

Abstract
Nucleoside reverse transcriptase inhibitors (NRTIs), essential components of combinational therapies used for treatment of human immunodeficiency virus-1, damage heart mitochondria. Here, we have shown mitochondrial compromise in H9c2 rat cardiomyocytes exposed for 16 passages (P) to the NRTIs zidovudine (AZT, 50μM) and didanosine (ddI, 50μM), and we have demonstrated protection from mitochondrial compromise in cells treated with 200μM 1-oxyl-2,2,6,6-tetramethyl-4-hydroxypiperidine (Tempol) or 200μM 1-hydroxy-4-[2-triphenylphosphonio)-acetamido]-2,2,6,6-tetramethylpiperidine (Tempol-H), along with AZT/ddI, for 16P. Exposure to AZT/ddI caused a moderate growth inhibition at P3, P5, P7, and P13, which was not altered by addition of Tempol or Tempol-H. Mitochondrial oxidative phosphorylation capacity was determined as uncoupled oxygen consumption rate (OCR) by Seahorse XF24 Analyzer. At P5, P7, and P13, AZT/ddI-exposed cells showed an OCR reduction of 8.8–57.2% in AZT/ddI-exposed cells, compared with unexposed cells. Addition of Tempol or Tempol-H, along with AZT/ddI, resulted in OCR levels increased by about 300% above the values seen with AZT/ddI alone. The Seahorse data were further supported by electron microscopy (EM) studies in which P16 cells exposed to AZT/ddI/Tempol had less mitochondrial pathology than P16 cells exposed to AZT/ddI. Western blots of P5 cells showed that Tempol and Tempol-H upregulated expression of mitochondrial uncoupling protein-2 (UCP-2). However, Complex I activity that was reduced by AZT/ddI, was not restored in the presence of AZT/ddI/Tempol. Superoxide levels were increased in the presence of AZT/ddI and significantly decreased in cells exposed to AZT/3TC/Tempol at P3, P7, and P10. In conclusion, Tempol protects against NRTI-induced mitochondrial compromise, and UCP-2 plays a role through mild uncoupling.
Keywords: mitochondria, antiretroviral therapy, electron microscopy, oxidative phosphorylation, superoxide, Seahorse extracellular flux analyzer
Abbreviations
- ARV
antiretroviral
- ATP
adenosine-5′-triphosphate
- AZT
zidovudine, 3′-azido-3′-deoxythymidine
- CAM
chloramphenicol
- DCFH
dichlorofluorescin diacetate
- ddI
didanosine, 2′,3′-dideoxyinosine
- EM
electron microscopy
- FCCP
carbonyl cyanide p-trifluoromethoxy-phenylhydroazone
- HIV-1
human immunodeficiency virus-1
- H9c2
rat cardiomyocyte cell line
- mtDNA
mitochondrial DNA
- NRTI
nucleoside reverse transcriptase inhibitor
- OCR
oxygen consumption rate
- OLIGO
oligomycin
- OXPHOS
oxidative phosphorylation
- P
passage(s)
- ROS
reactive oxygen species
- Tempol
1-oxyl-2,2,6,6-tetramethyl-4-hydroxypiperidine
- Tempol-H
1-hydroxy-4-[2-triphenylphosphonio)-acetamido]-2,2,6,6-tetramethylpiperidine
- RFU
relative fluorescence units
- UCP-2
uncoupling protein-2
It has been established that long-term use of combination antiretroviral therapy containing nucleoside reverse transcriptase inhibitors (NRTIs), used as therapy for human immunodeficiency virus-1 (HIV-1) infection, causes mitochondrial compromise that can lead to treatment limitation (Cossarizza and Moyle, 2004; Koczor and Lewis, 2010; Lewis, 2005). Heart, skeletal muscle, liver, and brain are particularly at risk, as they are organs requiring high levels of energy for normal functioning (Cossarizza and Moyle, 2004; Koczor and Lewis, 2010; Lewis, 2005). NRTI-based combination therapies must be given continuously in order to control HIV-1 infection, to attenuate the progression of AIDS-associated symptoms, and to limit mother-to-child transmission of HIV-1 during pregnancy. However, long-term treatment greatly increases the incidence of drug-related toxicities. Because NRTIs are likely to remain the backbone of HIV-1 treatment for the foreseeable future, searching for potential options to minimize NRTI-induced mitochondrial toxicity becomes important.
NRTI-induced mitochondrial damage is due both to drug incorporation into host mitochondrial DNA (mtDNA), which terminates DNA replication (Anderson et al., 2004; Hoschele, 2006), and drug inhibition of the mtDNA polymerase-γ (Koczor and Lewis, 2010; Lewis, 2005; Lewis et al., 2003). Thirteen respiratory chain polypeptides, which are critical for oxidative phosphorylation (OXPHOS), are encoded by mtDNA. In addition to the essential production of adenosine-5′- triphosphate (ATP) for energy, mitochondria are major sites for generation of cellular reactive oxygen species (ROS), and are direct targets for ROS damage (Drose and Brandt, 2012; Sena and Chandel, 2012). Because oxidative stresses play a major role in mitochondrial compromise, therapeutic strategies that reduce ROS and stimulate mitochondrial biogenesis may be successful in mitochondrial protection.
1-Oxyl-2,2,6,6-tetramethyl-4-hydrox ypiperidine (Tempol) is a cyclic nitroxide, a stable free radical that permeates the biological membrane with unique antioxidant properties (Soule et al., 2007). Tempol possesses peroxidase activity, exhibits a superoxide dismutase mimetic action to degrade superoxide anions, and may inhibit the Fenton reaction. Preclinical studies, using a variety of mouse models, have shown the potential for many clinical applications, including protection against ionizing radiation and DNA damage, cancer prevention and treatment, control of hypertension and weight gain, and prevention of drug treatment-related cardiomyopathy (Bartha et al., 2011; Soule et al., 2007; Wilcox, 2010). 1-Hydroxy-4-[2-triphenylphosphonio)-acetamido]-2,2,6,6-tetramethylpiperidine (Tempol-H) is the hydroxylamine metabolite of Tempol, and as such has similar antioxidant and radiation protective effects, but shows less cytotoxicity in both in vitro and in vivo models. In this study, we tested the hypothesis that both Tempol and Tempol-H may protect cardiomyocytes against the toxic effects of long-term NRTI-exposures.
For these investigations, we have employed H9c2 rat cardiomyocyte cells exposed during 16 passages (P) to the NRTIs zidovudine (AZT) and didanosine (ddI). Mitochondrial status, in the form of oxygen consumption rate (OCR) and extracellular acidification rate (ECAR), were evaluated by Seahorse XF24. In addition, mitochondrial morphological integrity was ascertained by electron microscopy (EM), levels of various mitochondria-related proteins were determined by Western blots, and superoxides were measured by hydroethidine fluorescence. All of these endpoints were applied to explore the possibility that NRTI-induced mitochondrial toxicity may be attenuated when H9c2 cells are cotreated with AZT/ddI and Tempol or Tempol-H.
MATERIALS AND METHODS
Cell culture, reagents and exposures
The H9c2 cell line, originally derived from embryonic rat heart tissue using selective serial passaging, was purchased from the American Tissue Type Collection (ATCC, Manassas, VA). Cells were maintained in Dulbecco's Modified Eagle Medium (DMEM from ATCC) with 10% fetal bovine serum. AZT, ddI, oligomycin (OLIGO), carbonyl-cyanide-4-(trifluoromethoxy)-phenlyhydrazone (FCCP), and rotenone (ROT) were all purchased from Sigma-Aldrich (St. Louis, MO). Tempol was purchased from Sigma-Aldrich and the reduced hydroxylamine form of Tempol (Tempol-H) was prepared as previously described (Krishna et al., 1991).
For the studies performed here, the doses used were consistent for all experiments, and some experiments were performed with either Tempol or Tempol-H, but not both. AZT and ddI were both used at 50μM, and Tempol and Tempol-H were both used at 200μM.
Cell growth
H9c2 cell proliferation was determined using the IncuCyte Live-Cell Imaging System (Essen BioScience, Inc., Ann Arbor, MI). Equal numbers (20,000) of H9c2 cells from differently exposed cells were plated in quadruplicate wells of a 24-well culture plate (Corning Incorporated, Corning, NY). The kinetics of cell growth were monitored over 96 h using the IncuCyte integrated confluence algorithm, where confluence is a surrogate for cell number. The drug treatments did not alter the passage length, and cells were consistently split twice a week.
Measurements of OCR by Seahorse XF24 Analyzer
Equal numbers of H9c2 cells, cultured for up to 16P in 0 or 50μM AZT/50μM ddI, with or without 200μM Tempol or 200μM Tempol-H, were seeded in quadruplicate wells of XF24 V7 cell culture plates (Seahorse Bioscience, North Billerica, MA). After 24 h, the medium was replaced with XF Assay Medium (Seahorse Bioscience), supplied with 0.9% glucose, 1mM sodium pyruvate, and 2mM glutamine. After 1 h of incubation in a non-CO2 incubator, OCR was measured using the Seahorse Bioscience XF24 Extracellular Flux Analyzer (Seahorse Bioscience) where cells were subjected in sequence to the following additions: (1) basal levels were measured with no additives; (2) 1μM OLIGO, which reversibly inhibits ATP Synthase and OXPHOS, was added to show glycolysis alone; (3) 0.3μM FCCP, a mitochondrial uncoupler, was added to induce maximal respiration; (4) 0.1μM ROT, a Complex I inhibitor and mitochondrial poison, was added to end the reaction. Three separate measurements were taken after each of the above reagents was added. Triplicate or quadruplicate experimental wells were examined and the results plotted by Seahorse software. Cell treatments for the Seahorse studies at P5, P7, and P13 were performed twice on separate occasions.
Western blot analysis
The methods for cell lysate preparation, protein concentration determination, and electrophoresis have been described previously (Liu et al., 2008). The primary antibodies used were: antiuncoupling protein-2 (UCP-2, ORIGENE, Rockville, MD); Mitoprofile Total OXPHOS Rodent Western Blot Antibody Cocktail (including subunit NDUFB8 of Complex I, 30 kDa subunit of Complex II, protein-2 of Complex III, subunit I of Complex IV, and alpha subunit of Complex V; Abcam, Cambridge, MA); anti-COX4 (Cytochrome C Oxidase IV; Abcam); and antiactin (Novus Biologicals, Littleton, CO). Blots were developed using ECLTM Prime Western Blotting Reagents (GE Healthcare, Buckinghamshire, UK). The Western blots shown are representative of at least two or three independent experiments, and quantification was accomplished using ImageJ software.
Morphology of H9c2 cell mitochondria determined by EM
Mitochondrial morphology was assessed by EM in cells exposed for 16P to the following conditions: no drug, 200μM Tempol, 50μM AZT/50μM ddI, or 200μM Tempol plus 50μM AZT/50μM ddI. Cells were grown in six-well plates, and at approximately 75% confluence, were fixed with 2% glutaraldehyde in 0.1M cacodylate buffer for 1 h at room temperature. The samples were kept at 4°C and further processed for EM (SAIC Laboratory, NCI-Frederick, MD). Images were captured randomly in a “Z” pattern and there were 10 photomicrographs (×30,000 magnification)/treatment group. The photos were coded for scoring by three independent investigators. The degree of mitochondrial and cellular pathology was evaluated using a scoring paradigm of 0 to 5, where 0 represents undamaged mitochondria and 5 represents the highest level of damage (Divi et al., 2005).
Scoring of EM photomicrographs
Scoring data were obtained using a paradigm where 0 = perfect and 5 = the most highly damaged mitochondria, as follows: (0) nearly all mitochondria possessed an intact membrane, compact, well-defined cristae, and a dark matrix, whereas an infrequent mitochondrion showed less definitive cristae and a lighter matrix (that were considered to be within normal limits), and the rough endoplasmic reticulum (if visible) was uniform and compressed; (+1) the majority of mitochondria were intact, and although a few possessed discontinuous membranes, no loss of cristae material, matrical density, nor irregularity in endoplasmic reticulum or Golgi apparatus was apparent; (+2) an increasing number of mitochondria were dissolved with membrane disruptions but with minimal loss of cristae and matrical density, whereas most of the remaining mitochondria looked essentially normal but did display disruption in endoplasmic reticulum and Golgi apparatus; (+3) the majority of mitochondria possessed membrane disruptions, partial loss of cristae material, and elevated matrical lucency along with widespread, distention, and increased lucency surrounding the mirochondria; (+4) nearly all mitochondria possessed dissolved membranes and loss of cristae and matrical structure, along with swelling, disorganization, and increased lucency of the surrounding structures; (+5) in many areas the mitochondrial membrane was completely dissolved and the cristae were fragmented and disorganized, in other areas, mitochondria containing no central architecture were apparent, and all other areas looked like those graded (+4). Each data point represents mean ± SE for 10 coded photomicrographs/group scored by three investigators. p < 0.01 for control versus AZT/ddI, Tempol versus AZT/ddI, and AZT/ddI versus AZT/ddI/Tempol.
Mitochondrial isolation and determination of Complex I activity
H9c2 cells, grown in T-75 flasks (n = 4/treatment group), were exposed for 3P and 7P as follows: control, 200μM Tempol, 50μM AZT/50μM ddI, or 200μM Tempol plus 50μM AZT/50μM ddI. Mitochondria were isolated from the harvested cells using the Mitochondrial Isolation Kit for Cultured Cells from Abcam. For measurement of Complex I activity, the negative control consisted of mitochondria isolated from cells treated for 5 days with 40μM chloramphenicol (CAM), an inhibitor of mitochondrial protein synthesis. The Complex I Enzyme Activity Microplate Assay Kit (Abcam) was used to determine Complex I activity according to the manufacturer's protocol (Abcam). A Multi-Plate Reader (Tecan System Inc., San Jose, CA) was used to read the plate at a wavelength of 450 nm. Mitochondria from each treatment group were assayed in three wells, and the results were reported as mean absorbance ± SE. The experiment was repeated twice.
Measurement of superoxides
Hydroethidine fluorescence was used to analyze for the presence of superoxides (Zhao et al., 2003). Cells, grown in 96-well plates, were washed twice with PBS and stained for 30 min at 37°C with 10μM hydroethidine for the detection of superoxide. The cells were subsequently washed twice with PBS and fluorescence was measured using a TECAN Multi-Plate Reader (Tecan System Inc.). The wavelengths used were 510/590 nm. Each sample was assayed in at least triplicate wells from a single cell treatment, and the results were reported as mean absorbance ± SE (n = 8 wells). The experiments were performed twice at P1, P3, P7, and P10.
Statistical analysis
The Seahorse analyzes, EMs, Western blots, and superoxide data were all presented as mean ± SE. Statistical comparisons employed Student's t-test or one-way ANOVA.
RESULTS
Tempol and Tempol-H Did Not Alter Cell Growth
In a previous study, we showed that H9c2 cells exposed for 96 hr to 50μM AZT/50μM ddI had about a 25% inhibition of cell growth (Liu et al., 2012). Here, we repeated the AZT/ddI studies and also evaluated effects of Tempol and Tempol-H on cell growth. In P3 cells followed for 96 h, 50μM AZT/50μM ddI inhibited cell growth by 35%. The addition of Tempol or Tempol-H alone did not affect cell growth (Supplement fig. 1), and the addition of Tempol or Tempol-H, along with AZT/ddI, did not change cell growth, compared with cells exposed to AZT/ddI alone. Supplementary figure 1 shows data from P3, but similar results were obtained in cell growth experiments at P5, P7, and P13 (data not shown).
OCR Measured by Seahorse in H9c2 Cardiomyocytes
Previously, using the Seahorse, we showed mitochondrial compromise in H9c2 cells exposed to 50μM AZT/50μM ddI for 39P (Liu et al., 2012). In these experiments, using the same doses of AZT/ddI, OCR was measured several times at passages between 1 and 16, with similar results. A representative Seahorse experiment is shown in Figure 1A, where cells at P5 were evaluated for basal activity (1–18 min), glycolysis alone (18–44 min), maximal mitochondrial uncoupling (44–79 min), and complete mitochondrial impairment (79–96 min).
FIG. 1.
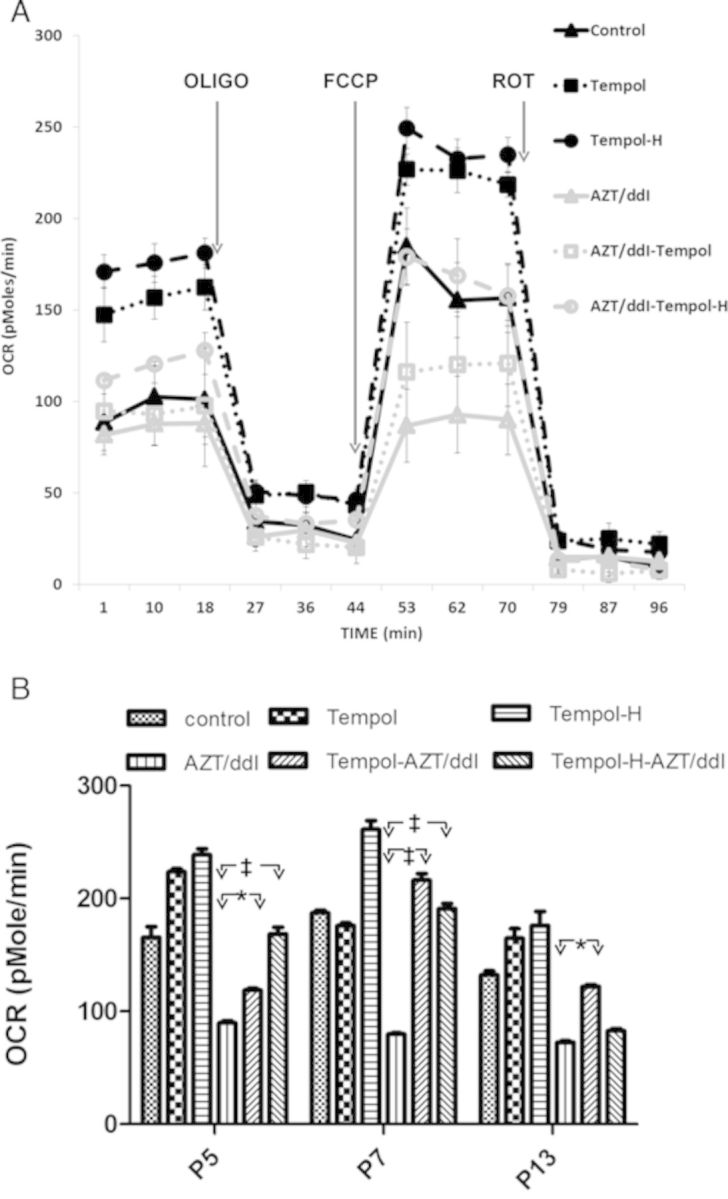
Tempol and Tempol-H (each at 200μM) stimulated OCR, and restored the reduction in OCR observed in cells grown in 50μM AZT plus 50μM ddI. (A) Representative Seahorse profile for cells at P5 (3–4 wells/sample) were incubated sequentially with: (1) no additives, to observe basal levels (1–20 min); (2) OLIGO, to visualize glycolysis (21–44 min); (3) FCCP, to observe maximal or uncoupled capacity (24–79 min); and (4) ROT, to end the reaction (80–96 min). (B) The average maximal uncoupled OCR values at P5, P7, and P13 for one experiment in which each data point represents mean ± SE, n = 9–12 wells. The data show that OCR values for unexposed controls were significantly (p < 0.01) higher than for cells grown in AZT/ddI alone at P5, P7, and P13. OCR values for AZT/ddI alone were significantly lower than values for AZT/ddI/Tempol or AZT/ddI/Tempol-H, at P5 and P7 (p < 0.05), and significantly lower than for AZT/ddI/Tempol (p < 0.01) but not AZT/ddI-Tempol-H, at P13. The data also show that Tempol and Tempol-H, given with no NRTI, increased OCR at P5 and P13 (p < 0.05), but only Tempol-H increased OCR significantly at P7 (*p < 0.01; ‡p < 0.05).
Figure 1B shows one of two experiments in which maximal uncoupled OCR values are presented at P5, P7, and P13, in cells exposed to no drug, Tempol, Tempol-H, AZT/ddI, AZT/ddI/Tempol, and AZT/ddI/Tempol-H. The figure shows that AZT/ddI alone significantly inhibited maximal uncoupled OCR at P5, P7, and P13 (p < 0.01), when compared with unexposed control cells. Figure 1B also shows that cells exposed to AZT/ddI/Tempol and AZT/ddI/Tempol-H had significantly higher OCR (p < 0.05) at P5 and P7, than cells exposed to AZT/ddI. At P13, cells exposed to AZT/ddI/Tempol had significantly higher OCR levels than cells exposed to AZT/ddI (p < 0.05), but cells exposed to AZT/ddI/Tempol-H did not show a similar significant increase.
Values for % inhibition and % recovery in the replicate experiments at P5, P7, and P13 are shown in Table 1. With AZT/ddI alone the maximal OCR reduction, compared with unexposed cells, was 8.8–57.2%. Table 1 also shows that the maximal OCR % recovery, in the presence of Tempol, was between 109–272%, and in the presence of Tempol-H it was 129–326%. Because these cells were at different passages, and because cell growth may have been different among the experiments, there is some inherent variability in the Seahorse data. However, the available data show that addition of Tempol and Tempol-H almost always reversed the inhibition of OCR seen in AZT/ddI-exposed cells.
TABLE 1.
AZT/ddI-Induced Inhibition of Maximal OCR (% Inhibition) Determined by Seahorse, and OCR Recovery and % Recovery in Cells Exposed to AZT/ddI/Tempol or AZT/ddI/Tempol-H, at P5, P7, and P13
| Passage | AZT/ddI | AZT/ddI/Tempol | AZT/ddI/Tempol-H | |||
|---|---|---|---|---|---|---|
| Inhibition | % Inhibition | Recovery | % Recovery | Recovery | % Recovery | |
| 5 (1)a | 166 versus 90 | 45.80 | 90 versus 119 | 132 | 90 versus 169 | 188 |
| 5 (2) | 171 versus 156 | 8.80 | 156 versus 198 | 127 | NRb | NRb |
| 7 (1) | 187 versus 80 | 57.20 | 80 versus 216 | 272 | 80 versus 261 | 326 |
| 7 (2) | 165 versus 84 | 49.40 | 84 versus 131 | 156 | 84 versus 145 | 173 |
| 13 (1) | 210 versus 188 | 10.50 | 188 versus 205 | 109 | 188 versus 242 | 129 |
| 13 (2) | 132 versus 73 | 44.70 | 73 versus 128 | 175 | 73 versus 196 | 268 |
a(1) and (2) refer to different experiments performed with separate batches of cells.
bNR, no recovery.
Tempol Protected Mitochondria from Morphological Damage
In these experiments, we used EM to examine the morphological integrity of H9c2 cell mitochondria at P16, and the results are shown in Figure 2. Figure 2A (top left) shows normal mitochondria in the unexposed controls. In contrast, in cells exposed to AZT/ddI (Fig. 2A, bottom left), there were multiple manifestations of mitochondrial pathology including: mitochondrial swelling, membrane breaks, loss of cristae, and increased lucency of the surrounding structures. In cells treated with Tempol alone (Fig. 2A, top right), there was minor loss of mitochondrial integrity, and in cells treated with AZT/ddI/Tempol (Fig. 2A, bottom right), mitochondrial morphology was improved, compared with cells exposed to AZT/ddI alone. Because EM is not quantitative, the visual impressions were confirmed by scoring the photomicrographs.
FIG. 2.

(A) EM photomicrographs showing mitochondrial morphology in P16 H9c2 cells exposed to no drug (top left), 200μM Tempol (top right), 50μM AZT/50μM ddI (bottom left), and AZT/dd/Tempol (bottom right). (B) Results of scoring data for the same cells, using a paradigm where 0 = perfect and 5 = the most highly damaged mitochondria, and the details are described in the Materials and Methods section. Each data point represents mean ± SE for 10 coded photomicrographs/group scored by three investigators (n = 30). ‡p < 0.01 for control versus AZT/ddI, Tempol versus AZT/ddI, and AZT/ddI versus AZT/ddI/Tempol.
Randomized and coded photomicrographs (n = 10/treatment group) were scored by three independent investigators using the scoring paradigm presented in the legend to Figure 2B. Consistent with previous findings (Liu et al., 2012), AZT/ddI exposure induced significant mitochondrial morphological damage, compared with the unexposed controls (control vs. AZT/ddI and Tempol vs. AZT/ddI both p < 0.01). In addition, the cells exposed to AZT/ddI/Tempol sustained significantly less mitochondrial damage than the cells exposed to AZT/ddI alone (p < 0.01).
Tempol and Tempol-H Stimulated UCP-2 Expression
Because the data obtained by Seahorse (Fig. 1A) showed that Tempol and Tempol-H, in the absence of NRTIs, increased the OCR values significantly (p < 0.05) at P5 and P13, compared with the unexposed control cells, we hypothesized that Tempol and Tempol-H may induce a mild mitochondrial uncoupling in H9c2 cells. To explore this possibility further, protein levels of mitochondrial UCP-2 were determined by Western blot in cells that had been exposed for 5P to no drug, Tempol, Tempol-H, AZT/ddI, AZT/ddI/Tempol, or AZT/ddI/Tempol-H (Fig. 3). Western blot values, normalized to COX4, showed a 3.2- and 2.5-fold increase in UCP-2 protein in cells exposed to Tempol and Tempol-H, compared with control cells (Fig. 3A, left panels). In addition, 1.8- and 1.7-fold increases were observed in cells exposed to AZT/ddI/Tempol or AZT/ddI/Tempol-H, respectively, compared with cells exposed to AZT/ddI alone (Fig. 3A, right panels). This experiment was repeated three times in cells at P5, and the mean ± SE for n = 3 are shown in Figure 4B, where the fold-increase is shown as a number inside of each bar.
FIG. 3.
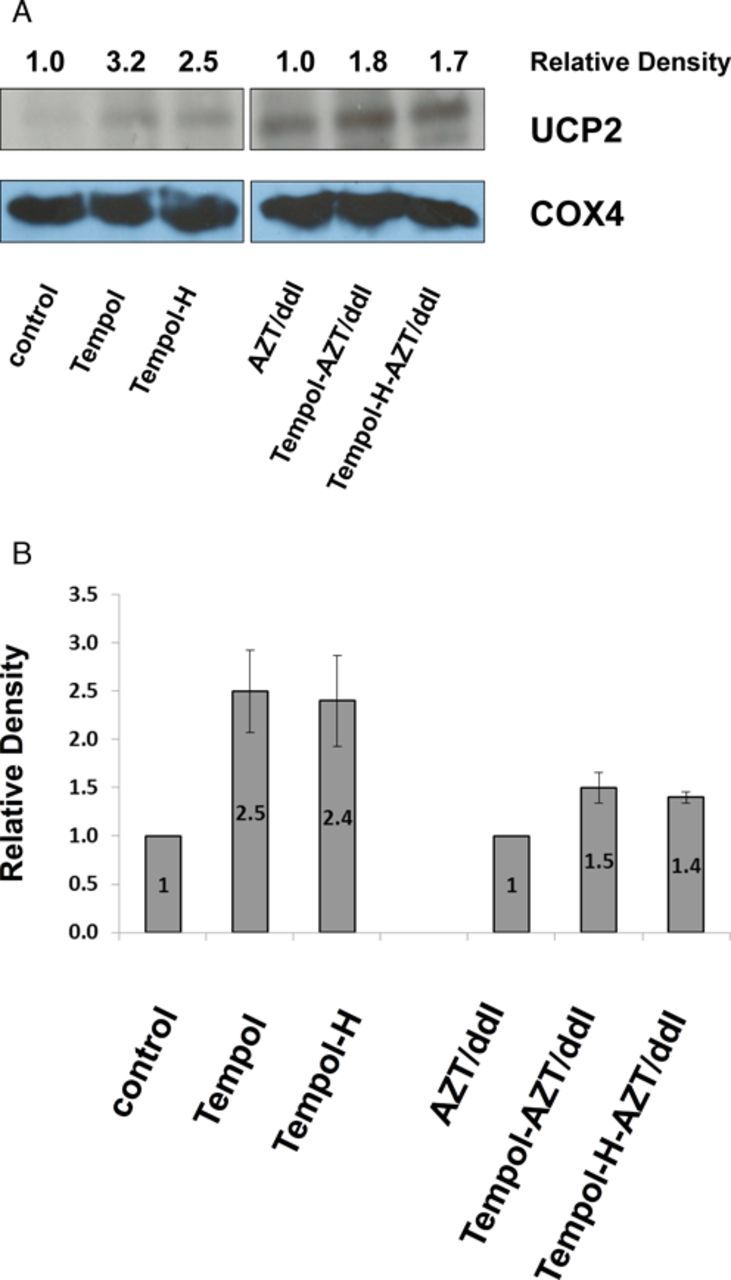
Tempol stimulated induction of UCP2. (A) Representative Western blot showing UCP-2 protein levels in P5 H9c2 cells. Quantitation was performed using ImageJ software, where each number represents the density of a specific band, normalized to the corresponding COX4 band, and expressed relative to the appropriate control (designated as 1). (B) Graph showing fold-increase (mean ± SE, n = 3 experiments) for UCP2 intensity in P5 cells, determined by Western blot. The unexposed cells (designated 1) were compared with cells exposed to Tempol or Tempol-H, and the AZT/ddI-exposed cells (designated 1) were compared with the cells exposed to AZT/ddI/Tempol and AZT/ddI/Tempol-H.
FIG. 4.

Mitochondrial Complex I protein level (A) and Complex I activity (B). (A) H9c2 cells, exposed to 50μM AZT or 50μM AZT/50μM ddI for 3P, 7P, and 16P, were evaluated by Western blot using OXPHOS protein antisera (see the Materials and Methods section). Quantitation was performed using ImageJ software, and each number represents the density of a specific band, normalized to the corresponding actin band, and expressed relative to the appropriate control (designated as 1). Western blots were repeated two times. (B) Mitochondria were isolated and Complex I activity was evaluated (using the Abcam Complex I Enzymatic Activity Microplate Assay) in H9c2 cells exposed as in Figure 5A at P3. The assay uses 40μM CAM as a negative control. Each data point represents mean ± SE (n = 3 wells), and the experiments were performed twice. Compared with unexposed or Tempol-exposed cells, significant decreases in Complex I activity were found in cells exposed to AZT/ddI (‡p < 0.01), and values did not change for cells exposed to AZT/ddI/Tempol.
Effect of NRTI Exposure on OXPHOS Protein Levels and Complex I Activity
Using Western blot with a monoclonal antibody mixture that contained sera specific for all of the OXPHOS proteins, we examined levels in cells exposed to no drug or AZT/ddI. The antibodies were specific for one or more subunits of each protein, including: Complex I subunit NDUFB8; Complex II, 30 kDa subunit; Complex III, Core protein-2; Complex IV, subunit I; and Complex V, α-subunit. Whereas most complexes were not altered in cells exposed to AZT/ddI (data not shown), the expression of Complex I NDUFB8 was reduced in cells exposed to AZT alone, and to AZT/ddI, at P3, P7, and P16 (Fig. 4A).
To explore this further, we evaluated Complex I enzymatic activity at P3 (Fig. 4B) and P7 (not shown), using a microplate assay. The control cells and Tempol-treated cells had similar levels of Complex I at P3 and P7, levels which were significantly higher than those found for AZT/ddI or AZT/ddI/Tempol (p <0.01 for both at both passages). Therefore, Complex I activity was reduced in cells exposed to AZT/ddI, and not rescued in cells treated with AZT/ddI/Tempol.
Tempol Reduced Superoxide Levels
Although under mild uncoupling conditions, mitochondria have an increased respiratory rate and reduced membrane potential, both of which may potentially decrease superoxide generation. To observe changes in active oxygen levels, and because Tempol is reported to mimic superoxide dismutase, we evaluated superoxide levels at P3, P7, and P10 in cells exposed as follows: control, Tempol, AZT/ddI, and AZT/ddI/Tempol. The results are shown in Figure 5. In cells exposed to AZT/ddI, there was a significant increase in superoxide (p < 0.01 at all passages), compared with unexposed cells. In addition, there was a significant reduction in superoxide levels in cells exposed to AZT/ddI/Tempol compared with cells exposed to AZT/ddI (p < 0.01 at all passages).
FIG. 5.

Effects of Tempol and AZT/ddI on cellular levels of superoxides. H9c2 cells at P3, P7, and P10, were plated and stained with hydroethidine to determine the levels of superoxide anions. The figure shows superoxide data, expressed as relative fluorescence units (RFU). The results are reported as mean ± SE (n = 3–4 wells). For all three passages: control versus AZT/ddI (‡p < 0.01); and AZT/ddI versus AZT/ddI/Tempol (‡p < 0.01).
DISCUSSION
The overall conclusion of this study is that Tempol and Tempol-H can be used to restore OXPHOS capacity in cells with mitochondria damaged by long-term exposure to AZT/ddI. In order to mimic the long-term human exposures, the AZT/ddI exposures in this study extended for 16P. Several of the mitochondria-related endpoints applied in these experiments, OXPHOS capacity in the form of OCR, mitochondrial morphology by EM, alteration in levels of various mitochondria-related proteins, and levels of superoxides, showed amelioration of AZT/ddI-induced toxicity when Tempol or Tempol-H was concomitantly incubated with the NRTIs. Complex I protein levels and activity were decreased with AZT/ddI, and the nitroxides did not increase Complex I activity, suggesting that Complex I levels were sufficient to allow function. The potential for the future application of this work depends on reproducing these findings in an appropriate whole animal model, but these preliminary data suggest that Tempol, or its metabolite Tempol-H, which are both nontoxic in humans, may be useful protective agents against NRTI-induced cardiac mitochondrial toxicity in HIV-1-infected patients.
Mitochondrial damage leads to decreased aerobic metabolism, imbalanced bioenergetics, and further cell/tissue injury and death. Thus, mitigation of mitochondrial damage is a critical goal for many preclinical and clinical studies. Most long-term clinical manifestations of NRTI-induced toxicity, including skeletal muscle- and cardio-myopathy, neuromuscular and cognitive impairment, hepatic steatosis, and possible lactic acidosis, are the result of mitochondrial compromise (Barret et al., 2003; Blanche et al., 1999, 2006; Cossarizza and Moyle, 2004; Koczor and Lewis, 2010). To date, there is no universal approach for mitochondrial protection, particularly as it relates to NRTI-induced cardiomyopathy.
We hypothesized that Tempol, being a stable free radical, might protect mitochondria against the oxidative damage known to result from NRTI therapy, and thereby decrease drug-induced long-term mitochondrial toxicity. In a previous study (Liu et al., 2012), we reported mitochondrial compromise, in the form of OCR impairment, in H9c2 rat cardiomyocytes exposed for 32 passages to the same doses of AZT/ddI used here. In this study, we also showed consistent reductions in maximal uncoupled OCR at P5, P7, and P13 in AZT/ddI-exposed cells, compared with the unexposed cells. However, in parallel cultures in which cells were exposed to Tempol or Tempol-H, along with AZT/ddI, for up to 16P, there were consistently increased levels of OCR at P5, P7, and P13, compared with cells exposed to AZT/ddI alone. In support of the Seahorse data, mitochondrial morphology was also analyzed in NRTI-exposed cells protected with Tempol, and Western blotting for UCP-2 suggested that the Tempol-induced protection may involve a mild mitochondrial uncoupling. Interestingly, qRT-PCR of UCP-2 was not different in the presence or absence of Tempol, suggesting that the upregulation of UCP-2 did not occur through an RNA-based mechanism. Furthermore, superoxide formation was attenuated in cells exposed to AZT/ddI plus Tempol, compared with cells exposed to AZT/ddI, at P3, P7, and P10. Taken together all these data suggest that Tempol may cause a mild mitochondrial uncoupling, which has been proposed as an important mechanism in reducing oxidative stress and altering cellular energy metabolism to protect tissues such as the heart from mitochondrial toxins (Cunha et al., 2011; Modriansky and Gabrielova, 2009).
Our observation that UCP-2 is upregulated by Tempol in H9c2 cardiomyocytes is consistent with previous experiments in vivo in a mouse model (Mitchell et al., 2003). The UCPs belong to the mitochondrial anion transporter superfamily located in the inner mitochondrial membrane. UCP-2, a recently-identified member of the UCP family, is essentially ubiquitous and has been shown to mediate proton conductance (Azzu et al., 2010; Baffy et al., 2011). Whereas the precise mechanism of activation and inhibition of UCP-2, and its physiological role, remains uncertain, considerable recent progress in understanding regulation in the UCP family implicates these proteins in adaptation to nutritional status and oxidative stress. Among many functions of UCP-2, cytoprotection is frequently documented and includes protection of many tissues (brain, pancreas, macrophages, and even cancer cells) against multiple types of damage (Azzu et al., 2010; Baffy, 2010 ; Baffy et al., 2011). In fact, many types of cancer have elevated levels of UCPs compared with their corresponding normal tissues. Furthermore, UCP-2 is linked to chemo-resistance of cancer cells (Baffy, 2010; Baffy et al., 2011).
A paper recently published by Branco et al. (2012) demonstrated different toxic responses to doxorubicin in H9c2 cells at multiple differentiation stages. They found that differentiated cells may accumulate more drug, have increased manganese superoxide dismutase levels and increased Bcl-xL levels, as well as altered responses to drug treatment. An interesting question for future studies will be to evaluate NRTI-induced mitochondrial toxicity and Tempol's protection in more differentiated H9c2 cells.
Finally, this study shows that Tempol and Tempol-H target cardiomyocyte mitochondria for protection against AZT/ddI-induced damage. Currently, we are applying Tempol in a patas monkey transplacental model of NRTI-induced toxicity, where mitochondrial integrity will be examined in fetal heart, skeletal muscle, brain, and liver. If successful in the monkey model, the next step would involve use of Tempol or Tempol-H to protect HIV-1-infected patients from NRTI-induced mitochondrial toxicities in the clinic.
SUPPLEMENTARY DATA
Supplementary data are available online at http://toxsci.oxfordjournals.org/.
FUNDING
Intramural Research Program of the National Institutes of Health, Center for Cancer Research, National Cancer Institute.
Supplementary Material
Acknowledgments
We wish to thank Alexandra Michalowski for statistical support.
Footnotes
Disclaimer: The content of this publication does not necessarily reflect the views or policies of the U.S. Department of Health and Human Services, nor does the mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
REFERENCES
- Anderson P. L., Kakuda T. N., Lichtenstein K. A. The cellular pharmacology of nucleoside- and nucleotide-analogue reverse-transcriptase inhibitors and its relationship to clinical toxicities. Clin. Infect. Dis. 2004;38:743–753. doi: 10.1086/381678. [DOI] [PubMed] [Google Scholar]
- Azzu V., Jastroch M., Divakaruni A. S., Brand M. D. The regulation and turnover of mitochondrial uncoupling proteins. Biochim. Biophys. Acta. 2010;1797:785–791. doi: 10.1016/j.bbabio.2010.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baffy G. Uncoupling protein-2 and cancer. Mitochondrion. 2010;10:243–252. doi: 10.1016/j.mito.2009.12.143. [DOI] [PubMed] [Google Scholar]
- Baffy G., Derdak Z., Robson S. C. Mitochondrial recoupling: A novel therapeutic strategy for cancer. Br. J. Cancer. 2011;105:469–474. doi: 10.1038/bjc.2011.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barret B., Tardieu M., Rustin P., Lacroix C., Chabrol B., Desguerre I., Dollfus C., Mayaux M. J., Blanche S. Persistent mitochondrial dysfunction in HIV-1-exposed but uninfected infants: Clinical screening in a large prospective cohort. AIDS. 2003;17:1769–1785. doi: 10.1097/00002030-200308150-00006. [DOI] [PubMed] [Google Scholar]
- Bartha E., Solti I, Szabo A., Olah G., Magyar K., Szabados E., Kalai T., Hideg K., Toth K., Gero D. Regulationof kinase cascade activation and heat shock protein expression bypoly(ADP-ribose) polymerase inhibition in doxorubicin-induced heart failure. J. Cardiovasc. Pharmacol. 2011;58:380–391. doi: 10.1097/FJC.0b013e318225c21e. [DOI] [PubMed] [Google Scholar]
- Blanche S., Tardieu M., Benhammou V., Warszawski J., Rustin P. Mitochondrial dysfunction following perinatal exposure to nucleoside analogues. AIDS. 2006;20:1685–1690. doi: 10.1097/01.aids.0000242814.42344.77. [DOI] [PubMed] [Google Scholar]
- Blanche S., Tardieu M., Rustin P., Slama A., Barret B., Firtion G., Ciraru-Vigneron N., Lacroix C., Rouzioux C., Mandelbrot L., et al. Persistent mitochondrial dysfunction and perinatal exposure to antiretroviral nucleoside analogues. Lancet. 1999;354:1084–1089. doi: 10.1016/S0140-6736(99)07219-0. [DOI] [PubMed] [Google Scholar]
- Branco A. F., Sampaio S. F., Moreira A. C., Holy J., Wallace K. B., Baldeiras I., Oliveira P. J., Sardao V. A. Differentiation-dependent Doxorubicin toxicity on H9c2 cardiomyoblasts. Cardiovasc. Toxicol. 2012;12:326–340. doi: 10.1007/s12012-012-9177-8. [DOI] [PubMed] [Google Scholar]
- Cossarizza A., Moyle G. Antiretroviral nucleoside and nucleotide analogues and mitochondria. AIDS. 2004;18:137–151. doi: 10.1097/00002030-200401230-00002. [DOI] [PubMed] [Google Scholar]
- Cunha F. M., Caldeira da Silva C. C., Cerqueira F. M., Kowaltowski A. J. Mild mitochondrial uncoupling as a therapeutic strategy. Curr. Drug Targets. 12:783–789. doi: 10.2174/138945011795528778. [DOI] [PubMed] [Google Scholar]
- Divi R. L., Leonard S. L., Kuo M. M., Walker B. L., Orozco C. C., Claire M. C., Jr, Nagashima K., Harbaugh S. W., Harbaugh J. W., Thamire C., et al. Cardiac mitochondrial compromise in 1-yr-old Erythrocebus patas monkeys perinatally-exposed to nucleoside reverse transcriptase inhibitors. Cardiovasc. Toxicol. 2005;5:333–346. doi: 10.1385/ct:5:3:333. [DOI] [PubMed] [Google Scholar]
- Drose S., Brandt U. Molecular mechanisms of superoxide production by the mitochondrial respiratory chain. Adv. Exp. Med. Biol. 2012;748:145–169. doi: 10.1007/978-1-4614-3573-0_6. [DOI] [PubMed] [Google Scholar]
- Hoschele D. Cell culture models for the investigation of NRTI-induced mitochondrial toxicity. Relevance for the prediction of clinical toxicity. Toxicol. In Vitro. 2006;20:535–546. doi: 10.1016/j.tiv.2005.11.007. [DOI] [PubMed] [Google Scholar]
- Koczor C. A., Lewis W. Nucleoside reverse transcriptase inhibitor toxicity and mitochondrial DNA. Expert. Opin. Drug Metab. Toxicol. 2010;6:1493–504. doi: 10.1517/17425255.2010.526602. [DOI] [PubMed] [Google Scholar]
- Krishna M. C., DeGraff W., Tamura S., Gonzalez F. J., Samuni A., Russo A., Mitchell J. B. Mechanisms of hypoxic and aerobic cytotoxicity of mitomycin C in Chinese hamster V79 cells. Cancer Res. 1991;51:6622–6628. [PubMed] [Google Scholar]
- Lewis W. Nucleoside reverse transcriptase inhibitors, mitochondrial DNA and AIDS therapy. Antivir. Ther. 2005;10(Suppl 2):M13–M27. [PubMed] [Google Scholar]
- Lewis W., Day B. J., Copeland W. C. Mitochondrial toxicity of NRTI antiviral drugs: An integrated cellular perspective. Nat. Rev. Drug Discov. 2003;2:812–822. doi: 10.1038/nrd1201. [DOI] [PubMed] [Google Scholar]
- Liu Y., Borchert G. L., Surazynski A., Phang J. M. Proline oxidase, a p53-induced gene, targets COX-2/PGE2 signaling to induce apoptosis and inhibit tumor growth in colorectal cancers. Oncogene. 2008;27:6729–6737. doi: 10.1038/onc.2008.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y., Nguyen P., Baris T. Z., Poirier M. C. Molecular analysis of mitochondrial compromise in rodent cardiomyocytes exposed long term to nucleoside reverse transcriptase inhibitors (NRTIs) Cardiovasc. Toxicol. 2012;12:123–134. doi: 10.1007/s12012-011-9148-5. [DOI] [PubMed] [Google Scholar]
- Mitchell J. B., Xavier S., DeLuca A. M., Sowers A. L., Cook J. A., Krishna M. C., Hahn S. M., Russo A. A low molecular weight antioxidant decreases weight and lowers tumor incidence. Free Radic. Biol. Med. 2003;34:93–102. doi: 10.1016/s0891-5849(02)01193-0. [DOI] [PubMed] [Google Scholar]
- Modriansky M., Gabrielova E. Uncouple my heart: The benefits of inefficiency. J. Bioenerg. Biomembr. 2009;41:133–136. doi: 10.1007/s10863-009-9212-z. [DOI] [PubMed] [Google Scholar]
- Sena L. A., Chandel N. S. Physiological roles of mitochondrial reactive oxygen species. Mol. Cell. 2012;48:158–167. doi: 10.1016/j.molcel.2012.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soule B. P., Hyodo F., Matsumoto K., Simone N. L., Cook J. A., Krishna M. C., Mitchell J. B. The chemistry and biology of nitroxide compounds. Free Radic. Biol. Med. 2007;42:1632–1650. doi: 10.1016/j.freeradbiomed.2007.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcox C.S. Effectsof tempol and redox-cycling nitroxides in models of oxidative stress. Pharmacol. Ther. 2010;126:119–145. doi: 10.1016/j.pharmthera.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H., Kalivendi S., Zhang H., Joseph J., Nithipatikom K., Vasquez-Vivar J., Kalyanaraman B. Superoxide reacts with hydroethidine but forms a fluorescent product that is distinctly different from ethidium: Potential implications in intracellular fluorescence detection of superoxide. Free Radic. Biol. Med. 2003;34:1359–1368. doi: 10.1016/s0891-5849(03)00142-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.