

Abstract
Evidence demonstrating that human exposure to various organophosphorus insecticides (OPs) is associated with neurobehavioral deficits in children continues to emerge. The present study focused on diazinon (DZ) and its active oxygen metabolite, diazoxon (DZO), and explored their ability to impair neurite outgrowth in rat primary hippocampal neurons as a mechanism of developmental neurotoxicity. Both DZ and DZO (0.5–10 μM) significantly inhibited neurite outgrowth in hippocampal neurons, at concentrations devoid of any cyototoxicity. These effects appeared to be mediated by oxidative stress, as they were prevented by antioxidants (melatonin, N-t-butyl-alpha-phenylnitrone, and glutathione ethyl ester). Inhibition of neurite outgrowth was observed at concentrations below those required to inhibit the catalytic activity of acetylcholinesterase. The presence of astrocytes in the culture was able to provide protection against inhibition of neurite outgrowth by DZ and DZO. Astrocytes increased neuronal glutathione (GSH) in neurons, to levels comparable to those of GSH ethyl ester. Astrocytes depleted of GSH by L-buthionine-(S,R)-sulfoximine no longer conferred protection against DZ- and DZO-induced inhibition of neurite outgrowth. The findings indicate that DZ and DZO inhibit neurite outgrowth in hippocampal neurons by mechanisms involving oxidative stress, and that these effects can be modulated by astrocytes and astrocyte-derived GSH. Oxidative stress from other chemical exposures, as well as genetic abnormalities that result in deficiencies in GSH synthesis and regulation, may render individuals more susceptible to these developmental neurotoxic effects of OPs.
Keywords: Organophosphorus insecticide, Diazinon, Diazoxon, Neurite outgrowth, Glutathione, Glial–neuronal interactions
1. Introduction
Diazinon (DZ) and its active metabolite diazoxon (DZO) are members of the widely used class of organophosphorus insecticides (OPs) (EPA, 2011). Children in communities in close proximity to crops where these insecticides are sprayed are exposed regularly to a variety of OPs, and may be at increased risk for adverse neurological effects. Recent studies link such exposures to various neurobehavioral deficits, such as attention deficit hyperactivity disorder and lowered I.Q. (Bouchard et al., 2010; Eskenazi et al., 2007; Rauh et al., 2011; Rohlman et al., 2011). While acute effects of OPs primarily results from acetylcholinesterase (AChE) inhibition and subsequent cholinergic overstimulation, increasing evidence suggests that these compounds can exert other non-cholinergic effects, including alterations in signal transduction, inhibition of DNA synthesis, and increases in oxidative stress (Adigun et al., 2010; Guizzetti et al., 2005; Lukaszewicz-Hussain, 2010; Slotkin et al., 2006).
Young animals are more sensitive to the acute systemic effects of OPs and to their effects on the CNS than adults (Pope and Liu, 1997; Won et al., 2001). Other studies have shown long-term effects of late gestational and neonatal exposures to OPs, with an emphasis on learning and memory (Icenogle et al., 2004; Levin et al., 2008; Roegge et al., 2006), as well as neural cell development and synaptic function (Slotkin et al., 2008). Of relevance is that such neurotoxic effects at levels appear to be independent from AChE inhibition (Rush et al., 2010; Sidiropoulou et al., 2009; Yang et al., 2008), suggesting that alternative neurotoxic mechanisms of these compounds may be involved in developmental neurotoxicity.
Several studies have pointed to oxidative stress as a potential mechanism of OP neurotoxicity (Giordano et al., 2007; Lukaszewicz-Hussain, 2010; Slotkin et al., 2005). The overproduction of reactive oxygen species (ROS) and reactive nitrogen species (RNS) results in cellular oxidative stress. This ultimately leads to deleterious effects on various macromolecules, including DNA, lipids, and proteins (Valko et al., 2007). Furthermore, oxidative stress is increasingly implicated in a variety of diseases, including several neurodegenerative diseases, such as Alzheimer’s disease and Parkinson’s disease (Barnham et al., 2004), as well as to neurodevelopmental disorders, including autism and schizophrenia (Chauhan et al., 2012; Do et al., 2009; Tang et al., 2013).
Cellular defenses against oxidative stress include enzymes such as superoxide dismutases, catalase, and glutathione peroxidases, as well as factors such as glutathione, ascorbic acid (vitamin C), α-tocopherol (vitamin E), and flavonoids (Valko et al., 2007). Nevertheless, these defense systems can be overwhelmed in times of acute and/or chronic stress, rendering the cell defenseless against free radicals and oxidative species. The brain is particularly vulnerable to oxidative stress due to its high oxygen consumption, oxidizable lipid content (i.e. polyunsaturated fatty acids), as well as relatively low levels of endogenous antioxidants (Barnham et al., 2004; Matés, 2000). Levels of antioxidants in the brain differ by region and cell type. Glial cells, for example, have a higher glutathione content than neuronal cells; ascorbate, however, appears to predominate in neurons (Rice and Russo-Menna, 1997).
Glutathione (GSH; γ-glutamyl-cysteinyl-glycine) is an abundant cellular thiol tripeptide, and one of the most prominent antioxidants in the CNS (Lu, 2013). GSH is a potent defender against ROS, due to its ability to non-enzymatically scavenge free radicals, as well as its role as a co-factor for glutathione peroxidases and glutathione transferases against reactive aldehyde and peroxide accumulation within the cell (Dringen, 2000). The dysregulation of GSH redox cycling, as well as genetic deficiencies in GSH-related enzymes have been shown to adversely affect neurodevelopment and play a role in various neurodegenerative diseases (Ballatori et al., 2009; Sian et al., 1994); furthermore, GSH has been shown to modulate neurotoxicity that results from several environmental chemicals, including OPs (Giordano et al., 2007, 2008, 2006).
The present study investigated the ability of DZ and its metabolite DZO to inhibit neurite outgrowth in primary rat hippocampal neurons and its underlying mechanisms. Results show that neuritogenesis is inhibited by OP-induced oxidative stress, and is antagonized by antioxidants and by co-culture with astrocytes, which enhance neuronal GSH content.
2. Materials and methods
2.1. Materials
Neurobasal-A medium, DMEM medium, fetal bovine serum (FBS), Hanks’ balanced salt solution (HBSS), GlutaMAX, anti-mouse Alexa fluor-488 secondary antibody, Hoechst 33342, 2,7′-dichlorofluorescin diacetate (H2 DCF-DA), Super-Signal West Pico Chemiluminescent Substrate (Pierce), papain, and gentamicin were from Invitrogen (Carlsbad, CA). Diazinon (DZ; 99.4%), diazinon-O-analog (diazoxon; DZO; 98%) and chlorpyrifos (CPF; 99.5%) were from Chem-Service (West Chester, PA). Poly-D-lysine, antibodies: peroxidase-conjugated anti-mouse IgG, mouse anti-beta-actin, horseradish peroxidase-conjugated anti-rabbit IgG, rabbit anti-fibronectin, rabbit anti-map-2, mouse anti-tau, goat serum, dimethyl sulfoxide (DMSO), hydrogen peroxide (H2 O2 ), 3-(4,5-dimethylthiazol-2-yl)-2,5 diphenyltetrazolium bromide (MTT), and N-t-butyl-alpha-phenylnitrone (PBN), glutathione ethyl-ester (GSHee), L-buthionine-(S,R)-sulfoximine (BSO), reduced L-glutathione (GSH), tris (2-carboxyethyl)-phosphine hydrochloride (TCEP), and naphthalene dicarboxaldehyde (NDA) were from Sigma–Aldrich (St. Louis, MO). Mouse β-III-tubulin antibody was from Millipore (Billerica, MA). Melatonin was from EMD Chemicals (Rockland, MA).
2.2. Preparation of fetal rat hippocampal neurons
Hippocampal neurons from embryonic day 21 Sprague–Dawley rat fetuses were prepared as previously described (Brewer et al., 1993; Guizzetti et al., 2008; Van De Mark et al., 2009). Neurons were freshly prepared and plated on poly-D-lysine-coated glass coverslips for 2 h in Neurobasal A–FBS (10%) medium to allow attachment. After 2 h, the neurons were washed once in HBSS and the medium was replaced with astrocyte-conditioned medium (ACM) for 24 h, after which neurons were treated as indicated for 24 h.
2.3. Treatments of hippocampal neurons
After 24 h in culture, neurons were treated with varying concentrations of DZ and DZO (0, 0.1, 1, 10 μM, in DMSO) for 24 h. For experiments where antioxidants were used, neurons were pre-treated with either melatonin (200 μM), N-t-butyl-alpha-phenylnitrone (PBN; 100 μM), or glutathione ethyl-ester (2.5 mM) for 3 h prior to two washes with HBSS, replacement of ACM, and treatment with DZ or DZO.
2.4. Preparation of astrocyte-conditioned medium (ACM)
Primary cultures of cortical astrocytes were prepared as previously described (Guizzetti and Costa, 1996). After 14 days in culture (with regular medium changes), flasks of confluent astrocytes were washed twice with PBS and “serum-free” DMEM–BSA (0.1%) medium was added for 24 h. This medium was considered astrocyte-conditioned. The ACM was collected from the flasks and centrifuged at 300 × g for 10 min at room temperature to pellet any cells or debris. ACM was used as the culture medium for the rest of the incubation period for all neurite outgrowth experiments in neurons.
2.5. Astrocyte-neuron co-cultures
To assess the potential for astrocytes to protect neurons from DZ- and DZO-induced inhibition of neurite outgrowth, an indirect co-culture model was used. This model provides a way to understand astrocyte–neuronal interactions in an in vitro system that more accurately reflects the in vivo processes. Hippocampal neurons were prepared as described, and plated on glass coverslips to which four paraffin beads were previously affixed to prevent their touching the astrocyte monolayer while allowing them to share the same medium. After 2 h incubation in Neurobasal A/FBS (10%) medium to allow neurons to attach, neurons were washed twice in HBSS and ACM was added, as previously described. After 24 h, the glass coverslips containing the neurons were inverted onto 24-well plates containing astrocytes, as described by Viviani et al. (1998). This astrocyte-neuron co-culture was treated with 10 μM DZ or DZO or vehicle control (0.1% DMSO) for 24 h. In some experiments, astrocytes were previously treated with L-buthionine-(S,R)-sulfoximine (BSO; 25 μM) for 24 h to deplete GSH levels.
2.6. Measurement of cell viability
Neuron viability was measured by the MTT assay, where 50 μL of MTT reagent (5 mg/mL) was added to each well after 24 h treatment with DZ or DZO. After 3 h at 37 °C, the medium was removed and the formazan reaction product was dissolved in 250 μL DMSO. Absorbance was read at 562 nm and results were expressed as mean percentage of viable cells relative to untreated controls.
2.7. Measurement of GSH levels
Total intracellular glutathione (GSH) levels were measured as previously described (Giordano et al., 2008). Briefly, neurons were homogenized in Locke’s buffer and an aliquot was taken to measure the protein concentration while a second aliquot was diluted (1:1) with 10% 5-sulfosalicylic acid (SSA). The SSA fraction was centrifuged at 13,400 × g for 5 min at 4 °C and the supernatant was used for GSH determination. Aliquots from the SSA fraction were added to a 96-well plate and pH was adjusted to 7.0 with 0.2 M N-ethylmorpholine/0.02 M KOH. Oxidized glutathione was reduced by adding 10 μL of 10 mM tris (2-carboxyethyl)-phosphine hydrochloride (TCEP) for 15 min at room temperature. The pH was then adjusted to 12.5 using 0.5N NaOH before adding naphthalene dicarboxaldehyde (NDA; 10 mM for 30 min). Finally, the samples were analyzed on a spectrofluorometric plate reader (Ex 472 nm and Em 528 nm). The total amount of GSH in the sample was expressed as nmol/mg protein determined from a standard curve obtained by plotting known amounts of GSH incubated in the same experimental conditions versus fluorescence. To assess the effect of astrocytes on neuronal GSH, after which astrocytes, plated on poly-D-lysine-coated inserts and cultured for 4 days, were added to the wells.
2.8. Quantitative morphological analysis of neurite outgrowth
After 24 h treatment, hippocampal neurons were fixed in 4% paraformaldehyde in HBSS and permeabilized in 0.1% Triton X-100. Neurons were labeled with an anti-β-III-tubulin isoform antibody followed by a fluorescein-conjugated secondary antibody (Alexa-488); the nuclei were stained with Hoechst 33342. Glass coverslips were then mounted on microscope slides. Only pyramidal neurons (which represent more than 90% of neurons in the cultures (Giordano et al., 2009b)) with three or more neurites, not touching any other cells/neurites were included for quantitative analysis. For each cell, the following parameters were measured: axon length (including the length of any branches originating from the axon); length of minor neurites; total number of neurites/cell. Neurite length was measured from the point of emergence at the cell body to the tip of each segment. The identity of the longest neurite as the axon, and of minor neurites as dendrites was verified by staining with the specific markers Tau and MAP-2 (microtubule-associated protein-2), as previously described (Guizzetti et al., 2008; Giordano et al., 2009b). Quantification of the morphological parameters was carried out using MetaMorph 6.1 analysis software. At least 30 cells per treatment were analyzed for each experiment.
2.9. Measurement of reactive oxygen species
Reactive oxygen species (ROS) formation was determined by fluorescence using 2,7′-dichlorofluorescin diacetate (DCFH2-DA), as previously shown (Giordano et al., 2006, 2007). Upon entering cells DCFH2-DA is de-esterified to DCFH2, which is then oxidized by ROS to form the fluorescent 2,7′-dichlorofluorescein (DCF). Neurons were plated in a black-welled 96-well plate (2 × 104 in 100 μL/well) previously coated overnight with 100 μg/mL poly-D-Lysine. After 2 h in Neurobasal A–FBS (10%) medium, cells were washed and switched to ACM as previously described. After 48 h in culture, cells were incubated for 30 min with 10 μM DCFH2-DA in HBSS. Afterwards, the probe was washed out and the neurons were treated with the appropriate chemical dilutions for the desired time point(s). After treatment, neurons were washed once in HBSS and the plate was read on a Perkin-Elmer spectrofluorimeter (excitation 488 nm, emission 530 nm).
2.10. Measurement of acetylcholinesterase (AChE) activity
AChE activity in neurons were measured as previously described (Li et al., 2000), using a microtiter plate assay based on the method of Ellman et al. (Ellman et al., 1961). Hippocampal neurons were plated in 6-well plates (1.2 × 106 per well) in Neurobasal A–FBS (10%) for 2 h, after which they were washed 1× with HBSS and switched to ACM. After 24 h, neurons were treated with DZ or DZO (1 or 10 μM) for an additional 24 h. Cell lysates were collected in a 0.1 M sodium phosphate buffer (pH 8.0). For duplicate assays, 50 μL of cell lysates was combined with 150 μL of the assay buffer containing 0.1 M sodium phosphate and 0.1 mM of DTNB (5,5′-dithiobis-2-nitrobenzoic acid). The kinetic assay was initiated by addition of acetylthiocholine (final concentration: 1 mM) and the reaction was continuously monitored for 10 min at room temperature. Absorbance was read at 412 nm in a Beckman DU-70 spectrophotometer. The amount of 5-thio-2-nitrobenzoate formed was calculated using an extinction coefficient of 13 600 M−1/cm. AChE activity was calculated as nmol/min/mg protein and presented as percent untreated control.
2.11. Statistical analysis
For neurite outgrowth measurements, the following parameters were measured for each cell using Metamorph software: longest neurite length (including the length of any branches originating from the neurite); length of minor neurites; and total number of neurites/cell. About 30 cells per treatment were analyzed in each experiment. Final data summaries are displayed as the mean ± SEM for 90–120 cells per treatment derived from at least three independent experiments, unless otherwise indicated. For ROS, GSH, and AChE measurements, at least three separate experiments were performed where all samples were completed in duplicate or triplicate. Results are reported as the mean percent of the control (±SEM) of results from at least three experiments. Statistical significance for all analyses is evaluated by one-way ANOVA followed by Bonferroni’s post-hoc test, except for GSH levels, which were analyzed with the non-parametric ANOVA Kruskal–Wallis followed by Dunn’s post-hoc test using GraphPad Prism software.
3. Results
3.1. DZ and DZO inhibit neurite outgrowth in hippocampal neurons
Primary hippocampal neurons were exposed to varying concentrations of DZ or DZO (0.1–10 μM) for 24 h in ACM. Both compounds caused a concentration-dependent decrease in longest neurite length, with significant inhibition of longest neurite outgrowth from 0.5 μM DZ or DZO (Figs. Fig. 11A and Fig. 22A). Chlorpyrifos (CPF) was used as a positive control (Fig. 1) because of ites reported ability to inhibit neurite outgrowth (Yang et al., 2008). No differences in minor neurite length were observed as a result of DZ exposure (Fig. 1B), while DZO (0.5–10 μM) decreased minor neurite length (Fig. 2B). No differences in number of neurites per cell were seen in neurons exposed to either DZ or DZO (Figs. Fig. 11C and Fig. 22C). Representative images of hippocampal neurons exposed to DZ, DZO or CPF are shown in Fig. 3. Neither DZ nor DZO affected hippocampal neuron viability, as assessed by the MTT assay (Table 1).
Fig. 1.

DZ inhibits neurite outgrowth in hippocampal neurons. Hippocampal neurons were plated in Neurobasal/FBS (10%) medium for 2 h before being washed and switched to ACM for 24 h. Neurons were then treated with varying concentrations of DZ for 24 h. Chlorpyrifos (CPF; 5 μM) was used as a positive control. Results are expressed as mean (±SEM) of 90–120 cells derived from at least three independent experiments. Significantly different from control, **p < 0.01, ***p < 0.001.
Fig. 2.

DZO inhibits neurite outgrowth in primary hippocampal neurons. Hippocampal neurons were plated in Neurobasal/FBS (10%) medium for 2 h before being washed and switched to ACM for 24 h. Neurons were then treated with varying concentrations of DZO for 24 h. Results are expressed as mean (±SEM) of 90–120 cells derived from at least three independent experiments. Significantly different from control, *p < 0.05, ***p < 0.001.
Fig. 3.

DZ and DZO inhibit neurite outgrowth in primary hippocampal neurons. Representative images from primary cultures exposed to different concentrations of DZ or DZO. (A) Control; (B) 5 μM CPF; (C) 0.5 μM DZ; (D) 10 μM DZ; (E) 0.5 μM DZO; (F) 10 μM DZO.
Table 1.
Viability of DZ or DZO-treated neurons.
| Viability
| ||
|---|---|---|
| Concentration (μM) | DZ | DZO |
| 0 | 100 | 100 |
| 1 | 108.9 ± 6.1 | 103.9 ± 2.8 |
| 10 | 100.1 ± 2.8 | 104.8 ± 0.8 |
Hippocampal neurons were treated with varying concentrations of either DZ or DZO for 24 h and cell viability was assessed by the MTT method. Results are expressed as percent of control, and represent the mean (±SEM) of three separate experiments where neurons were treated in triplicate.
3.2. Oxidative stress modulates neurite outgrowth in mechanism of DZ and DZO neurotoxicity
As DZ and DZO have been reported to cause oxidative stress in mouse cerebellar granule neurons (Giordano et al., 2007), we sought to determine whether they would have a similar effect in rat hippocampal neurons, and whether oxidative stress would be involved in inhibition of neurite outgrowth. Reactive oxygen species (ROS) production in primary hippocampal neurons was measured by fluorescence using 2,7′-dichlorofluorescin diacetate (DCFH2-DA). As shown in Fig. 4, both DZ and DZO (1 and 10 μM) caused an increase in ROS production in hippocampal neurons. OP-induced oxidative stress appears to be involved in the inhibition of neurite outgrowth caused by DZ and DZO. Indeed, pre-treatment of neurons for 3 h with various antioxidants (melatonin (200 μM); N-t-butyl-alpha-phenylnitrone (PBN; 100 μM); and glutathione ethyl-ester (GSHee; 2.5 mM)) completely prevented the inhibition of longest neurite outgrowth caused by 10 μM DZ or DZO (Table 2). The effect of DZO on minor neurite length was also antagonized by antioxidants (Table 2).
Fig. 4.
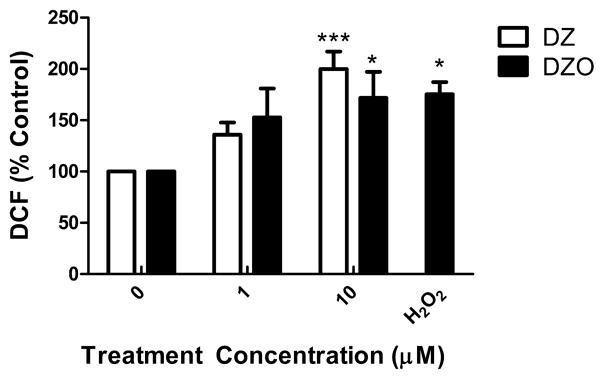
DZ and DZO increase ROS production in hippocampal neurons. Hippocampal neurons were plated in Neurobasal/FBS (10%) medium for 2 h before being washed and switched to ACM for an additional 46 h. Neurons were probed with 10 μM H2DCF-DA for 30 min, after which the probe was washed out and neurons were treated with DZ or DZO for 1 h. H2 O2 (20 μM) was used as a positive control. Results are expressed as mean (±SEM) of at least three independent experiments in which treatments were performed in triplicate. Significantly different from control, *p < 0.05, ***p < 0.001.
Table 2.
Effects of antioxidants on the inhibitory effects of DZ and DZO on neurite outgrowth in primary hippocampal neurons.
| Treatment | Longest neurite length (μm) | Minor neurite length (μm) | No. neurites/cell |
|---|---|---|---|
| Control | 139.6 ± 5.4 | 17.0 ± 0.5 | 4.6 ± 0.1 |
| Mel | 132.2 ± 10.8 | 18.0 ± 1.2 | 4.3 ± 0.2 |
| PBN | 116.5 ± 6.5 | 16.8 ± 0.7 | 4.6 ± 0.2 |
| GSHee | 129.7 ± 12.4 | 15.5 ± 0.9 | 5.1 ± 0.4 |
| DZ | 69.0 ± 5.9# | 14.4 ± 0.7 | 4.3 ± 0.1 |
| DZ + Mel | 122.2 ± 10.1 | 17.3 ± 1.0 | 4.5 ± 0.2 |
| DZ + PBN | 105.0 ± 9.6 | 17.2 ± 0.9 | 4.8 ± 0.2 |
| DZ + GSHee | 152.9 ± 10.2 | 16.1 ± 0.9 | 4.7 ± 0.2 |
| DZO | 86.8 ± 5.9# | 12.5 ± 0.6# | 4.2 ± 0.1 |
| DZO + Mel | 133.1 ± 9.4 | 13.8 ± 0.7 | 4.4 ± 0.2 |
| DZO + PBN | 121.8 ± 7.8 | 14.5 ± 0.6 | 4.6 ± 0.2 |
| DZO + GSHee | 155.1 ± 10.4 | 13.4 ± 0.9 | 5.1 ± 0.2 |
After 24 h in ACM, neurons were pre-treated with 200 μM melatonin (Mel), 100 μM PBN, or 2.5 mM glutathione ethyl-ester (GSHee) for 3 h prior to wash-out and treatment with 10 μM DZ or DZO for 24 h, as described in Section 2. Results represent the mean (±SEM) of 60–90 cells per treatment group.
Significantly different from untreated control, p < 0.001.
3.3. Astrocytes protect against DZ- and DZO-induced inhibition of neurite outgrowth by modulating neuronal glutathione content
To more closely resemble in vivo conditions, the effect of DZ and DZO on neurite outgrowth was assessed in the presence of astrocytes. Results of these experiments, shown in Fig. 5, indicate that the presence of astrocytes prevents the inhibition of neurite outgrowth caused by DZ or DZO. Since astrocytes have a greater antioxidant capacity than neurons, we hypothesized that astrocytes may prevent inhibition of neurite outgrowth by protecting against the oxidative conditions produced by DZ and DZO exposure. Astrocytes had been previously shown to protect neurons against the cytotoxicity of brominated flame retardants by increasing neuronal content of GSH (Giordano et al., 2009a). A similar mechanism appears to be occurring in case of neurite outgrowth. This was indeed the case. Levels of GSH in hippocampal neurons were 12.4 ± 1.2 nmol/mg protein, and increased to 20.2 ± 1.4 nmol/mg of protein in the presence of astrocytes (n = 3; p < 0.05). This increase is similar to that observed when neurons were treated for 3 h with 2.5 mM GSHee (22.8 ± 4.5 nmol/mg of protein).
Fig. 5.

Astrocytes protect against DZ- and DZO-induced inhibition of neurite outgrowth. After 24 h in ACM, neurons on glass coverslips were inverted onto astrocyte-containing wells and treated with DZ or DZO. In the presence of astrocytes DZ and DZO (1 or 10 μM) did not affect the length of the longest neurite. This is in contrast with the shown effect (at 10 μM) in the absence of astrocytes. Results are expressed as mean (±SEM) of at least 90 cells obtained from three independent experiments. Significantly different from the effect in the presence of astrocytes, **p < 0.01, ***p < 0.001.
To further confirm the role of GSH in the protective effect of astrocytes on DZ- and DZO-induced inhibition of neurite outgrowth, astrocytes were treated with 25 μM L-buthionine sulfoximine (BSO) for 24 h before culturing them with the neurons. BSO specifically inhibits γ-glutamylcysteine synthetase (EC 6.3.2.2; also known as glutamate cysteine ligase, GCL) the enzyme responsible for the first step of GSH biosynthesis (Anderson, 1998). Upon exposure to BSO, GSH levels in astrocytes decreased from 17.4 ± 0.9 to 5.3 ± 0.3 nmol/mg protein (n = 3; p < 0.05; Giordano et al., 2009b). As shown in Fig. 6, GSH-depleted astrocytes were not able to confer protection to neurons against DZ- and DZO-induced inhibition of neurite outgrowth as they had previously. Neurons cultured with BSO-treated astrocytes and exposed to 10 μM DZ or DZO for 24 h displayed significantly decreased longest neurite lengths, 62% and 65% of control levels, respectively (p < 0.001; Fig. 6A). This is in contrast to the lack of neurite outgrowth inhibition of neurons treated with DZ or DZO in the presence of astrocytes with their GSH levels intact (Fig. 5). Similarly, the inhibition of minor neurite length caused by DZO is no longer attenuated in the presence of astrocytes depleted of GSH (Fig. 6B).
Fig. 6.

Astrocytes depleted of GSH do not protect against DZ- and DZO-induced inhibition of neurite outgrowth. After 24 h in ACM, neurons were placed with astrocytes previously treated with 25 μM BSO for 24 h. Shown are measurements in neurons cultured with control astrocytes, or with BSO-treated astrocytes, and treated with 10 μM DZ or DZO in the presence of BSO-treated astrocytes. Results are expressed as mean (±SEM) of at least 90 cells obtained from three independent experiments. Significantly different from control, *p < 0.05, ***p < 0.001.
3.4. Effect of DZ and DZO AChE activity in hippocampal neurons
AChE activity in hippocampal neurons was measured after 24 h incubation with 1 or 10 μM DZ or DZO. DZO caused higher inhibition of AChE activity than DZ, as expected by the oxon form of the parent compound. Treatment with 1 or 10 μM DZ for 24 h resulted in a 20% and 43% decrease in AChE activity, while exposure to 1 or 10 μM DZO for 24 h resulted in a 55% and 85% decrease in AChE activity, respectively (Fig. 7). Mean AChE activity (±SEM) in untreated control neurons was 3.77 ± 0.55 nmol/min/mg protein.
Fig. 7.
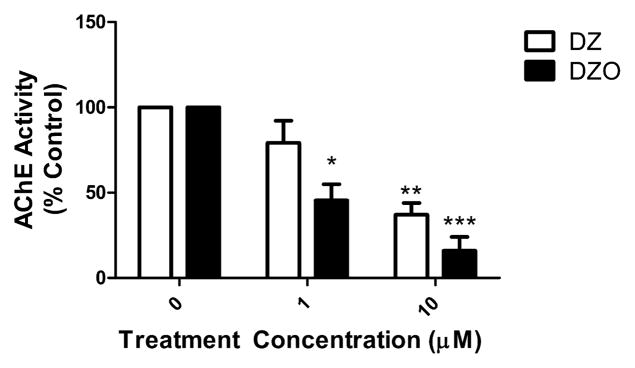
AChE activity in primary hippocampal neurons exposed to DZ or DZO. Newly harvested hippocampal neurons were plated in Neurobasal/FBS (10%) for 2 h before being switched to astrocyte-conditioned media (ACM) for an additional 24 h. Astrocytes were then treated for 24 h with DZ or DZO (1 or 10 μM). Results, expressed as percent of control, represent the mean (±SEM) of three independent experiments. Mean AChE activity (±SEM) in control neurons was 3.77 ± 0.55 nmol/min/mg protein. Significantly different from control, *p < 0.05, **p < 0.01, p < 0.001.
4. Discussion
The main findings in this study are that DZ and DZO inhibit neurite outgrowth in primary hippocampal neurons, that this inhibitory effect is due to oxidative stress, and that it is prevented by the presence of astrocytes. The finding that these OPs inhibit neurite outgrowth confirms and extends to primary rat hippocampal neurons previous results which showed inhibition of neurite outgrowth in mouse neuroblastoma cells (N2a cells; Axelrad et al., 2003; Flaskos et al., 2007; Sidiropoulou et al., 2009). Both DZ and DZO decreased the length of the longest neurite (identified as the axon by Tau staining) at concentrations as low as 0.5 μM. Another commonly used OP, chlorpyrifos (CPF), was used as a positive control as it had been shown to inhibit neurite outgrowth in different neuronal cell types (Sachana et al., 2001; Yang et al., 2008). We confirmed that CPF (5 μM) caused a significant inhibition of neurite outgrowth in hippocampal neurons, comparable to that caused by 10 μM DZ or DZO (Fig. 1A). In our morphological analysis, we also measured the length of the minor neurites, which were identified as dendrites by MAP-2 staining. Neither DZ nor the positive control CPF affected minor neurite length, while DZO had a small but significant effect starting at 0.5 μM (Figs. Fig. 11B and Fig. 22B). None of the OPs had any effect on the number of neurites per cell. All effects of DZ and DZO occurred at concentrations that were devoid of cytotoxicity (Table 1).
DZ and DZO, as well other OPs, have been shown to elicit oxidative stress in neuronal cells in vitro (Giordano et al., 2007; Slotkin and Seidler, 2009), and in brain of rodents upon in vivo exposure (Jafari et al., 2012; Yilmaz et al., 2012). In addition, we have recently found that oxidative stress is involved in the effect of DZ and DZO on astrocytic extracellular matrix proteins and on astrocyte–neuron interactions (Pizzurro et al., 2014). We thus examined whether these OPs would cause oxidative stress in hippocampal neurons, and whether this effect may be involved in the observed inhibition of neurite outgrowth. As shown in Fig. 4, both DZ and DZO increased ROS levels in hippocampal neurons. Moreover, three antioxidants (melatonin, PBN, and GSHee) successfully prevented inhibition of neurite outgrowth caused by DZ and DZO, including the decrease in minor neurite length caused by DZO (Table 2). These results suggest that DZ- and DZO-induced increase in oxidative stress in hippocampal neurons plays a primary role in their inhibition of neurite outgrowth.
It has been reported that astrocyte-conditioned medium protects against inhibition of neurite outgrowth by DZ in neuroblastoma cells (Harris et al., 2009). In the present study, primary hippocampal neurons (overall more “delicate” than neuroblastoma cell lines) were cultured in the presence of ACM, hence this experimental paradigm could not be directly tested. Other studies have shown that astrocytes can protect neurons against the toxicity of polybrominated diphenyl ethers (PBDEs) (Giordano et al., 2009a), ethanol (Watts et al., 2005), and the pesticides rotenone and paraquat (Rathinam et al., 2012). This indicates that, in a situation in which neurons and astrocytes are in close proximity (as in vivo), the latter can protect neurons from toxicity by various compounds. We found that when hippocampal neurons were cultured in the presence of astrocytes and the whole co-culture was exposed to DZ or DZO, the inhibitory effect on neurite outgrowth was prevented (Fig. 5).
Astrocytes are known to modulate neuronal functions and well-being by secreting a variety of factors (e.g. extracellular matrix proteins, growth factors), as well as by regulating various homeostatic mechanisms (e.g. glucose and neurotransmitter levels). Since DZ- and DZO-induced inhibition of neurite outgrowth appeared to be related to oxidative stress, we hypothesized that astrocytes may increase neuronal defenses against oxidative stress. Astrocytes have a higher content of the thiol-redox-capable peptide, GSH, than neurons (Rice and Russo-Menna, 1997; Giordano et al., 2008). This is attributed to nuclear factor erythroid-2-related factor 2 (Nrf-2)-driven gene activation that is preferential in astrocytes over neurons (Kraft et al., 2004; Lee et al., 2003). The synchronized increase in astrocytic GSH production and release is an essential component in the observed neuronal protection by Nrf2-activation in astrocytes, especially in response to oxidative stress (Calkins et al., 2010; Lee et al., 2003; Vargas and Johnson, 2009). Astrocytes devoid of Nrf2 (i.e. obtained from Nrf2−/− mice) not only have lower GSH levels than those with normal Nrf2 function, but are unable to induce Nrf2-dependent gene transcription and increase GSH after stimulation with the Nrf2-inducing agent, tert-butylhydroquinone (tBHQ) (Lee et al., 2003).
GSH is a crucial player in a multitude of cellular processes, and has a vital role in cellular defense against oxidative stress and insult (Ballatori et al., 2009). In the context of glial–neuronal interactions, astrocytes can increase GSH levels in neurons by providing the necessary precursors for GSH biosynthesis. Upon release from astrocytes, GSH is metabolized by γ-glutamyl transpeptidase to the dipeptide cysteinylglycine, from which cysteine is released by an ectopeptidase found on the surface of neuronal membranes. Cysteine is then taken up by neurons through the EAAC1 transporter (Ayoama et al., 2006), and utilized for GSH synthesis within the cell (Dringen et al., 1999; Giordano et al., 2009a; Sagara et al., 1993). In accordance with this mechanism, we found that the presence of astrocytes increased intracellular GSH in hippocampal neurons by about two-fold, an effect similar to that of the cell-permeable ethyl-ester form of glutathione (GSHee), which was shown to fully antagonize DZ and DZO effects on neurite outgrowth (Table 2). Further evidence for an astrocyte-mediated increase in neuronal GSH as an important mechanism of neuroprotection against DZ-and DZO-induced inhibition of neurite outgrowth, was provided by experiments in which astrocytes were pre-treated with L-buthionine sulfoximine (BSO), prior to co-culture with neurons and exposure to the OPs. BSO inhibits the enzyme that catalyzes the first and rate-limiting step of GSH biosynthesis, glutamate cysteine ligase (GCL) (Anderson, 1998; Berger et al., 1994), and has been shown to significantly reduce GSH levels in various in vitro models, including primary astrocyte cultures (Gabryel et al., 2005; Giordano et al., 2009b; Maryon et al., 2013). BSO-treated astrocytes were no longer able to confer protection to neurons under conditions of simultaneous exposure to DZ or DZO (Fig. 6).
Altogether, the findings of this study indicate that DZ and DZO alter neuronal outgrowth in hippocampal neurons by causing oxidative stress, and that astrocytes can provide protection against this effect primarily by enhancing GSH levels in neurons, though other mechanisms cannot be ruled out. It is known that GSH deficiency or shifts in the GSH/GSSG (glutathione disulfide) ratio results in increased susceptibility to oxidative stress and the subsequent oxidative damage (Ballatori et al., 2009). Chemical exposures that decrease GSH would be expected to enhance neuronal susceptibility to DZ and DZO and impair the protective functions of astrocytes. Polymorphisms in genes associated with GSH metabolism may also cause increased susceptibility to the effects of DZ and DZO on neuritogenesis, including polymorphisms of the Gclm (glutamate cysteine ligase modulatory subunit) gene, that are associated with low levels of GSH (Dalton et al., 2004; Nakamura et al., 2002). On the other hand, increasing GSH levels, e.g. by efforts upregulating Nrf2, may provide neuroprotection against these effects of DZ and DZO. Certain dietary components (e.g. flavonoids, sulfur-containing vegetables) can indeed increase GSH, as well as induce Nrf2-dependent transcription of other antioxidant-responsive genes (Bousova and Skalova, 2012; Vomhof-DeKrey and Picklo, 2012; Yanaka et al., 2009).
Both DZ and DZO decreased acetylcholinesterase (AChE) activity in hippocampal neurons, with DZO causing predictably higher levels of inhibition than the parent compound. Such significant inhibition of AChE activity by the parent compound was not expected due to the fact that the parent compound does not inhibit AChE directly. It is thought that any inhibition of AChE in the neurons is a result of the presence of DZO, either by bio-activation of DZ (Khokhar and Tyndale, 2012; Miksys and Tyndale, 2013), and/or by contamination of DZ with DZO. Inhibition of AChE may also be involved in DZ- and DZO-induced inhibition of neurite outgrowth. Earlier work had shown that AChE can regulate neurite outgrowth by a non-enzymatic mechanism (Layer et al., 1993; Munoz et al., 1999), and that AChE inhibition would results in decrease neurite outgrowth (Dupree and Bigbee, 1994). This has been attributed to the morphogenic action of AChE (Bigbee et al., 1999), which is independent of its catalytic action toward acetylcholine. Yang et al. (2008) concluded that CPF may affect neurite outgrowth in dorsal root ganglion neurons by interfering with the morphogenic activity of AChE; this was supported by the finding that CPF had no effect in neurons from AChE−/− mice, and that it inhibited neurite outgrowth at concentrations below those required to inhibit the catalytic activity of AChE. In agreement with the latter finding, we observed inhibition of neurite outgrowth at a concentration of DZ (0.5 μM) lower than that (1.0 μM) shown to be devoid of inhibitory activity toward AChE (Fig. 7). Though interferences with AChE may be involved in the effects of DZ and DZO on neurite outgrowth, the fact that such effects were completely antagonized by various antioxidants (melatonin, PBN), and by raising neuronal GSH levels (GSHee, co-culture with astrocytes), suggests that other mechanisms should also be considered.
5. Conclusions
Findings of this study indicate that the OP DZ and its active metabolite DZO, impair neuritogenesis in rat hippocampal neurons by a mechanism that involves induction of oxidative stress. The presence of astrocytes prevents inhibition of neurite outgrowth, as astrocytes are able to increase GSH levels in neurons. Neurite outgrowth is believed to be a most important event in brain development, and inhibition of neurite outgrowth is being utilized as an important indicator of developmental neurotoxicity (Radio and Mundy, 2008). The fact that human cells appear to be more sensitive than rodent cells to adverse effects on neurite outgrowth (Harrill et al., 2011), also suggests that the findings of the present study have potential direct relevance for human exposure to OPs.
Supplementary Material
Acknowledgments
This study was supported by the Center for Child Environmental Health Risk Research (EPA/NIH; P01ES009601) and the Environmental Toxicology and Pathology (EP/T) Training Grant (NIEHS; T32 ES007032-35). We thank Dr. Judit Marsillach-Lopez for her assistance with the AChE measurements, Collin White for assistance in the GSH and ROS measurements, and Dr. Terrance J. Kavanagh for helpful suggestions.
Footnotes
Conflict of interest
The authors declare no conflicts of interest.
Transparency document
The Transparency document associated with this article can be found in the online version.
References
- Adigun AA, Wrench N, Seidler FJ, Slotkin TA. Neonatal organophosphorus pesticide exposure alters the developmental trajectory of cell-signaling cascades controlling metabolism: differential effects of diazinon and parathion. Environ Health Perspect. 2010;118:210–215. doi: 10.1289/ehp.0901237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson ME. Glutathione: an overview of biosynthesis and modulation. Chem Biol Interact. 1998:111–112. 1–14. doi: 10.1016/s0009-2797(97)00146-4. [DOI] [PubMed] [Google Scholar]
- Axelrad JC, Howard CV, McLean WG. The effects of acute pesticide exposure on neuroblastoma cells chronically exposed to diazinon. Toxicology. 2003;185:67–78. doi: 10.1016/s0300-483x(02)00592-9. [DOI] [PubMed] [Google Scholar]
- Ayoama K, Su SW, Hamby AM, liu J, Chan J, Chen Y, et al. Neuronal glutathione deficiency and age-dependent neurodegeneration in the EAAC1 deficient mouse. Nat Neurosci. 2006;9:119–126. doi: 10.1038/nn1609. [DOI] [PubMed] [Google Scholar]
- Ballatori N, Krance SM, Notenboom S, Shi SJ, Tieu K, Hammond CL. Glutathione dysregulation and the etiology and progression of human diseases. Biol Chem. 2009;390:191–214. doi: 10.1515/BC.2009.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnham KJ, Masters CL, Bush AI. Neurodegenerative diseases and oxidative stress. Nat Rev Drug Discov. 2004;3:205–214. doi: 10.1038/nrd1330. [DOI] [PubMed] [Google Scholar]
- Berger SJ, Gosky D, Zborowska E, Willson JK, Berger NA. Sensitive enzymatic cycling assay for glutathione: measurements of glutathione content and its modulation by buthionine sulfoximine in vivo and in vitro in human colon cancer. Cancer Res. 1994;54:4077–4083. [PubMed] [Google Scholar]
- Bigbee JW, Sharma KV, Gupta JJ, Dupree JL. Morphogenic role for acetyl-cholinesterase in axonal outgrowth during neuronal development. Environ Health Perspect. 1999;107 (Suppl 1):81–87. doi: 10.1289/ehp.99107s181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouchard MF, Bellinger DC, Wright RO, Weisskopf MG. Attention-deficit/hyperactivity disorder and urinary metabolites of organophosphate pesticides. Pediatrics. 2010;125:E1270–E1277. doi: 10.1542/peds.2009-3058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bousova I, Skalova L. Inhibition and induction of glutathione S-transferases by flavonoids: possible pharmacological and toxicological consequences. Drug Metab Rev. 2012;44:267–286. doi: 10.3109/03602532.2012.713969. [DOI] [PubMed] [Google Scholar]
- Brewer GJ, Torricelli JR, Evege EK, Price PJ. Optimized survival of hippocampal-neurons in B27-supplemented neurobasal(Tm), a new serum-free medium combination. J Neurosci Res. 1993;35:567–576. doi: 10.1002/jnr.490350513. [DOI] [PubMed] [Google Scholar]
- Calkins MJ, Vargas MR, Johnson DA, Johnson JA. Astrocyte-specific over-expression of Nrf2 protects striatal neurons from mitochondrial complex II inhibition. Toxicol Sci. 2010;115:557–568. doi: 10.1093/toxsci/kfq072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chauhan A, Audhya T, Chauhan V. Brain region-specific glutathione redox imbalance in autism. Neurochem Res. 2012;37:1681–1689. doi: 10.1007/s11064-012-0775-4. [DOI] [PubMed] [Google Scholar]
- Dalton TP, Chen Y, Schneider SN, Nebert DW, Shertzer HG. Genetically altered mice to evaluate glutathione homeostasis in health and disease. Free Radic Biol Med. 2004;37:1511–1526. doi: 10.1016/j.freeradbiomed.2004.06.040. [DOI] [PubMed] [Google Scholar]
- Do KQ, Cabungcal JH, Frank A, Steullet P, Cuenod M. Redox dysregulation, neurodevelopment, and schizophrenia. Curr Opin Neurobiol. 2009;19:220–230. doi: 10.1016/j.conb.2009.05.001. [DOI] [PubMed] [Google Scholar]
- Dringen R. Metabolism and functions of glutathione in brain. Prog Neurobiol. 2000;62:649–671. doi: 10.1016/s0301-0082(99)00060-x. [DOI] [PubMed] [Google Scholar]
- Dringen R, Pfeiffer B, Hamprecht B. Synthesis of the antioxidant glutathione in neurons: supply by astrocytes of CysGly as precursor for neuronal glutathione. J Neurosci. 1999;19:562–569. doi: 10.1523/JNEUROSCI.19-02-00562.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupree JL, Bigbee JW. Retardation of neuroitic outgrowth and cytoskeletal changes accompany acetylcholinesterase inhibitor treatment in cultured rat dorsal root ganglion neurons. J Neurosci Res. 1994;39:567–575. doi: 10.1002/jnr.490390508. [DOI] [PubMed] [Google Scholar]
- Ellman GL, Courtney KD, Andres V, Featherstone RM. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem Pharmacol. 1961;7:88–95. doi: 10.1016/0006-2952(61)90145-9. [DOI] [PubMed] [Google Scholar]
- EPA (Environmental Protection Agency) Market Estimates. EPA; Washington DC: 2011. Pesticides Industry Sales and Usage: 2006–2007. [Google Scholar]
- Eskenazi B, Marks AR, Bradman A, Harley K, Barr DB, Johnson C, Morga N, Jewell NA. Organophosphate pesticide exposure and neurodevelopment in young Mexican-American children. Environ Health Perspect. 2007;115:792–798. doi: 10.1289/ehp.9828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flaskos J, Harris W, Sachana M, Munoz D, Tack J, Hargreaves AJ. The effects of diazinon and cypermethrin on the differentiation of neuronal and glial cell lines. Toxicol Appl Pharm. 2007;219:172–180. doi: 10.1016/j.taap.2006.10.033. [DOI] [PubMed] [Google Scholar]
- Gabryel B, Toborek T, Malecki A. Immunosuppressive immunophilin ligands attenuate damage in cultured rat astrocytes depleted of glutathione and exposed to simulated ischemia in vitro: comparison with N-acetylcysteine. Neurotoxicology. 2005;26:373–384. doi: 10.1016/j.neuro.2005.03.004. [DOI] [PubMed] [Google Scholar]
- Giordano G, Afsharinejad Z, Guizzetti M, Vitalone A, Kavanagh TJ, Costa LG. Organophosphorus insecticides chlorpyrifos and diazinon and oxidative stress in neuronal cells in a genetic model of glutathione deficiency. Toxicol Appl Pharm. 2007;219:181–189. doi: 10.1016/j.taap.2006.09.016. [DOI] [PubMed] [Google Scholar]
- Giordano G, Kavanagh TJ, Costa LG. Neurotoxicity of a polybrominated diphenyl ether mixture (DE-71) in mouse neurons and astrocytes is modulated by intracellular glutathione levels. Toxicol Appl Pharm. 2008;232:161–168. doi: 10.1016/j.taap.2008.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giordano G, Kavanagh TJ, Costa LG. Mouse cerebellar astrocytes protect cerebellar granule neurons against toxicity of the polybrominated diphenyl ether (PBDE) mixture DE-71. Neurotoxicology. 2009a;30:326–329. doi: 10.1016/j.neuro.2008.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giordano G, Pizzurro D, VanDeMark K, Guizzetti M, Costa LG. Manganese inhibits the ability of astrocytes to promote neuronal differentiation. Toxicol Appl Pharm. 2009b;240:226–235. doi: 10.1016/j.taap.2009.06.004. [DOI] [PubMed] [Google Scholar]
- Giordano G, White CC, McConnachie LA, Fernandez C, Kavanagh TJ, Costa LG. Neurotoxicity of domoic acid in cerebellar granule neurons in a genetic model of glutathione deficiency. Mol Pharmacol. 2006;70:2116–2126. doi: 10.1124/mol.106.027748. [DOI] [PubMed] [Google Scholar]
- Guizzetti M, Costa LG. Inhibition of muscarinic receptor-stimulated glial cell proliferation by ethanol. J Neurochem. 1996;67:2236–2245. doi: 10.1046/j.1471-4159.1996.67062236.x. [DOI] [PubMed] [Google Scholar]
- Guizzetti M, Moore NH, Giordano G, Costa LG. Modulation of neuritoge-nesis by astrocyte muscarinic receptors. J Biol Chem. 2008;283:31884–31897. doi: 10.1074/jbc.M801316200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guizzetti M, Pathak S, Giordano G, Costa LG. Effect of organophosphorus insecticides and their metabolites on astroglial cell proliferation. Toxicology. 2005;215:182–190. doi: 10.1016/j.tox.2005.07.004. [DOI] [PubMed] [Google Scholar]
- Harrill JA, Freudenrich TM, Robinette BL, Mundy WR. Comparative sensitivity of human and rat cultures to chemical-induced inhibition of neurite outgrowth. Toxicol Appl Pharmacol. 2011;256:268–280. doi: 10.1016/j.taap.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Harris W, Sachana M, Flaskos J, Hargreaves AJ. Neuroprotection from diazinon-induced toxicity in differentiating murine N2a neuroblastoma cells. Neurotoxicology. 2009;30:958–964. doi: 10.1016/j.neuro.2009.05.010. [DOI] [PubMed] [Google Scholar]
- Icenogle LM, Christopher NC, Blackwelder WP, Caldwell DP, Qiao D, Seidler FJ, Slotkin TA, Levin ED. Behavioral alterations in adolescent and adult rats caused by a brief subtoxic exposure to chlorpyrifos during neurulation. Neurotoxicol Teratol. 2004;26:95–101. doi: 10.1016/j.ntt.2003.09.001. [DOI] [PubMed] [Google Scholar]
- Jafari M, Salehi M, Ahmadi S, Asgari A, Abasnezhad M, Hajigholamali M. The role of oxidative stress in diazinon-induced tissues toxicity in Wistar and Norway rats. Toxicol Mech Methods. 2012;22:638–647. doi: 10.3109/15376516.2012.716090. [DOI] [PubMed] [Google Scholar]
- Khokhar JY, Tyndale RF. Rat brain CYP2B-enzymatic activation of chlorpyrifos to the oxon mediates cholinergic neurotoxicity. Toxicol Sci. 2012;126:325–335. doi: 10.1093/toxsci/kfs029. [DOI] [PubMed] [Google Scholar]
- Kraft AD, Johnson DA, Johnson JA. Nuclear factor E2-related factor 2-dependent antioxidant response element activation by tert-butylhydroquinone and sulforaphane occurring preferentially in astrocytes conditions neurons against oxidative insult. J Neurosci. 2004;24:1101–1112. doi: 10.1523/JNEUROSCI.3817-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Layer PG, Weikert T, Alber R. Cholinesterases regulate neurite growth of chick nerve cells in vitro by means of a non-enzymatic mechanism. Cell Tissue Res. 1993;273:219–226. doi: 10.1007/BF00312823. [DOI] [PubMed] [Google Scholar]
- Lee JM, Calkins MJ, Chan K, Kan YW, Johnson JA. Identification of the NF-E2-related factor-2-dependent genes conferring protection against oxidative stress in primary cortical astrocytes using oligonucleotide microarray analysis. J Biol Chem. 2003;278:12029–12038. doi: 10.1074/jbc.M211558200. [DOI] [PubMed] [Google Scholar]
- Levin E, Timofeeva O, Seidler F, Slotkin T. Long-term cognitive effects of low-level developmental organophosphate pesticide exposure: divergent effects of chlorpyrifos, diazinon and parathion. Neurotoxicol Teratol. 2008;30:251. doi: 10.1016/j.ntt.2007.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li WF, Costa LG, Richter RJ, Hagen T, Shih DM, Tward A, Lusis AJ, Furlong CE. Catalytic efficiency determines the in-vivo efficacy of PON1 for detoxifying organophosphorus compounds. Pharmacogenetics. 2000;10:767–779. doi: 10.1097/00008571-200012000-00002. [DOI] [PubMed] [Google Scholar]
- Lu SC. Glutathione synthesis. Biochim Biophys Acta. 2013;1830:3143–3153. doi: 10.1016/j.bbagen.2012.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukaszewicz-Hussain A. Role of oxidative stress in organophosphate insecticide toxicity – short review. Pestic Biochem Physiol. 2010;98:145–150. [Google Scholar]
- Maryon EB, Molloy SA, Kaplan JH. Cellular glutathione plays a key role in copper uptake mediated by human copper transporter 1. Am J Physiol Cell Physiol. 2013;304:C768–C779. doi: 10.1152/ajpcell.00417.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matés JM. Effects of antioxidant enzymes in the molecular control of reactive oxygen species toxicology. Toxicology. 2000;153:83–104. doi: 10.1016/s0300-483x(00)00306-1. [DOI] [PubMed] [Google Scholar]
- Miksys S, Tyndale RFP. 2011 CCNP Heinz Lehmann Award paper: cytochrome P450-mediated drug metabolism in the brain. J Psychiatry Neurosci. 2013;38:152–163. doi: 10.1503/jpn.120133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz FJ, Aldunate R, Inestrosa NC. Peripheral binding site is involved in the neurotrophis activity of acetylcholinesterase. NeuroReport. 1999;10:3621–3625. doi: 10.1097/00001756-199911260-00029. [DOI] [PubMed] [Google Scholar]
- Nakamura S, Kugiyama K, Sugiyama S, Miyamoto S, Koide S, Fukushima H, Honda O, Yoshimura M, Ogawa H. Polymorphism in the 5′-flanking region of human glutamate-cysteine ligase modifier subunit gene is associated with myocardial infarction. Circulation. 2002;105:2968–2973. doi: 10.1161/01.cir.0000019739.66514.1e. [DOI] [PubMed] [Google Scholar]
- Pizzurro DM, Dao K, Costa LG. Diazinon and diazoxon impair the ability of astrocytes to foster neurite outgrowth in primary hippocampal neurons. Toxicol Appl Pharmacol. 2014;274:372–382. doi: 10.1016/j.taap.2013.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pope CN, Liu J. Age-related differences in sensitivity to organophosphorus pesticides. Environ Toxicol Pharmacol. 1997;4:309–314. doi: 10.1016/s1382-6689(97)10029-1. [DOI] [PubMed] [Google Scholar]
- Radio NM, Mundy WR. Develpmental neurotoxicity testing in vitro: models for assessing chemical effects on neurite outgrowth. Neurotoxicology. 2008;29:361–376. doi: 10.1016/j.neuro.2008.02.011. [DOI] [PubMed] [Google Scholar]
- Rathinam ML, Watts LT, Narasimhan M, Riar AK, Mahimainathan L, Hender-son GI. Astrocyte mediated protection of fetal cerebral cortical neurons from rotenone and paraquat. Environ Toxicol Pharmacol. 2012;33:353–360. doi: 10.1016/j.etap.2011.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauh V, Arunajadai S, Horton M, Perera F, Hoepner L, Barr DB, Whyatt R. 7-year neurodevelopmental scores and prenatal exposure to chlorpyrifos, a common agricultural pesticide. Environ Health Perspect. 2011;119:1196–1201. doi: 10.1289/ehp.1003160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice ME, Russo-Menna I. Differential compartmentalization of brain ascorbate and glutathione between neurons and glia. Neuroscience. 1997;82:1213–1223. doi: 10.1016/s0306-4522(97)00347-3. [DOI] [PubMed] [Google Scholar]
- Roegge C, Timofeva O, Seidler F, Slotkin TA, Levin ED. Persisting effects of early postnatal diazinon exposure on emotional reactivity in rats. Neurotoxicol Teratol. 2006;28:709. [Google Scholar]
- Rohlman DS, Anger WK, Lein PJ. Correlating neurobehavioral performance with biomarkers of organophosphorous pesticide exposure. Neurotoxicology. 2011;32:268–276. doi: 10.1016/j.neuro.2010.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rush T, Liu XQ, Hjelmhaug J, Lobner D. Mechanisms of chlorpyrifos and diazinon induced neurotoxicity in cortical culture. Neuroscience. 2010;166:899–906. doi: 10.1016/j.neuroscience.2010.01.025. [DOI] [PubMed] [Google Scholar]
- Sachana M, Flaskos J, Alexaki E, Glynn P, Hargreaves AJ. The toxicity of chlorpyrifos towards differentiating mouse N2a neuroblastoma cells. Toxicol In Vitro. 2001;15:369–372. doi: 10.1016/s0887-2333(01)00038-8. [DOI] [PubMed] [Google Scholar]
- Sagara JI, Miura K, Bannai S. Maintenance of neuronal glutathione by glial cells. J Neurochem. 1993;61:1672–1676. doi: 10.1111/j.1471-4159.1993.tb09802.x. [DOI] [PubMed] [Google Scholar]
- Sian J, Dexter DT, Lees AJ, Daniel S, Agid Y, Javoyagid F, Jenner P, Marsden CD. Alterations in glutathione levels in Parkinsons-disease and other neurodegenerative disorders affecting basal ganglia. Ann Neurol. 1994;36:348–355. doi: 10.1002/ana.410360305. [DOI] [PubMed] [Google Scholar]
- Sidiropoulou E, Sachana M, Flaskos J, Harris W, Hargreaves AJ, Woldehiwet Z. Diazinon oxon affects the differentiation of mouse N2a neuroblastoma cells. Arch Toxicol. 2009;83:373–380. doi: 10.1007/s00204-008-0339-1. [DOI] [PubMed] [Google Scholar]
- Slotkin TA, Bodwell BE, Levin ED, Seidler FJ. Neonatal exposure to low doses of diazinon: Long-term effects on neural cell development and acetylcholine systems. Environ Health Perspect. 2008;116:340–348. doi: 10.1289/ehp.11005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slotkin TA, Levin ED, Seidler FJ. Comparative developmental neurotoxicity of organophosphate insecticides: effects on brain development are separable from systemic toxicity. Environ Health Perspect. 2006;114:746–751. doi: 10.1289/ehp.8828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slotkin TA, Oliver CA, Seidler FJ. Critical periods for the role of oxidative stress in the developmental neurotoxicity of chlorpyrifos and terbutaline, alone or in combination. Dev Brain Res. 2005;157:172–180. doi: 10.1016/j.devbrainres.2005.04.001. [DOI] [PubMed] [Google Scholar]
- Slotkin TA, Seidler FJ. Oxidative and excitatory mechanisms of developmental neurotoxicity: transcriptional profiles for chlorpyrifos, diazinon, dieldrin, and divalent nickel in PC12 cells. Environ Health Perspect. 2009;117:587–596. doi: 10.1289/ehp.0800251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang GM, Rios PG, Kuo SH, Akman HO, Rosoklija G, Tanji K, Dwork A, Schon EA, DiMauro S, Goldman J, Sulzer D. Mitochondrial abnormalities in temporal lobe of autistic brain. Neurobiol Dis. 2013;54:349–361. doi: 10.1016/j.nbd.2013.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valko M, Leibfritz D, Moncol J, Cronin MT, Mazur M, Telser J. Free radicals and antioxidants in normal physiological functions and human disease. Int J Biochem Cell Biol. 2007;39:44–84. doi: 10.1016/j.biocel.2006.07.001. [DOI] [PubMed] [Google Scholar]
- VanDeMark KL, Guizzetti M, Giordano G, Costa LG. Ethanol inhibits muscarinic receptor-induced axonal growth in rat hippocampal neurons. Alcohol Clin Exp Res. 2009;33:1945–1955. doi: 10.1111/j.1530-0277.2009.01032.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargas MR, Johnson JA. The Nrf2-ARE cytoprotective pathway in astrocytes. Expert Rev Mol Med. 2009;11:e17. doi: 10.1017/S1462399409001094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viviani B, Corsini E, Galli CL, Marinovich M. Glia increase degeneration of hippocampal neurons through release of tumor necrosis factor-alpha. Toxicol Appl Pharm. 1998;150:271–276. doi: 10.1006/taap.1998.8406. [DOI] [PubMed] [Google Scholar]
- Vomhof-DeKrey EE, Picklo MJ. The Nrf2-antioxidant response element pathway: a target for regulating energy metabolism. J Nutr Biochem. 2012;23:1201–1206. doi: 10.1016/j.jnutbio.2012.03.005. [DOI] [PubMed] [Google Scholar]
- Watts LT, Rathinam ML, Schenker S, Henderson GI. Astrocytes protect neurons from ethanol-induced oxidative stress and apoptotic death. J Neurosci Res. 2005;80:655–666. doi: 10.1002/jnr.20502. [DOI] [PubMed] [Google Scholar]
- Won YK, Liu J, Olivier K, Zheng Q, Pope CN. Age-related effects of chlorpyrifos on acetylcholine release in rat brain. Neurotoxicology. 2001;22:39–48. doi: 10.1016/s0161-813x(00)00009-7. [DOI] [PubMed] [Google Scholar]
- Yanaka A, Fahey JW, Fukumoto A, Nakayama M, Inoue S, Zhang SH, Tauchi M, Suzuki H, Hyodo I, Yamamoto M. Dietary sulforaphane-rich broccoli sprouts reduce colonization and attenuate gastritis in helicobacter pylori-infected mice and humans. Cancer Prev Res (Phila) 2009;2:353–360. doi: 10.1158/1940-6207.CAPR-08-0192. [DOI] [PubMed] [Google Scholar]
- Yang D, Howard A, Bruun D, Ajua-Alemanj M, Pickart C, Lein PJ. Chlorpyrifos and chlorpyrifos-oxon inhibit axonal growth by interfering with the morphogenic activity of acetylcholinesterase. Toxicol Appl Pharm. 2008;228:32–41. doi: 10.1016/j.taap.2007.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yilmaz N, Yilmaz M, Altuntas I. Diazinon-induced brain toxicity and protection by vitamins E plus C. Toxicol. Ind Health. 2012;28:51–57. doi: 10.1177/0748233711404035. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.