

Abstract
Ca2+ signaling includes store-operated Ca2+ entry (SOCE) following depletion of endoplasmic reticulum (ER) Ca2+ stores. On store depletion, the ER Ca2+ sensor STIM1 activates Orai1, the pore-forming unit of Ca2+-release-activated Ca2+ (CRAC) channels. Here, we show that Orai1 is regulated by serum- and glucocorticoid-inducible kinase 1 (SGK1), a growth factor-regulated kinase. Membrane Orai1 protein abundance, ICRAC, and SOCE in human embryonic kidney (HEK293) cells stably expressing Orai1 and transfected with STIM1 were each significantly enhanced by coexpression of constitutively active S422DSGK1 (by+81, +378, and+136%, respectively) but not by inactive K127NSGK1. Coexpression of the ubiquitin ligase Nedd4-2, an established negatively regulated SGK1 target, down-regulated SOCE (by −48%) and ICRAC (by −60%), an effect reversed by expression of S422DSGK1 (by +175 and +173%, respectively). Orai1 protein abundance and SOCE were significantly lower in mast cells from SGK1-knockout (sgk1−/−) mice (by −37% and −52%, respectively) than in mast cells from wild-type (sgk1+/+) littermates. Activation of SOCE by sarcoplasmic/endoplasmic reticulum Ca2+-ATPase-inhibitor thapsigargin (2 μM) stimulated migration, an effect significantly higher (by +306%) in S422DSGK1-expressing than in K127NSGK1-expressing HEK293 cells, and also significantly higher (by +108%) in sgk1+/+ than in sgk1−/− mast cells. SGK1 is thus a novel key player in the regulation of SOCE.—Eylenstein, A., Gehring, E.-M., Heise, N., Shumilina, E., Schmidt, S., Szteyn, K., Münzer, P., Nurbaeva, M. K., Eichenmüller, M., Tyan, L., Regel, I., Föller, M., Kuhl, D., Soboloff, J., Penner, R., Lang, R. Stimulation of Ca2+-channel Orai1/STIM1 by serum- and glucocorticoid-inducible kinase 1 (SGK1).
Keywords: CRAC, store-operated calcium channel, SOCE, degranulation, migration
Cytosolic Ca2+ activity regulates several fundamental processes, such as excitation-contraction, exocytosis, migration, cell proliferation, and cell death (1–4). Increases in cytosolic Ca2+ can be accomplished by both release of Ca2+ from intracellular stores and Ca2+ entry across the cell membrane (5). Emptying of intracellular Ca2+ stores stimulates the Ca2+-release-activated Ca2+ (CRAC) channel (6). The channel is currently thought to consist of tetrameric assemblies (7–9) of the pore-forming units Orai 1, 2, or 3 (10–13), which bind to their regulators STIM 1 or 2 (9, 14, 15). Orai proteins have been shown to function as CRAC current (ICRAC) in a wide variety of tissues (16, 17), including lymphocytes (6, 18) and mast cells (19, 20). Orai and STIM have also been proposed to associate with TRPC channels, which might also contribute to store-operated Ca2+ entry (SOCE; refs. 21–23).
Orai and STIM are regulated by receptor for activated protein kinase C-1 (RACK1; ref. 23), arachidonic acid (24), reactive oxygen species (25), and lipid rafts (26). STIM1 has been shown to be inhibited by phosphorylation (27). The mechanisms linking growth factor signaling to Orai activity have, however, remained ill-defined.
Previously, SGK1 has been reported to regulate a wide variety of carriers and channels (28). The SGK1-sensitive ion channels include the renal epithelial Na+ channel (ENaC; refs. 29, 30). SGK1 regulates ENaC in part by phosphorylation of the ubiquitin ligase Nedd4-2 (neuronal precursor cells expressed developmentally down-regulated; ref. 31), which otherwise ubiquitinates ENaC and thus prepares the channel protein for clearance from the cell membrane and subsequent degradation (32). Phosphorylation of Nedd4-2 fosters binding of Nedd4-2 to 14-3-3, thus preventing its interaction with the target proteins (33, 34). SGK1 has been shown to stimulate the epithelial Ca2+ channels TRPV5 (35) and TRPV6 (36). Moreover, lack of SGK1 decreases the antigen-induced Ca2+ entry into mast cells (37). The present study explored whether Orai1/STIM1-dependent SOCE and ICRAC are sensitive to the serum- and glucocorticoid-inducible kinase SGK1, a kinase stimulated by growth factors and involved in the regulation of cell survival (28).
MATERIALS AND METHODS
DNA constructs, HEK293 cells, and transfection
HEK293 cells stably transfected with Orai1 (38) were cultured in Dulbecco's MEM (Life Technologies, Inc.; Invitrogen, Carlsbad, CA, USA), containing 1 mg/ml glucose and maintained in the presence of G418 (0.5 mg/ml; Gibco, Darmstadt, Germany). The cells were transfected transiently with 1–2 μg DNA encoding STIM1 (14), the constitutively active SGK1 mutant S422DSGK1 (hSGK1SD in pIRES-EGFP or pCDNA3.1; ref. 39), the inactive mutant K127NSGK1 (hSGK1KN in pIRES-EGFP or pCDNA3.1; ref. 39), or the ubiquitin ligase E3 Nedd4-2 (in pRFP) using FuGene HD transfection reagent (Roche Diagnostics, Mannheim, Germany) according to the manufacturer's instructions. SiRNA (10 nM; Life Technologies, Inc.; Ambion, Austin, TX, USA) was transfected according to the manufacturer's instructions using Polyplus Interferin (PeqLab, Erlangen, Germany).
Animals
Bone marrow was obtained from 6- to 8-wk-old female and male SGK1-knockout (sgk1−/−) mice and their wild-type (sgk1+/+) littermates. Generation, breeding, and genotyping of the mice have been described earlier (40).
Culture of bone marrow-derived mast cells (BMMCs)
Femoral BMMCs from 6- to 8-wk-old sgk1+/+ and sgk1−/− mice were cultured for 4 wk in RPMI 1640 (Gibco) containing 10% fetal calf serum, 1% penicillin/streptomycin, 20 ng/ml interleukin-3 (R&D Systems, Hessen, Germany), and 100 ng/ml of the c-kit ligand stem cell factor (SCF; Peprotech, Rocky Hill, NJ, USA). BMMC maturation was confirmed by flow cytometry (FACS Calibur; Becton Dickinson, Franklin Lakes, NJ, USA; ref. 37).
Whole-cell lysates
For total protein analysis, cells were harvested with lysis buffer (50 mM Tris, 150 mM NaCl, 1% Triton X-100, 0.5% Na-deoxycholate, 0.4% β-mercaptoethanol, and protease inhibitor cocktail; Roche) 48 h after transfection. Clarified protein lysate was applied to a polyacrylamide gel and analyzed by Western blot.
Western blot analysis
Proteins of whole-cell lysates (50 μg) were used for Western blot analysis and incubated with primary antibody against Orai1 (1:1000, Millipore, Bedford, MA, USA; or 1:200, Protein Tech, Chicago, IL, USA), Nedd4-2 (1:1000; Abcam, Cambridge, UK), or tubulin (1:1000; Cell Signaling, Danvers, MA, USA). For detection, a secondary anti-rabbit IgG antibody conjugated with horse radish peroxidase (HRP; 1:2000; Cell Signaling) was used. The blots were stripped and reprobed with tubulin to verify equal loading. Antibody binding was detected with ECL detection reagent (Amersham, Freiburg, Germany). Bands were quantified with Quantity One Software (Bio-Rad, Munich, Germany).
Biotinylation of cells
After transfection, HEK293 cells were cultured for 48 h, incubated with EZ-Link-Sulfo-NHS-Biotin (Pierce Protein Research Products, Rockford, IL, USA) at a final concentration of 0.5 mg/ml for 30 min and finally solubilized in lysis buffer (20 mM Tris-HCl, pH 7.4; 5 mM MgCl2; 5 mM Na2HPO4; 0.1% SDS; 1 mM EDTA; 1 mM PMSF; and 80 mM sucrose) containing protease inhibitor cocktail. Labeled protein (800 μg) was collected by rotating the lysates overnight at 4°C with Neutravidin-coated agarose beads (Pierce Protein Research). After elution, samples were analyzed by Western blotting using the affinity purified rabbit anti-Orai1 (intracellular) antibody (1 μg/ml, Millipore).
Immunofluorescence
BMMCs (5×104) were pipetted onto poly-l-lysine (Sigma-Aldrich, Taufkirchen, Germany)-coated microscope slides and incubated at 37°C for 24 h. BMMCs or HEK293 cells were fixated by incubating the cells for 15 min at room temperature in acetone/methanol (1:1), followed by permeabilization with 0.5% Triton X-100/PBS for 10 min. After blocking with 5% BSA/PBS, the cells were incubated overnight at 4°C (BMMCs) or for 2 h at room temperature (HEK293) with goat anti-Orai1 (BMMCs; 1:100; Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA) or rabbit anti-Orai1 (HEK293 cells; 1:500; Millipore) antibody, respectively. BMMC slides were incubated with the secondary anti-goat-FITC antibody (1:500; Abcam), and HEK293 slides were incubated with FITC-conjugated goat anti-rabbit IgG (H+L; 1:500; Santa Cruz Biotechnology) for 1.5 h at room temperature. Nuclei were stained with DRAQ-5 dye (1:2000; Biostatus, Shepshed, UK) for 5–10 min at room temperature. The slides were mounted with ProLong Gold antifade reagent (Invitrogen). Images were taken on a Zeiss LSM 5 Exciter Confocal Laser Scanning Microscope (Carl Zeiss MicroImaging GmbH, Overkochen, Germany) with a water-immersion Plan-Neofluar ×63/1.3 NA DIC.
Measurement of intracellular Ca2+ by fluorescence spectrometry
HEK293 cells (5×105 to 1×106) or BMMCs (1×106) were centrifuged at 1500 rpm for 5 min at room temperature. Afterward, the cells were loaded with Fura-2/AM (5 μM) in the presence of 0.2 μg/ml pluronic acid (Biotium Inc., Hayward, CA, USA). The staining was done for 30 min at 37°C. The Ca2+ measurements were performed in initially divalent cation-free solution (HEK293 cells) containing (in mM) 135 NaCl, 10 CsCl, 2.8 KCl, 10 HEPES, 0.1 EGTA, and 10 glucose, pH 7.2 (41) or PBS containing 0.1 mM EGTA (BMMCs) by using a LS 55 fluorescence spectrometer (PerkinElmer, Wellesley, MA, USA). After recording the baseline for 50 s, 5 μM thapsigargin (Invitrogen) and, after 10 min, 2 mM Ca2+ were added to the cell suspension. Cells were excited alternatively between 340 and 380 nm, whereas the emission was measured at 509 nm.
Measurement of intracellular Ca2+ concentration by fluorescence microscopy
Fura-2/AM fluorescence was utilized to determine intracellular Ca2+ by fluorescence microscopy (42). Cells were excited alternatively at 340 and 380 nm through the objective (Fluar ×40/1.30 oil; Zeiss) of an inverted phase-contrast microscope (Axiovert 100; Zeiss). Emitted fluorescence intensity was recorded at 505 nm. Data were acquired using specialized computer software (Metafluor; Universal Imaging, Burbank, CA, USA). Changes in cytosolic Ca2+ concentrations were estimated from the 340/380-nm ratio. HEK293 cells or BMMCs were loaded with Fura-2/AM (5 μM; Molecular Probes; Invitrogen) for 20 min at 37°C, 5% CO2. SOCE was determined by extracellular Ca2+ removal and subsequent Ca2+ readdition in the presence of 2 μM thapsigargin (43). For quantification of the Ca2+ entry, the slope (Δ ratio/s) and peak (Δ ratio) were calculated following readdition of Ca2+.
For intracellular calibration purposes, 10 μM ionomycin was applied at the end of each experiment. Experiments were performed with Ringer solution containing (in mM) 125 NaCl, 5 KCl, 1.2 MgSO4, 2 CaCl2, 2 Na2HPO4, 32 HEPES, and 5 glucose, pH 7.4. To reach nominally Ca2+-free conditions, experiments were performed using Ca2+-free Ringer solution containing (in mM) 125 NaCl, 5 KCl, 1.2 MgSO4, 2 Na2HPO4, 32 HEPES, 0.5 EGTA, and 5 glucose, pH 7.4.
Measurement of intracellular Ca2+ with flow cytometry (FACS)
HEK293 cells were trypsinized and centrifuged, resuspended in Dulbecco's MEM, and loaded with 2.5 μM fura red AM (Invitrogen) for 30 min at 37°C. Then 1 μM fluo3 was added to the cells, followed by incubation for 30 min at 37°C. The cells were subsequently collected and resuspended in Ca2+- and Mg2+-free PBS containing 2 μM thapsigargin. Cells were analyzed for basal levels of intracellular free Ca2+ on a FACSCalibur flow cytometer (BD Biosciences, Erembodegem, Belgium). After 25 s, Ca2+ was added to the samples at a final concentration of 2 mM, and intracellular Ca2+ levels were monitored for 300 s. The ratio of fluo3 to fura red was analyzed using Cell Quest Pro software (BD Biosciences) and FlowJo software (Tree Star, Inc., Ashland, OR, USA).
Patch clamp
Patch-clamp experiments were performed at room temperature in voltage-clamp, fast whole-cell mode. Borosilicate glass pipettes (2–4 MΩ tip resistance; GC 150 TF-10; Clark Medical Instruments, Pangbourne, UK) manufactured by a microprocessor-driven DMZ puller (Zeitz, Augsburg, Germany) were used in combination with a MS314 electrical micromanipulator (MW; Märzhäuser, Wetzlar, Germany). The currents were recorded by an EPC-9 amplifier (Heka, Freiburg, Germany) using Pulse software (Heka) and an ITC-16 Interface (Instrutech, Port Washington, NY, USA). For ICRAC measurements, whole-cell currents were elicited by 200-ms square-wave voltage pulses from −100 to +80 mV in 20-mV steps, delivered from a holding potential of 0. Alternatively, the currents were recorded with 200-ms voltage ramps from −120 to +100 mV. Leak currents, determined as the currents at the very beginning of each experiment, immediately after reaching the whole-cell mode, were subtracted. All voltages were corrected for a liquid-junction potential of 15 mV. The currents were recorded with an acquisition frequency of 10 kHz and 3-kHz low-pass filtered.
For ICRAC measurements, cells were superfused with a bath solution containing (in mM) 140 NaCl, 5 KCl, 10 CaCl2, 20 glucose, and 10 HEPES/NaOH, pH 7.4. The patch-clamp pipettes were filled with an internal solution containing (in mM) 128 Cs-aspartate, 10 CsBAPTA, 8 MgCl2, 10 HEPES/CsOH, and 0.02 inositol 1,4,5-trisphosphate [Ins(1,4,5)P3], pH 7.2. Cs-BAPTA was from Invitrogen, all other chemicals from Sigma-Aldrich.
Migration
For migration assays, transwell inserts (BD Falcon; 353097) and BD BioCoat Matrigel Invasion Chambers (354480; BD Biosciences) with a pore diameter of 8 μm were used. The transwells were placed in a 24-well culture plate containing cell culture medium (750 μl) with or without 2 μM thapsigargin in the lower chamber. The upper chambers were filled with 500 μl cell culture medium containing HEK293 cells at a concentration of 5 × 104 cells/ml, with or without 2 μM thapsigargin. After an incubation time of 32 h at 37°C, migrated cells were analyzed by staining the migrated cells with DAPI. Beforehand, nonmigrated cells were removed by scrubbing with a cotton-tipped swab twice and washing with PBS. After 15 min fixation in 4% PFA, the membranes were removed with a scalpel and mounted with ProLong Gold antifade reagent (Invitrogen). To determine the total number of migrating cells, the slices were then viewed under the microscope, and the number of cells per field in representative areas was counted. Experiments were performed in triplicates.
For the migration of mast cells from sgk1+/+ and sgk1−/− mice, 4 × 105 cells/ml were used with or without 100 μM 2-aminoethoxydiphenyl borate (2-APB) and 2 μM thapsigargin. Cells were incubated for 3 h at 37°C and analyzed as described above.
Statistics
Data are provided as means ± se; n = number of independent experiments. All data were tested for significance using Student's unpaired 2-tailed t test, 1-sample t test, or ANOVA (Tukey's test), where applicable. Results with values of P < 0.05 were considered statistically significant.
RESULTS
In HEK293 cells stably expressing Orai1 and transiently transfected with STIM1, addition of the store-depleting SERCA inhibitor thapsigargin (5 μM) in nominally Ca2+-free solution was followed by rapid, transient increases in cytosolic Ca2+, as measured by fluorescent spectrometry (Fig. 1A). Subsequent addition of Ca2+ to the extracellular bath resulted in a rapid and sustained increase in cytosolic Ca2+ due to SOCE. Both slope and peak of SOCE were enhanced significantly by transient expression of the constitutively active mutant S422DSGK1 but not the inactive mutant K127NSGK1 (Fig. 1B).
Figure 1.
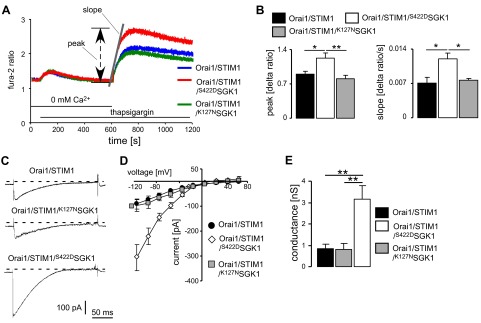
Effect of SGK1 on SOCE and ICRAC in HEK293 cells expressing Orai1 and STIM1. A) SOCE in HEK293 cells. Representative tracings of Fura-2 fluorescence ratio in fluorescence spectrometry during and after Ca2+ depletion and thapsigargin (5 μM) addition in HEK293 cells stably expressing Orai1, transfected with STIM1 (blue), STIM1/S422DSGK1 (red), and STIM1/K127NSGK1 (green). Slope (Δ ratio/s) and peak (Δ ratio) following readdition of Ca2+ were determined. B) Arithmetic means (n=5–6 independent experiments; 5×105 cells/experiment) of peak and slope of Ca2+ increase in HEK293 cells following readdition of Ca2+, determined by fluorescence spectrometry. C) Original ramp currents in HEK293 cells expressing Orai1 and transfected with STIM1, STIM1/K127NSGK1, or STIM1/S422DSGK1. D) Mean I/V relationships constructed from the currents elicited by the step pulses (n=5–6) of ICRAC in HEK293 cells. E) Mean conductance (n=5–6) calculated by linear regression of I/V curves between −115 and −35 mV in HEK293 cells. Values are means ± se. *P < 0.05, **P < 0.01; ANOVA.
The basal Fura-2 fluorescence ratio, reflecting resting intracellular Ca2+ concentration, in Orai1/STIM1-expressing cells was similar in control cells (1.46±0.17, n=5, 5×105 cells/experiment), cells expressing the constitutively active S422DSGK1 (1.39±0.04, n=6, 5×105 cells/experiment) and cells expressing the inactive mutant K127NSGK1 (1.29±0.06, n=6, 5×105 cells/experiment). Likewise, the increase of the Fura-2 fluorescence ratio due to Ca2+ release following thapsigargin (5 μM) treatment in the continued absence of extracellular Ca2+ was similar in Orai1/STIM1-expressing cells (Δ ratio=0.52±0.09, n=5, 5×105 cells/experiment), Orai1/STIM1-expressing cells additionally expressing S422DSGK1 (Δ ratio=0.51±0.12, n=6, 5×105 cells/experiment) and Orai1/STIM1-expressing cells additionally expressing K127NSGK1 (Δ ratio=0.65±0.06, n=6, 5×105 cells/experiment).
Similar results were obtained in a second series of experiments utilizing Fura-2 fluorescence imaging (data not shown). Both slope and peak of the increase in cytosolic Ca2+ activity following readdition of Ca2+ after store depletion with thapsigargin (2 μM) and exposure to Ca2+-free extracellular fluid were enhanced by transient expression of STIM1. The additional expression of S422DSGK1 led again to a significant further enhancement of Ca2+ entry. In contrast, the coexpression of K127NSGK1 again failed to significantly modify Ca2+ entry.
The alterations of SOCE were paralleled by corresponding changes in ICRAC (Fig. 1C–E). ICRAC in HEK293 cells expressing both STIM1 and Orai1 was increased significantly by coexpression of S422DSGK1, but not by K127NSGK1.
The observed increases in Ca2+ entry could be due to either activation of Ca2+ channels or increases in channel protein abundance. According to confocal microscopy, the protein abundance of Orai1 in the plasma membrane of Orai1-expressing HEK293 cells was increased significantly following transfection with STIM1. A significant further increase of Orai1 protein abundance was observed following additional transfection with S422DSGK1 but not following additional transfection with K127NSGK1 (Fig. 2A, B). Western blot analysis revealed that total Orai1 protein abundance was enhanced by transfection with S422DSGK1, but not by K127NSGK1 (Fig. 2C). Furthermore, biotinylation studies revealed that S422DSGK1 increased the Orai1 protein abundance in the plasma membrane of HEK293 cells (Fig. 2D).
Figure 2.
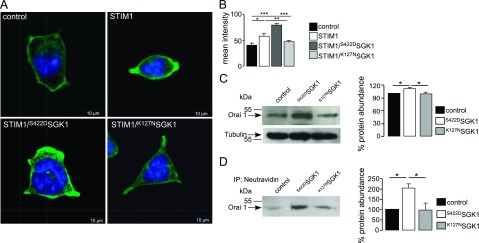
Effect of SGK1 on Orai1 protein abundance of HEK293 cells expressing Orai1 and STIM1. A) Confocal microscopy of Orai1 abundance in HEK293 cells stably expressing Orai1. Shown are Orai1-expressing HEK293 cells transfected with control plasmid (control), with STIM1, STIM1 and the active mutant S422DSGK1, or STIM1 and the inactive mutant K127NSGK1. Nuclei are stained with DRAQ5 (blue); Orai1 with FITC-conjugated secondary antibody (green). B) Arithmetic means of Orai1 protein abundance in the cell membrane, estimated by confocal microscopy, in Orai1-expressing HEK293 cells transfected with control plasmid, STIM1, STIM1/S422DSGK1, or STIM1/K127NSGK1 (n=5 independent experiments; 13 cells/experiment). C) Western blot analysis of whole-cell lysate protein of Orai1. Left panel: representative experiment showing Orai1 band and after stripping the Tubulin control band. Right panel: arithmetic means (n=4). D) Western blot analysis of biotinylated Orai1 in HEK293 cells. Orai1 was precipitated by NeutrAvidin beads binding to Biotin. Arrow indicates Orai1 band. Left panel: representative experiment. Right panel: arithmetic means (n=6). Values are means ± se. *P < 0.05, **P < 0.01, ***P < 0.001; ANOVA.
We suggest that the increased Orai1 protein abundance resulted from decreased retrieval and degradation of channel proteins. SGK1 is known to phosphorylate the ubiquitin ligase Nedd4-2, which ubiquitinates several channel and carrier proteins in the plasma membrane and thus initiates their degradation (44, 45). SGK1-dependent phosphorylation results in binding of Nedd4-2 to the scaffolding protein 14-3-3, which prevents the interaction of Nedd4-2 with target proteins (44). Therefore, we examined the relationship between Orai1 and Nedd4-2. To this end, the Orai1 protein abundance of HEK293 cells stably expressing Orai1 was determined following transfection with control plasmid, with Nedd4-2, and with Nedd4-2 and S422DSGK1 (Fig. 3). Nedd4-2 transfection led to a marked decrease in Orai1 protein abundance, an effect that was reversed by additional expression of S422DSGK1 (Fig. 3A, B). Conversely, silencing of Nedd4-2 by siRNA, which decreased Nedd4-2 protein abundance by up to 60%, significantly elevated the expression of Orai1 (Fig. 3C). Furthermore, expression of Nedd4-2 led to a decrease in Orai1 abundance, an effect reversed by coexpression with S422DSGK1 (Fig. 3D). These changes in Orai1 protein abundance were paralleled by corresponding alterations in SOCE.
Figure 3.
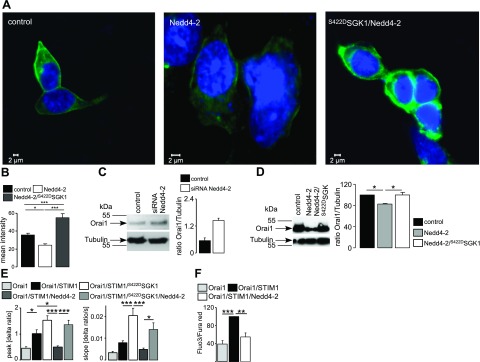
Effect of ubiquitin ligase Nedd4-2 on Orai1 protein abundance and SOCE in HEK293 cells expressing Orai1 and STIM1. A) Confocal microscopy of Orai1 protein abundance in HEK293 cells stably expressing Orai1, transfected with control plasmid, Nedd4-2, or Nedd4-2/S422DSGK1. B) Arithmetic means of Orai1 protein abundance, determined by confocal microscopy in HEK293 cells stably expressing Orai1, transfected with control plasmid, Nedd4-2, or Nedd4-2/S422DSGK1 (n=4 independent experiments; 9 cells/experiment). C) Orai1 protein abundance of whole-lysate protein in HEK293 cells transfected with scrambled siRNA (control) or siRNA for Nedd4-2. Left panel: representative blot. Right panel: arithmetic means (n=4). D) Orai1 protein abundance of whole-cell lysate protein in HEK293 cells transfected with control plasmid, Nedd4-2, or Nedd4-2 and S422DSGK1. Left panel: representative blot. Right panel: arithmetic means (n=5). E) Arithmetic means (n=14–27) of peak and slope increase of Fura-2 fluorescence ratio in cells expressing Orai1 alone, Orai1/STIM1, Orai1/STIM1/S422DSGK1, Orai1/STIM1/Nedd4-2, or Orai1/STIM1/Nedd4-2/S422DSGK1. F) Geometric means (n=6) of change in the fluo3/fura red fluorescence ratio for HEK293 control cells (light shaded bar), STIM1-transfected cells (solid bar), and cells transfected with STIM1 and Nedd4-2 (dark shaded bar). Values are means ± se. *P < 0.05, **P < 0.01, ***P < 0.001; ANOVA.
Ca2+ entry into HEK293 cells stably expressing Orai1 was significantly enhanced by additional transfection with STIM1 and was significantly decreased by additional expression of Nedd4-2 (Fig. 3E). The effect of SOCE was confirmed by patch-clamp analysis showing decreased ICRAC after cotransfection of Nedd4-2 in Orai1- and STIM1-expressing HEK293 cells. ICRAC conductances assessed between −115 and −35 mV were significantly (P<0.002) lower in Orai1/STIM1/Nedd4-2-expressing cells (0.83±0.11 nS, n=26) than in cells expressing Orai1/STIM1 alone (2.09±0.46 nS, n=22). Cotransfection of S422DSGK1 with Nedd4-2 partially restored ICRAC (conductance 1.44±0.17 nS, n=21; significantly different from Orai1/STIM1/Nedd4-2-expressing cells, P<0.004). Down-regulation of SOCE following transfection of Nedd4-2 was also observed in FACS analysis (Fig. 3F).
To determine whether SGK1-dependent regulation is relevant for native Orai1 protein abundance in nontransfected cells, Orai1 expression of BMMCs was determined by immunocytochemistry. As illustrated in Fig. 4A, B, Orai1 protein abundance was significantly lower in BMMCs from sgk1−/− mice than in BMMCs from sgk1+/+ mice. Orai1 expression was not uniform in mast cells, but the average abundance of Orai1 was clearly and significantly weaker in sgk1−/− than in sgk1+/+ BMMCs. Accordingly, the rate of Ca2+ entry following store depletion was significantly smaller in sgk1−/− BMMCs than in sgk1+/+ BMMCs (Fig. 4C, D).
Figure 4.

Orai1 protein abundance and SOCE in BMMCs from sgk1+/+ and sgk1−/−mice. A) Confocal microscopy of Orai1 protein abundance in sgk1+/+BMMCs (left panel) and sgk1−/− BMMCs (right panel). Nuclei are blue (DRAQ5); Orai1 is green (FITC-conjugated antibody). B) Arithmetic means of Orai1 protein abundance, estimated by confocal microscopy (n=5 independent experiments; 16 cells/experiment). C) Representative tracings of Fura-2 fluorescence ratio in sgk1+/+BMMCs and sgk1−/−BMMCs during and after Ca2+ depletion in the presence of thapsigargin (5 μM). D) Arithmetic means (n=5 independent experiments; 5×105 cells/experiment) of peak (Δ ratio) and slope (Δ ratio/s) of Fura-2 fluorescence ratio increase following readdition of Ca2+ in sgk1+/+BMMCs (open bars) and sgk1−/−BMMCs (solid bars), measured by fluorescence spectrometry. Values are means ± se. *P < 0.05, **P < 0.01; t test.
A further series of experiments assessed the effect of SGK1-dependent regulation of Orai1 on cell migration, which was estimated from translocation of cells from one chamber to another across a membrane with a pore diameter of 8 μm. In Orai1-transfected HEK293 cells stably expressing STIM1, migration was stimulated by thapsigargin (2 μM). Both, prior to and following thapsigargin treatment, the number of migrated cells was significantly higher when cells were cotransfected with S422DSGK1 (171±26 and 323±30, respectively) compared to cotransfection with K127NSGK1 (39±7 and 80±19, respectively) or even without transfection (75±10 and 221±26, respectively; Fig. 5A). Similar observations were made in HEK293 cells stably expressing Orai1 after transfection with STIM1, STIM1/S422DSGK1, and STIM1/K127NSGK1 (data not shown).
Figure 5.

Influence of SGK1 on the migration of Orai1-expressing HEK293 cells and BMMCs. A) Arithmetic means (n=6) of the normalized migration of HEK293 cells expressing STIM1 alone; STIM1 and Orai1; STIM1, Orai1, and S422DSGK1; or STIM1, Orai1, and K127NSGK1 in the absence or presence of 2 μM thapsigargin. B) Arithmetic means (n=3–4 mice/group) of normalized migration of mast cells from sgk1−/− mice and from their sgk1+/+ littermates in the absence or the presence of 2 μM thapsigargin or additional treatment with the SOCE inhibitor 2-APB. Values are means ± se. *P < 0.05, **P < 0.01, ***P < 0.001 between indicated groups; #P < 0.05, ##P < 0.01, ###P < 0.001 vs. corresponding control group; ANOVA.
The effect of SGK1 on migration was further determined by analysis of sgk1−/− and sgk1+/+ BMMCs. Both prior to and following the administration of thapsigargin, significantly fewer sgk1−/− BMMCs (10±1 and 24±1, respectively) crossed the membrane in transwell chambers than sgk1+/+ BMMCs (21±2 and 49±5, respectively; Fig. 5B). After adding the SOCE inhibitor 2-APB, the migration rate was significantly reduced (22±1 in sgk1+/+ and 10±1 in sgk1−/−). Migration of sgk1+/+ BMMCs was similar to migration of HEK cells with transfection of either STIM1 or Orai1 alone, but was less than the migration of HEK cells overexpressing both Orai1 and STIM1.
DISCUSSION
The present study reveals a powerful influence of the serum- and glucocorticoid-inducible kinase SGK1 on store-operated Ca2+ entry. We show that transfection of HEK293 cells with constitutively active S422DSGK1, but not with inactive K127NSGK1, enhances SOCE by increasing the CRAC current. Orai1 protein abundance in the cell membrane and SOCE were significantly lower in BMMCs sgk1−/− mice compared to the corresponding sgk1+/+ mice.
Additional studies disclose the underlying mechanism. We demonstrate that Orai1 is a target of the ubiquitin ligase Nedd4-2, which previously has been shown to ubiquitinate, and thus regulate, the ENaC (45–47) and further channels and carriers (48–51). As shown in other cells, SGK1 phosphorylates Nedd4-2, leading to binding of the phosphorylated protein to a heterodimeric protein complex composed of 14-3-3β and 14-3-3ε (52). Nedd4-2 bound to 14-3-3 is thus unable to ubiquitinate its targets (53, 54). Accordingly, phosphorylation of Nedd4-2 by SGK1 is followed by decreased ubiquitination and subsequent degradation of the channel protein.
SGK1 may modify cytosolic Ca2+ activity not only by up-regulation of Orai1. SGK1 has previously been shown to increase the activity of the Ca2+-permeable TRP channels TRPV5 (55) and TRPV6 (56). Moreover, SGK1 up-regulates a wide variety of K+ channels (57, 58). Activation of those channels is expected to hyperpolarize the cell membrane and thus enhance the driving force for Ca2+ entry through several Ca2+ channels.
SGK1 expression (59, 60) and activity (61) are stimulated by increased cytosolic Ca2+ activity. Thus, at least in theory, SGK1 could serve as an amplifier of Ca2+ entry.
SGK1 is functionally relevant for cell migration, which has previously been shown to critically depend on Ca2+ signaling (62). Cell migration is stimulated by emptying intracellular Ca2+ stores with thapsigargin, and this is augmented by expression of Orai1, STIM1, and constitutively active SGK1, but not by coexpression of inactive SGK1. We further show that thapsigargin stimulates migration in BMMCs from sgk1+/+ mice, an effect markedly decreased in BMMCs from sgk1−/− mice. The residual difference of migration between sgk1+/+ and sgk1−/− BMMCs in the presence of the Ca2+ channel blocker 2-APB may point to some additional, Orai1-independent effect of SGK1 on migration.
SGK1-dependent regulation of SOCE might further be functionally relevant for degranulation. As shown previously (37), antigen-induced Ca2+ entry, activation of Ca2+-activated K+ channels, and degranulation were all markedly depressed in BMMCs from sgk1−/− mice. Moreover, the anaphylactic reaction, which critically depends on mast cell function (63), was strongly impaired in those mice (37). However, those earlier studies did not define the SGK1-sensitive Ca2+ entry mechanism involved.
The role of SGK1 in the regulation of Orai1/STIM1 is presumably not restricted to degranulation or migration. SGK1 is ubiquitously expressed (28), and Orai1 and STIM1 have been identified in a wide variety of tissues (16, 17, 64, 65) including those of the immune system (6, 18, 66, 67).
SGK1 activity is triggered by PI3 kinase and phosphoinositide-dependent kinase PDK1 signaling (28), which is stimulated by growth factors (68–70). Along those lines, Ca2+ entry into mast cells is stimulated similarly by PDK1 (71). Thus, the SGK1-dependent regulation of Orai1 establishes a previously unknown link between growth factor receptor-mediated signaling and control of SOCE. SGK1 might be up-regulated in tumor cells (28, 48, 72, 73) and Orai1 (74–76) as well as SGK1 (77–79) have been shown to be critically important in the regulation of cell proliferation. The SGK1-dependent regulation of Orai1 might thus participate in the machinery underlying tumor cell proliferation and migration.
In summary, SGK1 is of critical importance for the regulation of Orai1/STIM1, thereby indirectly modulating the numerous ICRAC-dependent functions, including excitation, exocytosis, migration, cell proliferation, and cell death. In the present work, this role was specifically demonstrated for migration. However, since STIM1, Orai1, and SGK1 are ubiquitously expressed, our findings are presumably relevant for diverse functions in multiple cell types. In fact, SGK1 has been shown to participate in a multitude of physiological and pathophysiological mechanisms (28), which may in part prove to be secondary to Orai regulation.
Acknowledgments
The authors acknowledge the technical assistance of E. Faber and the preparation of the manuscript by T. Loch and L. Subasic.
This study was supported by the Deutsche Forschungsgemeinschaft (GK 1302, SFB 773).
REFERENCES
- 1. Burgoyne R. D. (2007) Neuronal calcium sensor proteins. generating diversity in neuronal Ca2+ signalling. Nat. Rev. Neurosci. 8, 182–193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Orrenius S., Zhivotovsky B., Nicotera P. (2003) Regulation of cell death. the calcium-apoptosis link. Nat. Rev. Mol. Cell. Biol. 4, 552–565 [DOI] [PubMed] [Google Scholar]
- 3. Roderick H. L., Cook S. J. (2008) Ca2+ signalling checkpoints in cancer: remodelling Ca2+ for cancer cell proliferation and survival. Nat. Rev. Cancer 8, 361–375 [DOI] [PubMed] [Google Scholar]
- 4. Salter R. D., Watkins S. C. (2009) Dendritic cell altered states: what role for calcium? Immunol. Rev. 231, 278–288 [DOI] [PubMed] [Google Scholar]
- 5. Parekh A. B., Penner R. (1997) Store depletion and calcium influx. Physiol. Rev. 77, 901–930 [DOI] [PubMed] [Google Scholar]
- 6. Cahalan M. D., Zhang S. L., Yeromin A. V., Ohlsen K., Roos J., Stauderman K. A. (2007) Molecular basis of the CRAC channel. Cell Calcium 42, 133–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ji W., Xu P., Li Z., Lu J., Liu L., Zhan Y., Chen Y., Hille B., Xu T., Chen L. (2008) Functional stoichiometry of the unitary calcium-release-activated calcium channel. Proc. Natl. Acad. Sci. U. S. A. 105, 13668–13673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mignen O., Thompson J. L., Shuttleworth T. J. (2008) Orai1 subunit stoichiometry of the mammalian CRAC channel pore. J. Physiol. 586, 419–425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Penna A., Demuro A., Yeromin A. V., Zhang S. L., Safrina O., Parker I., Cahalan M. D. (2008) The CRAC channel consists of a tetramer formed by Stim-induced dimerization of Orai dimers. Nature 456, 116–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Prakriya M., Feske S., Gwack Y., Srikanth S., Rao A., Hogan P. G. (2006) Orai1 is an essential pore subunit of the CRAC channel. Nature 443, 230–233 [DOI] [PubMed] [Google Scholar]
- 11. Putney J. W., Jr. (2007) New molecular players in capacitative Ca2+ entry. J. Cell Sci. 120, 1959–1965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Vig M., Peinelt C., Beck A., Koomoa D. L., Rabah D., Koblan-Huberson M., Kraft S., Turner H., Fleig A., Penner R., Kinet J. P. (2006) CRACM1 is a plasma membrane protein essential for store-operated Ca2+ entry. Science 312, 1220–1223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yeromin A. V., Zhang S. L., Jiang W., Yu Y., Safrina O., Cahalan M. D. (2006) Molecular identification of the CRAC channel by altered ion selectivity in a mutant of Orai. Nature 443, 226–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Peinelt C., Vig M., Koomoa D. L., Beck A., Nadler M. J., Koblan-Huberson M., Lis A., Fleig A., Penner R., Kinet J. P. (2006) Amplification of CRAC current by STIM1 and CRACM1 (Orai1). Nat. Cell Biol. 8, 771–773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhang S. L., Yu Y., Roos J., Kozak J. A., Deerinck T. J., Ellisman M. H., Stauderman K. A., Cahalan M. D. (2005) STIM1 is a Ca2+ sensor that activates CRAC channels and migrates from the Ca2+ store to the plasma membrane. Nature 437, 902–905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Peel S. E., Liu B., Hall I. P. (2008) ORAI and store-operated calcium influx in human airway smooth muscle cells. Am. J. Respir. Cell. Mol. Biol. 38, 744–749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shin D. M., Muallem S. (2008) Skeletal muscle dressed in SOCs. Nat. Cell Biol. 10, 639–641 [DOI] [PubMed] [Google Scholar]
- 18. Feske S. (2007) Calcium signalling in lymphocyte activation and disease. Nat. Rev. Immunol. 7, 690–702 [DOI] [PubMed] [Google Scholar]
- 19. Baba Y., Nishida K., Fujii Y., Hirano T., Hikida M., Kurosaki T. (2008) Essential function for the calcium sensor STIM1 in mast cell activation and anaphylactic responses. Nat. Immunol. 9, 81–88 [DOI] [PubMed] [Google Scholar]
- 20. Vig M., DeHaven W. I., Bird G. S., Billingsley J. M., Wang H., Rao P. E., Hutchings A. B., Jouvin M. H., Putney J. W., Kinet J. P. (2008) Defective mast cell effector functions in mice lacking the CRACM1 pore subunit of store-operated calcium release-activated calcium channels. Nat. Immunol. 9, 89–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dietrich A., Kalwa H., Gudermann T. (2010) TRPC channels in vascular cell function. Thromb. Haemost. 103, 262–270 [DOI] [PubMed] [Google Scholar]
- 22. Liao Y., Plummer N. W., George M. D., Abramowitz J., Zhu M. X., Birnbaumer L. (2009) A role for Orai in TRPC-mediated Ca2+ entry suggests that a TRPC. Orai complex may mediate store and receptor operated Ca2+ entry. Proc. Natl. Acad. Sci. U. S. A. 106, 3202–3206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Woodard G. E., Lopez J. J., Jardin I., Salido G. M., Rosado J. A. (2010) TRPC3 regulates agonist-stimulated Ca2+ mobilization by mediating the interaction between type I inositol 1,4,5-trisphosphate receptor, RACK1, and Orai1. J. Biol. Chem. 285, 8045–8053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Shuttleworth T. J. (2009) Arachidonic acid, ARC channels, and Orai proteins. Cell Calcium 45, 602–610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bogeski I., Kummerow C., Al Ansary D., Schwarz E. C., Koehler R., Kozai D., Takahashi N., Peinelt C., Griesemer D., Bozem M., Mori Y., Hoth M., Niemeyer B. A. (2010) Differential redox regulation of ORAI ion channels. a mechanism to tune cellular calcium signaling. Sci. Signal 3, ra24 [DOI] [PubMed] [Google Scholar]
- 26. Pani B., Singh B. B. (2009) Lipid rafts/caveolae as microdomains of calcium signaling. Cell Calcium 45, 625–633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Smyth J. T., Petranka J. G., Boyles R. R., DeHaven W. I., Fukushima M., Johnson K. L., Williams J. G., Putney J. W., Jr. (2009) Phosphorylation of STIM1 underlies suppression of store-operated calcium entry during mitosis. Nat. Cell Biol. 11, 1465–1472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lang F., Bohmer C., Palmada M., Seebohm G., Strutz-Seebohm N., Vallon V. (2006) (Patho)physiological significance of the serum- and glucocorticoid-inducible kinase isoforms. Physiol. Rev. 86, 1151–1178 [DOI] [PubMed] [Google Scholar]
- 29. Chen S. Y., Bhargava A., Mastroberardino L., Meijer O. C., Wang J., Buse P., Firestone G. L., Verrey F., Pearce D. (1999) Epithelial sodium channel regulated by aldosterone-induced protein sgk. Proc. Natl. Acad. Sci. U. S. A. 96, 2514–2519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Naray-Fejes-Toth A., Canessa C., Cleaveland E. S., Aldrich G., Fejes-Toth G. (1999) sgk is an aldosterone-induced kinase in the renal collecting duct. Effects on epithelial na+ channels. J. Biol. Chem. 274, 16973–16978 [DOI] [PubMed] [Google Scholar]
- 31. Debonneville C., Flores S. Y., Kamynina E., Plant P. J., Tauxe C., Thomas M. A., Munster C., Chraibi A., Pratt J. H., Horisberger J. D., Pearce D., Loffing J., Staub O. (2001) Phosphorylation of Nedd4-2 by Sgk1 regulates epithelial Na(+) channel cell surface expression. EMBO J. 20, 7052–7059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Staub O., Gautschi I., Ishikawa T., Breitschopf K., Ciechanover A., Schild L., Rotin D. (1997) Regulation of stability and function of the epithelial Na+ channel (ENaC) by ubiquitination. EMBO J. 16, 6325–6336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bhalla V., Daidie D., Li H., Pao A. C., LaGrange L. P., Wang J., Vandewalle A., Stockand J. D., Staub O., Pearce D. (2005) Serum- and glucocorticoid-regulated kinase 1 regulates ubiquitin ligase neural precursor cell-expressed, developmentally down-regulated protein 4–2 by inducing interaction with 14-3-3. Mol. Endocrinol. 19, 3073–3084 [DOI] [PubMed] [Google Scholar]
- 34. Ichimura T., Yamamura H., Sasamoto K., Tominaga Y., Taoka M., Kakiuchi K., Shinkawa T., Takahashi N., Shimada S., Isobe T. (2005) 14-3-3 proteins modulate the expression of epithelial Na+ channels by phosphorylation-dependent interaction with Nedd4-2 ubiquitin ligase. J. Biol. Chem. 280, 13187–13194 [DOI] [PubMed] [Google Scholar]
- 35. Embark H. M., Setiawan I., Poppendieck S., van de Graaf S. F., Boehmer C., Palmada M., Wieder T., Gerstberger R., Cohen P., Yun C. C., Bindels R. J., Lang F. (2004) Regulation of the epithelial Ca2+ channel TRPV5 by the NHE regulating factor NHERF2 and the serum and glucocorticoid inducible kinase isoforms SGK1 and SGK3 expressed in Xenopus oocytes. Cell. Physiol. Biochem. 14, 203–212 [DOI] [PubMed] [Google Scholar]
- 36. Sopjani M., Kunert A., Czarkowski K., Klaus F., Laufer J., Foller M., Lang F. (2010) Regulation of the Ca(2+) channel TRPV6 by the kinases SGK1, PKB/Akt, and PIKfyve. J. Membr. Biol. 233, 35–41 [DOI] [PubMed] [Google Scholar]
- 37. Sobiesiak M., Shumilina E., Lam R. S., Wölbing F., Matzner N., Kaesler S., Zemtsova I. M., Lupescu A., Zahir N., Kuhl D., Biedermann T., Lang F. (2009) Impaired mast cell activation in gene targeted mice lacking the serum- and glucocorticoid-inducible kinase SGK1. J. Immunol. 183, 4395–4402 [DOI] [PubMed] [Google Scholar]
- 38. Zhou Y., Mancarella S., Wang Y., Yue C., Ritchie M., Gill D. L., Soboloff J. (2009) The short N-terminal domains of STIM1 and STIM2 control the activation kinetics of Orai1 channels. J. Biol. Chem. 284, 19164–19168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lang F., Klingel K., Wagner C. A., Stegen C., Warntges S., Friedrich B., Lanzendorfer M., Melzig J., Moschen I., Steuer S., Waldegger S., Sauter M., Paulmichl M., Gerke V., Risler T., Gamba G., Capasso G., Kandolf R., Hebert S. C., Massry S. G., Broer S. (2000) Deranged transcriptional regulation of cell-volume-sensitive kinase hSGK in diabetic nephropathy. Proc. Natl. Acad. Sci. U. S. A. 97, 8157–8162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wulff P., Vallon V., Huang D. Y., Volkl H., Yu F., Richter K., Jansen M., Schlunz M., Klingel K., Loffing J., Kauselmann G., Bosl M. R., Lang F., Kuhl D. (2002) Impaired renal Na(+) retention in the sgk1-knockout mouse. J. Clin. Invest. 110, 1263–1268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Liao Y., Erxleben C., Yildirim E., Abramowitz J., Armstrong D. L., Birnbaumer L. (2007) Orai proteins interact with TRPC channels and confer responsiveness to store depletion. Proc. Natl. Acad. Sci. U. S. A. 104, 4682–4687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Shumilina E., Lam R. S., Wölbing F., Matzner N., Zemtova I. M., Sobiesiak M., Mahmud H., Sausbier U., Biedermann T., Ruth P., Sausbier M., Lang F. (2008) Blunted IgE-mediated activation of mast cells in mice lacking the Ca2+-activated K+ channel KCa3.1. J. Immunol. 180, 8040–8047 [DOI] [PubMed] [Google Scholar]
- 43. Bird G. S., DeHaven W. I., Smyth J. T., Putney J. W., Jr. (2008) Methods for studying store-operated calcium entry. Methods 46, 204–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bhalla V., Soundararajan R., Pao A. C., Li H., Pearce D. (2006) Disinhibitory pathways for control of sodium transport. regulation of ENaC by SGK1 and GILZ. Am. J. Physiol. Renal Physiol. 291, F714–F721 [DOI] [PubMed] [Google Scholar]
- 45. Snyder P. M. (2009) Down-regulating destruction. phosphorylation regulates the E3 ubiquitin ligase Nedd4-2. Sci. Signal. 2, e41 [DOI] [PubMed] [Google Scholar]
- 46. Krueger B., Haerteis S., Yang L., Hartner A., Rauh R., Korbmacher C., Diakov A. (2009) Cholesterol depletion of the plasma membrane prevents activation of the epithelial sodium channel (ENaC) by SGK1. Cell. Physiol. Biochem. 24, 605–618 [DOI] [PubMed] [Google Scholar]
- 47. Flores S. Y., Debonneville C., Staub O. (2003) The role of Nedd4/Nedd4-like dependant ubiquitylation in epithelial transport processes. Pflügers Arch. 446, 334–338 [DOI] [PubMed] [Google Scholar]
- 48. Lang F., Perrotti N., Stournaras C. (2010) Colorectal carcinoma cells–regulation of survival and growth by SGK1. Int. J. Biochem. Cell. Biol. 42, 1571–1575 [DOI] [PubMed] [Google Scholar]
- 49. Boehmer C., Laufer J., Jeyaraj S., Klaus F., Lindner R., Lang F., Palmada M. (2008) Modulation of the voltage-gated potassium channel Kv1.5 by the SGK1 protein kinase involves inhibition of channel ubiquitination. Cell. Physiol. Biochem. 22, 591–600 [DOI] [PubMed] [Google Scholar]
- 50. Klaus F., Palmada M., Lindner R., Laufer J., Jeyaraj S., Lang F., Boehmer C. (2008) Up-regulation of hypertonicity-activated myo-inositol transporter SMIT1 by the cell volume-sensitive protein kinase SGK1. J. Physiol. 586, 1539–1547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Rajamanickam J., Palmada M., Lang F., Boehmer C. (2007) EAAT4 phosphorylation at the SGK1 consensus site is required for transport modulation by the kinase. J. Neurochem. 102, 858–866 [DOI] [PubMed] [Google Scholar]
- 52. Liang X., Butterworth M. B., Peters K. W., Walker W. H., Frizzell R. A. (2008) An obligatory heterodimer of 14-3-3beta and 14-3-3epsilon is required for aldosterone regulation of the epithelial sodium channel. J. Biol. Chem. 283, 27418–27425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Nagaki K., Yamamura H., Shimada S., Saito T., Hisanaga S., Taoka M., Isobe T., Ichimura T. (2006) 14-3-3 Mediates phosphorylation-dependent inhibition of the interaction between the ubiquitin E3 ligase Nedd4-2 and epithelial Na+ channels. Biochemistry 45, 6733–6740 [DOI] [PubMed] [Google Scholar]
- 54. Liang X., Peters K. W., Butterworth M. B., Frizzell R. A. (2006) 14-3-3 isoforms are induced by aldosterone and participate in its regulation of epithelial sodium channels. J. Biol. Chem. 281, 16323–16332 [DOI] [PubMed] [Google Scholar]
- 55. Palmada M., Poppendieck S., Embark H. M., van de Graaf S. F., Boehmer C., Bindels R. J., Lang F. (2005) Requirement of PDZ domains for the stimulation of the epithelial Ca2+ channel TRPV5 by the NHE regulating factor NHERF2 and the serum and glucocorticoid inducible kinase SGK1. Cell. Physiol. Biochem. 15, 175–182 [DOI] [PubMed] [Google Scholar]
- 56. Bohmer C., Palmada M., Kenngott C., Lindner R., Klaus F., Laufer J., Lang F. (2007) Regulation of the epithelial calcium channel TRPV6 by the serum and glucocorticoid-inducible kinase isoforms SGK1 and SGK3. FEBS. Lett. 581, 5586–5590 [DOI] [PubMed] [Google Scholar]
- 57. Laufer J., Boehmer C., Jeyaraj S., Knuwer M., Klaus F., Lindner R., Palmada M., Lang F. (2009) The C-terminal PDZ-binding motif in the Kv1.5 potassium channel governs its modulation by the Na+/H+ exchanger regulatory factor 2. Cell. Physiol. Biochem. 23, 25–36 [DOI] [PubMed] [Google Scholar]
- 58. Lang F., Artunc F., Vallon V. (2009) The physiological impact of the serum and glucocorticoid-inducible kinase SGK1. Curr. Opin. Nephrol. Hypertens. 18, 439–448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Klingel K., Warntges S., Bock J., Wagner C. A., Sauter M., Waldegger S., Kandolf R., Lang F. (2000) Expression of cell volume-regulated kinase h-sgk in pancreatic tissue. Am. J. Physiol. Gastrointest Liver Physiol. 279, G998–G1002 [DOI] [PubMed] [Google Scholar]
- 60. Taruno A., Niisato N., Marunaka Y. (2008) Intracellular calcium plays a role as the second messenger of hypotonic stress in gene regulation of SGK1 and ENaC in renal epithelial A6 cells. Am. J. Physiol. Renal Physiol. 294, F177–F186 [DOI] [PubMed] [Google Scholar]
- 61. Imai S., Okayama N., Shimizu M., Itoh M. (2003) Increased intracellular calcium activates serum and glucocorticoid-inducible kinase 1 (SGK1) through a calmodulin-calcium calmodulin dependent kinase kinase pathway in Chinese hamster ovary cells. Life Sci. 72, 2199–2209 [DOI] [PubMed] [Google Scholar]
- 62. Becchetti A., Arcangeli A. (2010) Integrins and ion channels in cell migration. implications for neuronal development, wound healing and metastatic spread. Adv. Exp. Med. Biol. 674, 107–123 [DOI] [PubMed] [Google Scholar]
- 63. Bradding P., Wulff H. (2009) The K+ channels K(Ca)3.1 and K (v) 1.3 as novel targets for asthma therapy. Br. J. Pharmacol. 157, 1330–1339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Roberts-Thomson S. J., Peters A. A., Grice D. M., Monteith G. R. (2010) ORAI-mediated calcium entry. mechanism and roles, diseases and pharmacology. Pharmacol Ther. 127, 121–130 [DOI] [PubMed] [Google Scholar]
- 65. Robinson L. J., Blair H. C., Barnett J. B., Zaidi M., Huang C. L. (2010) Regulation of bone turnover by calcium-regulated calcium channels. Ann. N. Y. Acad. Sci. 1192, 351–357 [DOI] [PubMed] [Google Scholar]
- 66. Lioudyno M. I., Kozak J. A., Penna A., Safrina O., Zhang S. L., Sen D., Roos J., Stauderman K. A., Cahalan M. D. (2008) Orai1 and STIM1 move to the immunological synapse and are up-regulated during T cell activation. Proc. Natl. Acad. Sci. U. S. A. 105, 2011–2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Schaff U. Y., Dixit N., Procyk E., Yamayoshi I., Tse T., Simon S. I. (2010) Orai1 regulates intracellular calcium, arrest, and shape polarization during neutrophil recruitment in shear flow. Blood 115, 657–666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Katso R., Okkenhaug K., Ahmadi K., White S., Timms J., Waterfield M. D. (2001) Cellular function of phosphoinositide 3-kinases. implications for development, homeostasis, and cancer. Annu. Rev. Cell. Dev. Biol. 17, 615–675 [DOI] [PubMed] [Google Scholar]
- 69. Rudd C. E., Taylor A., Schneider H. (2009) CD28 and CTLA-4 coreceptor expression and signal transduction. Immunol. Rev. 229, 12–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Segal R. A. (2003) Selectivity in neurotrophin signaling. theme and variations. Annu. Rev. Neurosci. 26, 299–330 [DOI] [PubMed] [Google Scholar]
- 71. Shumilina E., Zemtsova I. M., Heise N., Schmid E., Eichenmuller M., Tyan L., Rexhepaj R., Lang F. (2010) Phosphoinositide-dependent kinase PDK1 in the regulation of Ca2+ entry into mast cells. Cell. Physiol. Biochem. 26, 699–706 [DOI] [PubMed] [Google Scholar]
- 72. Rotte A., Bhandaru M., Foller M., Biswas R., Mack A. F., Friedrich B., Rexhepaj R., Nasir O., Ackermann T. F., Boini K. M., Kunzelmann K., Behrens J., Lang F. (2009) APC sensitive gastric acid secretion. Cell. Physiol. Biochem. 23, 133–142 [DOI] [PubMed] [Google Scholar]
- 73. Dehner M., Hadjihannas M., Weiske J., Huber O., Behrens J. (2008) Wnt signaling inhibits Forkhead box O3a-induced transcription and apoptosis through up-regulation of serum- and glucocorticoid-inducible kinase 1. J. Biol. Chem. 283, 19201–19210 [DOI] [PubMed] [Google Scholar]
- 74. Baryshnikov S. G., Pulina M. V., Zulian A., Linde C. I., Golovina V. A. (2009) Orai1, a critical component of store-operated Ca2+ entry, is functionally associated with Na+/Ca2+ exchanger and plasma membrane Ca2+ pump in proliferating human arterial myocytes. Am. J. Physiol. Cell Physiol. 297, C1103–C1112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Berra-Romani R., Mazzocco-Spezzia A., Pulina M. V., Golovina V. A. (2008) Ca2+ handling is altered when arterial myocytes progress from a contractile to a proliferative phenotype in culture. Am. J. Physiol. Cell Physiol. 295, C779–C790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Faouzi M., Hague F., Potier M., Ahidouch A., Sevestre H., Ouadid-Ahidouch H. (2011) Down-regulation of Orai3 arrests cell cycle progression and induces apoptosis in breast cancer cells but not in normal breast epithelial cells. J. Cell. Physiol. 226, 542–551 [DOI] [PubMed] [Google Scholar]
- 77. Amato R., D'Antona L., Porciatti G., Agosti V., Menniti M., Rinaldo C., Costa N., Bellacchio E., Mattarocci S., Fuiano G., Soddu S., Paggi M. G., Lang F., Perrotti N. (2009) Sgk1 activates MDM2-dependent p53 degradation and affects cell proliferation, survival, and differentiation. J. Mol. Med. 87, 1221–1239 [DOI] [PubMed] [Google Scholar]
- 78. Sherk A. B., Frigo D. E., Schnackenberg C. G., Bray J. D., Laping N. J., Trizna W., Hammond M., Patterson J. R., Thompson S. K., Kazmin D., Norris J. D., McDonnell D. P. (2008) Development of a small-molecule serum- and glucocorticoid-regulated kinase-1 antagonist and its evaluation as a prostate cancer therapeutic. Cancer Res. 68, 7475–7483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Wang K., Gu S., Nasir O., Foller M., Ackermann T. F., Klingel K., Kandolf R., Kuhl D., Stournaras C., Lang F. (2010) SGK1-dependent intestinal tumor growth in APC-deficient mice. Cell. Physiol. Biochem. 25, 271–278 [DOI] [PubMed] [Google Scholar]