardial infarction. Heart Lung Circ. 2009;18:171–180. doi: 10.1016/j.hlc.2008.09.007. doi:10.1016/j.hlc.2008.09.007. [DOI] [PubMed] [Google Scholar]
- 5.Meyer GP, Wollert KC, Lotz J, Steffens J, Lippolt P, Fichtner S, et al. Intracoronary bone marrow cell transfer after myocardial infarction: eighteen months' follow-up data from the randomized, controlled BOOST (BOne marrOw transfer to enhance ST-elevation infarct regeneration) trial. Circulation. 2006;113:1287–1294. doi: 10.1161/CIRCULATIONAHA.105.575118. doi:10.1161/CIRCULATIONAHA.105.575118. [DOI] [PubMed] [Google Scholar]
- 6.Seckl JR, Walker BR. Minireview: 11beta-hydroxysteroid dehydrogenase type 1- a tissue-specific amplifier of glucocorticoid action. Endocrinology. 2001;142:1371–1376. doi: 10.1210/endo.142.4.8114. doi:10.1210/en.142.4.1371. [DOI] [PubMed] [Google Scholar]
- 7.Hadoke PW, Iqbal J, Walker BR. Therapeutic manipulation of glucocorticoid metabolism in cardiovascular disease. Br J Pharmacol. 2009;156:689–712. doi: 10.1111/j.1476-5381.2008.00047.x. doi:10.1111/j.1476-5381.2008.00047.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wamil M, Seckl JR. Inhibition of 11beta-hydroxysteroid dehydrogenase type 1 as a promising therapeutic target. Drug Discov Today. 2007;12:504–520. doi: 10.1016/j.drudis.2007.06.001. doi:10.1016/j.drudis.2007.06.001. [DOI] [PubMed] [Google Scholar]
- 9.Mazancova K, Kopecky M, Miksik I, Pacha J. 11beta-Hydroxysteroid dehydrogenase in the heart of normotensive and hypertensive rats. J Steroid Biochem Mol Biol. 2005;94:273–277. doi: 10.1016/j.jsbmb.2005.02.003. doi:10.1016/j.jsbmb.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 10.Slight SH, Ganjam VK, Gomez-Sanchez CE, Zhou MY, Weber KT. High affinity NAD(+)-dependent 11 beta-hydroxysteroid dehydrogenase in the human heart. J Mol Cell Cardiol. 1996;28:781–787. doi: 10.1006/jmcc.1996.0072. doi:10.1006/jmcc.1996.0072. [DOI] [PubMed] [Google Scholar]
- 11.Hadoke PW, Christy C, Kotelevtsev YV, Williams BC, Kenyon CJ, Seckl JR, et al. Endothelial cell dysfunction in mice after transgenic knockout of type 2, but not type 1, 11beta-hydroxysteroid dehydrogenase. Circulation. 2001;104:2832–2837. doi: 10.1161/hc4801.100077. doi:10.1161/hc4801.100077. [DOI] [PubMed] [Google Scholar]
- 12.Christy C, Hadoke PW, Paterson JM, Mullins JJ, Seckl JR, Walker BR. 11beta-hydroxysteroid dehydrogenase type 2 in mouse aorta: localization and influence on response to glucocorticoids. Hypertension. 2003;42:580–587. doi: 10.1161/01.HYP.0000088855.06598.5B. doi:10.1161/01.HYP.0000088855.06598.5B. [DOI] [PubMed] [Google Scholar]
- 13.Small GR, Hadoke PW, Sharif I, Dover AR, Armour D, Kenyon CJ, et al. Preventing local regeneration of glucocorticoids by 11beta-hydroxysteroid dehydrogenase type 1 enhances angiogenesis. Proc Natl Acad Sci USA. 2005;102:12165–12170. doi: 10.1073/pnas.0500641102. doi:10.1073/pnas.0500641102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sunderkotter C, Nikolic T, Dillon MJ, Van Rooijen N, Stehling M, Drevets DA, et al. Subpopulations of mouse blood monocytes differ in maturation stage and inflammatory response. J Immunol. 2004;172:4410–4417. doi: 10.4049/jimmunol.172.7.4410. [DOI] [PubMed] [Google Scholar]
- 15.Nahrendorf M, Swirski FK, Aikawa E, Stangenberg L, Wurdinger T, Figueiredo JL, et al. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J Exp Med. 2007;204:3037–3047. doi: 10.1084/jem.20070885. doi:10.1084/jem.20070885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.van Amerongen MJ, Harmsen MC, van Rooijen N, Petersen AH, van Luyn MJ. Macrophage depletion impairs wound healing and increases left ventricular remodeling after myocardial injury in mice. Am J Pathol. 2007;170:818–829. doi: 10.2353/ajpath.2007.060547. doi:10.2353/ajpath.2007.060547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Morimoto H, Takahashi M, Izawa A, Ise H, Hongo M, Kolattukudy PE, et al. Cardiac overexpression of monocyte chemoattractant protein-1 in transgenic mice prevents cardiac dysfunction and remodeling after myocardial infarction. Circ Res. 2006;99:891–899. doi: 10.1161/01.RES.0000246113.82111.2d. doi:10.1161/01.RES.0000246113.82111.2d. [DOI] [PubMed] [Google Scholar]
- 18.Leor J, Rozen L, Zuloff-Shani A, Feinberg MS, Amsalem Y, Barbash IM, et al. Ex vivo activated human macrophages improve healing, remodeling, and function of the infarcted heart. Circulation. 2006;114:I94–I100. doi: 10.1161/CIRCULATIONAHA.105.000331. doi:10.1161/CIRCULATIONAHA.105.000331. [DOI] [PubMed] [Google Scholar]
- 19.Hafezi-Moghadam A, Simoncini T, Yang Z, Limbourg FP, Plumier JC, Rebsamen MC, et al. Acute cardiovascular protective effects of corticosteroids are mediated by non-transcriptional activation of endothelial nitric oxide synthase. Nat Med. 2002;8:473–479. doi: 10.1038/nm0502-473. doi:10.1038/nm0502-473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Morrison J, Reduto L, Pizzarello R, Geller K, Maley T, Gulotta S. Modification of myocardial injury in man by corticosteroid administration. Circulation. 1976;53:I200–I204. [PubMed] [Google Scholar]
- 21.Roberts RD, deMello V, Sobel BE. Deleterious effects of methylprednisolone in patients with myocardial infarction. Circulation. 1976;53:I204–I206. [PubMed] [Google Scholar]
- 22.Scheuer DA, Mifflin SW. Chronic corticosterone treatment increases myocardial infarct size in rats with ischemia-reperfusion injury. Am J Physiol. 1997;272:R2017–R2024. doi: 10.1152/ajpregu.1997.272.6.R2017. [DOI] [PubMed] [Google Scholar]
- 23.Cupps TR, Fauci AS. Corticosteroid-mediated immunoregulation in man. Immunol Rev. 1982;65:133–155. doi: 10.1111/j.1600-065x.1982.tb00431.x. doi:10.1111/j.1600-065X.1982.tb00431.x. [DOI] [PubMed] [Google Scholar]
- 24.Galon J, Franchimont D, Hiroi N, Frey G, Boettner A, Ehrhart-Bornstein M, et al. Gene profiling reveals unknown enhancing and suppressive actions of glucocorticoids on immune cells. FASEB J. 2002;16:61–71. doi: 10.1096/fj.01-0245com. doi:10.1096/fj.01-0245com. [DOI] [PubMed] [Google Scholar]
- 25.Chapman KE, Coutinho AE, Gray M, Gilmour JS, Savill JS, Seckl JR. The role and regulation of 11beta-hydroxysteroid dehydrogenase type 1 in the inflammatory response. Mol Cell Endocrinol. 2009;301:123–131. doi: 10.1016/j.mce.2008.09.031. doi:10.1016/j.mce.2008.09.031. [DOI] [PubMed] [Google Scholar]
- 26.Kotelevtsev Y, Holmes MC, Burchell A, Houston PM, Schmoll D, Jamieson P, et al. 11beta-hydroxysteroid dehydrogenase type 1 knockout mice show attenuated glucocorticoid-inducible responses and resist hyperglycemia on obesity or stress. Proc Natl Acad Sci USA. 1997;94:14924–14929. doi: 10.1073/pnas.94.26.14924. doi:10.1073/pnas.94.26.14924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Virag JI, Murry CE. Myofibroblast and endothelial cell proliferation during murine myocardial infarct repair. Am J Pathol. 2003;163:2433–2440. doi: 10.1016/S0002-9440(10)63598-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kido M, Du L, Sullivan CC, Li X, Deutsch R, Jamieson SW, et al. Hypoxia-inducible factor 1-alpha reduces infarction and attenuates progression of cardiac dysfunction after myocardial infarction in the mouse. J Am Coll Cardiol. 2005;46:2116–2124. doi: 10.1016/j.jacc.2005.08.045. doi:10.1016/j.jacc.2005.08.045. [DOI] [PubMed] [Google Scholar]
- 29.Yau TM, Fung K, WEisel RD, Fujii T, Mickle DAG, Li R. Enhanced myocardial angiogenesis by gene transfer with transplanted cells. Circulation. 2001;104:I218–I222. doi: 10.1161/hc37t1.094896. doi:10.1161/hc37t1.094896. [DOI] [PubMed] [Google Scholar]
- 30.Ren G, Michael LH, Entman ML, Frangogiannis NG. Morphological characteristics of the microvasculature in healing myocardial infarcts. J Histochem Cytochem. 2002;50:71–79. doi: 10.1177/002215540205000108. [DOI] [PubMed] [Google Scholar]
- 31.Grass TM, Lurie DI, Coffin JD. Transitional angiogenesis and vascular remodeling during coronary angiogenesis in response to myocardial infarction. Acta Histochem. 2006;108:293–302. doi: 10.1016/j.acthis.2006.09.005. doi:10.1016/j.acthis.2006.09.005. [DOI] [PubMed] [Google Scholar]
- 32.Longenecker JP, Kilty LA, Johnson LK. Glucocorticoid inhibition of vascular smooth muscle cell proliferation: influence of homologous extracellular matrix and serum mitogens. J Cell Biol. 1984;98:534–540. doi: 10.1083/jcb.98.2.534. doi:10.1083/jcb.98.2.534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li A, Dubey S, Varney ML, Dave BJ, Singh RK. IL-8 directly enhanced endothelial cell survival, proliferation, and matrix metalloproteinases production and regulated angiogenesis. J Immunol. 2003;170:3369–3376. doi: 10.4049/jimmunol.170.6.3369. [DOI] [PubMed] [Google Scholar]
- 34.Kocher AA, Schuster MD, Bonaros N, Lietz K, Xiang G, Martens TP, et al. Myocardial homing and neovascularization by human bone marrow angioblasts is regulated by IL-8/Gro CXC chemokines. J Mol Cell Cardiol. 2006;40:455–464. doi: 10.1016/j.yjmcc.2005.11.013. doi:10.1016/j.yjmcc.2005.11.013. [DOI] [PubMed] [Google Scholar]
- 35.Sun J, Li SH, Liu SM, Wu J, Weisel RD, Zhuo YF, et al. Improvement in cardiac function after bone marrow cell therapy is associated with an increase in myocardial inflammation. Am J Physiol Heart Circ Physiol. 2009;296:H43–H50. doi: 10.1152/ajpheart.00613.2008. doi:10.1152/ajpheart.00613.2008. [DOI] [PubMed] [Google Scholar]
- 36.Fraccarollo D, Galuppo P, Schraut S, Kneitz S, van Rooijen N, Ertl G, et al. Immediate mineralocorticoid receptor blockade improves myocardial infarct healing by modulation of the inflammatory response. Hypertension. 2008;51:905–914. doi: 10.1161/HYPERTENSIONAHA.107.100941. doi:10.1161/HYPERTENSIONAHA.107.100941. [DOI] [PubMed] [Google Scholar]
- 37.Gilmour JS, Coutinho AE, Cailhier JF, Man TY, Clay M, Thomas G, et al. Local amplification of glucocorticoids by 11 beta-hydroxysteroid dehydrogenase type 1 promotes macrophage phagocytosis of apoptotic leukocytes. J Immunol. 2006;176:7605–7611. doi: 10.4049/jimmunol.176.12.7605. [DOI] [PubMed] [Google Scholar]
- 38.Thieringer R, Le Grand CB, Carbin L, Cai TQ, Wong B, Wright SD, et al. 11 Beta-hydroxysteroid dehydrogenase type 1 is induced in human monocytes upon differentiation to macrophages. J Immunol. 2001;167:30–35. doi: 10.4049/jimmunol.167.1.30. [DOI] [PubMed] [Google Scholar]
- 39.Frantz S, Bauersachs J, Ertl G. Post-infarct remodelling: contribution of wound healing and inflammation. Cardiovasc Res. 2009;81:474–481. doi: 10.1093/cvr/cvn292. doi:10.1093/cvr/cvn292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Frangogiannis NG, Smith CW, Entman ML. The inflammatory response in myocardial infarction. Cardiovasc Res. 2002;53:31–47. doi: 10.1016/s0008-6363(01)00434-5. doi:10.1016/S0008-6363(01)00434-5. [DOI] [PubMed] [Google Scholar]
- 41.Paterson JM, Holmes MC, Kenyon CJ, Carter R, Mullins JJ, Seckl JR. Liver-selective transgene rescue of hypothalamic-pituitary-adrenal axis dysfunction in 11beta-hydroxysteroid dehydrogenase type 1-deficient mice. Endocrinology. 2007;148:961–966. doi: 10.1210/en.2006-0603. doi:10.1210/en.2006-0603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Harris HJ, Kotelevtsev Y, Mullins JJ, Seckl JR, Holmes MC. Intracellular regeneration of glucocorticoids by 11beta-hydroxysteroid dehydrogenase (11beta-HSD)-1 plays a key role in regulation of the hypothalamic-pituitary-adrenal axis: analysis of 11beta-HSD-1-deficient mice. Endocrinology. 2001;142:114–120. doi: 10.1210/endo.142.1.7887. doi:10.1210/en.142.1.114. [DOI] [PubMed] [Google Scholar]
- 43.Nossuli TO, Lakshminarayanan V, Baumgarten G, Taffet GE, Ballantyne CM, Michael LH, et al. A chronic mouse model of myocardial ischemia-reperfusion: essential in cytokine studies. Am J Physiol Heart Circ Physiol. 2000;278:H1049–H1055. doi: 10.1152/ajpheart.2000.278.4.H1049. [DOI] [PubMed] [Google Scholar]
- 44.Cavalcanti DM, Lotufo CM, Borelli P, Ferreira ZS, Markus RP, Farsky SH. Endogenous glucocorticoids control neutrophil mobilization from bone marrow to blood and tissues in non-inflammatory conditions. Br J Pharmacol. 2007;152:1291–1300. doi: 10.1038/sj.bjp.0707512. doi:10.1038/sj.bjp.0707512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Savill JS, Henson PM, Haslett C. Phagocytosis of aged human neutrophils by macrophages is mediated by a novel “charge-sensitive" recognition mechanism. J Clin Invest. 1989;84:1518–1527. doi: 10.1172/JCI114328. doi:10.1172/JCI114328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ruiz LM, Bedoya G, Salazar J, Garcia de OD, Patino PJ. Dexamethasone inhibits apoptosis of human neutrophils induced by reactive oxygen species. Inflammation. 2002;26:215–222. doi: 10.1023/a:1019714618068. doi:10.1023/A:1019714618068. [DOI] [PubMed] [Google Scholar]
- 47.Kakio T, Matsumori A, Ono K, Ito H, Matsushima K, Sasayama S. Roles and relationship of macrophages and monocyte chemotactic and activating factor/monocyte chemoattractant protein-1 in the ischemic and reperfused rat heart. Lab Invest. 2000;80:1127–1136. doi: 10.1038/labinvest.3780119. [DOI] [PubMed] [Google Scholar]
- 48.Loke P, Nair MG, Parkinson J, Guiliano D, Blaxter M, Allen JE. IL-4 dependent alternatively-activated macrophages have a distinctive in vivo gene expression phenotype. BMC Immunol. 2002;3:7. doi: 10.1186/1471-2172-3-7. doi:10.1186/1471-2172-3-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008;8:958–969. doi: 10.1038/nri2448. doi:10.1038/nri2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu X, Huang Y, Pokreisz P, Vermeersch P, Marsboom G, Swinnen M, et al. Nitric oxide inhalation improves microvascular flow and decreases infarction size after myocardial ischemia and reperfusion. J Am Coll Cardiol. 2007;50:808–817. doi: 10.1016/j.jacc.2007.04.069. doi:10.1016/j.jacc.2007.04.069. [DOI] [PubMed] [Google Scholar]
- 51.Lambert JM, Lopez EF, Lindsey ML. Macrophage roles following myocardial infarction. Int J Cardiol. 2008;130:147–158. doi: 10.1016/j.ijcard.2008.04.059. doi:10.1016/j.ijcard.2008.04.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fadok VA, Bratton DL, Konowal A, Freed PW, Westcott JY, Henson PM. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. J Clin Invest. 1998;101:890–898. doi: 10.1172/JCI1112. doi:10.1172/JCI1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cleutjens JP, Verluyten MJ, Smiths JF, Daemen MJ. Collagen remodeling after myocardial infarction in the rat heart. Am J Pathol. 1995;147:325–338. [PMC free article] [PubMed] [Google Scholar]
- 54.Pitt B, White H, Nicolau J, Martinez F, Gheorghiade M, Aschermann M, et al. Eplerenone reduces mortality 30 days after randomization following acute myocardial infarction in patients with left ventricular systolic dysfunction and heart failure. J Am Coll Cardiol. 2005;46:425–431. doi: 10.1016/j.jacc.2005.04.038. doi:10.1016/j.jacc.2005.04.038. [DOI] [PubMed] [Google Scholar]
- 55.Hermanowski-Vosatka A, Balkovec JM, Cheng K, Chen HY, Hernandez M, Koo GC, et al. 11beta-HSD1 inhibition ameliorates metabolic syndrome and prevents progression of atherosclerosis in mice. J Exp Med. 2005;202:517–527. doi: 10.1084/jem.20050119. doi:10.1084/jem.20050119. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.