

1. Introduction
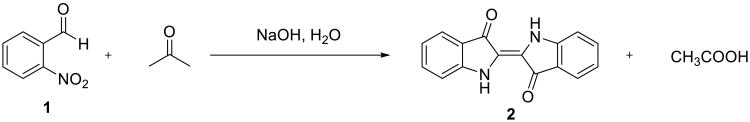
Water is the lingua franca of life on our planet and is the solvent of choice for Nature to carry out her syntheses.1 In contrast, our methods of making complex organic molecules have taken us far away from the watery milieu of biosynthesis. Indeed, it is fair to say that most organic reactions commonly used both in academic laboratories and in industry fail in the presence of water or oxygen. As a direct consequence of our attempts to mimick Nature's way of making new chemical bonds, we learned to rely on highly reactive nucleophilic and electrophilic reagents to gain control of the chemical reactivity and to channel chemical reactions down a desired pathway. The requirement for the protection of all protic functional groups, such as alcohols and amines, is another corollary of our reliance on these energetic species. Nevertheless, chemical transformations in aqueous solvents are not new to organic chemists. On the contrary, they have attracted attention of scientists for many years: the first use of water for an organic reaction could be dated back to Wöhler's synthesis of urea from ammonium cyanate.2 From a true organic synthesis perspective, the earliest example could be the synthesis of indigo by Baeyer and Drewsen in 1882 (Scheme 1).3 In their synthesis, a suspension of o-nitrobenzaldehyde 1 in aqueous acetone was treated with a solution of sodium hydroxide. The immediate formation of the characteristic blue color of indigo 2 ensued, and the product subsequently precipitated.
Scheme 1.
Water possesses many unique physical and chemical properties: large temperature window in which it remains in the liquid state, extensive hydrogen bonding, high heat capacity, large dielectric constant, and optimum oxygen solubility to maintain aquatic life forms. These distinctive properties are the consequence of the unique structure of water.4,5 The structure and properties of water have been studied by scientists representing almost all fields of knowledge, and new theoretical models continue to emerge.6,7 Water is also known to enhance the rates and to affect the selectivity of a wide variety of organic reactions.8,9
In spite of these potential advantages, water is still not commonly used as a sole solvent for organic synthesis, at least in part because most organic compounds do not dissolve in water to a significant extent, and solubility is generally considered a prerequisite for reactivity: “corpora non agunt nisi soluta” (substances do not react unless dissolved). Consequently, in the many examples of “aqueous reactions” organic co-solvents are employed in order to increase the solubility of organic reactants in water.9,10 Alternatively, hydrophilicity of the reactants is increased by the introduction of polar functional groups, again to make the resulting compound at least partially water soluble.11 However, these manipulations tend to diminish and even negate the advantages of low cost, simplicity of reaction conditions, ease of workup, and product isolation that water has over traditional solvents. Therefore, the currently burgeoning field of organic synthesis in aqueous media encompasses a large family of reactions. The solubility of reacting species and products can range from complete to partial to practically none, so that reaction mixtures can be both homogeneous and heterogeneous. The amount of water can also range widely, from substoichiometric quantities to a large volume in which the reactants are suspended or dissolved. Several terms have been used in the literature to describe reactions in aqueous millieu. In water, in the presence of water, and on water are commonly found in the recent publications and are often used interchangeably to describe reactions that proceed under very different conditions.12,13 There is also a growing number of examples micellar catalysis in the presence of non-ionic surfactants, such as Triton X-100 and PTS (a tocopherol-based amphiphile).14-18
In this review, we attempt to survey organic transformations that benefit from being performed on water under the conditions defined by Sharpless and co-workers: when insoluble reactant(s) are stirred in aqueous emulsions or suspensions without the addition of any organic co-solvents. In many cases, it is impossible to ascertain whether the reaction is occuring in or on water, but as long as the reaction mixture remains heterogeneous and the overall process appears to benefit from it (either in terms of increased reaction rate or enhanced selectivity), it qualifies.
The ‘on water’ moniker reflects the defining attribute of these reactions: the lack of solubility of the reactant(s) in water. A considerable rate acceleration is often observed in reactions carried out under these conditions over those in organic solvents.19 Furthermore, in many cases a significant rate increase of on water reactions over reactions carried out neat indicates that rate acceleration is not merely a consequence of increased concentration of the reacting species. Naturally, the degree of on water acceleration varies between different reaction classes, and even when it is modest, there are other advantages to carrying out reactions in this manner. Firstly, water is an excellent heat sink due to its large heat capacity, making exothermic processes safer and more selective, especially when they are carried out on large scale. Secondly, reactions of water-insoluble substrates usually lead to the formation of water-insoluble products. In such cases, product isolation simply involves filtration of solid products (or phase separation in case of liquids). Finally, the growing list of examples wherein reactions performed on water are not only faster but also more selective (whether chemo-, regio-, or enantio-) underscores the significant potential for process intensification for reactions performed on water.
Although claims of the ecological advantages and “greenness” of water are almost invariably found in the opening paragraphs of reports describing aqueous reactions, they should be taken with a grain of salt. The low cost, relative abundance, and inherent safety of water notwithstanding, the environmental impact of a process is determined by many factors, such as the efficiency of the reaction in terms of atom economy,20 the nature of solvents used in the reaction workup, the residual concentration of regulated organic compounds and metal catalysts remaining in the aqueous waste, and the costs of its clean up or disposal.21,22 The mere finding that a process performs as well in water as it does in an organic solvent tells us little about its potential environmental impact.
The field of aqueous organic synthesis has been regularly and comprehensively reviewed.9,10,23-27 In addition, recent reviews focusing on microwave assisted organic synthesis in water,28 reactions in near-critical water,29 and biocatalysis in water30 have been published. Accordingly, these topics are not covered in the present review.
2. On Water Reactions
2.1. Diels–Alder Reactions
The Diels–Alder cycloaddition is a powerful synthetic transformation that has been used prolifically by organic chemists, and the effect of aqueous solvents on this large family of reactions was studied as early as in 1939, when Hopff and Rautenstrauch disclosed that Diels–Alder reactions could be carried out efficiently in an “aqueous dispersion.”31 Most reactions described in their patent were carried out in the presence of dispersing or emulsifying agents.
In 1980, Rideout and Breslow reported that both rate enhancement and excellent selectivity could be achieved for certain Diels–Alder reactions when they were performed in dilute aqueous solutions.32 The authors studied the reaction of cyclopentadiene 3 (0.4 mM) with butenone 4 (12.1 and 25.5 mM) in water (Scheme 2) and followed its progress by UV-Vis spectrometry. The second-order rate constants for the reaction in different solvents were obtained and are listed in Table 1. The reaction was accelerated more than a 700-fold in water vs. the aprotic non-polar organic solvent, 2,2,4-trimethylpentane. The rate in methanol, a protic polar organic solvent, was intermediate, but closer to that in the hydrocarbon solution. The effect of different additives (lithium chloride, guanidinium chloride, and cyclodextrins) was also examined. Although 4.86 M guanidinium chloride did not significantly affect the rate, the reaction was accelerated in 4.86 M lithium chloride solution.
Scheme 2.
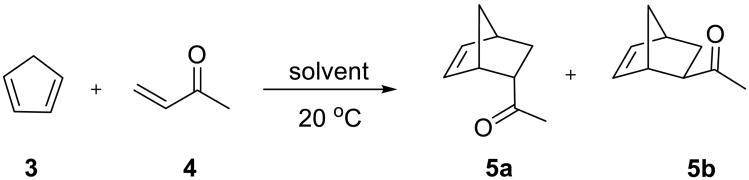
Table 1.
Rate constants for the Diels–Alder reaction of 3 and 4 in different solvents.32
| Solvent | k2 × 105, M-1s-1 |
|---|---|
| 2,2,4-trimethylpentane | 5.94 ± 0.3 |
| Methanol | 75.5 |
| H2O | 4400 ± 70 |
| H2O (4.86 M LiCl) | 10,800 |
The authors also examined two other Diels–Alder reactions. The cycloaddition of cyclopentadiene with acrylonitrile showed a small rate increase on changing the solvent from 2,2,4-trimethylpentane to methanol but was significantly accelerated in water. The reaction of anthracene-9-carbinol with N-ethylmaleimide was fastest in water; it proceeded slower in polar solvents than in solutions of non-polar hydrocarbons. The observed rate increase in water was explained by the hydrophobic effect, i.e. the propensity of hydrophobic molecules to associate in order to minimize their contact surface with water.33-35
In a subsequent study,36 the same authors demonstrated that in addition to the enhanced rate of the reaction, the endo vs. exo selectivity of the cycloaddition was also much higher in water than in ethanol or in the absence of a solvent (Table 2). These reactions were performed at a higher concentration (0.15 M in both reactants), so the reaction mixture was heterogeneous – an emulsion in water. This could be the first example of an on water reaction for which a significant rate enhancement and a much higher selectivity were achieved simply by using water to support the reaction of two insoluble substances.
Table 2.
| solvent | endo/exo ratio (5a/5b) |
|---|---|
| neata | 3.85 |
| ethanol | 8.5 |
| H2O | 21.4 |
Excess butenone was used
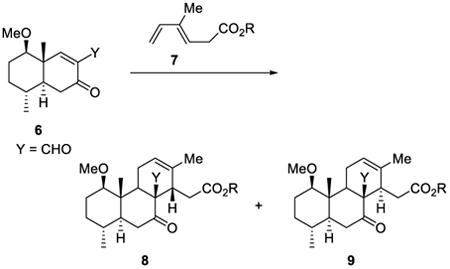
Another early example of an aqueous heterogeneous Diels–Alder reaction was reported by Grieco and co-workers in 1983.37,38 Their study of the cycloaddition of enal 6 with acyclic diene 7 that contained a carboxylic acid functionality proceeded significantly faster and with higher selectivity when the substrates were suspended in water (Table 3).
Table 3. Cycloaddition of enal 6 with diene 7.
| |||||
|---|---|---|---|---|---|
|
| |||||
| R | solvent | [7], M | time, h | yield, % | ratio 8/9 |
| Et | benzene | 1 | 288 | 52 | 0.85 |
| Et | neat | – | 144 | 69 | 1.3 |
| Et | H2O | 1 | 168 | 82 | 1.3 |
| Na | H2O | 1 | 8 | 83 | 2.0 |
| Na | H2O | 2 | 5 | 100 | 3.0 |
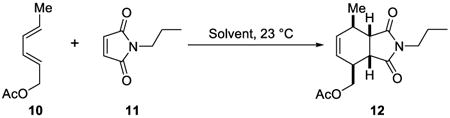
In 2005, Sharpless and co-workers demonstrated the on water effect on the Diels–Alder reaction of trans, trans-2,4-hexadienyl acetate 10 and N-propylmaleimide 11 (Table 4).19 For example, the reaction in toluene took 144 h, in methanol 48 h, and still required 10 h when no solvent was used to obtain similar product yields (81-82%), whereas pure product was isolated in 81% yield after 8 h of stirring on water.
Table 4.
Diels–Alder Reaction of 10 and 11 in different solvents.
| ||
|---|---|---|
|
| ||
| solvent | time to completion (h) | yield of 12, (%) |
| toluene | 144 | 79 |
| CH3CN | >144 | 43 a |
| CH3OH | 48 | 82 |
| neat | 10 | 82 |
| H2O | 8 | 81 |
after chromatographic separation
Pizzo and coworkers studied an inverse electron-demand hetero Diels–Alder reaction on water.39 (E)-3-diazenylbut-2-enes 13 reacted with a variety of alkenes to yield dihydropyridazines 15(a+b), favoring the endo-isomer (Scheme 3). The on water reaction gave a higher yield, although the endo:exo selectivity did not improve.
Scheme 3.

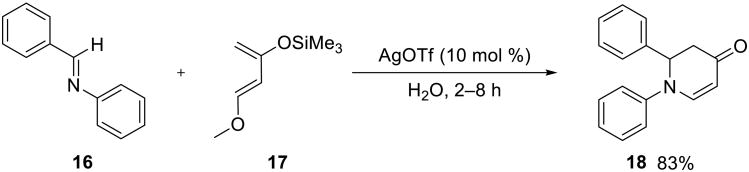
In another example of a hetero Diels–Alder reaction, Kobayashi and co-workers found that silver triflate activated imines 16 towards the cycloaddition with Danishefsky's diene 17 on water, providing ready access to substituted dihydro-4-pyridones 18 in excellent yields (Scheme 4).40 A one–pot three– component variant of this aza Diels–Alder reaction, where the imine was formed in situ from the amine and the aldehyde, also proceeded smoothly on water.
Scheme 4.
2.2. 1,3-Dipolar Cycloadditions
The diversity of available dipoles and dipolarophiles has made 1,3-dipolar cycloadditions41 broadly useful for the construction of complex molecular architectures, ranging from natural products to polymeric materials.42,43 The beneficial effects of water on several classes of dipolar cycloadditions have been documented.44-46
A recent example from Novartis describes a simple and elegant synthesis of cyanotriazoles from organic azides and 2-chloroacrylonitrile 20, a cyanoacetylene equivalent (Scheme 5).47 The challenge in carrying out these cycloadditions is that 20 can polymerize under both acidic and basic conditions. Upon the reaction of 20 with organic azides such as 19, the intermediate triazoline loses HCl, which increases the acidity of the reaction medium. When an organic solvent is used, HCl remains in the reaction mixture and causes polymerization of 20, thereby decreasing the yield of the triazole. In contrast, when the reactants are heated together on water, the azide 19 and the acrylonitrile 20 form the organic phase, while water constitutes the other phase. During the course of the reaction, the generated HCl is continuously extracted into the aqueous phase, thus reducing the propensity of 20 to polymerize. In addition, the reaction rate is highest in the two-phase system.47
Scheme 5.

The 1,3-dipolar cycloaddition of organic azides with alkynes occupies a special place in the family of Huisgen cycloaddition processes. Although both azides and alkynes are highly energetic species, they are kinetically stable and are quite inert to most common organic functional groups. Their cycloaddition is strongly thermodynamically favored (ΔHo = –45-60 kcal/mol), but has a relatively high kinetic barrier (ca. 26 kcal/mol for methyl azide and propyne48), thus rendering the reaction very slow at room temperature for unactivated reactants. However, even in the absence of transition metal catalysts, electron-deficient acetylenes may be sufficiently reactive dipolarophiles. Thus, acetylene dicarboxylates and organic azides react readily and cleanly on water. As shown in Scheme 6, azido alcohols 23 and 26 were obtained from the isomeric diepoxides 22 and 25 by opening the latter with azide anion in water containing a catalytic amount of ammonium chloride. The azido alcohols were formed regioselectively and were subsequently submitted to the reaction with diethyl acetylenedicarboxylate on water. The product bis-triazoles 24 and 27, which are crystalline solids, were isolated by simple filtration.49
Scheme 6.

The on water 1,3-dipolar cycloadditions of organic azides and electron-deficient alkynes (both terminal and internal alkynoates were used) were studied on water by Ju and co-workers.50 The reactions appeared to be facile even at room temperature and proceeded to completion in 6-12 h. Remarkably, the authors isolated regioisomerically pure 1,4-disubstituted 1,2,3-triazoles 29 and 30 (Scheme 7) in 82% and 94% yield, respectively.
Scheme 7.

The 1,3-dipolar cycloaddition reactions of phthalazinium ylides reported by Butler and coworkers are noteworthy because they involve at least one solid reactant. For example, phthalazinium-2-dicyanomethanide 31, which is a solid compound insoluble in water, reacted with methyl methacrylate 32,51 substituted styrenes 33,52 or 2-buteneone 3452 (Scheme 8), providing dihydropyrrolophthalazines 35-37, respectively, in high yield when the reaction was performed in a 9:1 water-acetonitrile mixture.
Scheme 8.

Based on the observed rate enhancement, dipolarophiles were grouped into two classes, ‘water-normal’ and ‘water-super’. Ethers, sulfones, nitriles, styrenes, and aryl acetylenes showed less than a 20-fold rate enhancement on water compared to the homogeneous solution conditions, and thus were ‘normal.’ Enones and ynones showed greater than a 45-fold (often a 100-fold) rate enhancement and, accordingly, were placed in the ‘super’ category.
A ‘solid-solid’ on water reaction of the same dipole with insoluble dipolarophiles also efficiently proceeded on water (Scheme 9).51 However, heating was required for the reactions of diphenylacetylene 39 and p-chlorophenyl butenone 40.
Scheme 9.

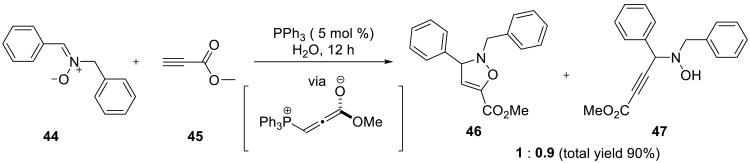
Cycloadditions of nitrones with allenolates, which were generated from propiolates and a catalytic amount of triphenylphosphine or tertiary amine, were performed on water (Scheme 12).53,54 Thus, nitrone 44 reacted with methyl propiolate 45, providing the approximately 1:1 mixture of the trisubstituted isoxazoline 46 and hydroxylamine 47 in 90% combined yield. The yields were very low in toluene, benzene and dichloromethane.
Scheme 12.
With internal alkynes, such as methyl heptynoate 48, only the 1,3-dipolar cycloaddition product, such as oxazoline 49, was formed (Scheme 13). This reaction failed in toluene and dichloromethane and proceeded poorly on pure water. However, performing the reaction in 3M aqueous solution of lithium chloride significantly improved its efficiency (Table 5).
Scheme 13.
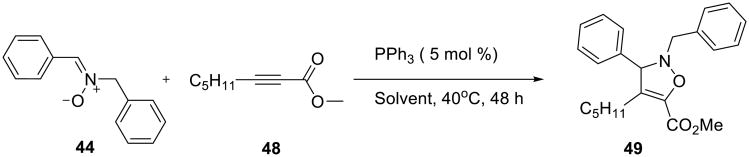
Table 5.
No product was observed after 24 h Scheme 14.
In another example of a 1,3-dipolar cycloaddition reaction, perfluorophenyl diazomethane, generated in situ from 2,4,6-triisopropyl-N′-(perfluorobenzyl)benzenesulfonylhydrazine 50 by the Bamford–Stevens reaction, reacted on water with α,β-unsaturated esters 51 or nitriles yielding the corresponding pyrazolines 52 (Scheme 14).55 Although the reaction could be carried out in tetrahydrofuran, the on water process gave virtually quantitative yields. The insoluble products were easily isolated by filtration.
Scheme 14.

Bala and Hailes reported the intermolecular 1,3-dipolar cycloaddition reactions of nitrile oxides, which were generated in situ by halogenation/dehydrohalogenation of oximes 53, with olefins. Benzopyrans, quinolines and fused isoxazolines 54 (Scheme 15) were prepared in excellent yields.56 Although these annulations were slower on water than in mixed solvent systems (e.g. THF/H2O), the facility of product isolation from the on water reactions was a practical advantage: no starting materials remained, and the isoxazoline products precipitated from the reaction mixture.
Scheme 15.

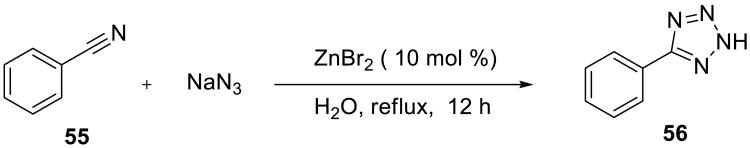
Demko and Sharpless developed a practical synthesis of tetrazoles 56 and tetrazole analogs of α-amino acids from aryl nitriles 55 and sodium azide on water (Scheme 16).57 Among several zinc salts that were examined, zinc(II) bromide was identified as the most effective catalyst. The mechanism of these reactions was examined computationally.58,59
Scheme 16.
2.3. Cycloadditions of Azodicarboxylates
Reactions of azodicarboxylates with unsaturated hydrocarbons are powerful C–N bond forming processes. Such reactions are often highly exergonic due to the energetic nature of the azo functionality and, as a result, are reliable and versatile. A striking example of an on water accelerated 2σ+2σ+2π cycloaddition reaction between quadricyclane 57 and dimethyl azodicarboxylate 5860 (Scheme 17) was described by Sharpless and co-workers.19 This transformation usually requires prolonged reaction times and heating when carried out in an organic solvent or under neat conditions.61,62 Thus, in homogeneous organic solutions this reaction took from 18 h (in methanol) to over 5 days (in toluene) to reach completion (Table 6). In contrast, it proceeded in ca. 10 min on water at room temperature.
Scheme 17.
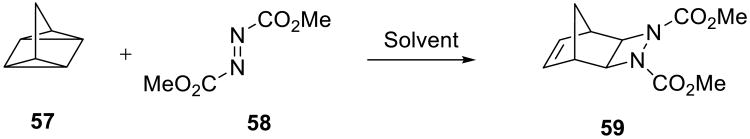
Table 6.
| solvent | time to completion |
|---|---|
| toluene | >120 h |
| EtOAc | >120 h |
| CH3CN | 84 h |
| CH2Cl2 | 72 h |
| DMSO | 36 h |
| CH3OH | 18 h |
| neat | 48 h |
| H2O | 10 min |
The cycloaddition of quadricyclane and dimethyl azodicarboxylate was also examined in methanol-water mixtures. As illustrated in Table 7, the reaction proceeded to completion in 4 h in a homogeneous solution of 3:1 methanol/water, while in pure methanol it took 18 h. Upon increasing the proportion of water in the solvent from 25% to 50%, at which point the reaction mixture became heterogenous, the reaction was significantly accelerated, and it proceeded to completion in 10 min instead of 4 h.
Table 7.
Solvent effects on the reaction of 57 with 58.
| solvent | time to completion |
|---|---|
| CH3OH/H2O (3:1, homogeneous) | 4 h |
| CH3OH/H2O (1:1, heterogeneous) | 10 min |
| CH3OH/H2O (1:3, heterogeneous) | 10 min |
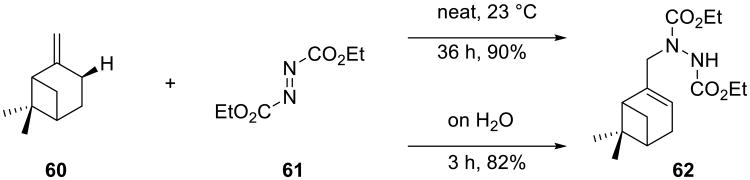
Ene reactions of azodicarboxylates are convenient methods for the allylic amination of olefins.63,64 Sharpless and co-workers disclosed that the ene reaction of diethyl azodicarboxylate 61 and β-pinene 60 proceeded very fast ‘on water’: product formation was complete within three hours, as compared to the 36 h for the neat reaction (Scheme 18).19,65
Scheme 18.
Furthermore, the ene reaction of cyclohexene 63 with bis(trichloroethyl) azodicarboxylate 64 (Scheme 19) was also found to be significantly accelerated on water.19 Even though the azo reagent is a solid compound, the product was obtained in 91% yield after the mixture was vigorously stirred on water for 8 h at 50 °C. In contrast, performing the reaction neat at 50 °C or in benzene at 80 °C resulted in lower yields even after prolonged heating (36 h, Table 8).
Scheme 19.
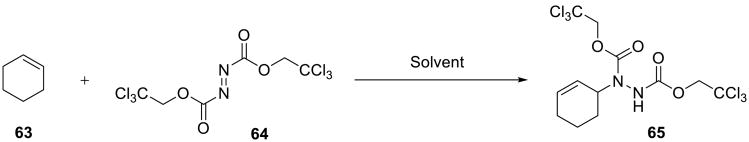
Table 8.
| solvent | t, °C | time, h | yield, % |
|---|---|---|---|
| benzene | 80 | 24 | 70 |
| neat | 50 | 36 | 62 |
| H2O | 50 | 8 | 91 |
2.4. Claisen Rearrangement
In 1987, Gajewski, Ganem, Carpenter and co-workers reported a detailed experimental and mechanistic study of the rearrangement of chorismic acid to prephenic acid, an important step in the shikimic acid pathway.54 The accelerating effect of polar solvents on the rate of Claisen rearrangements was noted and was subsequently studied on the model carboxylate 66 by Grieco and Gajewski (Scheme 20).66,67 The first-order rate constants for Claisen rearrangement of this carboxylate-containing allyl vinyl ether were determined in different solvents (Table 9). The reaction in water was over 20 times faster than in polar organic solvents, and more than two orders of magnitude faster than the analogous rearrangement of the methyl ester in a non-polar organic solvent, cyclohexane.
Scheme 20.

Table 9.
| solvent | rate (k × 10-5 s-1) | |
|---|---|---|
| 1 | methanol | 0.79 |
| 2 | 3:1 methanol : H2O | 1.6 |
| 3 | 1:1 methanol : H2O | 4.6 |
| 4 | 1:3 methanol : H2O | 11 |
| 5 | CF3CH2OH a | 2.6 |
| 6 | 1:3 CF3CH2OH : H2O | 6.9 |
| 7 | 1:3 DMSO : H2O | 3.0 |
| 8 | H2O | 18 |
0.1 M solution of pyridine
The rearrangement of aromatic substrates was examined in 2005 by Sharpless and co-workers on allyl naphthyl ether 68 (Scheme 21). The reactions were performed in different solvents as well as neat.19 Although absolute reaction rates were not determined, the rearranged product 69 was obtained in almost quantitative yield on water. The yields were much lower when the reaction was carried out in toluene, dimethylformamide, acetonitirle, or methanol (Table 10).
Scheme 21.

Table 10.
| Solvent | Yield (%) |
|---|---|
| Toluene | 16 |
| DMF | 21 |
| CH3CN | 27 |
| CH3OH | 56 a |
| Neat | 73 |
| H2O | >99 |
small amount of 4-chloro-1-napthol was also observed.
Nicolaou and coworkers reported a water-accelerated Claisen rearrangement and Diels–Alder cascade sequence in their synthesis of gambogin, a cytotoxic natural product isolated from the evergreen plants of the genus Garcinia. The authors noted the unprecedented facility of the Claisen rearrangement using the model substrate 70 (Scheme 22), which proceeded at room temperature in ethanol/water mixture. The quantitative conversion of intermediate 72 to the key gambogin precursor 73 was then accomplished by the Claisen rearrangement/Diels–Alder sequence in 2:1 water/methanol at 65 °C.68
Scheme 22.

2.5. Passerini and Ugi Reactions
Multicomponent reactions provide rapid access to chemical diversity by combining several reactants into densely functionalized molecules.69-71 Among the commonly used multi-component reactions, Ugi and Passerini reactions are easily among the most versatile and widely utilized transformations. Pirrung and Sarma72,73 examined these reactions in aqueous solutions and reported a significant increase in their rate and efficiency compared to organic solvents. For example, in the Passerini reaction shown in Scheme 23, the amide 77 was obtained in nearly quantitative isolated yield in water, and the reaction time was considerably shorter (3.5 h) than in dichloromethane (18 h, 50% yield, Table 11).
Scheme 23.
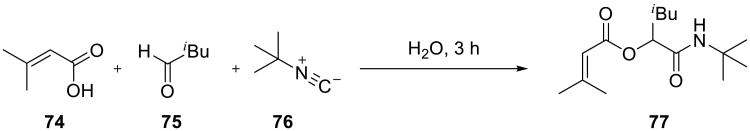
Table 11.
| solvent | time, h | conversion, % |
|---|---|---|
| CH2Cl2 | 18 | 50 |
| CH3OH | 24 | - a |
| DMF | 24 | 15 |
| H2O | 3.5 | 100 |
no progress was observed
The addition of lithium chloride or glucose further accelerated the reaction. It is also noteworthy that these reactions showed inverse temperature dependence. Thus, the rate was 11% higher at 4 °C vs. 25°C, while increasing the temperature to 50°C slowed the reaction by 44%.
Evaluation of the rates of Ugi reactions of α,β-unsaturated acids 74 (Scheme 24) revealed a near 50-fold acceleration on water in comparison to methanol.72,73 These transformations provided a practical and high-yielding route to the target amides 79 that did not require high pressure equipment or extended reaction times.
Scheme 24.

The utility of Ugi reactions of β-aminoacids performed on water were demonstrated by synthesis of strained β-lactams (Scheme 25).73 These reactions did not proceed in methanol, tetrahydrofuran, or dichloromethane. In contrast, β-lactam derivatives, such as 81, were obtained in good yield after 72 h when reactions were performed on water.
Scheme 25.
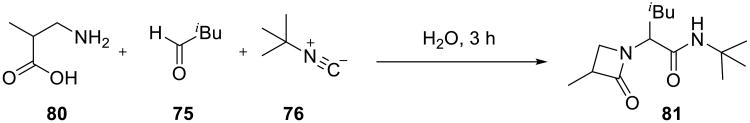
The authors employed the on-water accelerated Passerini and Ugi reactions for the construction of diverse libraries of β-lactams from simple building blocks shown in Table 12.
Table 12.
| Acids |
|
| Aldehydes |
|
| Isonitriles |
|
| Amines |
|
|
| |
| β-Amino acids |
|
Blackwell and co-workers applied water-accelerated Ugi reactions to the construction of small-molecule macroarray systems on planar cellulose supports.74 These solid-supported processes were also accelerated by water, enabling facile synthesis of small molecule arrays with excellent purity.
Vigalok and Shapiro demonstrated that long-chain (branched and linear) aliphatic and aromatic aldehydes could be oxidized on water using air or oxygen (Scheme 26) to yield the corresponding carboxylic acids in high yields.75 No catalysts or activators were required. No reaction was observed in organic solvents such as methanol and dichloromethane. The addition of even 5% (v/v) of dioxane completely suppressed the reaction.
Scheme 26.

Carboxylic acids formed in this oxidation process were directly utilized in Passerini reactions, wherein aldehydes served as precursors of both carbonyl and ester functions. When water-insoluble starting materials were used (i.e., the on water conditions), these pseudo three-component reactions produced only Passerini products, such as 85. In contrast, when water soluble components were used (i.e. when the reaction was performed in water), α-hydroxyamide products 86 were isolated (Scheme 27). The on water reactions were considerably faster than the homogeneous processes, and the rate was dependent on the stirring speed.
Scheme 27.

2.6. Nucleophilic opening of three-membered rings
Nucleophilic openings of three-membered rings, such as epoxides and aziridines, are important and reliable methods for making carbon–heteroatom bonds because the competing elimination processes are stereoelectronically disfavored.76 As a result, ring-opened products are commonly obtained in high yield. As already illustrated in section 2.2 (Scheme 8), the nucleophilic opening of epoxides with azide anion proceeds efficiently on water to generate a variety of useful azide-containing products.
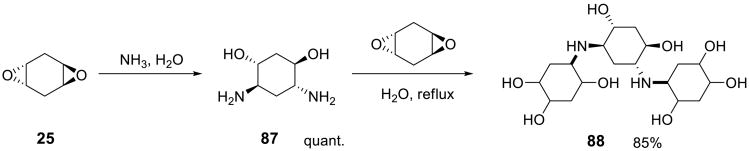
1,4-Cyclohexadiene diepoxide 25 also reacts with ammonia to form aminoalcohol 87 in quantitative yield (Scheme 28) which can further react with another molecule of the epoxide, furnishing 88.
Scheme 28.
The nucleophilic opening of cyclohexadiene monoepoxide 89 with substituted piperazine 90 (Scheme 29) was evaluated in organic solvents, on water, and under neat conditions.19 The on water reaction was fast and high-yielding (88%, 12 h), while it took 3 days in ethanol to produce the same yield of the aminoalcohol 91 (Table 13).
Scheme 29.
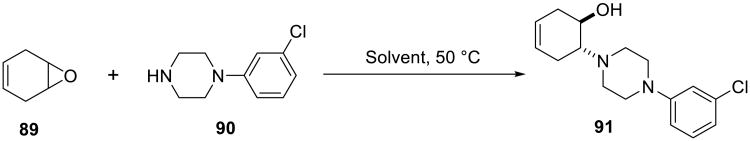
Table 13.
| entry | solvent | time, h | yield, % |
|---|---|---|---|
| 1 | Toluene | 120 | <10 |
| 2 | EtOH | 72 | 89 |
| 3 | Neat | 60 | 76 |
| 4 | H2O | 12 | 88 |
Efficient aminolysis of a series of epoxides by aliphatic and aromatic amines was accomplished on water by Saidi and Azizi (Scheme 30).77 The aminoalcohol products were obtained in high yields without any catalyst or organic co-solvent. α-Amino alcohols 93 were synthesized under these mild conditions with high selectivity and in excellent yields.
Scheme 30.

Similarly high yields were obtained in the reaction of styrene oxide with anilines, although the reaction was not regioselective. The opening of styrene oxide with diethylamine proceeded well in water (92% yield), was less efficient in ethanol (50% yield), and failed in dichloromethane, acetonitrile, hexane, and diethyl ether (Scheme 31 and Table 14).
Scheme 31.
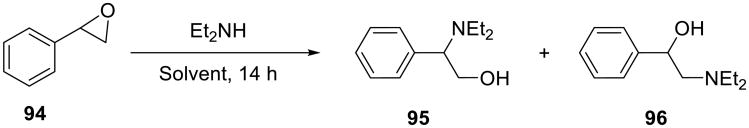
Table 14.
| solvent | yield of 95 + 96, % | 95 : 96 |
|---|---|---|
| neat | - a | - |
| CH2Cl2 | - a | - |
| CH3CN | - a | - |
| hexane | - a | - |
| ehanol | 50 | 24 : 76 |
| diethyl ether | - a | - |
| H2O | 92 | 45 : 55 |
No product observed
The nucleophilic opening of epoxides with dithiocarbamate anions generated in situ from amines and carbon disulfide gave β-dithiocarbamate derivatives, such as 99 in excellent yield (Scheme 32).78 In most cases, yields were slightly higher or comparable to those obtained in dimethylformamide/LiClO4 system.
Scheme 32.
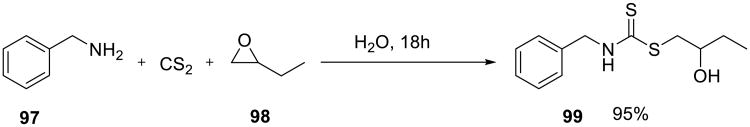
Nucleophilic openings of other strained ring systems may also benefit from the on water conditions. For example, even unactivated aziridines, such as cyclohexyl imine 100, readily reacted with buffered sodium azide to produce 1,2-amino azide in 90% yield (Scheme 33). Similarly, the reaction of this aziridine with hydrazine proceeded rapidly in water at ambient temperature.65
Scheme 33.

Scheme 34 illustrates the benefits of the on water conditions on the nucleophilic opening of the aziridinium ions derived from α-amino β-chloroesters 103.79-81 A significant improvement in both yield and regioselectivity was attained upon increasing the water content of the reaction mixture, with best results obtained in pure water. Reactions of aziridinum ions are particularly well suited to the aqueous environment, because these three-membered ring intermediates bear a positive charge balanced by the anionic counter ion which serves as the leaving group. When this leaving group is chloride or sulfonate, the favorable effects of water are most apparent, probably a result of the strong protic solvation of the harder anions in water.82
Scheme 34.

Kobayashi and coworkers reported asymmetric ring opening of meso-epoxides 106 using 1 mol% of Sc(OSO3C12H25)3 and 1.2 mol% of a chiral bipyridine ligand in water. The reaction provided β-amino alcohols 107 in high yields and with excellent enantioselectivity (Scheme 35).83
Scheme 35.

Vilotijevic and Jamison recently reported an impressive example of the effect of water on epoxide-opening cascade reactions (Scheme 36).84 Such transformations of polyepoxides into ladder polyethers via a “domino” cascade of epoxide-opening events had been proposed by Nakanishi to explain the biosynthesis of marine polyether natural products. However, there was no experimental evidence to support this hypothesis. In fact, in organic solvents these epoxide openings are generally disfavored. In stark contrast, water promoted the desired opening cascade as long as one templating tetrahydropyrane was present in the polyepoxide chain. Thus, diepoxide 108 and triepoxide 110 (Scheme 36) were readily converted into the corresponding fused tetrahydropyranes 109 and 111 in 60% and 53% yield, respectively, maintaining absolute stereocontrol. This is a superb example of the effect that water can have on the selectivity of an organic reaction.
Scheme 36.

A study by Schreiner and coworkers reported a thiourea-based organocatalyst 112 that promoted aqueous epoxide ring opening reactions (Scheme 37).85 The catalyst was designed to mimic the active site components of the enzyme epoxide hydrolase. Comparison of the reaction in dichloromethane and in water showed a prominent effect of the latter on the yield of the opening reaction. The effect of the catalyst itself was marginal (cf. 59% yield in the absence of the catalyst vs. 68% when 10 mol% of 112 was used). Examples with propyelene oxide and cyclohexane epoxide with seven different amine nucleophiles were reported. Reaction in D2O was slower by ca. 20%. The authors explained this observation by the higher viscosity of deuterated water.
Scheme 37.

Cravotto and co-workers investigated effects of ultrasonication on organic reactions. In a recent example from their laboratories, regioselective opening of epoxides under simultaneous microwave irradiation and ultrasonication was reported.86 The opening reactions with sodium azide were clean and fast, and provided the azidoalcohols in good yields.
2.7. Nucleophilic substitution reactions
An example of an efficient nucleophilic substitution reaction was reported by Finn, Sharpless and co-workers.87,88 The dichloro thiabicyclooctane 116 was treated with sodium azide or ammonia to produce diazide 117 and diamine 118, respectively (Scheme 38). Treatment of the same electrophile with NaSCN at different temperatures produced either isothiocyanate 119 (25 °C) or cyanate 120 (100 °C)
Scheme 38.

An interesting example of on water nucleophilic substitution of ferrocenyl alcohols 121 by carbon nucleophiles was reported by Cozzi and Zoli (Scheme 39).89 The reaction proceeded without the addition of Lewis or Bronsted acid catalysts or surfactants. Variously substituted indoles, pyrroles, thiophenols, and imidazoles readily participated in the reaction. With the exception of the most electron-deficient nucleophiles, the substituted ferrocenes were obtained in good yields (45–95%), and virtually no racemization was observed. Although the on water reactions were not directly compared with those in organic solvents, the practical aspects of this substitution should make it useful for the synthesis of diversely substituted chiral ferrocenes.
Scheme 39.

In the subsequent study,90 the authors rationally selected electrophile/nucleophile/solvent combinations taking into account the electrophilicity parameters introduced by Mayr.91-93 The selection of benzylic alcohols that participated in the substitution reaction was significantly extended, as was the selection of the nucleophiles (Scheme 40).
Scheme 40.

2.7. Transformations Catalyzed by Transition Metals
The stereotypical notion that organometallic reagents are not compatible with water stems from the extreme basicity and rapid hydrolysis of organolithium and organomagnesium compounds, the organometallic reagents on which most organic chemists have been educated. However, many organometallic compounds of transition metals are not as highly reactive with water and are, in fact, fully compatible with the aqueous environments because the hydrolysis of their carbon–metal bond is kinetically disfavored. With their accessible d-orbitals, transition metals can selectively interact with the “soft” nucleophilic (π) and electrophilic (π*) orbitals of alkynes, olefins, and arenes, paying relatively little attention to “hard” nucleophilic groups that may be present at whatever concentration in the aqueous solution. This property is responsible for the extensive and expanding family of aqueous-phase transformations mediated by transition metals. These include oxidations, reductions, carbon– carbon and carbon–heteroatom bond-forming reactions. All of these topics have been recently reviewed,23,94,95 and therefore only several illustrative transformations that exhibit the on water reactivity enhancements will be covered in the section below.
Transformations of acetylenes catalyzed by transition metals figure prominently in this field.96 Among those, reactions of copper(I) acetylides date back as far as 1869, when Glaser discovered the oxidative coupling of terminal alkynes.97,98 The scope of the reaction was extended in the subsequent studies by Baeyer,99 Straus,100,101 Reppe,102 and Eglinton.103 Heterocoupling reactions of copper(I) acetylides that do not involve oxygen have been reported by Castro and Stephens (with aryl halides),104,105 and Cadiot and Chodkiewicz (with bromoalkynes).106,107 It is noteworthy that many of these reactions are performed, and actually require, water as a (co-)solvent. This observation may be explained, at least in part, by the propensity of copper(I) acetylides to engage in the multitude of σ- and π-interactions, resulting in the formation of polynuclear clusters108 which are generally not very reactive. Indeed, once a copper acetylide is prepared by the conventional organometallic means and is isolated, it becomes insoluble and unreactive unless its polymeric structure is disrupted, either by strongly coordinating ligands or by organolithium/organomagnesium reagents. Water appears to be an ideal solvent capable of supporting copper(I) acetylides in their reactive state, especially when they are formed in situ. The copper(I)-catalyzed azide-alkyne cycloaddition (CuAAC) reaction is a striking example of the unique reactivity of copper(I) acetylides in water. When the acetylide is generated by the in situ reduction of a copper(II) salt (or from copper metal) in water with or without an organic co-solvent, as reported by the groups of Fokin and Sharpless,109 its reaction with organic azides cleanly proceeds in nearly quantitative yields, does not require any ligands, nor does the reaction mixture need to be protected from oxygen. In contrast, when the reaction is performed in organic solvents using copper(I) iodide as the catalyst, as reported independently by the group of Meldal,110 the addition of N-diisopropylethyl amine is required, and the reaction is plagued by the formation of oxidative coupling byproducts unless the alkyne is bound to a solid support (Scheme 41).
Scheme 41.

The very broad scope, compatibility with most functional groups, exclusive regioselectivity, and operational simplicity have placed CuAAC among the most widely utilized click processes.49 Its mechanism has been examined both computationally48,111,112 and through the studies of the kinetics,113,114 and its general features and applications have been reviewed.115-117
Another example of the aqueous transition metal-catalyzed alkyne chemistry is the aldehyde– alkyne–amine (A3) coupling reaction and its asymmetric variants, developed by Li and co-workers.118 Thus, copper(I) acetylides added to imines that were generated in situ from aldehydes and anilines.119 These three-component asymmetric (AA3) coupling reactions were catalyzed by chiral copper(I)-bis(oxazolinyl)pyridine complexes (Scheme 42) and produced optically active propargyl amines 139 in good yield and enantioselectivity.
Scheme 42.

In a subsequent report, the same authors described that the A3 coupling could be catalyzed by gold salts (Scheme 43).120 Although not asymmetric, this synthesis allows preparpation of propargyl amines (140) in excellent yields in water at 100 °C with as little as 0.25 mol % of AuBr3 catalyst. The reaction was significantly faster in water than in dimethylformamide or toluene. Aromatic and aliphatic aldehydes and amines participated in the reaction, and nearly quantitative yields were obtained in most cases. Different gold(I) and gold(III) salts, e.g. AuBr3, AuCl3, AuI, and AuCl, were effective catalysts.
Scheme 43.

The catalytic activity of gold, silver and copper salts was also examined in the coupling reactions of α-alkoxyaldehydes 142, alkynes and amines (Scheme 44).121 Gold(I) was the most effective catalyst in this reaction, providing propargylamines in good yields and moderate diastereoselectivities. Silver catalysts were superior when α-alkyl-substituted aldehydes were used.
Scheme 44.

A variation of the Au(III)-catalyzed A3-coupling reaction was reported by Yan and Liu (Scheme 45).122 Pyridine-2-carboxaldehyde 144 was condensed with terminal alkynes and amines to form 1,3-disubstituted indolizines 145 in generally good yields.
Scheme 45.

Another example of a gold-catalyzed on water synthesis of heterocycles from alkynes was reported by Che. The tricyclic pyrrolo[1,2-a]quinolines 147 were obtained from N-alkynyl anilines 146 and alkynes in the presence of a Au(I) catalyst (Scheme 46).123 The reaction was also examined in organic solvents, but water was superior to all of them. Several examples of the pyrrolo[1,2-a]quinolines products 148-151 illustrate the scope of the process.
Scheme 46.


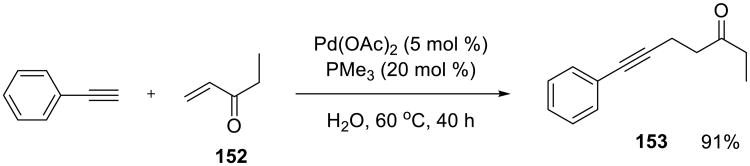
Pd-catalyzed 1,4-additions of terminal alkynes to α,β-unsaturated ketones 152 were reported to proceed well on water by Chen and Li (Scheme 47).124 The process was experimentally simple, could be performed in the presence of oxygen, leading to a wide range of γ,δ-alkynyl ketones 153.
Scheme 47.
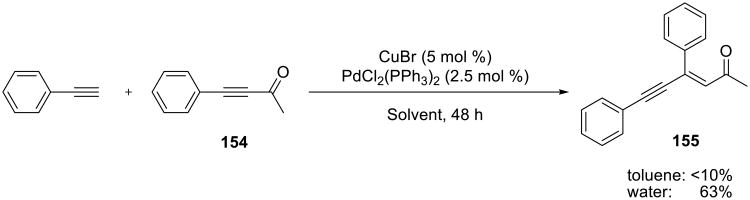
A combination of Pd(II) and Cu(I) catalysts efficiently promoted the addition of terminal alkynes to ynones 154 (Scheme 48).125 Although yields were only moderate on water, the reaction did not proceed at all in toluene. The addition of terminal alkynes to methyl propiolates proceeded in 48-88% yield at 60 °C in 25-40 h, and only E olefins were isolated.
Scheme 48.
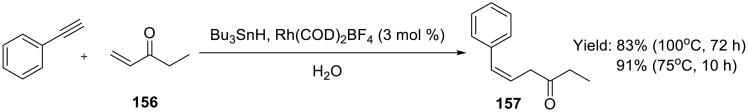
A one-pot rhodium-catalyzed hydrostannylation-conjugate addition reaction was developed by Li and Wu (Scheme 49).126 Inverse temperature dependence, i.e. lower yield at higher temperature, was observed.
Scheme 49.
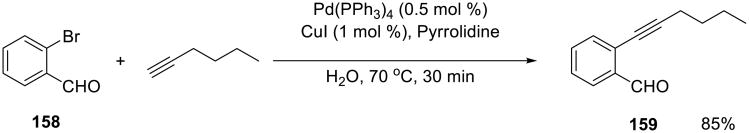
Bhattacharya and Sengupta described exceedingly facile on water Sonogashira reactions127 of aryl iodides and bromides. The reactions were fast (30 min at 70°C and 3 h at room temperature) and high-yielding (Scheme 50). The reactions of aryl iodides were facilitated by the addition of diisopropylethylamine, and those of aryl bromides proceeded best in the presence of pyrrolidine. It is noteworthy that the addition of organic co-solvents (e.g. dimethylformamide) significantly retarded the rate of the reaction and led to the formation of side products, underscoring the importance of the heterogeneous aqueous conditions.
Scheme 50.
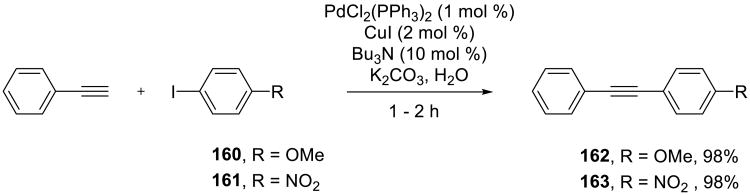
In another account of the on water Sonogashira reaction by Beletskaya and co-workers, diaryl alkynes, such as 162–163, were synthesized from aryl iodides and phenylacetylene (Scheme 51).128 The reaction was slightly slower in aqueous DMF and failed in the dioxane-water and acetonitrile-water mixtures.
Scheme 51.
The inertness and relatively high solubility of boronic acids in water makes them particularly suitable reagents for aqueous transformations. The boronic acid Mannich reaction, discovered by Petasis and co-workers,129,130 and Suzuki-Miyaura couplings131 in aqueous solvents are well known.
The aqueous Suzuki-Miyaura reactions are often performed at the elevated temperature and in the presence of water-soluble catalysts.132-134 A recent example reported by Buchwald and Anderson is an exception. The authors developed a highly-active water-soluble palladium catalyst based on the water-soluble phosphine 164 and utilized it in Suzuki-Miyaura and Sonogashira coupling reactions.135
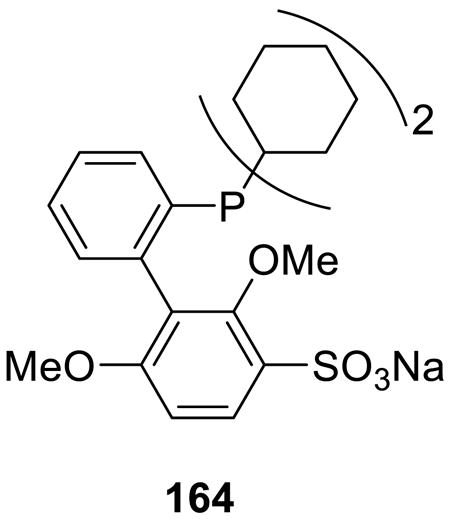
Aryl halides readily reacted with boronic acids at room temperature in the presence of this catalyst in water (Scheme 52), whereas the reactions required 14 h at 100 °C when performed in acetonitrile, n-butanol, or dimethylformamide. Excellent substrate scope, both with respect to the halide and the boronic acid, was reported.
Scheme 52.
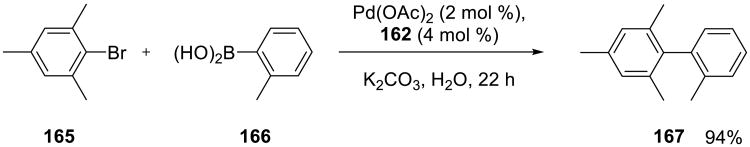
SanMartin, Domínguez, and co-workers examined catalytic efficiency of a series of N–C–N pincer palladium complexes 168–170 in Suzuki-Miyaura, Mizoroki-Heck, and Sonogashira reactions.132,134 These complexes were also very soluble in water, especially at the elevated temperature used for the reactions. The Suzuki-Miyaura couplings proceeded efficiently at 100 °C in water using these catalysts. These reactions proceeded as well in organic solvents. On the other hand, the copper-free Sonogashira reactions of aryl iodides and terminal acetylenes performed better in pyrrolidine at 100 °C than in water in the presence of catalyst 170.134
Highly efficient hydrosilylation of terminal alkynes 173-177 by triethyl silane was performed on water using a Pt catalyst (Scheme 53).136 The optimal catalyst, which contained the divinyl disiloxane ligand 171 and bis(diphenylphosphinomethylene)butylamine 172 performed well across the board and selectively provided E-vinylsilanes in excellent yields.
Scheme 53.

Wang and Liao reported that the Doyle-Kirmse [2,3]-sigmatropic rearrangements of sulfonium ylides readily proceeded on water (Scheme 54)137 in nearly quantitative yields in the presence of 0.5 mol % of a rhodium(II) carboxylate catalyst. Allyl 179 and propargyl 180 sulfides readily participated in the reaction. Solid reactants were pre-dissolved in toluene before they were added to the reaction mixture. Moderate enantioselectivity (up to 68% ee) was observed when chiral rhodium(II) catalysts were employed.
Scheme 54.

Afonso and coworkers demonstrated that selective intramolecular C-H insertion reactions of rhodium carbenes can be performed on water (Scheme 55) using Rh(II) carboxylates if the substrate was sufficiently hydrophobic.138
Scheme 55.
Yorimitsu and co-workers developed an efficient Ni-catalyzed alkylation of aldehydes with alkylboranes on water (Scheme 56). Ketones 185 were readily obtained on water at room temperature without a base additive.139 In contrast, the transformation failed in toluene.
Scheme 56.

Shinokubo and co-workers reported a three-component rhodium catalyzed coupling of aryl boronic acids with internal alkynes and acrylates. Substituted dienes 189 (Scheme 57) were obtained in good yields under the on water conditions,140 whereas the 1,4-addition of the boronic acid to the acrylate was observed in dioxane-water mixtures (product 190), and the simple Heck-type products 191 were isolated from the biphasic reactions performed in diisopropyl ether-water mixtures. Water soluble ligands did not affect the reaction.
Scheme 57.

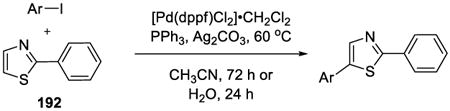
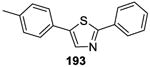
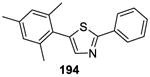
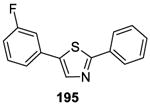
A general on water method for the direct arylation of thiazoles 192 (Table 15)was disclosed by Greaney and co-workers.141 Although comparable yields were obtained under the solvent-free conditions, the on water process operationally simple and product isolation was easy. The reaction exhibited excellent substrate scope, as illustrated by the examples in Table 15.
Table 15. Arylation of 2-phenylthiazole 192.
| ||
|---|---|---|
|
| ||
| product | yield, % | |
|
| ||
| CH3CN | H2O | |
|
74 | 90 |
|
0 | >99 |
|
65 | >99 |
|
0 | >99 |
|
|
0 | >99 |
Using the same on water protocol, thiazoles, benzoxazoles, benzimidazoles, and thiophenes (198-201) were arylated in good yields.
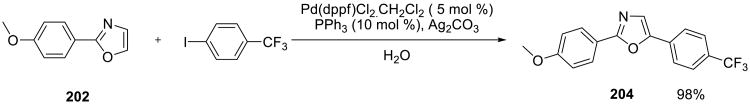
A high-yielding and selective arylation of oxazoles 202 was accomplished on water using Pd(dppf)Cl2 catalyst (Scheme 58).142 Both electron-rich and electron-deficient aryl iodides participated in the reaction, the 5-arylated oxazole products were obtained in high yields. The reaction was utilized in the synthesis of two oxazole-containing natural products, balsoxin and texaline.
Scheme 58.
SanMartin, Dominguez, and co-workers utilized CuI/cyclohexane-1,2-diamine catalyst to arylate thiols with aryl halides. A variety of diaryl sulfides 208-211 were obtained in good yield (Scheme 59).143
Scheme 59.

2.8. Metal-free Carbon-Carbon Bond-Forming Processes
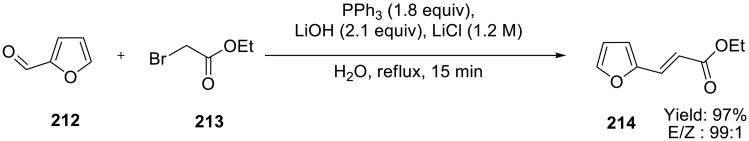
One-pot Wittig reactions of α-bromoacetate 213 (or bromoacetonitrile) with aldehydes 212 (Scheme 60) were carried out in refluxing aqueous 1.2 M LiCl in the presence of LiOH and PPh3.144 Good to excellent yields were obtained with varying E/Z ratio.
Scheme 60.
Bergdahl and co-workers studied Wittig reactions of stabilized ylides 216 (Table 16) on water.145 Higher yields of dienes 217 were obtained on water compared to most organic solvents or ionic liquids, although the E/Z selectivity varied. Aldehydes containing both electron-donating and electron-withdrawing groups participated in the reaction. The addition of lithium chloride increased the yields while a surfactant, sodium dodecylsulfonate, had little effect on the outcome of the process.
Table 16.

| ||
|---|---|---|
|
| ||
| solvent | yield, % | E/Z ratio |
| methanol | 94 | 67 : 33 |
| CH2Cl2 | 78 | 84 : 16 |
| toluene | 59 | 91 : 9 |
| CCl4 | 71 | 91 : 9 |
| THF | 33 | 92 : 8 |
| CH3CN | 41 | 90 : 10 |
| H2O | 88 | 84 : 16 |
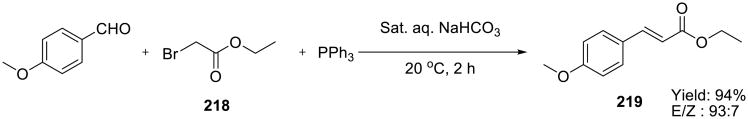
Wittig reactions of ylides formed in situ from PPh3 and α-bromoesters 218 with aldehydes were also successfully carried out in saturated aqueous NaHCO3 solutions (Scheme 61).
Scheme 61.
Li and co-workers described an on water dehydrogenative coupling reaction. The reaction of indoles 220 with 1,4-benzoquinone 221 readily occurred without a catalyst (Scheme 62).146 Yields were good to excellent in all cases, and water outperformed all other solvents as well as the neat conditions (Table 17). The reactants were insoluble in water and formed aqueous suspensions when the reaction mixtures were vigorously stirred.
Scheme 62.

Table 17.
No product observed
When the reaction was carried out in the presence of excess of indole, bis(indolyl)-1,4-quinones 230 (Scheme 63) were obtained in excellent yield.
Scheme 63.


A three-component coupling of aldehydes with propiolates to produce propargylic enol ethers 227 (Scheme 64) was reported by Tellado et al.54 Reactions between terminal alkynoates 226 and aldehydes 225 were catalyzed by tertiary amines, such as quinine, or triphenylphosphine, and addition of LiCl further enhanced the rate of the reaction and the overall yield
Scheme 64.

2.9. Bromination Reactions
One of the most remarkable on water processes was reported in 1955 by Guss and Rosenthal.147 They showed that bromohydrins could be prepared simply by vigorously stirring the olefin substrates with N-bromosuccinimide in water (Scheme 65). The product bromohydrins cleanly separated from the water phase, while the succinimide by-product remains in it. A number of olefins were efficiently oxidized in this manner. These authors also reported that NBS could be precipitated from the crude aqueous layer (50% recovery) simply by adding bromine to it.
Scheme 65.
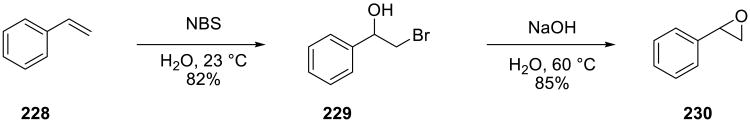
Guss and Rosenthal also showed that the corresponding epoxides could be accessed by heating the product bromohydrins in aqueous NaOH solution.147 This is an efficient method for the synthesis of racemic epoxides, and is especially valuable when the products are acid-sensitive.
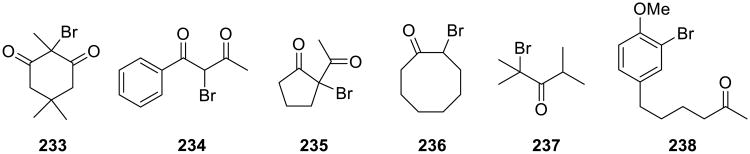
Bromination of 1,3-diketones, β-ketoesters, cyclic ketones, aryl alkyl and dialkyl ketones were accomplished by Iskra and colleagues on water using aqueous H2O2–HBr system (Scheme 66).148 Reactions were carried out at room temperature and monobrominated products were obtained in high yield. The gradual addition of both peroxide and hyhdrobromic acid considerably increased the yield. Comparison with a commonly used organic solvent showed that reactions went much faster under on water conditions. A variety of monobrominated products (233-238) were accessed by this method.
Scheme 66.

The same authors also studied benzylic brominations with N-bromosuccinimide. These Wohl– Ziegler-type reactions149 are normally performed in boiling carbon tetrachloride. However, Iskra and colleagues showed that these brominations could be accomplished on water with good to excellent yield (Scheme 67).150 Only ambient light (or a 40W incandescent bulb) was used to initiate the reaction. For example, benzylmethylketone 239 was selectively brominated at the benzylic carbon. However, the authors also reported that particularly activated aromatic substrates suffered from the competing aromatic bromination: 4-methylanisol 241 was ortho-brominated in 82% yield. The succinimide byproduct is soluble in water, whereas the brominated product forms a separate phase, making its separation easy.
Scheme 67.

Stavber and coworkers reported an example of bromination where a specific site selectivity (Scheme 68) was achieved by performing the reaction in water.151 Under the solvent-free conditions, α-bromination of the ortho-substituted benzophenone 243 to form the α-bromomethyl product 244 was the dominant pathway, whereas on water the aromatic ring was brominated, yielding 245.
Scheme 68.

2.10. Oxidations and reductions
Water is a relatively red-ox inert molecule. In addition to being difficult to oxidize, it is supportive of a variety of chemical and electrochemical oxidants as well as transition metal catalysts. Hence it could be a useful solvent for oxidations and reductions of organic compounds. Indeed, aqueous organic oxidations and reductions are some of the oldest fields in catalysis, and their comprehensive coverage is well beyond the scope of the present review. Accordingly, only a handful of recent examples which meet the on water criteria are mentioned below. For the additional coverage, the reader is referred to recent review literature.95,152,153
In a recent report, Malkov and Bourhani developed a catalyzed epoxidation of allylic alcohols 246 with the in situ generated vanadium catalyst (Scheme 69).154 These single atom transfers were successfully performed on water at 0 °C, and the yields of epoxides 247 were moderate to good. While these epoxidations also proceeded in toluene, the aqueous reactions were ligand-accelerated155 and only required a stoichiometric amount of the ligand with respect to vanadium.
Scheme 69.

Li and co-workers disclosed a catalyst-free on water oxidation of aromatic silyl enol ethers 249. The substrates were converted to α-hydroxy ketones 250 (Scheme 70) in good to excellent yields simply by stirring on water in the presence of air.156 These oxidations failed in most organic solvents as well as under neat conditions (Table 18). Several representative examples of the obtained products 252-256 illustrate the scope of this method.
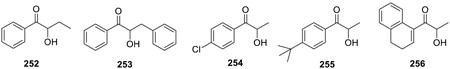
Scheme 70.
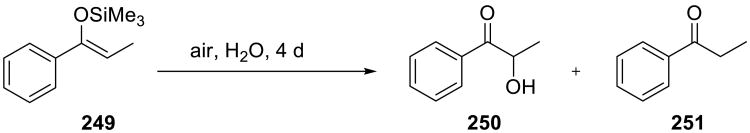
Table 18.
| Solvent | Yield (%, 336:337) |
|---|---|
| THF | - a |
| Acetone | - a |
| Benzene | - a |
| EtOH | - a |
| EtOH-H2O (1:1) | 25 : 35 |
| EtOH-H2O (1:9) | 74 : 16 |
| Neat | - a |
| H2O | 86 : 9 |
No product was observed
Tour and Price reported a unique way for functionalizing single walled carbon nanotubes in water (Scheme 71).157 A variety of substituted anilines 257 were activated with isoamyl nitrite to decorate the surface of the nanotubes 258.
Scheme 71.

Tris(trimethylsilyl) silane was used to reduce several organohalides 259, with 2-mercaptoethanol as co-catalyst under on water conditions (Scheme 72).158 Although both water soluble (2,2′-azobis(2-amidinopropane, AAPH) as well as water insoluble (1,1′-azobis(cyclohezanecarbonitrile), ACCN) azocompounds were used as initiators, ACCN was found to be the better choice. Yields were excellent (75% -100%) in a broad range of substrates. (Me3Si)3SiH did not suffer from any side reactions with water.
Scheme 72.

This method was subsequently used in several radical transformations of both hydrophobic and hydrophilic substrates.159 Radical cyclizations (Scheme 73), hydrosilylations, and reductions of azides were successfully performed. The addition of 2-mercaptoethanol was required for the reactions of hydrophilic substrates.
Scheme 73.
Hydrosilylation of alkynes proceeded smoothly under these conditions, and the E/Z selectivity of the reaction was slightly higher on water (Scheme 74).
Scheme 74.

An efficient and chemoselective transfer hydrogenation reaction on water was reported by Xiao and coworkers (Scheme 75).160 Iridium catalysts produced best results. The on water reactions were fast and high-yielding, and diversely substituted aryl and heteroaryl aldehydes 267–272 participated in the reaction. Aliphatic and α,β-unsaturated aldehydes were also reduced, but the reactions required heating to 80 °C.
Scheme 75.

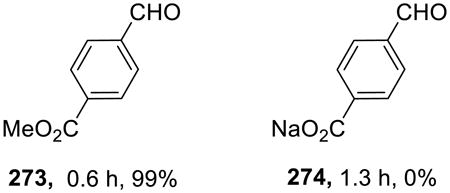
The requirement for the heterogeneous on water conditions was demonstrated in a comparative study of aldehydes 273 and 274. The water-insoluble ester 273 was readily and quantitatively reduced to the corresponding alcohol, whereas the carboxylate salt 274 did not react at all.
Reactions of aliphatic aldehydes were complicated by the competing aldol condensation. Therefore, enolizable aldehydes required slow addition to suppress this undesired pathway. Transfer hydrogenation reactions in water have been recently reviewed by Xiao.161
3. Theoretical studies
To date, there is no unifying theory that could explain the effects of heterogeneous aqueous conditions on organic reactions. The possibility of the water–organic interface acting as a catalyst for the on water reactions is very enticing. An alternative explanation is that even most insoluble organic compounds can enter the aqueous phase, thus effectively turning the ‘on water’ reactions into highly diluted homogeneous processes. Rate acceleration in homogeneous aqueous solution has been attributed to a variety of effects such as hydrophobic aggregation,34,35,162 cohesive energy density,163-167 or destabilization of the reactants vs. the transition state.33,168,169 Breslow has invoked solution-phase hydrophobic effects to explain the high endo selectivity of certain Diels–Alder reactions in aqueous suspension and solution.170 Engberts made a fundamental point by providing evidence that, in cycloaddition reactions, hydrophobic destabilization will have a considerably greater impact on the reactants than on the transition state.169,171 The importance of hydrogen bonding in the acceleration of Diels–Alder reactions in aqueous solution is supported by both experimental172,173 and theoretical174 studies. An excellent review on the structure and properties of water, which helps rationalize rate enhancements observed for organic reactions in aqueous media, has been published recently.175
Lubineau and Pirrung invoked the concept of cohesive energy density to explain the observed rate acceleration of heterogeneous aqueous reactions. Cohesive energy of a solvent is the energy required to remove a molecule from its nearest neighbors in the bulk, leading to the creation of a cavity, factored by the volume of the molecule removed. Water, primarily due to the extensive internal hydrogen bonding and small size, possesses high cohesive energy density (550 cal/cm3). The cohesive energy of water corresponds to the internal pressure of ca. 22 kbar, hence the reactions which have negative activation volume and are accelerated with pressure should respond in a similar way to the aqueous conditions. The early experiments to test this theory were reported by Lubineau in 1986163 and in the subsequent full account.176 The aldol reaction of benzaldehyde and silyl enol ether 279 gave similar outcome in organic solvent at high pressure (10 kbar) and in aqueous medium without pressure which is very different than the outcome of the reaction in organic solvent at atmospheric pressure (Table 19).
Table 19.
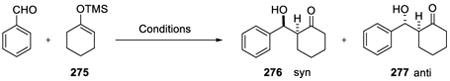
| ||||
|---|---|---|---|---|
|
| ||||
| Entry | Solvent | Reaction conditions | Yield (%) | 276 : 277 |
| 1 | CH2Cl2 | 20°C, 2d, TiCl4 (1 equiv.) | 82 | 25:75 |
| 2 | CH2Cl2 | 60°C, 9d, 10000 atm pressure | 90 | 75:25 |
| 3 | H2O | 20°C, 5d | 23 | 85:15 |
Although hydrophobic effect and CED models may explain rate accelerations in some aqueous reactions, they require that organic reactants have certain solubility to enter the aqueous phase, however minute the amounts may be. Additionally, an attempt to correlate the reaction rates with cohesive energy density of the solvent for a bimolecular reaction (dimerization of cyclopentadiene, a negative volume of activation reaction) and a unimolecular reaction (dissociation of the dimer of triphenylmethyl radical, a positive volume of activation reaction) failed, leading to the conclusion that the concept of cohesive pressure is useful only for reactions of neutral, non-polar molecules in non-polar solvents. In reactions involving polar molecules, or reactions in polar solvents, the contribution of cohesive pressure is simply negligibly small comparing to the solvation interactions.177
The unique features of the water-organic interface can offer an alternative explanation of the reactivity of particularly hydrophobic organic substrates on water. Recently, Jung and Marcus proposed a model which suggests that interactions of hydrophobic organic molecules with water surface may be responsible for the rate enhancement observed on water.178 This theory is based on the experimental evidence that the surface of water has as much as 25% free hydrogen bonds (i.e. hydroxyl groups that are not involved in hydrogen bonding).6,7 Using density functional theory models, the authors showed that interactions of the unbound hydroxyls with organic reactants and, more importantly, with the transition state, are the key factors responsible for the rate enhancements observed in on water reactions. Simple kinetic considerations demonstrated that the reaction on water should be faster than reactions under homogeneous and neat conditions. The difference in solvation pattern in cases of on and in water conditions is shown in Scheme 76.
Scheme 76.
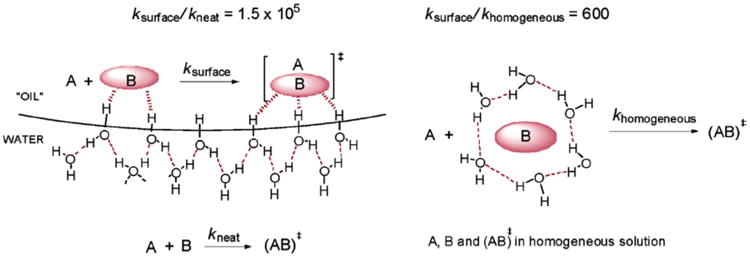
Cartoon representation of the differences in solvation patterns on water and in water.178
Further DFT studies of the reaction between quadricyclane and DMAD demonstrated that there is an increase in the number of hydrogen bonds between the water surface and the reactants and the transition state (shown as red dashed lines in Scheme 77). The Jung-Marcus proposal is that these additional hydrogen bonds stabilize the transition state and therefore explain the experimentally observed rate acceleration.
Scheme 77.
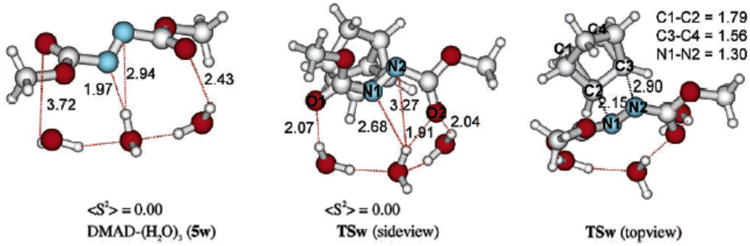
Difference in the hydrogen bonding patterns of DMAD and the transition state TSw.178
Energy calculation of each species under neat and on water conditions showed that the transition state of the on water reaction (TSw, Scheme 78) is stabilized by 7.5 kcal/mol than the corresponding transition state in absence of water (TS1). Based on these calculations, the authors also predicted that a reaction between quadricyclane and acetylene dicarboxylate would not show the on water effect. This prediction has been confirmed experimentally.
Scheme 78.
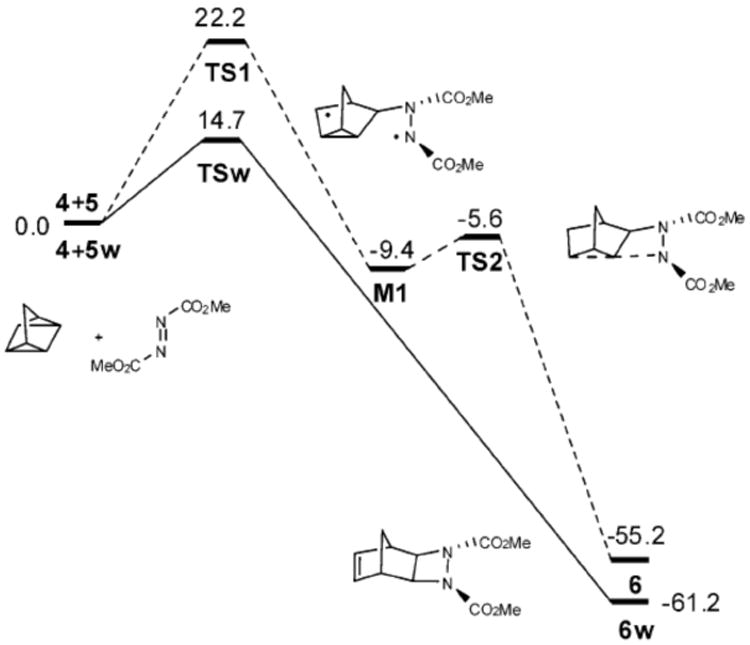
Energetics of the DMAD-quadricyclane reaction (in kcal/mol).178
4. Concluding remarks
Performing organic reactions in water with substrates that are not soluble seems counterintuitive at first. We hope that this survey convinced the reader that attempting such heterogeneous reactions in water is a worthwhile endeavor. Apart from discovering new or improving existing organic transformations, it will surely give us a glimpse into the fascinating and still poorly understood world of water. Like a growing child, organic synthesis on water still surprises us by most unexpected questions and observations. Freeing it from the stereotypes and constraints with which we grew up will propel it a prominent place in the arsenal of tools of synthetic organic chemists. Perhaps one day water will become the most used solvent in synthesis, and the ones that are considered common and conventional today will take an honorable place on the rare chemicals shelf.
Acknowledgments
We thank The Skaggs Institute for Chemical Biology and Pfizer Inc. for the support of our research. AC acknowledges a postdoctoral fellowship from Pfizer.
References
- 1.Ball P. H2O: A Biography of Water. Phoenix Press; London: 2000. [Google Scholar]
- 2.Wöhler F. Ann Phys. 1828;37:330. [Google Scholar]
- 3.Baeyer A, Drewsen V. Ber. 1882;15:2856. [Google Scholar]
- 4.Head-Gordon T, Hura G. Chem Rev. 2002;102 doi: 10.1021/cr0006831. [DOI] [PubMed] [Google Scholar]
- 5.Bellissent-Funnel MC, Done JC. Hydrogen Bond Networks. Kluwer Academic Publications; Boston: 1994. [Google Scholar]
- 6.Moore FG, Richmond GL. Acc Chem Res. 2008;41:739. doi: 10.1021/ar7002732. [DOI] [PubMed] [Google Scholar]
- 7.Shen YR, Ostroverkhov V. Chem Rev. 2006;106:1140. doi: 10.1021/cr040377d. [DOI] [PubMed] [Google Scholar]
- 8.Li CJ, Chan TH. Organic reactions in aqueous media. Wiley; New York: 1997. [Google Scholar]
- 9.Grieco PA, editor. Organic Synthesis in Water. Blackie; London: 1998. [Google Scholar]
- 10.Lindström UM. Chem Rev. 2002;102:2751. doi: 10.1021/cr010122p. [DOI] [PubMed] [Google Scholar]
- 11.Itami K, Yoshida JI. Chemical Record. 2002;2:213. doi: 10.1002/tcr.10021. [DOI] [PubMed] [Google Scholar]
- 12.Brogan AP, Dickerson TJ, Janda KD. Angew Chem Int Ed. 2006;45:8100. doi: 10.1002/anie.200601392. [DOI] [PubMed] [Google Scholar]
- 13.Hayashi Y. Angew Chem Int Ed. 2006;45:8103. doi: 10.1002/anie.200603378. [DOI] [PubMed] [Google Scholar]
- 14.Lipshutz BH, Aguinaldo GT, Ghorai S, Voigtritter K. Org Lett. 2008;10:1325. doi: 10.1021/ol800028x. [DOI] [PubMed] [Google Scholar]
- 15.Lipshutz BH, Chung DW, Rich B. Org Lett. 2008;10:3793. doi: 10.1021/ol801471f. [DOI] [PubMed] [Google Scholar]
- 16.Lipshutz BH, Ghorai S, Aguinaldo GT. Adv Synth Catal. 2008;350:953. [Google Scholar]
- 17.Lipshutz BH, Taft BR. Org Lett. 2008;10:1329. doi: 10.1021/ol702755g. [DOI] [PubMed] [Google Scholar]
- 18.Lipshutz BH, Abela AR. Org Lett. 2008;10:5329. doi: 10.1021/ol801712e. [DOI] [PubMed] [Google Scholar]
- 19.Narayan S, Muldoon J, Finn MG, Fokin VV, Kolb HC, Sharpless KB. Angew Chem Int Ed. 2005;44:3275. doi: 10.1002/anie.200462883. [DOI] [PubMed] [Google Scholar]
- 20.Trost BM. Angew Chem Int Ed. 1995;34:259. [Google Scholar]
- 21.Blackmond DG, Armstrong A, Coombe V, Wells A. Angew Chem Int Ed. 2007;46:3798. doi: 10.1002/anie.200604952. [DOI] [PubMed] [Google Scholar]
- 22.Wei W, Keh C, Li CJ, Varma R. Clean Technologies and Environmental Policy. 2004;6:250. [Google Scholar]
- 23.Lindström UM, editor. Organic Reactions in Water: Principles, Strategies and Applications. Blackwell; Oxford: 2007. [Google Scholar]
- 24.Kobayashi S, Manabe K. Acc Chem Res. 2002;35:209. doi: 10.1021/ar000145a. [DOI] [PubMed] [Google Scholar]
- 25.Li CJ. Chem Rev. 1993;93:2023. [Google Scholar]
- 26.Li CJ. Chem Rev. 2005;105:3095. doi: 10.1021/cr030009u. [DOI] [PubMed] [Google Scholar]
- 27.Li CJ, Chen L. Chem Soc Rev. 2006;35:68. doi: 10.1039/b507207g. [DOI] [PubMed] [Google Scholar]
- 28.Dallinger D, Kappe CO. Chem Rev. 2007;107:2563. doi: 10.1021/cr0509410. [DOI] [PubMed] [Google Scholar]
- 29.Liotta CL, Hallett JP, Pollet P, Eckert CA. In: Organic Reactions in Water: Principles, Strategies and Applications. Lindström UM, editor. Blackwell; Oxford: 2007. pp. 256–300. [Google Scholar]
- 30.Nakamura K, Matsuda T. In: Organic Reactions in Water: Principles, Strategies and Applications. Lindström UM, editor. Blackwell; Oxford: 2007. pp. 301–349. [Google Scholar]
- 31.Hopff H, Rautenstrauch CW. US 2,262,002. 1939 [Google Scholar]
- 32.Rideout DC, Breslow R. J Am Chem Soc. 1980;102:7816. [Google Scholar]
- 33.Blokzijl W, Engberts JBFN. Angew Chem Int Ed. 1993;32:1545. [Google Scholar]
- 34.Breslow R. Acc Chem Res. 1991;24:159. [Google Scholar]
- 35.Breslow R. In: Structure and Reactivity in Aqueous Solution. Cramer CJ, Truhlar DG, editors. Vol. 568. American Chemical Society; Washington, DC: 1994. pp. 291–302. [Google Scholar]
- 36.Rideout DC, Moitra U, Breslow R. Tetrahedron Lett. 1983;24:1901. [Google Scholar]
- 37.Grieco PA, Garner P, He Z. Tetrahedron Lett. 1983;24:1897. [Google Scholar]
- 38.Grieco PA, Yoshida K, Garner P. J Org Chem. 1983;48:3137. [Google Scholar]
- 39.Attanasi OA, Crescentinia LD, Filipponea P, Fringuelli F, Mantellinia F, Matteuccib M, Piermattib O, Pizzo F. Helv Chim Acta. 2001;84:513. [Google Scholar]
- 40.Loncaric C, Manabe K, Kobayashi S. Adv Synth Catal. 2003;345:475. [Google Scholar]
- 41.Huisgen R. Angew Chem Int Ed. 1963;2:565. [Google Scholar]
- 42.Padwa A, editor. 1,3-dipolar cycloaddition chemistry. Wiley; New York: 1984. [Google Scholar]
- 43.Padwa A, Pearson WH, editors. Synthetic applications of 1,3 dipolar cycloaddition chemistry toward heterocycles and natural products. Wiley; New York: 2002. [Google Scholar]
- 44.Wijnen JW, Steiner RA, Engberts JBFN. Tetrahedron Lett. 1995;36:5389. [Google Scholar]
- 45.van Mersbergen D, Wijnen JW, Engberts JBFN. J Org Chem. 1998;63:8801. [Google Scholar]
- 46.Rispens T, Engberts JBFN. J Org Chem. 2003;68:8520. doi: 10.1021/jo0349330. [DOI] [PubMed] [Google Scholar]
- 47.Portmann R. WO 9802423. 1998 [Google Scholar]
- 48.Himo F, Lovell T, Hilgraf R, Rostovtsev VV, Noodleman L, Sharpless KB, Fokin VV. J Am Chem Soc. 2005;127:210. doi: 10.1021/ja0471525. [DOI] [PubMed] [Google Scholar]
- 49.Kolb HC, Finn MG, Sharpless KB. Angew Chem Int Ed. 2001;40:2004. doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 50.Li Z, Seo TS, Ju J. Tetrahedron Lett. 2004;45:3143. [Google Scholar]
- 51.Butler RN, Coyne AG, Moloney EM. Tetrahedron Lett. 2007;48:3501. [Google Scholar]
- 52.Butler RN, Coyne AG, Cunningham WJ, Moloney EM, Burke LA. Helv Chim Acta. 2005;88:1611. [Google Scholar]
- 53.Gonzalez-Cruz D, Tejedor D, de Armas P, Morales EQ, Garcia-Tellado F. Chem Comm. 2006:2798. doi: 10.1039/b606096j. [DOI] [PubMed] [Google Scholar]
- 54.Gonzalez-Cruz D, Tejedor D, de Armas P, Garcia-Tellado F. Chem Eur J. 2007;13:4823. doi: 10.1002/chem.200700227. [DOI] [PubMed] [Google Scholar]
- 55.Pang W, Zhu SF, Jiang HF, Zhu SZ. J Fluorine Chem. 2007;128:1379. [Google Scholar]
- 56.Bala K, Hailes HC. Synthesis. 2005:3423. [Google Scholar]
- 57.Demko ZP, Sharpless KB. J Org Chem. 2001;66:7945. doi: 10.1021/jo010635w. [DOI] [PubMed] [Google Scholar]
- 58.Himo F, Demko ZP, Noodleman L, Sharpless KB. J Am Chem Soc. 2002;124:12210. doi: 10.1021/ja0206644. [DOI] [PubMed] [Google Scholar]
- 59.Himo F, Demko ZP, Noodleman L, Sharpless KB. J Am Chem Soc. 2003;125:9983. doi: 10.1021/ja030204q. [DOI] [PubMed] [Google Scholar]
- 60.Rieber N, Alberts J, Lipsky JA, Lemal DM. J Am Chem Soc. 1969;91:5668. [Google Scholar]
- 61.Platz R, Fuchs W, Rieber N, Samuel UR, Jung J. DE 2615878. 1977 [Google Scholar]
- 62.Platz R, Fuchs W, Rieber N, Jung J. DE 2742034. 1979 [Google Scholar]
- 63.Brimble MA, Heathcock CH. J Org Chem. 1993;58:5261. [Google Scholar]
- 64.Leblanc Y, Zamboni R, Bernstein MA. J Org Chem. 1991;56:1971. [Google Scholar]
- 65.Narayan S, Fokin VV, Sharpless KB. In: Organic Reactions in Water: Principles, Strategies and Applications. Lindström UM, editor. Blackwell; Oxford: 2007. pp. 350–365. [Google Scholar]
- 66.Gajewski JJ, Jurayj J, Kimbrough DR, Gande ME, Ganem B, Carpenter BK. J Am Chem Soc. 1987;109:1170. [Google Scholar]
- 67.Brandes E, Grieco PA, Gajewski JJ. J Org Chem. 1989;54:515. [Google Scholar]
- 68.Nicolaou KC, Xu H, Wartmann M. Angew Chem Int Ed. 2005;44:756. doi: 10.1002/anie.200462211. [DOI] [PubMed] [Google Scholar]
- 69.Domling A. Current Opinion in Chemical Biology. 2002;6:306. doi: 10.1016/s1367-5931(02)00328-9. [DOI] [PubMed] [Google Scholar]
- 70.Hulme C. Multicomponent Reactions. 2005:311. [Google Scholar]
- 71.Ugi I, Werner B. Methods and Reagents for Green Chemistry. 2007:3. [Google Scholar]
- 72.Sarma KD, Pirrung MC. J Am Chem Soc. 2003;126:444. doi: 10.1021/ja038583a. [DOI] [PubMed] [Google Scholar]
- 73.Pirrung MC, Das Sarma K. Tetrahedron. 2005;61:11456. [Google Scholar]
- 74.Lin Q, O'Neill JC, Blackwell HE. Org Lett. 2005;7:4455. doi: 10.1021/ol051684o. [DOI] [PubMed] [Google Scholar]
- 75.Shapiro N, Vigalok A. Angew Chem Int Ed. 2008;47:2849. doi: 10.1002/anie.200705347. [DOI] [PubMed] [Google Scholar]
- 76.Fokin VV, Wu P. In: Aziridines and Epoxides in Organic Synthesis. Yudin AK, editor. Wiley-VCH; Weinheim: 2006. pp. 443–477. [Google Scholar]
- 77.Azizi N, Saidi MR. Org Lett. 2005;7:3649. doi: 10.1021/ol051220q. [DOI] [PubMed] [Google Scholar]
- 78.Saidi MR, Ziyaei-Halimjani A. Can J Chem. 2006;84:1515. [Google Scholar]
- 79.Chuang TH, Sharpless KB. Org Lett. 1999;1:1435. doi: 10.1021/ol990256d. [DOI] [PubMed] [Google Scholar]
- 80.Chuang TH, Sharpless KB. Org Lett. 2000;2:3555. doi: 10.1021/ol000221+. [DOI] [PubMed] [Google Scholar]
- 81.Chuang TH, Sharpless KB. Helv Chim Acta. 2000;83:1734. [Google Scholar]
- 82.Gajewski JJ. J Am Chem Soc. 2001;123:10877. doi: 10.1021/ja010600d. [DOI] [PubMed] [Google Scholar]
- 83.Azoulay Sp, Manabe K, Kobayashi S. Org Lett. 2005;7:4593. doi: 10.1021/ol051546z. [DOI] [PubMed] [Google Scholar]
- 84.Vilotijevic I, Jamison TF. Science. 2007;317:1189. doi: 10.1126/science.1146421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kleiner CM, Schreiner PR. Chem Comm. 2006:4315. doi: 10.1039/b605850g. [DOI] [PubMed] [Google Scholar]
- 86.Palmisano G, Tagliapietra S, Barge A, Binello A, Boffa L, Cravotto G. Synlett. 2007:2041. [Google Scholar]
- 87.Converso A, Burow K, Marzinzik A, Sharpless KB, Finn MG. J Org Chem. 2001;66:4386. doi: 10.1021/jo015632y. [DOI] [PubMed] [Google Scholar]
- 88.Converso A, Saaidi PL, Sharpless KB, Finn MG. J Org Chem. 2004;69:7336. doi: 10.1021/jo0489869. [DOI] [PubMed] [Google Scholar]
- 89.Cozzi PG, Zoli L. Green Chem. 2007;9:1292. [Google Scholar]
- 90.Cozzi PG, Zoli L. Angew Chem Int Ed. 2008;47:4162. doi: 10.1002/anie.200800622. [DOI] [PubMed] [Google Scholar]
- 91.Mayr H, Ofial AR. Pure Appl Chem. 2005;77:1807. [Google Scholar]
- 92.Mayr H, Ofial AR. J Phys Org Chem. 2008;21:584. [Google Scholar]
- 93.Nigst TA, Westermaier M, Ofial AR, Mayr H. Eur J Org Chem. 2008:2369. [Google Scholar]
- 94.Carril M, SanMartin R, Dominguez E. Chem Soc Rev. 2008;37:639. doi: 10.1039/b709565c. [DOI] [PubMed] [Google Scholar]
- 95.Sheldon RA. In: Organic Reactions in Water: Principles, Strategies and Applications. Lindström UM, editor. Blackwell; Oxford: 2007. pp. 215–235. [Google Scholar]
- 96.Chen L, Li CJ. Adv Synth Cat. 2006;348:1459. [Google Scholar]
- 97.Glaser C. Chemische Berichte. 1869;2:422. [Google Scholar]
- 98.Glaser C. Ann Chem Pharm. 1870;154:159. [Google Scholar]
- 99.Baeyer A. Ber Dtsch Chem Ges. 1885;18:674. [Google Scholar]
- 100.Straus F, Kollek L. Ber Dtsch Chem Ges. 1926;59B:1664. [Google Scholar]
- 101.Straus F. Justus Liebigs Ann Chem. 1905:190. [Google Scholar]
- 102.Reppe W, et al. Justus Liebigs Ann Chem. 1955;596:6. [Google Scholar]
- 103.Eglinton G, Galbraith AR. Chem & Ind (London) 1956:737. [Google Scholar]
- 104.Castro CE, Stephens RD. J Org Chem. 1963;28:3313. [Google Scholar]
- 105.Castro CE, Stephens RD. J Org Chem. 1963;28:2163. [Google Scholar]
- 106.Chodkiewicz W. Ann Chim (Paris)[13] 1957;2:819. [Google Scholar]
- 107.Chodkiewicz W, Cadiot P, Willemart A. Compt Rend. 1957;245:2061. [Google Scholar]
- 108.Mykhalichko BM, Temkin ON, Mys'kiv MG. Russ Chem Rev. 2001;69:957. [Google Scholar]
- 109.Rostovtsev VV, Green LG, Fokin VV, Sharpless KB. Angew Chem Int Ed. 2002;41:2596. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 110.Tornøe CW, Christensen C, Meldal M. J Org Chem. 2002;67:3057. doi: 10.1021/jo011148j. [DOI] [PubMed] [Google Scholar]
- 111.Ahlquist M, Fokin VV. Organometallics. 2007;26:4389. [Google Scholar]
- 112.Straub BF. Chem Comm. 2007:3868. doi: 10.1039/b706926j. [DOI] [PubMed] [Google Scholar]
- 113.Rodionov VO, Fokin VV, Finn MG. Angew Chem Int Ed. 2005;44:2210. doi: 10.1002/anie.200461496. [DOI] [PubMed] [Google Scholar]
- 114.Rodionov VO, Presolski SI, Diaz DD, Fokin VV, Finn MG. J Am Chem Soc. 2007;129:12705. doi: 10.1021/ja072679d. [DOI] [PubMed] [Google Scholar]
- 115.Bock VD, Hiemstra H, van Maarseveen JH. Eur J Org Chem. 2006:51. [Google Scholar]
- 116.Moses JE, Moorhouse AD. Chem Soc Rev. 2007;36:1249. doi: 10.1039/b613014n. [DOI] [PubMed] [Google Scholar]
- 117.Wu P, Fokin VV. Aldrichimica Acta. 2007;40:7. [Google Scholar]
- 118.Wei C, Li Z, Li CJ. Synlett. 2004:1472. [Google Scholar]
- 119.Wei CM, Li CJ. J Am Chem Soc. 2002;124:5638. doi: 10.1021/ja026007t. [DOI] [PubMed] [Google Scholar]
- 120.Wei CM, Li CJ. J Am Chem Soc. 2003;125:9584. doi: 10.1021/ja0359299. [DOI] [PubMed] [Google Scholar]
- 121.Huang BS, Yao XQ, Li CJ. Adv Synth Cat. 2006;348:1528. [Google Scholar]
- 122.Yan B, Liu Y. Org Lett. 2007;9:4323. doi: 10.1021/ol701886e. [DOI] [PubMed] [Google Scholar]
- 123.Liu XY, Che CM. Angew Chem Int Ed. 2008;47:3805. doi: 10.1002/anie.200800160. [DOI] [PubMed] [Google Scholar]
- 124.Chen L, Li CJ. Chem Comm. 2004:2362. doi: 10.1039/b407936a. [DOI] [PubMed] [Google Scholar]
- 125.Chen L, Li CJ. Tetrahedron Lett. 2004;45:2771. [Google Scholar]
- 126.Wu W, Li CJ. Lett Org Chem. 2004;1:122. [Google Scholar]
- 127.Bhattacharya S, Sengupta S. Tetrahedron Lett. 2004;45:8733. [Google Scholar]
- 128.Bumagin NA, Sukhomlinova Ll, Luzikova EV, Tolstaya TP, Beletskaya IP. Tetrahedron Lett. 1996;37:897. [Google Scholar]
- 129.Petasis NA, Akritopoulou I. Tetrahedron Lett. 1993;34:583. [Google Scholar]
- 130.Petasis NA, Zavialov IA. J Am Chem Soc. 1997;119:445. [Google Scholar]
- 131.Miyaura N, Suzuki A. Chem Rev. 1995;95:2457. [Google Scholar]
- 132.Churruca F, SanMartin R, Tellitu I, Domínguez E. Synlett. 2005:3116. [Google Scholar]
- 133.Inés B, Moreno I, SanMartin R, Domínguez E. The Journal of Organic Chemistry. 2008;73:8448. doi: 10.1021/jo8016633. [DOI] [PubMed] [Google Scholar]
- 134.Inés B, SanMartin R, Churruca Ft, Domínguez E, Urtiaga MK, Arriortua MaI. Organometallics. 2008;27:2833. [Google Scholar]
- 135.Anderson KW, Buchwald SL. Angew Chem Int Ed. 2005;44:6173. doi: 10.1002/anie.200502017. [DOI] [PubMed] [Google Scholar]
- 136.Wu W, Li CJ. Chem Comm. 2003:1668. [Google Scholar]
- 137.Liao MY, Wang JB. Green Chem. 2007;9:184. [Google Scholar]
- 138.Candeias NR, Gois PMP, Afonso CAM. J Org Chem. 2006;71:5489. doi: 10.1021/jo060397a. [DOI] [PubMed] [Google Scholar]
- 139.Hirano K, Yorimitsu H, Oshima K. Adv Synth Catal. 2006;348:1543. [Google Scholar]
- 140.Kurahashi T, Shinokubo H, Osuka A. Angew Chem Int Ed. 2006;45:6336. doi: 10.1002/anie.200602585. [DOI] [PubMed] [Google Scholar]
- 141.Turner GL, Morris JA, Greaney MF. Angew Chem Int Ed. 2007;46:7996. doi: 10.1002/anie.200702141. [DOI] [PubMed] [Google Scholar]
- 142.Ohnmacht SA, Mamone P, Culshaw AJ, Greaney MF. Chem Comm. 2008:1241. doi: 10.1039/b719466h. [DOI] [PubMed] [Google Scholar]
- 143.Carril M, SanMartin R, Dominguez E, Tellitu I. Chem Eur J. 2007;13:5100. doi: 10.1002/chem.200601737. [DOI] [PubMed] [Google Scholar]
- 144.Wu JL, Yue CY. Synth Commun. 2006;36:2939. [Google Scholar]
- 145.El-Batta A, Jiang CC, Zhao W, Anness R, Cooksy AL, Bergdahl M. J Org Chem. 2007;72:5244. doi: 10.1021/jo070665k. [DOI] [PubMed] [Google Scholar]
- 146.Zhang HB, Liu L, Chen YJ, Wang D, Li CJ. Eur J Org Chem. 2006:869. [Google Scholar]
- 147.Guss CO, Rosenthal R. J Am Chem Soc. 1955;77:2549. [Google Scholar]
- 148.Podgorsek A, Stavber S, Zupan M, Iskra J. Green Chem. 2007;9:1212. [Google Scholar]
- 149.Djerassi C. Chem Rev. 1948;43:271. doi: 10.1021/cr60135a004. [DOI] [PubMed] [Google Scholar]
- 150.Podgorsek A, Stavber S, Zupan M, Iskra J. Tetrahedron Lett. 2006;47:1097. [Google Scholar]
- 151.Pravst I, Zupan M, Stavber S. Tetrahedron Lett. 2006;47:4707. [Google Scholar]
- 152.Carraro M, Sartorel A, Scorrano G, Carofiglio T, Bonchio M. Synthesis. 2008:1971. [Google Scholar]
- 153.Herrerias CI, Yao X, Li Z, Li CJ. Chemical Reviews (Washington, DC, United States) 2007;107:2546. doi: 10.1021/cr050980b. [DOI] [PubMed] [Google Scholar]
- 154.Bourhani Z, Malkov AV. Chem Comm. 2005:4592. doi: 10.1039/b509436d. [DOI] [PubMed] [Google Scholar]
- 155.Berrisford DJ, Bolm C, Sharpless KB. Angew Chem Int Ed. 1995;34:1059. [Google Scholar]
- 156.Li HJ, Zhao JL, Chen YJ, Liu L, Wang D, Li CJ. Green Chem. 2005;7:61. [Google Scholar]
- 157.Price BK, Tour JM. J Am Chem Soc. 2006;128:12899. doi: 10.1021/ja063609u. [DOI] [PubMed] [Google Scholar]
- 158.Postigo A, Ferreri C, Navacchia ML, Chatgilialoglu C. Synlett. 2005:2854. [Google Scholar]
- 159.Postigo A, Kopsov S, Ferreri C, Chatgilialoglu C. Org Lett. 2007;9:5159. doi: 10.1021/ol7020803. [DOI] [PubMed] [Google Scholar]
- 160.Wu XF, Liu JK, Li XH, Zanotti-Gerosa A, Hancock F, Vinci D, Ruan JW, Xiao JL. Angew Chem Int Ed. 2006;45:6718. doi: 10.1002/anie.200602122. [DOI] [PubMed] [Google Scholar]
- 161.Wu XF, Xiao JL. Chem Comm. 2007:2449. doi: 10.1039/b618340a. [DOI] [PubMed] [Google Scholar]
- 162.Rideout DC, Breslow R. J Am Chem Soc. 1980;102:7816. [Google Scholar]
- 163.Lubineau A. J Org Chem. 1986;51:2142. [Google Scholar]
- 164.Lubineau A, Meyer E. Tetrahedron. 1988;44:6065. [Google Scholar]
- 165.Desimoni G, Faita G, Righetti PP, Toma L. Tetrahedron. 1990;46:7951. [Google Scholar]
- 166.Gajewski JJ. J Org Chem. 1992;57:5500. [Google Scholar]
- 167.Gajewski JJ, Brichford NL. In: Structure and reactivity in aqueous solution. Cramer CJ, Truhlar DG, editors. Vol. 568. 1994. pp. 229–42. [Google Scholar]
- 168.Blokzijl W, Engberts JBFN. In: Structure and Reactivity in Aqueous Solution. Cramer CJ, Truhlar DG, editors. Vol. 568. American Chemical Society; Washington, DC: 1994. pp. 303–317. [Google Scholar]
- 169.Meijer A, Otto S, Engberts JBFN. J Org Chem. 1998;63:8989. [Google Scholar]
- 170.Breslow R, Maitra U, Rideout D. Tetrahedron Lett. 1983;24:1901. [Google Scholar]
- 171.Blokzijl W, Engberts JBFN. J Am Chem Soc. 1992;114:5440. [Google Scholar]
- 172.Otto S, Blokzijl W, Engberts JBFN. J Org Chem. 1994;59:5372. [Google Scholar]
- 173.van der Wel GK, Wijnen JW, Engberts JBFN. J Org Chem. 1996;61:9001. doi: 10.1021/jo9614248. [DOI] [PubMed] [Google Scholar]
- 174.Chandrasekhar J, Shariffskul S, Jorgensen WL. J Phys Chem B. 2002;106:8078. [Google Scholar]
- 175.Engberts JBFN. In: Organic Reactions in Water: Principles, Strategies and Applications. Lindström UM, editor. Blackwell; Oxford: 2007. pp. 29–59. [Google Scholar]
- 176.Lubineau A, Auge J, Queneau Y. Synthesis. 1994:741. [Google Scholar]
- 177.Reichardt C. Solvents and Solvent Effects in Organic Chemistry. Wiley-VCH; Weinheim: 2003. pp. 221–225. [Google Scholar]
- 178.Jung YS, Marcus RA. J Am Chem Soc. 2007;129:5492. doi: 10.1021/ja068120f. [DOI] [PubMed] [Google Scholar]