

Summary
De novo protein synthesis is necessary for long-lasting modifications in synaptic strength and dendritic spine dynamics that underlie cognition1. Fragile X syndrome, characterized by intellectual disability and autistic behaviors, holds promise for revealing the molecular basis for these long-term changes in neuronal function. Loss-of-function of FMRP, the fragile X mental retardation protein, results in defects in synaptic plasticity and cognition in many models of the disease. FMRP is a polyribosome-associated RNA binding protein that regulates the synthesis of a set of plasticity-related proteins by stalling ribosomal translocation on target mRNAs. The recent identification of mRNA targets of FMRP and its upstream regulators, and the use of small molecules to stall ribosomes in the absence of FMRP, have the potential to be translated into novel therapeutic avenues for the treatment of FXS.
Introduction
Many forms of long-term synaptic and spine morphological plasticity require rapid protein synthesis for their expression while the requirement for new transcription is temporally delayed, suggesting that the translation of existing mRNAs is fundamental to the expression of synaptic plasticity. One of the most intriguing examples of a disease characterized by defects in complex behavior and cognition in humans, and defects in synaptic plasticity and circuit function in mouse models, is fragile X syndrome (FXS), which is caused by loss-of-function of the RNA binding protein FMRP (reviewed in2,3). Mouse models of FXS are characterized by widespread defects in synaptic plasticity4, and both mice and humans have been found to have increased numbers of long, thin dendritic spines relative to the mushroom-shaped spines characteristic of stronger, more mature synapses5,6. Recent studies have shown marked alterations in spine dynamics, including an accelerated turnover rate and a failure to respond to input7,8. Notably, several protein synthesis-dependent forms of plasticity lose their dependence on new translation in the absence of FMRP9,10, suggesting that plasticity-related proteins are already present in adequate levels. Plasticity and spine dynamics are essential for establishing and maintaining normal synaptic communication and for supporting larger groups of neurons that function together in circuits linked by these synaptic contacts. Defects in several neuronal circuits have been described due to absence of FMRP11. These studies underscore the importance of FMRP for normal brain function.
Elucidating FMRP function at the molecular level is key to unraveling its role in more complex processes at the level of synapses, circuits, and behavior. FMRP is expressed in all neurons, but not in mature glia12, and within the neuron FMRP is enriched in the cell body, though some is present in granules in dendrites as well as axons during development13. The vast majority of FMRP is associated with polyribosomes14,15 in complexes that contain several stalled ribosomes in addition to its target mRNAs16. The major function of FMRP is to repress translation of specific mRNAs and it is widely believed that loss of this form of translational control leads to the deficits seen in human FXS patients and disease models. However, more global effects on translation may be a secondary consequence of dysregulation of the primary mRNA targets of FMRP. Thus, elucidating the molecular details of the function of FMRP in regulating translation as well as identifying primary and secondary changes in protein expression that result from its absence is of critical importance. In addition, activity-dependent mechanisms by which FMRP is regulated are central to understand the both the pathology of FXS as well as how protein synthesis regulates synaptic strength in healthy neurons. This review focuses on presenting and evaluating recent experiments addressing these issues and how recent progress might inform therapeutic development for treatment of individuals with FXS.
FMRP as a translational regulator
Shortly after the human FMR1 gene was cloned17 it was recognized that the encoded protein, FMRP, harbors three canonical RNA binding domains18. Although almost all cases of FXS are caused by complete loss of expression of FMRP due to transcriptional silencing, a single patient exists with a point mutation in the second KH domain (KH2) that converts a key hydrophobic amino acid in the heart of the RNA binding pocket to a bulky charged group, referred to as I304N19,20. Polysome fractionation of I304N FXS lymphoblastoid cells21 and brain from an I304N knock-in mouse model22 revealed that the function of the KH2 RNA-binding domain is essential for normal polyribosome association of FMRP. Furthermore, characterization of endogenous I304N-FMRP in mouse brain demonstrated that the mutation results in loss of RNA binding by FMRP22, strongly suggesting that the association of FMRP with polysomes, mediated by RNA binding, is a critical aspect of its function. Coupled with studies demonstrating that impaired memory in an FXS model can be rescued using translational inhibitors23, it is likely that understanding translational control by FMRP will inform both the disease and normal neuronal biology.
Mammalian KH domains adopt a very consistent fold necessary for sequencespecific recognition of single-stranded RNA motifs of 4–7 nucleotides24. As FMRP harbors two KH domains, one of which (KH2) binds with high affinity to an RNA ligand with both sequence and structural determinants25, coupled with reports of the association of FMRP with a subset of mRNAs26,27 it seemed likely that FMRP bound to specific RNA targets through these motifs. The pursuit of this putative set of FMRP-specific mRNA targets has been reviewed elsewhere28 but has recently culminated in surprising findings.
The mRNA targets of FMRP
Recent studies have identified in vivo RNA binding sites for RNA binding proteins using UV irradiation to capture endogenous interactions by introducing a covalent crosslink between the protein and bound RNA29. After reducing the size of the bound RNA “tags” the RNABP:RNA complex can be stringently purified, usually by immunoprecipitation, and the small RNA ligands cloned and sequenced by high throughput methods. Application of this technique to capture RNA binding by FMRP in mouse brain was performed at P11-P25, a critical period for plasticity. This analysis identified a robust set of 842 FMRP target mRNAs, which were markedly enriched in both pre- and postsynaptic proteins16.
The postsynaptic targets of FMRP include about 30% of the postsynaptic density (PSD) proteome and include NMDA receptor subunits, the mGluR5 receptor, and many proteins that comprise the NMDA- and mGluR5-receptor interactomes16. For example, the NMDAR subunits NR1, 2A, 2B, and 3A, PSD-93 and PSD-95, SAPAP1-4, Shank1-3, Homer1, SynGAP1 and neuroligins 1-3 are FMRP targets. In contrast, FMRP does not appear to regulate expression of the AMPA receptor interactome. None of the 4 AMPAR subunits, none of the 8 stargazin/TARP/Cacng family, neither cornichon homolog (Cnih2/3), Shisa9/CKAMP-44, EphrinB2 nor the newly identified Gsg1L30 is an FMRP target. This is one example of selective mRNA regulation by FMRP to control expression of specific complexes and pathways within neurons that may have important consequences for synaptic function.
Postsynaptic targets of FMRP were expected based on synaptic plasticity deficits and the EM localization of FMRP to dendrites; however, the observation that FMRP may also regulate as much as 30% of the pre-synaptic proteome was surprising. This includes the large scaffolding proteins bassoon and piccolo, the neurexin family of cell surface adhesion molecules, synapsins 1 and 2, the synaptic vesicle glycoproteins SV2A and SV2B, subunits of the clathrin-associated adaptor complex AP-2, synaptotagmins 1, 7, and 11, unc13, the syntaxin-binding proteins 1 (unc18) and 5 (tomosyn), SNAP-25, and many voltage-gated calcium channels. In addition, several kinesins, which deliver cargo to presynaptic terminals, are FMRP targets (Kif1a/b/c, Kif5a/c, Kif3 and Kif21a/b). A presynaptic role for FMRP (reviewed by31) is supported by recent studies in hippocampus32–34, cortex35 and amygdala36. A need for rapid synthesis of presynaptic proteins for the formation and maintenance of new synapses has been elegantly described37. The recent report of enhanced vesicle recycling and enlarged readily-releasable and reserved vesicle pools found at CA3-CA1 synapses in Fmr1 KO mice33 is therefore consistent with the idea that FMRP might normally limit these processes by repressing presynaptic protein synthesis. The presence of FMRP in presynaptic or axonal FMRP-containing granules supports this model13,38.
A role for a general translational increase in fragile X syndrome
Although FMRP directly regulates the translation of a specific subset of target mRNAs, several studies have demonstrated that secondary alterations in general translation may accompany the primary changes due directly to loss of FMRP association with specific mRNAs. In studies using rabbit reticulocyte lysate assays it was shown that FMRP caused a dose-dependent inhibition of the translation of brain poly(A) RNA39,40 and translation was increased in cortical synaptoneurosome preparations from Fmr1 knockout mice as assayed by 35S-methionine labeling41,42. These in vitro studies are supported by several types of protein synthesis assays in cell cultures from FXS patients and brain slices from Fmr1 knockout mice41,43,44. Perhaps most importantly, in vivo studies with L-[1-14C] leucine have shown that Fmr1 knockout mice exhibit increased rates of brain protein synthesis 45.
It is impossible to ascertain whether the increased protein synthesis is due exclusively to release of translational repression on specific FMRP targets alone. However, the magnitude of the increased protein synthesis seems inconsistent with an exclusive translation of FMRP target mRNAs. FMRP target mRNAs represent about 5% of all mRNAs and are not particularly abundant; mRNAs for abundant housekeeping genes and ribosomal proteins are not generally associated with FMRP16. Moreover, FMRP is not expressed in mature glia and therefore should not directly affect protein synthesis in these cells, which may explain why increased protein synthesis is more apparent in areas of high neuronal cell body density relative to glia45. Thus, the increase in translation of specific FMRP target mRNAs due to its absence would have to be substantial to comprise 25% of all new protein synthesis.
Suggestions of specificity in increased protein synthesis do exist; radio-IP assays in labeled hippocampal slices have shown a 25% increase in target CaMKIIα synthesis with no change in the non-target GAPDH46. However, once again the magnitude of this increase appears inadequate to explain the increased protein synthesis in Fmr1 knockout mice and suggests that a global increase in translation due to long-term loss of FMRP may be a secondary outcome of loss of direct repression. In support of this possibility, no global increase in 35S-methionine incorporation was observed due to acute loss of polysome association by all three FXRP family members using an RNA decoy approach despite specific increases in FMRP targets16. In addition, it is notable that FMRP normally represses expression of the NMDAR and mGluR5 receptors, scaffolding proteins, and several components of the downstream extracellular signal-regulated kinase (ERK) and mammalian target of rapamycin complex 1 (mTORC1) pathways, as well as several potentially limiting initiation and elongation factors. Excess production of these and other proteins in the absence of FMRP could lead to secondary increases in protein synthesis.
Outcome of loss of translational repression
Once a high confidence list of FMRP mRNA targets and an equally robust list of non-targets controlled for length and neuronal abundance were identified it was possible to link mRNA binding with a functional outcome. Using a brain polyribosome-programmed in vitro translation system it was shown quantitatively that ribosome stalling occurs on FMRP target transcripts16 (Figure 1). Two genetic models for FMRP loss-of-function and an acute loss-of-function model were used to demonstrate that absence of FMRP resulted in relief of ribosome stalling and increased synthesis of the target proteins. Thus, the simplest model predicting the outcome of loss of translational repression by FMRP in vivo is an increase in the synthesis of its specific mRNA targets resulting in higher than normal levels of the encoded proteins, although compensatory mechanisms such as turnover may restore normal levels of some. Because increased levels of proteins are not always pathogenic, it is necessary to identify which of the many target proteins contribute to FXS pathogenesis. A logical first step is to use either Western blot or more global proteomic approaches to identify protein level changes in FXS models. Many such experiments have been performed with often-conflicting results, including reports of decreased levels of FMRP targets. In this section we review this data in light of the idea that an increase in global protein synthesis may accompany loss of FMRP.
Figure 1. FMRP and small molecules act to stall ribosomal elongation.
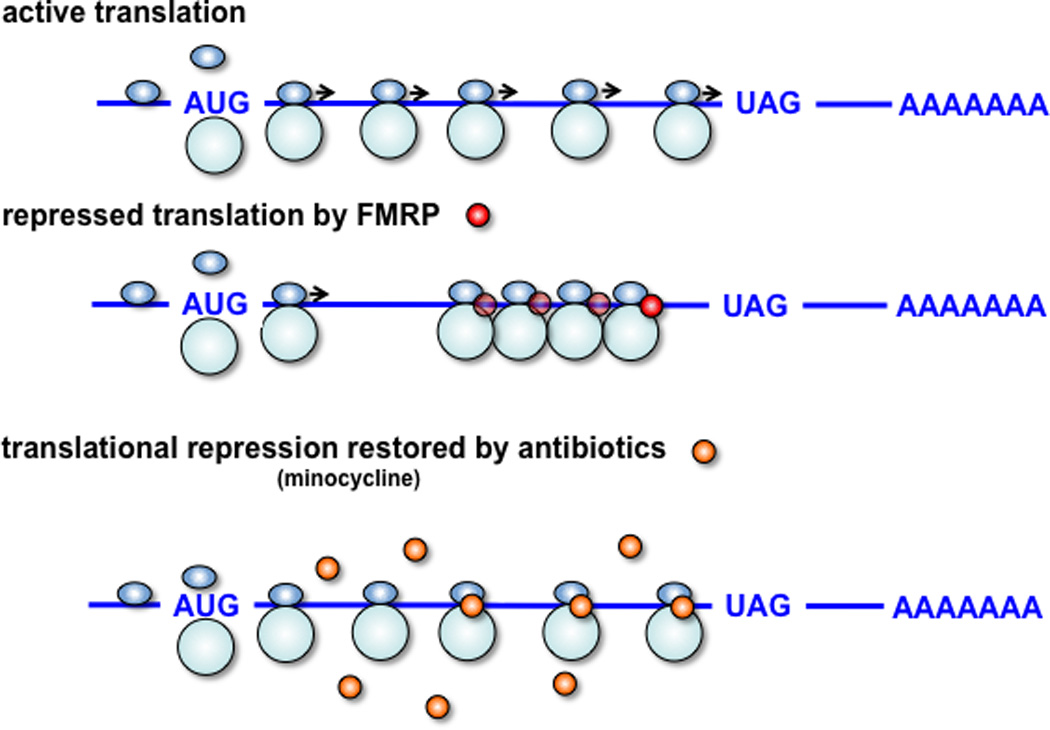
Upper panel, active translation: mRNAs are translated into protein by translocating ribosomes (40S and 60S subunits are shown in light blue) that assemble at the start codon (AUG) and dissociate at the stop codon (for example, UAG). Middle panel, FMRP-repressed translation: FMRP inhibits ribosomal translocation on specific mRNAs in a complex consisting of target mRNA and several stacked or condensed ribosomes. As the stoichiometry of FMRP to ribosomes to target mRNAs is not known, a minimum of one FMRP molecule (red sphere) is depicted in the stalled complex, recognizing the possibility that additional FMRP molecules (illustrated by transparent red spheres) may exist in the stalled complex. Lower panel, minocycline-repressed translation: Minocycline, a tetracycline analog, (orange spheres) and many other small molecules inhibit translation by interfering with ribosomal translocation in different ways. Because of the similarity in proposed action between FMRP and such small molecules it is possible that they might partially replace FMRP functions lost in fragile X syndrome and be therapeutically beneficial.
Western blots have been used to detect increases in FMRP target proteins in its absence. Perhaps the best example of this is the FMRP target MAP1B. In dfmr1-null flies that have only one FMRP paralog a robust bidirectional regulation in MAP1B levels is evident due to loss or overexpression of dfmr147. In mice, in which three potentially redundant paralogs exist, a 75% increase in MAP1B is observed in Fmr1 knockout hippocampal slices10 and a 30% increase in seen in total brain extract48. Increased levels of other FMRP target proteins also have been observed in mice including CaMKIIα10,48, the protein tyrosine phosphatase STEP49, the potassium channels Kv3.150 and Kv4.251 and the upstream regulator of phosphoinositide 3 kinase (PI3K), PI3K enhancer (PIKE)52. Examination of 17 known PSD proteins in purified PSD preparations revealed many FMRP targets are increased at either 2 weeks or 2 months of age in either neocortex or hippocampus including SAPAPs1-3 (dlgap1-3), Shanks 1 and 3, and NMDA receptor subunits53. In human samples, the target CYFIP2 was increased in FXS patients relative to normal controls54.
However, in many cases steady state levels of FMRP target proteins appear to be unchanged in the absence of FMRP. Although this might be explained by the age or tissue examined (see examples in12,53), activation state, or increased protein turnover that offsets increased synthesis, recent examples of apparently decreased levels of FMRP targets (an 8–15% decrease in FMRP targets NR1, NR2A, NR2B, and SAPAP355, a decrease in SYNGAP1 and NR2B12, as well as altered levels of non-targets further muddy the correlation between association of an mRNA with FMRP and increased protein synthesis in FMRP’s absence. These findings underscore the importance of complementary proteomic approaches in understanding the functional outcome of FMRP loss-of-function.
Functions encoded by FMRP-bound transcripts suggest new therapeutic targets
High throughput methods including HITS-CLIP have demonstrated that RNABPs can regulate functionally related sets of target RNAs in the mammalian nervous system29. Ingenuity functional pathway analysis suggests that this is true for the FMRP-regulated target transcripts as well16. Such analysis may be of special benefit considering that components of these pathways may be overexpressed in FXS, focusing efforts on their inhibition. Pathway analysis suggests that glutamate receptor signaling and synaptic plasticity might be impacted by loss of FMRP. These findings are consistent with and extend reports that in the absence of FMRP protein synthesis is exaggerated56 that either genetic or pharmacologic inhibition of mGluR5 rescues phenotype in Fmr1 knockout mice43,57, and that mGluR antagonists show evidence of clinical efficacy58. In addition to mGluR antagonists, the identification of the NMDAR pathway as an FMRP target is significant as NMDAR antagonists are available that might be considered for clinical evaluation as either single agents or in conjunction with mGluR5 antagonists.
mGluRs and NMDARs mediate changes in synaptic plasticity by signaling through the PI3K/Akt/mTORC1 and MEK/ERK pathways to increase cap-dependent translation59. Recent data suggests that FMRP may also regulate the activity of these translational control pathways directly. For example, the 6th ranked FMRP target is PIKE also known as centaurin-gamma1), which is overexpressed in the Fmr1 knockout mouse, resulting in elevated PI3K signaling to the mTORC1 pathway52. Moreover, FMRP target mRNAs that encode phosphatase and tensin homolog deleted from chromosome 10 (PTEN), neurofibromatosis 1 (NF1), and tuberin (TSC2), all of which negatively regulate mTORC1 signaling, suggest a critical role for FMRP in regulating the balance of cap-dependent translation. In addition, ERK1, all three eIF4G isoforms, eEF2 and eEF1, Ago1/2 and Dicer are FMRP targets. Therefore, it is possible that loss of FMRP may have secondary effects on neuronal translation. As will be discussed below, these findings add support to the idea that pharmacologic agents that act on the mTORC1 and ERK pathways may be worth considering therapeutically60,61.
FMRP target mRNAs also encode proteins involved in the regulation of cAMP levels, including adenylate cyclases and phosphodiesterases16. These findings are consistent with observations that dfmr1-null flies, Fmr1 knockout mice62 and human patients63 have altered basal or stimulated cAMP levels. In addition, a large number of proteins regulating small GTPases are targeted by FMRP, including at least 11 GTPase activating proteins (GAPs) and 12 guanine nucleotide exchange factors (GEFs). The striking enrichment of these proteins as FMRP targets is significant as some have been found to regulate the actin cytoskeleton in an activity-dependent manner in neurons64 and perturbing their function alters dendritic spine morphology and is linked with neurologic disease65. A prominent example is the target SYNGAP1, which is stimulated by NMDARs to regulate ERK66 and is a both a cause of non-syndromic mental retardation and strongly linked to autism67.
Receptors that regulate FMRP and their potential as therapeutics for FXS
Experiments examining synaptic plasticity in Fmr1 knockout mice were the first to identify potential molecular targets for therapeutics to treat individuals with FXS. The initial studies largely focused on protein synthesis-dependent forms of long-lasting synaptic plasticity because of the role of FMRP as a translational repressor. The first breakthrough was the finding that hippocampal metabotropic glutamate receptor-dependent long-term depression (mGluR-LTD), which requires dendritic protein synthesis68, is enhanced in Fmr1 knockout mice69. This gave rise to the "mGluR theory"56, which posited that abnormal synaptic function and aberrant behavior in FXS was due to exaggerated mGluR-dependent protein synthesis. Consistent with this theory, developmental defects and aberrant behavior in dfmr1 flies were reversed by the mGluR5 antagonist MPEP70. Moreover, genetic reduction of mGluR5 was shown to correct abnormal dendritic spine morphology, exaggerated protein synthesis, enhanced mGluR-LTD, and behavioral phenotypes displayed by Fmr1 knockout mice43. More recently, it was shown that acute treatment of young adult Fmr1 knockout mice with the selective mGluR5 inhibitor CTEP can reverse exaggerated protein synthesis, enhanced mGluR-LTD, and susceptibility to audiogenic seizures and chronic treatment reverses abnormal dendritic spine morphology and cognitive deficits57. These studies are particularly important when considering therapeutic treatments for FXS individuals because CTEP corrected FXS-associated phenotypes after their onset. There are numerous other examples demonstrating that blocking group I mGluRs can correct a broad array of neuronal, synaptic, and behavioral phenotypes associated with FXS2.
Fmr1 knockout mice also have impaired γ-aminobutyric acid (GABA) receptor function that is likely caused by decreased expression of GABAA receptor subunits and/or enzymes that synthesize GABA71–73, suggesting that inhibitory synaptic transmission is decreased in FXS. Indeed, in dfmr1 null flies, GABA treatment rescued multiple phenotypes, including abnormal behavior74, and it was demonstrated recently that GABAA receptor agonists reduce audiogenic seizures in Fmr1 knockout mice75. Moreover, exaggerated protein synthesis, susceptibility to audiogenic seizures, and repetitive behavior are normalized in Fmr1 knockout mice treated with arbaclofen, a GABAB receptor agonist76. These studies are particularly intriguing because they have been translated to FXS individuals, where arbaclofen improved social avoidance, one of the main clinical symptoms of the syndrome77. It remains to be determined whether GABAB receptor agonists can reverse FXS-associated phenotypes via the dampening of excessive mGluR5-mediated protein synthesis.
The therapeutic targets for FXS mentioned above are neurotransmitter receptors, which are the most common targets for the treatment neurological and psychiatric disorders. The advantage of targeting neurotransmitter receptors is that there are likely to be fewer peripheral side effects in individuals treated with the compounds. However, G-protein coupled receptors such as group I mGluRs regulate a plethora of downstream neuronal signaling pathways in the brain, and thus, receptor-targeted compounds could inhibit pathways that are not impacted in FXS individuals. Recent studies have begun to delineate the signaling pathways that couple group I mGluRs to the translation machinery, providing a new set of potential therapeutic targets for treatment of FXS.
Translational control pathways and their potential as therapeutics for FXS
Two protein kinase signaling pathways that regulate translation initiation, mTORC1 and ERK, have been implicated in the pathophysiology of FXS. mTORC1 phosphorylates eIF4E-binding protein (4E-BP) and p70 S6 kinase 1 (S6K1), both of which bind Raptor78. Unphosphorylated 4E-BPs bind tightly to eIF4E, whereas 4E-BPs phosphorylated by mTORC1 do not, thereby permitting eIF4E to bind eIF4G to form the eIF4F initiation complex allowing initiation to proceed. mTORC1 also impacts initiation by phosphorylating S6K1, which then phosphorylates downstream targets such as ribosomal protein S6 and eIF4B. Importantly, phosphorylation of eIF4B by S6K1 increases the helicase activity of eIF4A, as does the interaction of eIF4E with eIF4G. ERK phosphorylates the kinase Mnk1 that subsequently phosphorylates eIF4E, which is correlated with enhanced translation (Figure 2a79,80).
Figure 2. Signaling Pathways Controlling Translation in Normal and FXS Model Mice.
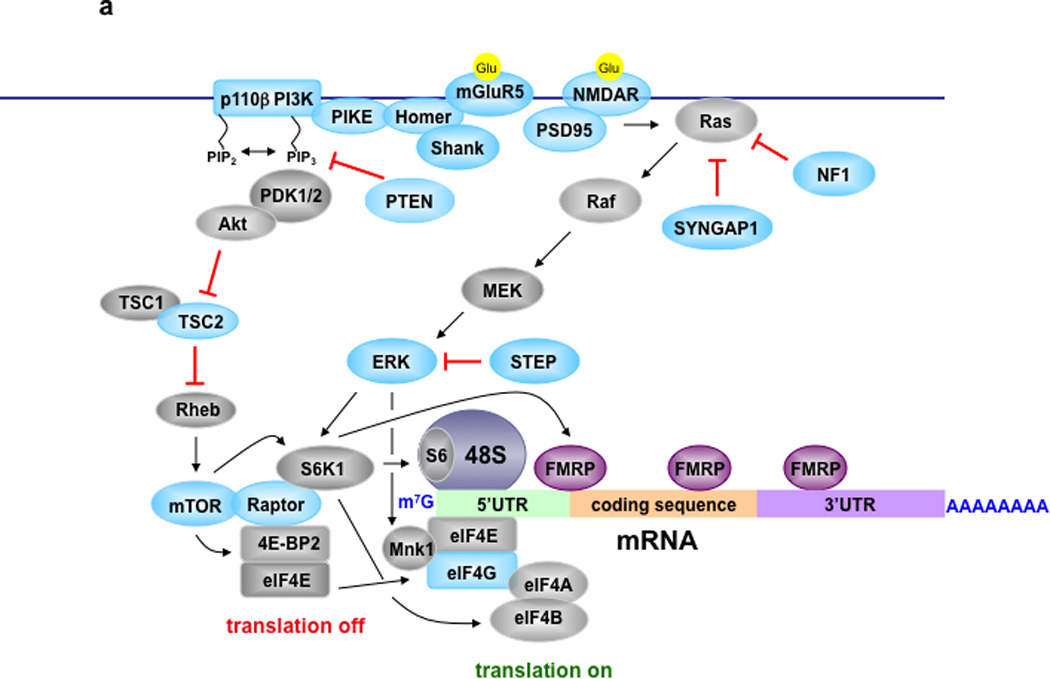
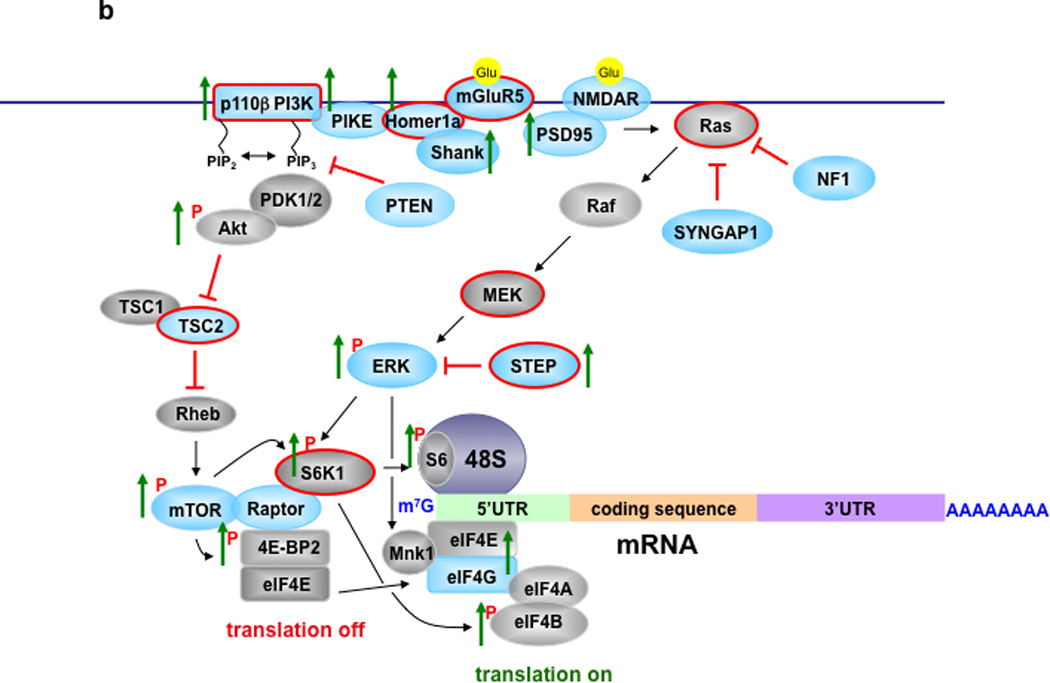
A) Translational control in neurons of normal mice. Metabotropic glutamate receptor 5 (mGluR5) and NMDA receptors are coupled to PI3K/Akt/mTORC1 and MEK/ERK signaling to regulate cap-dependent translation (for detailed reviews see59–61,78–80). Blue colored proteins are those whose mRNAs are FMRP targets. B) Translational control in neurons of FXS model mice that lack FMRP. Green arrows indicate either total or phosphorylated levels (indicated by red P) of proteins that have been reported to increase in either FXS model mice or cells from FXS individuals41,42,44,48,49,52,54,81. Proteins with red lines around them indicate those that have been successfully targeted either pharmacologically or genetically to reverse molecular, morphological, synaptic, and/or behavioral phenotypes in cellular and/or animal models of FXS41–44,46,49,52,57,70,74,81,83.
It was reported that Fmr1 knockout mice exhibit increased eIF4E-eIF4G interactions and phosphorylation of S6K1 in the hippocampus52, which is indicative of excessive mTORC1 signaling. What is the upstream signaling that couples mGluR5 to mTORC1? It appears to involve disrupted Homer scaffolds and increased PI3K signaling. In Fmr1 knockout mice mGluR5 was less associated with long Homer isoforms and more associated with the shorter Homer1a81. In addition, genetic deletion of Homer1a in Fmr1 knockout mice reduced increased eIF4F complex formation, exaggerated protein synthesis, and susceptibility to audiogenic seizures81. Interestingly, Homer interacts with PIKE, which, as mentioned above, is an FMRP target mRNA16 and whose protein expression is increased in Fmr1 knockout mice41,52. Moreover, the expression of the p110β subunit of PI3K, which also is an FMRP target mRNA16 is elevated in Fmr1 knockout mice. Inhibitors of PI3K corrected exaggerated protein synthesis and abnormal dendritic spine morphology in Fmr1 knockout mice41 as well as exaggerated protein synthesis in lymphoblastoid cells from FXS patients42. All together, these findings indicate that disrupted Homer scaffolds and excessive PI3K signaling contribute to exaggerated protein synthesis, as well as molecular, cellular, and behavioral phenotypes in FXS.
Because PI3K is typically an upstream regulator of mTORC160, one might predict that disruption of mTORC1 would correct multiple aspects of FXS. However, acute treatment with rapamycin to disrupt mTORC1 does not correct exaggerated protein synthesis and audiogenic seizures displayed by Fmr1 knockout mice46. In contrast, it recently was demonstrated that genetic deletion of S6K1 can correct exaggerated protein synthesis, enhanced mGluR-LTD, and several behavioral phenotypes displayed by Fmr1 knockout mice44. How can one reconcile these seemingly contradictory findings? A simple explanation is that chronic treatments of Fmr1 knockout mice with rapamycin are required to reduce exaggerated protein synthesis. Because both PIKE and the p110β subunit of PI3K are elevated in Fmr1 knockout mice, it is likely that PIKE/PI3K triggers the excessive activation of PDK1/2, and subsequently mTORC1 (Figure 2). However, in addition to mTORC1, S6K1 receives direct stimulatory input from PDK182, as well as ERK79. Thus, S6K1 activity could be stimulated by either PDK1 or ERK, the former being consistent with the observation of elevated PIKE/p110β levels in FXS mice52 and latter being consistent with the observation that hypersensitivity to ERK contributes to exaggerated protein synthesis and audiogenic seizures in Fmr1 knockout mice46. Moreover, lovastatin, which interferes with Ras activation, recently was shown to prevent ERK activation, exaggerated protein synthesis, and susceptibility to audiogenic seizures in Fmr1 knockout mice83. Thus, S6K1 is a potential integrator of signaling from both increased levels of PI3K and hypersensitivity to ERK in FXS (Figure 2b).
Tuberous sclerosis complex (TSC) is an autosomal dominant disorder caused by mutations in either hamartin (TSC1) or TSC284. Because inactivation of TSC1/2 upregulates mTORC1 signaling, one would predict that TSC model mice, similar to Fmr1 knockout mice, would exhibit enhanced translation. However, it was shown that TSC2 mutant mice have decreased translation in the hippocampus that can be blocked by rapamycin85, which is likely due to feedback inhibition of pathways upstream of mTORC160 and/or upregulation of the unfolded protein response86. Moreover, the TSC model mice, in contrast to Fmr1 knockout mice, displayed impaired mGluR-LTD. Finally, when TSC model mice were bred with Fmr1 knockout mice, protein synthesis, mGluR-LTD, and cognitive phenotypes that were present in both lines of mutant mice were normalized85. As mentioned earlier, TSC2 mRNA is a target of FMRP16, so it is possible that TSC2 protein expression is altered in Fmr1 knockout mice. An additional unknown is the molecular signaling responsible for the normalization of the phenotypes displayed by the TSC/FXS double mutant mice.
The mRNAs that encode PIKE, PI3K, and TSC2 are all associated with FMRP and thus, their protein products are potential therapeutic targets for FXS (Figure 2b). Other signaling molecules that may be targets include striatal-enriched protein tyrosine phosphatase (STEP)49 and GSK3β87,88 whose transcripts are also FMRP targets16. Thus, downstream signaling pathways that are impacted by the reduction of STEP and GSK3β and whether they interact with the mTORC1 and ERK pathways in FXS remains to be elucidated.
The idea that the lack of FMRP results in exaggerated mGluR-dependent protein synthesis is the leading theory for the molecular basis of FXS, and both genetic reduction43 and pharmacological inhibition of mGluR557 reduced exaggerated protein synthesis and a broad range of phenotypes displayed by Fmr1 knockout mice. However, it also was reported that mGluR-LTD is protein synthesis-independent in Fmr1 knockout mice9,10. Moreover, activation of group 1 mGluRs is uncoupled from the activation of PI3K41,89, mTORC152,81,89, and ERK10, the translation of FMRP target mRNAs, including MAP1B10, p110β41, and STEP49, the translation of Arc81, and general protein synthesis41,46. These findings suggest that the loss of FMRP limits the dynamic range for mGluR-dependent protein synthesis and that dampening mGluR-dependent translational control, via either an mGluR antagonist or by targeting downstream effectors such as PI3K and S6K1 is required to reset translational homeostasis in Fmr1 knockout mice (Figure 2b).
Novel therapy for fragile X syndrome directed at its molecular function rather than upstream or downstream targets
Although targeting signaling pathways has the advantage of being downstream of neurotransmitter receptors, many of these kinases, including PI3K and ERK, are still far enough upstream from the translational machinery that their inhibition could impact signaling cascades unaffected in FXS. Thus, if excessive translation of FMRP target mRNAs and/or exaggerated general protein synthesis is the causative factor in FXS, then the direct targeting of the translational machinery might be the most effective way to reset translational homeostasis. Evidence for the efficacy of this approach was first provided by studies in dfmr1-null flies, which showed that the general protein synthesis inhibitors cycloheximide and puromycin rescue impaired long-term memory23. Thus, direct targeting of the translational machinery may be a means for reversing abnormal behaviors associated with FXS.
There is speculation that exaggerated translation is causative not only for FXS, but also for other ASDs60,61, and there is evidence for a genetic link between ASD and the cap-binding translation initiation factor eIF4E90. Moreover, microdeletions in the Prader-Willi/Angelman critical region contain the gene for CYFIP1, which encodes a non-canonical eIF4E-binding protein that also binds to FMRP91. Importantly, it recently was shown that transgenic mice overexpressing eIF4E exhibit increased eIF4F complex formation, exaggerated protein synthesis, enhanced mGluR-LTD in the striatum and hippocampus, and aberrant autistic-like behaviors, all of which were reversed by 4EGI-1, an inhibitor of eIF4E-eIF4G interactions92. These findings have provided direct evidence for the idea that exaggerated protein synthesis can cause synaptic and behavioral aberrations associated with ASD. Moreover, because Fmr1 knockout mice exhibit increased eIF4E-eIF4G interactions52,81, these findings suggest that exaggerated protein synthesis, enhanced mGluR-LTD, and FXS-associated behaviors in Fmr1 knockout mice might be reversible by 4EGI-1.
As discussed above, FMRP functions to stall ribosomal translocation on its target mRNAs, suggesting that compounds that inhibit translation elongation might alleviate phenotypes associated with the loss of FMRP (Figure 1). Many small molecules, primarily used as antibiotics because they stall the translocation of bacterial ribosomes, already have been tested in relevant assays. Interestingly, minocycline, a second generation tetracycline analog, reversed abnormal synaptic structure, spine morphology, and aberrant behaviors exhibited by Fmr1 knockout mice93 and dfmr1 null flies94. Minocycline was tested in these models because it inhibits matrix metalloproteinase-9 (MMP-9), which is overexpressed in Fmr1 KO mice93 and flies94. However, one has to wonder whether all or part of the effect of minocycline is due to its ability to slow down ribosomes, taking the edge off excess translation as previously suggested16. Minocycline is an antibiotic used in humans that is quite lipid-soluble and crosses the blood brain barrier well95. Minocycline and other tetracyclines inhibit protein synthesis via A-site occupation and stall mammalian ribosomes with 7–20% of the efficacy with which they inhibit bacterial translation96. Preliminary open-label add-on clinical trials with minocycline have shown positive results for cognitive and behavioral measures in 20 FXS patients97, which warrants further research on minocycline and other inhibitors of ribosomal elongation.
Summary and Conclusions
Knowledge concerning the function of FMRP and how its loss alters receptor-mediated signaling and translational control leading to aberrant dendritic spine dynamics, synaptic plasticity, circuit function, and complex behavior in FXS has expanded exponentially in the last decade. Nonetheless, questions central to the development of therapeutics for treating individuals with FXS remain unanswered. Traditional methods of drug discovery for brain disorders focus on neurotransmitter receptors with good reason, and there is considerable optimism that pharmaceuticals targeting metabotropic glutamate receptors and GABA receptors will provide benefits for FXS patients. In addition, the identification of the mRNA targets of FMRP that encode synaptic proteins and components of signaling pathways involved translational control, many of which overlap with autism and other neurodevelopmental disorders, have provided a rich list of potential therapeutic targets for FXS. However, this plethora of targets implies that one should be very cautious in inferring the normal function of FMRP from studying the chronic loss-of-function state in Fmr1 mutant animals and cell lines from individuals with FXS. We argue that the main function of FMRP is to regulate protein synthesis and as such, FXS should be considered a disease of dysregulated translation. Finally, because the lack of FMRP appears to alter the expression of a multitude of proteins involved in translational control and synaptic function, we propose that a rational approach for effective treatment of FXS would be to reset translational homeostasis by targeting initiation and/or elongation, thereby translating translation to the clinic.
Acknowledgements
We would like to apologize to our colleagues whose work we were unable to reference due to space limitations. This work was supported by NIH grants HD040647 (J.C.D.) and NS047384 (E.K.), and CDRMP Award W81-XWH-11-1-0389 from the U.S. Department of Defense (E.K.).
References
- 1.Alberini CM. The role of protein synthesis during the labile phases of memory: revisiting the skepticism. Neurobiol Learn Mem. 2008;89:234–246. doi: 10.1016/j.nlm.2007.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bhakar AL, Dolen G, Bear MF. The pathophysiology of fragile X (and what it teaches us about synapses) Annu Rev Neurosci. 2012;35:417–443. doi: 10.1146/annurev-neuro-060909-153138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Santoro MR, Bray SM, Warren ST. Molecular mechanisms of fragile X syndrome: a twenty-year perspective. Annu Rev Pathol. 2012;7:219–245. doi: 10.1146/annurev-pathol-011811-132457. [DOI] [PubMed] [Google Scholar]
- 4.Gatto CL, Broadie K. The fragile X mental retardation protein in circadian rhythmicity and memory consolidation. Mol Neurobiol. 2009;39:107–129. doi: 10.1007/s12035-009-8057-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Comery TA, et al. Abnormal dendritic spines in fragile X knockout mice: maturation and pruning deficits. Proc Natl Acad Sci U S A. 1997;94:5401–5404. doi: 10.1073/pnas.94.10.5401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nimchinsky EA, Oberlander AM, Svoboda K. Abnormal development of dendritic spines in FMR1 knock-out mice. J Neurosci. 2001;21:5139–546.. doi: 10.1523/JNEUROSCI.21-14-05139.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cruz-Martin A, Crespo M, Portera-Cailliau C. Delayed stabilization of dendritic spines in fragile X mice. J Neurosci. 2010;30:7793–7803. doi: 10.1523/JNEUROSCI.0577-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pan F, Aldridge GM, Greenough WT, Gan WB. Dendritic spine instability and insensitivity to modulation by sensory experience in a mouse model of fragile X syndrome. Proc Natl Acad Sci U S A. 2010;107:17768–17773. doi: 10.1073/pnas.1012496107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nosyreva ED, Huber KM. Metabotropic receptor-dependent long-term depression persists in the absence of protein synthesis in the mouse model of fragile X syndrome. J Neurophysiol. 2006;95:3291–3295. doi: 10.1152/jn.01316.2005. [DOI] [PubMed] [Google Scholar]
- 10.Hou L, et al. Dynamic translational and proteasomal regulation of fragile X mental retardation protein controls mGluR-dependent long-term depression. Neuron. 2006;51:441–454. doi: 10.1016/j.neuron.2006.07.005. [DOI] [PubMed] [Google Scholar]
- 11.Harlow EG, et al. Critical period plasticity is disrupted in the barrel cortex of FMR1 knockout mice. Neuron. 2010;65:385–398. doi: 10.1016/j.neuron.2010.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Till SM, et al. Altered maturation of the primary somatosensory cortex in a mouse model of fragile X syndrome. Hum Mol Genet. 2012;21:2143–2156. doi: 10.1093/hmg/dds030. [DOI] [PubMed] [Google Scholar]
- 13.Christie SB, Akins MR, Schwob JE, Fallon JR. The FXG: a presynaptic fragile X granule expressed in a subset of developing brain circuits. J Neurosci. 2009;29:1514–1524. doi: 10.1523/JNEUROSCI.3937-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stefani G, Fraser CE, Darnell JC, Darnell RB. Fragile X mental retardation protein is associated with translating polyribosomes in neuronal cells. J Neurosci. 2004;24:7272–7276. doi: 10.1523/JNEUROSCI.2306-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Khandjian EW, et al. Biochemical evidence for the association of fragile X mental retardation protein with brain polyribosomal ribonucleoparticles. Proc Natl Acad Sci U S A. 2004;101:13357–13362. doi: 10.1073/pnas.0405398101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Darnell JC, et al. FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell. 2011;146:247–261. doi: 10.1016/j.cell.2011.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Verkerk AJ, et al. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell. 1991;65:905–914. doi: 10.1016/0092-8674(91)90397-h. [DOI] [PubMed] [Google Scholar]
- 18.Siomi H, Siomi MC, Nussbaum RL, Dreyfuss G. The protein product of the fragile X gene, FMR1, has characteristics of an RNA-binding protein. Cell. 1993;74:291–298. doi: 10.1016/0092-8674(93)90420-u. [DOI] [PubMed] [Google Scholar]
- 19.De Boulle K, et al. A point mutation in the FMR-1 gene associated with fragile X mental retardation. Nat Genet. 1993;3:31–35. doi: 10.1038/ng0193-31. [DOI] [PubMed] [Google Scholar]
- 20.Lewis HA, et al. Sequence-specific RNA binding by a Nova KH domain: implications for paraneoplastic disease and the fragile X syndrome. Cell. 2000;100:323–332. doi: 10.1016/s0092-8674(00)80668-6. [DOI] [PubMed] [Google Scholar]
- 21.Feng Y, et al. FMRP associates with polyribosomes as an mRNP, and the I304N mutation of severe fragile X syndrome abolishes this association. Mol Cell. 1997;1:109–118. doi: 10.1016/s1097-2765(00)80012-x. [DOI] [PubMed] [Google Scholar]
- 22.Zang JB, et al. A mouse model of the human Fragile X syndrome I304N mutation. PLoS Genet. 2009;5:e1000758. doi: 10.1371/journal.pgen.1000758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bolduc FV, Bell K, Cox H, Broadie KS, Tully T. Excess protein synthesis in Drosophila fragile X mutants impairs long-term memory. Nat Neurosci. 2008;11:1143–1145. doi: 10.1038/nn.2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ramos A, Hollingworth D, Pastore A. The role of a clinically important mutation in the fold and RNA-binding properties of KH motifs. RNA. 2003;9:293–298. doi: 10.1261/rna.2168503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Darnell JC, et al. Kissing complex RNAs mediate interaction between the Fragile-X mental retardation protein KH2 domain and brain polyribosomes. Genes Dev. 2005;19:903–918. doi: 10.1101/gad.1276805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ashley CT, Wilkinson KD, Reines D, Warren ST. FMR-1 protein: conserved RNP family domains and selective RNA binding. Science. 1993;262:563–566. doi: 10.1126/science.7692601. [DOI] [PubMed] [Google Scholar]
- 27.Corbin F, et al. The fragile X mental retardation protein is associated with poly(A)+ mRNA in actively translating polyribosomes. Hum Mol Genet. 1997;6:1465–1472. doi: 10.1093/hmg/6.9.1465. [DOI] [PubMed] [Google Scholar]
- 28.Darnell JC, Richter JD. Cytoplasmic RNA-binding proteins and the control of complex brain function. Cold Spring Harb Perspect Biol. 2012;4:a012344. doi: 10.1101/cshperspect.a012344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Darnell RB. HITS-CLIP: panoramic views of protein-RNA regulation in living cells. Wiley Interdiscip Rev RNA. 2010;1:266–286. doi: 10.1002/wrna.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shanks NF, et al. Differences in AMPA and kainate receptor interactomes facilitate identification of AMPA receptor auxiliary subunit GSG1L. Cell Rep. 2012;1:590–598. doi: 10.1016/j.celrep.2012.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Akins MR, Berk-Rauch HE, Fallon JR. Presynaptic translation: stepping out of the postsynaptic shadow. Front Neural Circuits. 2009;3:17. doi: 10.3389/neuro.04.017.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hanson JE, Madison DV. Presynaptic FMR1 genotype influences the degree of synaptic connectivity in a mosaic mouse model of fragile X syndrome. J Neurosci. 2007;27:4014–4018. doi: 10.1523/JNEUROSCI.4717-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Deng PY, Sojka D, Klyachko VA. Abnormal presynaptic short-term plasticity and information processing in a mouse model of fragile X syndrome. J Neurosci. 2011;31:10971–10982. doi: 10.1523/JNEUROSCI.2021-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang J, Hou L, Klann E, Nelson DL. Altered hippocampal synaptic plasticity in the FMR1 gene family knockout mouse models. J Neurophysiol. 2009;101:2572–2580. doi: 10.1152/jn.90558.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gibson JR, Bartley AF, Hays SA, Huber KM. Imbalance of neocortical excitation and inhibition and altered UP states reflect network hyperexcitability in the mouse model of fragile X syndrome. J Neurophysiol. 2008;100:2615–2626. doi: 10.1152/jn.90752.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Suvrathan A, Hoeffer CA, Wong H, Klann E, Chattarji S. Characterization and reversal of synaptic defects in the amygdala in a mouse model of fragile X syndrome. Proc Natl Acad Sci U S A. 2010;107:11591–11596. doi: 10.1073/pnas.1002262107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sebeo J, et al. Requirement for protein synthesis at developing synapses. J Neurosci. 2009;29:9778–9793. doi: 10.1523/JNEUROSCI.2613-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 38.Antar LN, Li C, Zhang H, Carroll RC, Bassell GJ. Local functions for FMRP in axon growth cone motility and activity-dependent regulation of filopodia and spine synapses. Mol Cell Neurosci. 2006;32:37–48. doi: 10.1016/j.mcn.2006.02.001. [DOI] [PubMed] [Google Scholar]
- 39.Laggerbauer B, Ostareck D, Keidel EM, Ostareck-Lederer A, Fischer U. Evidence that fragile X mental retardation protein is a negative regulator of translation. Hum Mol Genet. 2001;10:329–338. doi: 10.1093/hmg/10.4.329. [DOI] [PubMed] [Google Scholar]
- 40.Li Z, et al. The fragile X mental retardation protein inhibits translation via interacting with mRNA. Nucleic Acids Res. 2001;29:2276–2283. doi: 10.1093/nar/29.11.2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gross C, et al. Excess phosphoinositide 3-kinase subunit synthesis and activity as a novel therapeutic target in fragile X syndrome. J Neurosci. 2010;30:10624–10638. doi: 10.1523/JNEUROSCI.0402-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gross C, Bassell GJ. Excess protein synthesis in FXS patient lymphoblastoid cells can be rescued with a p110beta-selective inhibitor. Mol Med. 2012;18:336–345. doi: 10.2119/molmed.2011.00363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dolen G, et al. Correction of fragile X syndrome in mice. Neuron. 2007;56:955–962. doi: 10.1016/j.neuron.2007.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bhattacharya A, et al. Genetic Removal of p70 S6 Kinase 1 Corrects Molecular, Synaptic, and Behavioral Phenotypes in Fragile X Syndrome Mice. Neuron. 2012;76:325–337. doi: 10.1016/j.neuron.2012.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Qin M, Kang J, Burlin TV, Jiang C, Smith CB. Postadolescent changes in regional cerebral protein synthesis: an in vivo study in the FMR1 null mouse. J Neurosci. 2005;25:5087–5095. doi: 10.1523/JNEUROSCI.0093-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Osterweil EK, Krueger DD, Reinhold K, Bear MF. Hypersensitivity to mGluR5 and ERK1/2 leads to excessive protein synthesis in the hippocampus of a mouse model of fragile X syndrome. J Neurosci. 2010;30:15616–15627. doi: 10.1523/JNEUROSCI.3888-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang YQ, et al. Drosophila fragile X-related gene regulates the MAP1B homolog Futsch to control synaptic structure and function. Cell. 2001;107:591–603. doi: 10.1016/s0092-8674(01)00589-x. [DOI] [PubMed] [Google Scholar]
- 48.Zalfa F, et al. The fragile X syndrome protein FMRP associates with BC1 RNA and regulates the translation of specific mRNAs at synapses. Cell. 2003;112:317–327. doi: 10.1016/s0092-8674(03)00079-5. [DOI] [PubMed] [Google Scholar]
- 49.Goebel-Goody SM, et al. Genetic manipulation of STEP reverses behavioral abnormalities in a fragile X syndrome mouse model. Genes Brain Behav. 2012 doi: 10.1111/j.1601-183X.2012.00781.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Strumbos JG, Brown MR, Kronengold J, Polley DB, Kaczmarek LK. Fragile X mental retardation protein is required for rapid experience-dependent regulation of the potassium channel Kv3.1b. J Neurosci. 2010;30:10263–10271. doi: 10.1523/JNEUROSCI.1125-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lee HY, et al. Bidirectional regulation of dendritic voltage-gated potassium channels by the fragile X mental retardation protein. Neuron. 2011;72:630–642. doi: 10.1016/j.neuron.2011.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sharma A, et al. Dysregulation of mTOR signaling in fragile X syndrome. J Neurosci. 2010;30:694–702. doi: 10.1523/JNEUROSCI.3696-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schutt J, Falley K, Richter D, Kreienkamp HJ, Kindler S. Fragile X mental retardation protein regulates the levels of scaffold proteins and glutamate receptors in postsynaptic densities. J Biol Chem. 2009;284:25479–25487. doi: 10.1074/jbc.M109.042663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hoeffer CA, et al. Altered mTOR signaling and enhanced CYFIP2 expression levels in subjects with fragile X syndrome. Genes Brain Behav. 2012;11:332–341. doi: 10.1111/j.1601-183X.2012.00768.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Krueger DD, Osterweil EK, Chen SP, Tye LD, Bear MF. Cognitive dysfunction and prefrontal synaptic abnormalities in a mouse model of fragile X syndrome. Proc Natl Acad Sci U S A. 2011;108:2587–2592. doi: 10.1073/pnas.1013855108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bear MF, Huber KM, Warren ST. The mGluR theory of fragile X mental retardation. Trends Neurosci. 2004;27:370–377. doi: 10.1016/j.tins.2004.04.009. [DOI] [PubMed] [Google Scholar]
- 57.Michalon A, et al. Chronic pharmacological mGlu5 inhibition corrects fragile X in adult mice. Neuron. 2012;74:49–56. doi: 10.1016/j.neuron.2012.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Berry-Kravis E, et al. A pilot open label, single dose trial of fenobam in adults with fragile X syndrome. J Med Genet. 2009;46:266–271. doi: 10.1136/jmg.2008.063701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Klann E, Dever TE. Biochemical mechanisms for translational regulation in synaptic plasticity. Nat Rev Neurosci. 2004;5:931–942. doi: 10.1038/nrn1557. [DOI] [PubMed] [Google Scholar]
- 60.Hoeffer CA, Klann E. mTOR signaling: at the crossroads of plasticity, memory and disease. Trends Neurosci. 2010;33:67–75. doi: 10.1016/j.tins.2009.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kelleher R, Jr, Bear MF. The autistic neuron: troubled translation? Cell. 2008;135:401–406. doi: 10.1016/j.cell.2008.10.017. [DOI] [PubMed] [Google Scholar]
- 62.Kelley DJ, et al. The cyclic AMP cascade is altered in the fragile X nervous system. PLoS One. 2007;2:e931. doi: 10.1371/journal.pone.0000931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Berry-Kravis E, Sklena P. Demonstration of abnormal cyclic AMP production in platelets from patients with fragile X syndrome. Am J Med Genet. 1993;45:81–87. doi: 10.1002/ajmg.1320450120. [DOI] [PubMed] [Google Scholar]
- 64.Li J, Pelletier MR, Perez Velazquez JL, Carlen PL. Reduced cortical synaptic plasticity and GluR1 expression associated with fragile X mental retardation protein deficiency. Mol Cell Neurosci. 2002;19:138–151. doi: 10.1006/mcne.2001.1085. [DOI] [PubMed] [Google Scholar]
- 65.Newey SE, Velamoor V, Govek EE, Van Aelst L. Rho GTPases, dendritic structure, and mental retardation. J Neurobiol. 2005;64:58–74. doi: 10.1002/neu.20153. [DOI] [PubMed] [Google Scholar]
- 66.Rumbaugh G, Adams JP, Kim JH, Huganir RL. SynGAP regulates synaptic strength and mitogen-activated protein kinases in cultured neurons. Proc Natl Acad Sci U S A. 2006;103:4344–4351. doi: 10.1073/pnas.0600084103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Pinto D, et al. Functional impact of global rare copy number variation in autism spectrum disorders. Nature. 2010;466:368–372. doi: 10.1038/nature09146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Huber KM, Kayser MS, Bear MF. Role for rapid dendritic protein synthesis in hippocampal mGluR-dependent long-term depression. Science. 2000;288:1254–1257. doi: 10.1126/science.288.5469.1254. [DOI] [PubMed] [Google Scholar]
- 69.Huber KM, Gallagher SM, Warren ST, Bear MF. Altered synaptic plasticity in a mouse model of fragile X mental retardation. Proc Natl Acad Sci U S A. 2002;99:7746–7750. doi: 10.1073/pnas.122205699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.McBride SM, et al. Pharmacological rescue of synaptic plasticity, courtship behavior, and mushroom body defects in a Drosophila model of fragile X syndrome. Neuron. 2005;45:753–764. doi: 10.1016/j.neuron.2005.01.038. [DOI] [PubMed] [Google Scholar]
- 71.El Idrissi A, et al. Decreased GABA(A) receptor expression in the seizure-prone fragile X mouse. Neurosci Lett. 2005;377:141–146. doi: 10.1016/j.neulet.2004.11.087. [DOI] [PubMed] [Google Scholar]
- 72.D'Hulst C, et al. Decreased expression of the GABAA receptor in fragile X syndrome. Brain Res. 2006;1121:238–245. doi: 10.1016/j.brainres.2006.08.115. [DOI] [PubMed] [Google Scholar]
- 73.Adusei DC, Pacey LK, Chen D, Hampson DR. Early developmental alterations in GABAergic protein expression in fragile X knockout mice. Neuropharmacology. 2010;59:167–171. doi: 10.1016/j.neuropharm.2010.05.002. [DOI] [PubMed] [Google Scholar]
- 74.Chang S, et al. Identification of small molecules rescuing fragile X syndrome phenotypes in Drosophila. Nat Chem Biol. 2008;4:256–263. doi: 10.1038/nchembio.78. [DOI] [PubMed] [Google Scholar]
- 75.Heulens I, D'Hulst C, Van Dam D, De Deyn PP, Kooy RF. Pharmacological treatment of fragile X syndrome with GABAergic drugs in a knockout mouse model. Behav Brain Res. 2012;229:244–249. doi: 10.1016/j.bbr.2012.01.031. [DOI] [PubMed] [Google Scholar]
- 76.Henderson C, et al. Reversal of disease-related pathologies in the fragile X mouse model by selective activation of GABA(B) receptors with arbaclofen. Sci Transl Med. 2012;4:152ra128. doi: 10.1126/scitranslmed.3004218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Berry-Kravis EM, et al. Effects of STX209 (arbaclofen) on neurobehavioral function in children and adults with fragile X syndrome: a randomized, controlled, phase 2 trial. Sci Transl Med. 2012;4:152ra127. doi: 10.1126/scitranslmed.3004214. [DOI] [PubMed] [Google Scholar]
- 78.Richter JD, Klann E. Making synaptic plasticity and memory last: mechanisms of translational regulation. Genes Dev. 2009;23:1–11. doi: 10.1101/gad.1735809. [DOI] [PubMed] [Google Scholar]
- 79.Klann E, Antion MD, Banko JL, Hou L. Synaptic plasticity and translation initiation. Learn Mem. 2004;11:365–372. doi: 10.1101/lm.79004. [DOI] [PubMed] [Google Scholar]
- 80.Costa-Mattioli M, Sossin WS, Klann E, Sonenberg N. Translational control of long-lasting synaptic plasticity and memory. Neuron. 2009;61:10–26. doi: 10.1016/j.neuron.2008.10.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ronesi JA, et al. Disrupted Homer scaffolds mediate abnormal mGluR5 function in a mouse model of fragile X syndrome. Nat Neurosci. 2012;15:431–40. S1. doi: 10.1038/nn.3033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Romanelli A, Martin KA, Toker A, Blenis J. p70 S6 kinase is regulated by protein kinase Czeta and participates in a phosphoinositide 3-kinase-regulated signalling complex. Mol Cell Biol. 1999;19:2921–2928. doi: 10.1128/mcb.19.4.2921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Osterweil EK, et al. Lovastatin corrects excess protein synthesis and prevents epileptogenesis in a mouse model of fragile X syndrome. Neuron. 2013;77:243–250. doi: 10.1016/j.neuron.2012.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kwiatkowski DJ, Manning BD. Tuberous sclerosis: a GAP at the crossroads of multiple signaling pathways. Hum Mol Genet. 2005;14(Spec No. 2):R251–R258. doi: 10.1093/hmg/ddi260. [DOI] [PubMed] [Google Scholar]
- 85.Auerbach BD, Osterweil EK, Bear MF. Mutations causing syndromic autism define an axis of synaptic pathophysiology. Nature. 2011;480:63–68. doi: 10.1038/nature10658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ozcan U, et al. Loss of the tuberous sclerosis complex tumor suppressors triggers the unfolded protein response to regulate insulin signaling and apoptosis. Mol Cell. 2008;29:541–551. doi: 10.1016/j.molcel.2007.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Mines MA, Yuskaitis CJ, King MK, Beurel E, Jope RS. GSK3 influences social preference and anxiety-related behaviors during social interaction in a mouse model of fragile X syndrome and autism. PLoS One. 2010;5:e9706. doi: 10.1371/journal.pone.0009706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Guo W, et al. Inhibition of GSK3beta improves hippocampus-dependent learning and rescues neurogenesis in a mouse model of fragile X syndrome. Hum Mol Genet. 2011 doi: 10.1093/hmg/ddr501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ronesi JA, Huber KM. Homer interactions are necessary for metabotropic glutamate receptor-induced long-term depression and translational activation. J Neurosci. 2008;28:543–547. doi: 10.1523/JNEUROSCI.5019-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Neves-Pereira M, et al. Deregulation of EIF4E: a novel mechanism for autism. J Med Genet. 2009;46:759–765. doi: 10.1136/jmg.2009.066852. [DOI] [PubMed] [Google Scholar]
- 91.Napoli I, et al. The fragile X syndrome protein represses activity-dependent translation through CYFIP1, a new 4E-BP. Cell. 2008;134:1042–1054. doi: 10.1016/j.cell.2008.07.031. [DOI] [PubMed] [Google Scholar]
- 92.Santini E, et al. Exaggerated translation causes synaptic and behavioural aberrations associated with autism. Nature. 2013;493:411–415. doi: 10.1038/nature11782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Bilousova TV, et al. Minocycline promotes dendritic spine maturation and improves behavioural performance in the fragile X mouse model. J Med Genet. 2009;46:94–102. doi: 10.1136/jmg.2008.061796. [DOI] [PubMed] [Google Scholar]
- 94.Siller SS, Broadie K. Neural circuit architecture defects in a Drosophila model of Fragile X syndrome are alleviated by minocycline treatment and genetic removal of matrix metalloproteinase. Dis Model Mech. 2011;4:673–685. doi: 10.1242/dmm.008045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kim HS, Suh YH. Minocycline and neurodegenerative diseases. Behav Brain Res. 2009;196:168–179. doi: 10.1016/j.bbr.2008.09.040. [DOI] [PubMed] [Google Scholar]
- 96.Budkevich TV, El'skaya AV, Nierhaus KH. Features of 80S mammalian ribosome and its subunits. Nucleic Acids Res. 2008;36:4736–4744. doi: 10.1093/nar/gkn424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Paribello C, et al. Open-label add-on treatment trial of minocycline in fragile X syndrome. BMC Neurol. 2010;10:91. doi: 10.1186/1471-2377-10-91. [DOI] [PMC free article] [PubMed] [Google Scholar]