

Genotype-phenotype correlations for most monogenic neurological disorders are incompletely understood. Fu et al. draw upon data on 615 HPRT mutations, including 130 new cases, and their relationship to Lesch-Nyhan disease severity. The effect of mutations on hypoxanthine-guanine phosphoribosyltransferase activity accounts for much but not all of the phenotypic variability.
Keywords: Lesch-Nyhan disease, genotype–phenotype correlations, neurogenetics
Abstract
Establishing meaningful relationships between genetic variations and clinical disease is a fundamental goal for all human genetic disorders. However, these genotype–phenotype correlations remain incompletely characterized and sometimes conflicting for many diseases. Lesch-Nyhan disease is an X-linked recessive disorder that is caused by a wide variety of mutations in the HPRT1 gene. The gene encodes hypoxanthine-guanine phosphoribosyl transferase, an enzyme involved in purine metabolism. The fine structure of enzyme has been established by crystallography studies, and its function can be measured with very precise biochemical assays. This rich knowledge of genetic alterations in the gene and their functional effect on its protein product provides a powerful model for exploring factors that influence genotype–phenotype correlations. The present study summarizes 615 known genetic mutations, their influence on the gene product, and their relationship to the clinical phenotype. In general, the results are compatible with the concept that the overall severity of the disease depends on how mutations ultimately influence enzyme activity. However, careful evaluation of exceptions to this concept point to several additional genetic and non-genetic factors that influence genotype–phenotype correlations. These factors are not unique to Lesch-Nyhan disease, and are relevant to most other genetic diseases. The disease therefore serves as a valuable model for understanding the challenges associated with establishing genotype–phenotype correlations for other disorders.
Introduction
Understanding how abnormalities in specific genes cause disease is a fundamental goal in human genetics. Identifying benign sequence variants and linking specific pathological mutations with the clinical manifestations of a disease can be helpful for understanding variations in the clinical spectrum of the disease, and for elucidating the pathogenesis of different clinical features of a complex syndrome. Lesch-Nyhan disease historically has served as a model for exploring genotype–phenotype correlations (Jinnah et al., 2000, 2004). Lesch-Nyhan disease was the first neurogenetic disorder for which the responsible gene was identified (Jolly et al., 1983). The gene is designated HPRT1, and it is among the most intensively studied genes in human genetics (Keebaugh et al., 2006). The gene has only one functional messenger RNA transcript that encodes an enzyme involved in purine metabolism known as hypoxanthine-guanine phosphoribosyltransferase (HGprt). The disorder is inherited in an X-linked recessive manner, which means that the influence of genetic alterations of a single allele can be evaluated independent from a second allele.
Many different disease-causing mutations have been reported for the HPRT1 gene (www.lesch-nyhan.org). Spread through nearly the whole gene are missense mutations, nonsense mutations, splicing mutations, small and large coding and non-coding deletions or insertions, partial duplications, non-coding regulatory mutations, and more complex changes. A comprehensive study of genotype–phenotype correlations published more than a decade ago concluded that the most relevant factor for provoking disease was the effect of the HPRT1 mutation on residual HGprt enzyme activity (Jinnah et al., 2000). However, some apparent exceptions to this rule have been reported. In recent years, many new disease-causing mutations have been described, and there have been considerable advances in understanding the consequences of these mutations on the structure and biochemical kinetics of the HGprt enzyme (Duan et al., 2004; Fu and Jinnah, 2012).
There also has been increased appreciation for the need to accurately describe and quantify different aspects of the clinical phenotype, and a growing understanding of how the phenotype evolves with age. The classical clinical phenotype of Lesch-Nyhan disease is defined by uric acid overproduction, motor dysfunction, intellectual disability, and behavioural problems including recurrent self-injury (Lesch and Nyhan, 1964; Nyhan et al., 1965; Jinnah et al., 2006). However, there also are attenuated clinical variants in which some of these clinical features are clinically insignificant or even absent (Emmerson and Thompson, 1973; Puig et al., 2001; Jinnah et al., 2010; Torres et al., 2011). Collectively, patients with attenuated phenotypes are designed Lesch-Nyhan variants. The mildest variant is described by isolated overproduction of uric acid. These patients do not have clinically overt neurological or behavioural abnormalities, and most often are described as having HGprt-related hyperuricaemia (HRH). In between the two extreme phenotypes of Lesch-Nyhan disease and HRH is a spectrum of phenotypes with varying degrees of neurological and behavioural abnormalities, designated HGprt-related neurological dysfunction (HND). The patients with HND suffer from overproduction of uric acid along with some neurological or behavioural difficulties, but they do not exhibit the self-injurious behaviours seen in classic Lesch-Nyhan disease.
The present review provides a summary of 615 cases with disease-causing mutations, including 485 previously reported cases and 130 new cases. It also summarizes our current knowledge of the influence of these mutations on HGprt structure and function, and the relevance of this information for understanding the molecular pathogenesis of the disease and diagnostic testing. The analysis provides some valuable lessons for understanding challenges associated with establishing meaningful genotype–phenotype correlations in other neurogenetic disorders.
The HPRT1 gene and HGprt enzyme
The human HPRT1 gene is located on the X chromosome in the region q26-27 (Patel et al., 1984; Stout and Caskey, 1985; Melton, 1987; Keebaugh et al., 2006). It contains eight introns and nine exons that encode a single mature messenger RNA of ∼1.6 kb. The coding region contains 654 nucleotides and is translated into HGprt, a protein with a molecular weight of 24.6 kD. Aside from cleavage of the initial methionine from the protein and acetylation of the newly exposed alanine (Wilson et al., 1983), there are no known post-translational modifications. However, there are two slowly progressive alterations including spontaneous oxidation of C23 and deamidation of N107 to D107 (Johnson et al., 1982; Wilson et al., 1982; Keough et al., 1991).
The crystal structure of HGprt has been determined in different forms, including its free form (Keough et al., 2005), liganded with its inosine monophosphate or guanosine monophosphate (GMP) products (Eads et al., 1994; Xu et al., 1997), or complexed with a transition state analogue and pyrophosphate (Shi et al., 1999). The enzyme exhibits a core structure that consists of five parallel beta-sheets flanked by four alpha helices. HGprt assembles spontaneously as either a dimer or tetramer of identical subunits depending on the ionic strength of the solvent (Holden and Kelley, 1978; Johnson et al., 1979). The subunits are not covalently bound, but instead are linked by a network of hydrogen bonds, salt bridges, and hydrophobic interactions. Figure 1 shows a schematic predicted from the crystal structures. The subunits are designated A to D, and the most extensive interactions occur at the A–B interface (amino acids 23–28, 70–101 and 198–204) and the A–C interface (amino acids 7–23 and 38–51). The A–D interface involving amino acids 84-90 is minor. Most of the amino acids at the interface between subunits are far from the catalytic site.
Figure 1.

Ribbon diagram for human HGprt. (A) A monomer with guanosine monophosphate in the open loop state (PDB: 1hmp). (B) A monomer with transition-state analogue and PPi in the closed loop state (PDB: 1bzy). (A and B) The PPi loop is shown in red, the PRPP loop is shown in blue, the C-terminal hood domain is shown in green, and the long flexible loop is shown in yellow. K166 is displayed in purple and the ligands are black. (C) The tetrameric structure of HGprt with each of the four subunits in a different colour (A in grey, B in blue, C in red and D in green).
HGprt is one of a family of phosphoribosyltransferase enzymes that uses the high-energy phosphate bonds of phosphoribosylpyrophosphate (PRPP) rather than ATP to catalyze its reaction (Fox and Kelley, 1971; Musick, 1981). HGprt drives two similar reactions in the recycling pathway for purines. One involves the pyrophosphorylation of hypoxanthine into inosine monophosphate, a precursor for both adenine nucleotides (ATP, ADP, AMP) and guanine nucleotides (GTP, GDP, GMP). The other involves the pyrophosphorylation of guanine directly into GMP (Fig. 2). The reaction catalyzed by HGprt has been reported to proceed via an ordered mechanism, in which PRPP binds first and induces a conformational change for subsequent purine binding, followed by the release of pyrophosphate and nucleotide (Salerno and Giacomello, 1979).
Figure 2.
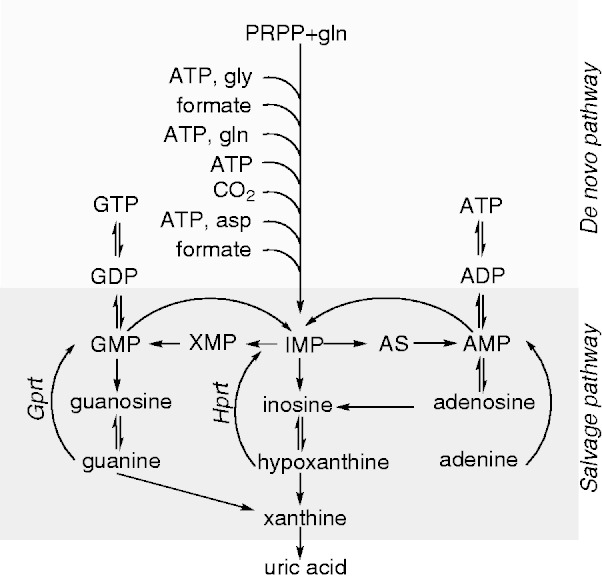
Purine metabolism.
AS = adenylosuccinate; asp = aspartic acid; ATP = adenosine triphosphate; GDP = guanosine diphosphate; gln = glutamine; gly = glycine; GMP = guanosine monophosphate; Gprt = guanine phosphoribosyltransferase; GTP = guanosine triphosphate; Hprt = hypoxanthine phosphoribosyltransferase; IMP = inosine monophosphate; XMP = xanthine monophosphate. In this diagram, HGprt is shown separately at its two distinct functional sites (Hprt and Gprt).
The catalytic reaction involves four domains of the HGprt protein including three loops that form the catalytic cleft and a long flexible loop that covers the reaction (Fig. 1). The PRPP binding loop between amino acids 130–140 is highly conserved across the phosphoribosyltransferase family, and serves to orient the ribose-phosphate moiety of PRPP in the catalytic cleft. Four consecutive amino acids from positions 68–71 constitute the PPi loop and also exhibit high structural homology across the phosphoribosyltransferase family. The PPi loop binds to the pyrophosphate moiety of PRPP in the catalytic cleft. A magnesium ion coordinates binding of hydroxyl groups and the 1-α-phosphate group of PRPP with other amino acids in active site. The hood domain at the carboxy terminus orients the purine ligand to PRPP bound within the cleft. Amino acid K166 influences substrate specificity toward the exocyclic 6-oxo group of the purine ring, and alterations here influence activity towards hypoxanthine, guanine, or xanthine. A long flexible loop from amino acids 100–128 exhibits open or closed conformations, moving up to 25 Å to shield the catalytic cleft from solvent during catalysis.
Studies of the crystal structure of HGprt have revealed multiple conformational changes during the catalytic cycle (Eads et al., 1994; Balendiran et al., 1999; Shi et al., 1999; Wang et al., 2001; Keough et al., 2005). The PRPP binding loop is relatively dynamic and opens when a 5’-phosphate group enters. This triggers a trans–cis isomerization between peptide bonds of L68–K69 to adopt a structure for optimal orientation of the ribose ring and pyrophosphate group. The binding of PRPP induces a rearrangement of the K166 side chain interaction, which turns to position the purine base via the 6-oxo group. The diacidic peptide E134–D135 of the PRPP binding loop moves to coordinate with the ribose ring, and the hood domain containing F187 folds over the ligands via stacking interactions. The long flexible loop then closes over the bound substrates. After the reaction, the flexible loop opens to liberate pyrophosphate followed by the nucleotide (Duan et al., 2004).
The precise knowledge of the structure and function of HGprt provides a rich background for delineating how mutations affect enzyme function and cause clinical manifestations. The four functional domains of the protein and the known subunit interactions lead to questions regarding their relative roles in disease pathogenesis. In addition, the dual roles of HGprt in recycling hypoxanthine and guanine raise questions regarding how mutations may affect one or both functions, and which function may be relevant for which aspects of the clinical phenotype.
The spectrum of the clinical phenotype
The classical phenotype of Lesch-Nyhan disease
The classical clinical phenotype of Lesch-Nyhan disease is relatively stereotyped. This phenotype includes overproduction of uric acid and an unusual neurobehavioural syndrome (Lesch and Nyhan, 1964; Nyhan et al., 1965). The overproduction of uric acid results in elevated serum levels of uric acid and increased excretion of uric acid in the urine (Torres et al., 2011). Because uric acid is near its limit of solubility in the body, it tends to precipitate in vulnerable body regions. Precipitation in the urogenital system, where it is concentrated by excretion, results in nephrolithiasis and associated problems such as urinary obstruction and renal failure. Precipitation in the subcutaneous tissues due to temperature gradients leads to solid masses known as tophi. Precipitation in the joints of the hands and feet with subsequent inflammation elicited by phagocytosis of the crystals by polymorphonuclear cells leads to gouty arthritis.
The neurobehavioural phenotype includes severe and recurrent self-injurious behaviours such as self-biting, self-hitting, eye poking, and others (Nyhan, 1976; Anderson and Ernst, 1994; Robey et al., 2003; Schretlen et al., 2005). Self-injurious behaviour is universal in Lesch-Nyhan disease. It usually emerges before 4 years of age, but may be delayed until the second decade of life. The neurobehavioural phenotype also includes severe motor handicap that resembles dystonic cerebral palsy (Watts et al., 1982; Jinnah et al., 2006). Motor function is dominated by generalized dystonia, a movement disorder that often is confused with spasticity or chorea. The motor disorder is sufficiently severe that all require wheelchairs and substantial assistance with activities of daily living. Most patients also have mild or moderate intellectual disability with IQ scores in the 60–80 range, but severe mental retardation is uncommon (Anderson et al., 1992; Matthews et al., 1995; Solan et al., 1997; Schretlen et al., 2001).
The milder Lesch-Nyhan variants
In the milder variant phenotypes, some clinical features are absent or sufficiently mild that they may escape clinical detection (Emmerson and Thompson, 1973; Puig et al., 2001; Jinnah et al., 2010; Torres et al., 2011). The mildest clinical phenotype of HRH includes only overproduction of uric acid and its associated problems. These patients do not have clinically overt neurological or behavioural abnormalities, although many have minor motor clumsiness or mild cognitive impairments that can be detected with appropriate neurological or psychometric testing.
In between the severe Lesch-Nyhan disease phenotype and the mild HRH phenotype is a broad spectrum of phenotypes with varying degrees of neurological and behavioural abnormalities known as HND (Jinnah et al., 2010). Patients with HND also suffer from overproduction of uric acid. They do not exhibit the self-injurious behaviours seen in classic Lesch-Nyhan disease, although they may have impulsivity and other undesirable behaviours. The motor features of HND vary from mild and clinically insignificant clumsiness to severe handicap similar to classic Lesch-Nyhan disease. Most also have some cognitive impairment, most notably difficulties with attention and concentration. Across the spectrum of HGprt deficiency, there is a rough correlation between the severity of motor and cognitive dysfunction (Jinnah et al., 2010), although rare cases may combine severe motor disability with little or no cognitive impairment.
Since Lesch-Nyhan disease is inherited in an X-linked recessive manner, the vast majority of cases are males. However, several manifesting females have been reported (Hooft et al., 1968; Hara et al., 1982; Ogasawara et al., 1989; Van Bogaert et al., 1992; Yukawa et al., 1992; Yamada et al., 1994; Aral et al., 1996; De Gregorio et al., 2000; Inokuchi et al., 2004; De Gregorio et al., 2005; Rinat et al., 2005; Jinnah et al., 2006, 2010; Sebesta et al., 2008). Eight of the manifesting females had the typical Lesch-Nyhan disease phenotype, and two were milder variants. The clinical features of the disease do not appear to be significantly influenced by sex. The clinical phenotype of these females does not result from haploinsufficiency. For those in whom the molecular defect was determined, all had a mutation in one allele together with skewed inactivation of the other allele. For females with the classic phenotype, skewed inactivation is thought to be complete, with corresponding severe loss of HGprt enzyme activity.
Limitations of clinical ascertainment
Although patients are clustered into three distinct sub-groups (Lesch-Nyhan disease, HND and HRH), the clinical spectrum actually is more continuous (Jinnah et al., 2010; Torres et al., 2011). Each of the major clinical problems associated with Lesch-Nyhan disease exhibits continuous grades of severity (Fig. 3). The existence of varying grades of severity means that some patients may fall between groups, and assignment to one group is sometimes arbitrary. For example, there are many cases initially published as HRH because of the absence of any overt neurological deficits. However, in some of these cases, careful neurological re-evaluation revealed mild impairments in motor skills (Jinnah et al., 2010). The cumulative severity of dystonia affecting all body regions can be estimated by the Fahn-Marsden dystonia rating scale, which goes from 0 (no dystonia) to 140 (severest dystonia). Some authors define a score of <5 as normal and compatible with HRH, to account for some minor clumsiness that may occur in otherwise normal children. Others have suggested a higher cut-off score of <10, highlighting the arbitrary nature of sub-grouping cases based on a disorder with a continuous spectrum of severity.
Figure 3.
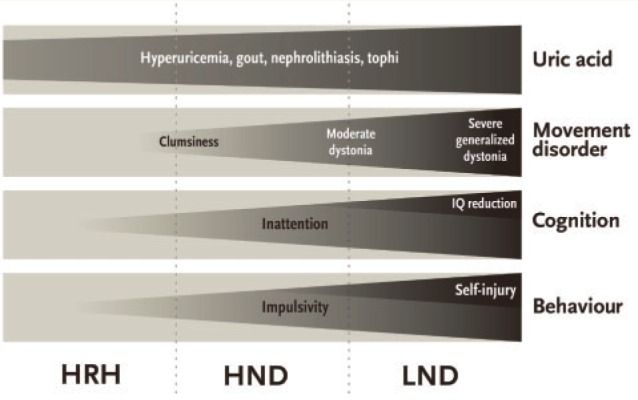
Clinical spectrum in Lesch-Nyhan disease and its variants. The four major aspects of the clinical phenotype are shown in grey boxes, with overall severity and frequency depicted by tapering triangles. LND = Lesch-Nyhan disease.
Cognitive disability also varies considerably. Some cases considered cognitively normal based on adequate school performance had more obvious deficits on formal neuropsychological testing (Schretlen et al., 2001; Jinnah et al., 2010). In other cases, specific cognitive impairments have been documented, despite global IQ scores suggestive of ‘normal’ intelligence. In some of these cases, the selective cognitive deficits have been sufficiently severe to impair normal social or occupational activities. The classification of these cases as HRH or HND depends on the rigor of neuropsychological testing, criteria used to define normal intelligence, and whether selective but severe deficits in particular cognitive domains are considered clinically relevant.
Even the occurrence of self-injury can lead to diagnostic uncertainty. In many classic Lesch-Nyhan disease cases, self-injury is severe and obvious with serious tissue damage, and misclassification is unlikely (Marmattom, 2005). However in some cases, self-injury is very mild. An example is a case with a single small abrasion on one thumb caused by a ‘habit’ of rapidly putting his hand to his mouth when excited or stressed (Jinnah et al., 2006). The abrasion resulted from repeated contact of the thumb with the teeth, but he never bit the thumb and there was no bleeding. The family did not view this behaviour as self-injurious, although the clinical team questioned it. Other examples are cases with isolated episodes of self-injury occurring once or twice in a lifetime, often triggered by some stressful event, such as a painful kidney stone. There also are several reports of Lesch-Nyhan disease variants without overt self-injury but multiple ‘accidental’ injuries from ‘bad wheelchair driving’ (Jinnah et al., 2010). Because wheelchair accidents leading to multiple injuries are not typical of other patients with similar motor disabilities, the accidental nature of the injuries might be questioned. Obviously, the classification of patients as Lesch-Nyhan disease or HND according to the presence or absence of self-injury depends on the criteria used to define ‘self injury’.
In addition to the problem of clinical ascertainment, it is important to acknowledge that HGprt deficiency causes a developmental disorder in which the clinical features unfold over time. Normal motor skills vary dramatically with age, with the emergence of dystonic movements and associated disability most often occurring gradually during the first few years of life. Some problems can be difficult to measure in infants. One example involves the c.500 G>T mutation (p.R167M), which was reported to be associated with markedly different clinical phenotypes in three members of one family (Sarafoglou et al., 2010). However, the motor and cognitive differences can be partly attributed to comparing toddlers with older adults (Fu and Jinnah, 2011). There are several additional cases in which motor skills were considered normal in the first month of life, but obvious motor delay became apparent within the first year (Kim et al., 1997; Mak et al., 2000; Zamora et al., 2007; Jurecka et al., 2008; Kim et al., 2009). Cognitive skills also change dramatically with age, and cognitive deficits are difficult to document for infants and very young children. The emergence of self-injurious behaviour also is age-dependent. The average age of onset is 3.1 years but the range varies from <1 year to >20 years (Jinnah et al., 2006). The varying age at onset means that distinguishing Lesch-Nyhan disease from variant cases is subject to error, especially for cases reported as children. Several cases initially classified as HND because of the absence of self-injurious behaviour (Fujimori et al., 1991), were reclassified as Lesch-Nyhan disease when self-injury emerged later (Fujimori et al., 1994).
Finally, the clinical phenotype can reflect the quality of care and education available. Patients with Lesch-Nyhan disease are difficult to work with because of their behavioural abnormalities, and they often present both physical and social challenges in school. As a result, educational opportunities and socialization sometimes are limited, and the result is low scores on standardized tests of cognitive function and sometimes inappropriate social behaviour. The management of physical disabilities also is challenging, with dystonic movement limiting mobility and behavioural problems that inhibit physiotherapy to prevent the consequences of disuse, such as development of contractures. Thus, the clinical phenotype of an adult with Lesch-Nyhan disease or its variants can reflect an interaction between the biology of the disease and psychosocial factors that may modify the disease over time.
Any attempt to correlate the severity of the clinical phenotype with mutations in HPRT1 or biochemical measurements of HGprt function must be done with careful attention to the rigor of neurological and psychological assessments, the age of the patient at evaluation, and the quality of care and education. Inadequate clinical assessments readily lead to misclassification of the phenotype, and assessments in very young children may reflect an incompletely evolved phenotype. Careful clinical assessments are essential starting points for understanding genotype–phenotype correlations.
The heterogeneity of clinically relevant mutations
Types of mutations
Tables 1–5 provide lists of mutations for 130 new cases from unrelated families referred to one of the centres of the Lesch-Nyhan Disease International Study Group for testing. Mutations were determined at each test site according to published methods (Jinnah et al., 2000; Torres et al., 2000; Ea et al., 2009; Yamada et al., 2011a). Most mutations were determined by reverse-transcriptase with PCR and direct sequencing of the resultant complementary DNA, using the reference sequence NM_000194.2. When complementary DNA could not be obtained, or contained an apparent deletion, genomic DNA was evaluated. Precise breakpoints in genomic DNA were not always determined as part of clinical diagnostic testing. Although all are new cases, some were found to have the same mutations previously described for other unrelated cases. All cases were males, except for one. The list includes 39 missense mutations, 13 nonsense mutations, 26 splicing mutations, 36 deletions, nine duplications, five indels and one with normal coding region. The female had a missense mutation with skewed X-inactivation.
Table 1.
New cases with missense mutations
| Case | Phenotype | Mutation | Result | Other cases reported |
|---|---|---|---|---|
| CF | LND | c.47G>T | p.G16V | Yes |
| NS | HRH | c.59A>T | p.D20V | Yes |
| AC | LND | c.65T>G | p.F22C | Yes |
| MD | HND | c.85G>C | p.A29P | Novel |
| AP | HND | c.115C>G | p.H39D | Yes |
| AC | LND | c.118G>A | p.G40R | Yes |
| OA | LND | c.125T>C | p.I42T | Yes |
| MG | LND | c.130G>C | p.D44H | Novel |
| LA | HND | c.143G>A | p.R48H | Yes |
| AD | HND | c.143G>A | p.R48H | Yes |
| LN159, DP | LND | c.145C>T | p.L49F | Yes |
| MB | LND | c.146T>G | p.L49R | Novel |
| CM; SM | LND | c.148G>A | p.A50T | Novel |
| ML; AL | LND | c.158T>G | p.V53G | Novel |
| NS | HRH | c.179A>G | p.H60R | Yes |
| YG | HRH | c.200T>C | p.V67A | Novel |
| RT | LND | c.209G>A | p.G70E | Yes |
| LN139F, O | LND | c.220T>G | p.F74V | Yes |
| LN139C, H4755, B | LND | c.293A>G | p.D98G | Yes |
| HPRT Rumania | HND | c.295T>G | p.F99V | Yes |
| MC | HRH | c.374T>C | p.L125S | Novel |
| TP | LND | c.379G>T | p.G127V | Novel |
| EG | LND | c.392T>G | p.L131W | Novel |
| PA | HRH | c.404A>G | p.D135G | Yes |
| NG | HND | c.475A>G | p.K159E | Yes |
| LN167, RT | LND | c.476A>C | p.K159T | Novel |
| OB | HRH | c.485G>C | p.S162T | Novel |
| TA | LND | c.530A>T | p.D177V | Yes |
| LN139A, G8081, JG | LND | c.539G>A | p.G180E | Yes |
| LN216, KD | LND | c.541T>C | p.F181L | Yes |
| India II | LND | c.550C>A | p.P184T | Novel |
| OA; MA | HND | c.554A>T | p.D185V | Novel |
| MB | HND | c.554A>T | p.D185V | Novel |
| LN203C, NP | LNV | c.584A>C | p.Y195S | Yes |
| PB; AB | LND | c.595T>G | p.F199V | Yes |
| DG; MG | HND | c.597C>G | p.F199L | Novel |
| LN155, KH | LND | c.611A>T | p.H204L | Novel |
| BW | LND | c.611A>G | p.H204R | Yes |
| MD | LND | c.635G>A | p.G212E | Yes |
This table includes a summary of previously unreported cases. In some cases, the molecular mutation is novel, while in others it is similar to a previously reported case. Previously reported cases are listed at www.lesch-nyhan.org. Abbreviations follow standard nomenclature for genetic diseases.
Table 2.
New cases with nonsense mutations
| Case | Phenotype | Mutation | Result | Other cases reported |
|---|---|---|---|---|
| NL | LND | c.84T>A | p.Y28* | Novel |
| JA | LND | c.151C>T | p.R51* | Yes |
| LN131, AJ | LND | c.151C>T | p.R51* | Yes |
| LN208 | LND | c.151C>T | p.R51* | Yes |
| SB | LND | c.151C>T | p.R51* | Yes |
| OS; MS | LND | c.430C>T | p.Q144* | Yes |
| JF | LND | c.454C>T | p.Q152* | Novel |
| LN148, SC | LND | c.508C>T | p.R170* | Yes |
| LN150, DP | LND | c.508C>T | p.R170* | Yes |
| LN169, LSZ | LND | c.508C>T | p.R170* | Yes |
| LN171, KM | LND | c.508C>T | p.R170* | Yes |
| TF; LS | LND | c.508C>T | p.R170* | Yes |
| VV | LND | c.508C>T | p.R170* | Yes |
This table includes a summary of previously unreported cases. In some cases, the molecular mutation is novel, while in others it is similar to a previously reported case. Previously reported cases are listed at www.lesch-nyhan.org. Abbreviations follow standard nomenclature for genetic diseases.
Table 3.
New cases with splicing mutations
| Case | Phenotype | Mutation | Result | Other cases reported |
|---|---|---|---|---|
| HH | LND | c.27+1G>A | Splice error | Yes |
| LN128, H6214, DC | LND | c.27+5G>A | Splice error | Yes |
| TH | LND | c.135-1G>A | Splice error | Novel |
| LN175 | LND | c.135-2A>G | Exon 3 excluded | Novel |
| LN132, JC | LND | c.318+1G>T | Exon 3 excluded | Yes |
| LN204, AJZ | LND | c.384+1G>T | Exon 4 excluded | Yes |
| LN122, R | LND | c.384+1G>T | Exon 4 excluded | Yes |
| BP | LND | c.385-1G>A | Exon 5 excluded | Yes |
| MP | LND | c.402+1G>A | Splice error | Yes |
| VP | LND | c.402+5G>A | Splice error | Novel |
| LN174 | LND | c.403-1G>C | Exon 5 excluded | Yes |
| LN203B, JS; DS | LNV | c.485+5G>A | Exon 6 excluded | Yes |
| AQ | LND | c.486-1G>A | Splice error | Novel |
| LN126, BM | LND | c.486-3C>G | Exon 7 excluded | Novel |
| LN158, JCG | LND | c.532+1G>A | Exon 7 excluded | Yes |
| LN217, TM | LND | c.532+1G>A | Exon 7 excluded | Yes |
| PMK; WBK | LND | c.532+2G>T | Exon 7 excluded | Novel |
| LN139B, H8475, H | LND | c.532+5G>A | Exon 7 excluded | Yes |
| NS | LND | c.532+5G>T | Exon 7 excluded | Novel |
| BV | LND | c.533-13T>G | Exon 8 excluded | Novel |
| KD | LND | c.533-1G>A | Splice error | Yes |
| LN172, LN203A, MB; LB | LND | c.609+6T>G | Exon 8 excluded | Yes |
| LN162, II | LND | c.610-1G>A | 17 bp of exon 9 excluded | Yes |
| FG | LND | c.610-1G>A | Splice error | Yes |
| GD | LND | c.610-1G>T | Splice error | Novel |
| LN136, KB | LND | c.610-2A>T | 610–626 bp of exon 9 excluded | Yes |
This table includes a summary of previously unreported cases. In some cases, the molecular mutation is novel, whereas in others it is similar to a previously reported case. Previously reported cases are listed at www.lesch-nyhan.org. Abbreviations follow standard nomenclature for genetic diseases.
Table 4.
New cases with deletion mutations
| Case | Phenotype | Mutation | Result | Other cases reported |
|---|---|---|---|---|
| ML | LND | 5'UTR deleted | No protein | Novel |
| LN130, ML | LND | Exon 1 deleted | 1 exon deleted | Yes |
| LN139D, H2339, JM, JHG, LN160 | LND | Exon 1 deleted | 1 exon deleted | Yes |
| LN161, OLH | LND | Exon 1 deleted | Macro del | Yes |
| LN201, JL | LND | Exon 1 deleted | Macro del | Yes |
| LN215, WM | LND | Exon 1 deleted | Macro del | Yes |
| Leganes | LND | Exon 1 deleted | 1 exon deleted | Yes |
| Colombia | LND | c.53delA | Frame shift in exon 2 | Novel |
| FM | LND | c.82_84delTAT | p.Y28del | Yes |
| LN170, RS | LND | c.105_108delGTTT | Frame shift in exon 2 | Novel |
| LN211, CN | LND | c.124_127delATTA | Frame shift in exon 2 | Novel |
| JC, AC | LND | c.131delA | Frame shift in exon 2 | Novel |
| LN139E, H4672, H | LND | Exons 2–3 deleted | 2 exons deleted | Yes |
| LN137, RC | LND | Exons 2–4 deleted | 3 exons deleted | Novel |
| FM | LND | c.212delG | Frame shift in exon 3 | Novel |
| LN165, EV | LND | c.269_270delAT | Frame shift in exon 3 | Yes |
| Bilbao | LND | c.275_298del | 16 bp deleted in exon 3 | Novel |
| LN205, RC | LND | c.288_289delTG | Frame shift in exon 3 | Novel |
| LN209, JB | LND | c.289_290delGT | Frame shift in exon 3 | Yes |
| LN164 | LND | c.337delG | Frame shift in exon 4 | Yes |
| DA | LND | Exon 4 deleted | 1 exon deleted | Yes |
| LN213, DR | LND | Exons 4–6 deleted | 3 exons deleted | Yes |
| GB | LND | Exons 4–8 deleted | No protein | Yes |
| ML | LND | Exons 4–9 deleted | No protein | Yes |
| LN124, ES | LND | c.402+1_402+4delGTAA | Splice error, frame shift in exon 5 | Novel |
| LN218, DD | LND | c.402+5delG | Splice error, frame shift in exon 5 | Novel |
| CA; AA | LND | c.512delG | Frame shift in exon 7 | Novel |
| VW | LND | Exon 7 deleted | 1 exon deleted | Yes |
| LN144, CS | LND | Exons 7–9 deleted | 3 exons deleted | Yes |
| PL | LND | Exon 8 deleted | 1 exon deleted | Yes |
| LN145, MP | LND | c.577delC | Frame shift in exon 8 | Novel |
| LD | LND | c.585_586delTA | Frame shift in exon 8 | Novel |
| LN146, KB | LND | c.6095_6098delGATT | Splice error, frame shift in exon 8 | Novel |
| LN127, GQ | LND | c.643_664del | Exon 9 excluded | Novel |
| Las Palmas | LND | Exon 9 deleted | 1 exon deleted | Yes |
| LN152, RR | LND | Exons 1–9 deleted | 9 exons deleted | Yes |
This table includes a summary of previously unreported cases. In some cases, the molecular mutation is novel, while in others it is similar to a previously reported case. Previously reported cases are listed at www.lesch-nyhan.org. Abbreviations follow standard nomenclature for genetic diseases, except for large deletions where actual breakpoints were not defined.
Table 5.
Other new cases with mutations
| Case | Phenotype | Mutation | Result | Other cases reported |
|---|---|---|---|---|
| Duplication | ||||
| Valencia | LND | c.50_51dupAT | Frame shift in exon 2 | Novel |
| Brussels | LND | c.212dupG | Frame shift in exon 3 | Yes |
| LN133, AC | LND | c.212dupG | Frame shift in exon 3 | Yes |
| LN135 | LND | c.212dupG | Frame shift in exon 3 | Yes |
| LN149, NY | LND | c.212dupG | Frame shift in exon 3 | Yes |
| LN153, DC | LND | c.212dupG | Frame shift in exon 3 | Yes |
| LN173, CD | LND | c.212dupG | Frame shift in exon 3 | Yes |
| DD | LND | c.212dupG | Frame shift in exon 3 | Yes |
| BT | LND | c.297dupT | Frame shift in exon 3 | Yes |
| Others | ||||
| LN210, G | LND | c.-4_4delinsGCC | Frame shift in exon 1 | Novel |
| DK | NA | c.52_54delinsTAA | p.D18* | Novel |
| GB | HND | c.238_239delinsTT | p.D80F | Yes |
| ID | LND | c.320_326delinsCTTTTTTAT | Frame shift in exon 4 | Novel |
| NS | LND | c.456delinsTT | Frame shift | Novel |
| NL | LND | normal** | No enzyme activity | Novel |
| Female | ||||
| LN143 | LND | c.[580G>T]; [XIC] | p.D194Y; non-random inactivation of other allele | Novel |
This table includes a list of previously unreported cases. In some cases, the molecular mutation is novel, whereas in others it is similar to a previously reported case. Previously reported cases are listed at www.lesch-nyhan.org. Abbreviations follow standard nomenclature for genetic diseases.
XIC = non-random X-chromosome inactivation.
Double asterisk denotes presumed non-coding mutation in regulatory regions of the gene.
Table 6 summarizes these 130 new cases combined with 485 cases reported through to the end of December 2012. Together, the mutations were quite heterogeneous. Single base substitutions were most common, with 381 cases accounting for 62.0% of the total. Of the substitutions, 242 (39.3%) were missense mutations producing a single amino acid substitution, 90 (14.6%) caused splicing errors, and 49 (8.0%) were nonsense codons leading to premature termination of protein translation. Deletions were the second most common type of mutation, with 158 cases representing 25.7% of the total. Approximately half were large intragenic deletions with loss of one or more exons, whereas the other half were smaller deletions of up to seven bases. Ten deletions affected splice sites predicted to cause splicing errors. There were also 40 partial gene duplications (6.5%) and 26 (4.2%) other mutations including 13 complex deletions, six non-coding sequence alterations, four insertions, two double mutations, and one translocation. Ten females were reported with a mutation in one allele and skewed inactivation of the other allele.
Table 6.
Summary of new and previously reported HPRT1 mutations
| Mutation | LND (n = 461) | LNV (n = 142) | NA (n = 12) | Total (n = 615) | % of total |
|---|---|---|---|---|---|
| Single base substitutions | 255 | 121 | 5 | 381 | 62.0 |
| Missense | 131 | 107 | 4 | 242 | |
| Nonsense | 47 | 1 | 1 | 49 | |
| Splice error | 77 | 13 | 0 | 90 | |
| Deletions | 146 | 6 | 6 | 158 | 25.7 |
| Coding sequences | 138 | 5 | 5 | 148 | |
| Splice error | 8 | 1 | 1 | 10 | |
| Duplications | 37 | 3 | 0 | 40 | 6.5 |
| Coding sequences | 35 | 3 | 0 | 38 | |
| Splice error | 2 | 0 | 0 | 2 | |
| Others | 15 | 10 | 1 | 26 | 4.2 |
| Translocations | 1 | 0 | 0 | 1 | |
| Regulatory changes | 2 | 4 | 0 | 6 | |
| Double mutations | 0 | 2 | 0 | 2 | |
| Insertions | 3 | 1 | 0 | 4 | |
| Complex deletions | 9 | 3 | 1 | 13 | |
| Females | 8 | 2 | 0 | 10 | 1.6 |
| % of total | 75.0 | 23.1 | 2.0 | 100 |
This table includes a list of all cases with HPRT1 mutations including the new cases from Tables 1–5 combined with all previously reported cases. A complete list of mutations is available at www.lesch-nyhan.org.
NA = not available; LND = Lesch-Nyhan Disease; LNV = Lesch-Nyhan variant.
Locations of mutations
Although the mutations are widely distributed throughout the HPRT1 gene, there are some isolated hot spots, where the same mutation has arisen multiple times in unrelated patients. Two of the most common hot spots are c.151C>T (n = 20) and c.508 C>T (n = 19). Both of these mutations result in the conversion of a codon encoding arginine to a stop signal (p.R51* and p.R170*), and both probably arise through a similar mechanism involving methylation of cytosine in CpG motifs, with subsequent deamination of 5-methylcytosine to produce thymine (O'Neill and Finette, 1998). Another hot spot mutation is insertion of an extra guanine nucleotide in the string of guanine nucleotides from positions 212 (c.212dupG), leading to a frame shift and premature stop (n = 15). Here the molecular mechanism probably involves insertion of an extra base due to strand slippage.
Although most mutations affect coding regions or splice sites, there were 12 mutations reported to affect non-coding regions among patients where a clinical phenotype consistent with Lesch-Nyhan disease or an attenuated variant was confirmed by demonstrating biochemical evidence for reduced HGprt activity. Some of these mutations may affect regulatory regions of the gene and reduce messenger RNA transcription or stability. In fact, three cases were reported to have promoter region deletions up to 33 Kb from the start site, resulting in no detectable messenger RNA (Taniguchi et al., 2011; Yamada et al., 2011b). Another two cases had intronic mutations, in which a single base substitution (c.485+2775T>A; and c.402+1229A>G) formed a functional splice site creating a pseudo-exon (Sege-Peterson et al., 1992; Corrigan et al., 2011). A third case was reported to have a translocation, involving intron 3 (Mizunuma et al., 2004; Yamada et al., 2007). Six more cases had unknown, presumably non-coding mutations with reduced messenger RNA and enzyme activity (Dawson et al., 2005; Garcia et al., 2008; Nguyen et al., 2012). Thus some cases have mutations that are difficult to detect with methods that focus on exons and splice junctions.
Genotype–phenotype correlations
Correlations with types of genetic mutations
Overall, 461 cases (75%) were associated with the typical clinical phenotype of Lesch-Nyhan disease, whereas 142 cases (23%) were associated with the less severe variant phenotypes. Twelve cases (2%) could not be clinically categorized due to insufficient clinical information in the original reports. As previously noted, placing patients into these clinical subgroups based on published reports is imprecise. The literature varies considerably in how carefully the clinical phenotype was ascertained, and some cases were reported at a very young age before the full clinical phenotype had time to develop. Despite these caveats, the large number of reported cases is sufficient to permit recognition of some general genotype–phenotype trends.
A comparison of Lesch-Nyhan disease versus Lesch-Nyhan variant reveals that specific categories of mutations are not equally represented in both groups. Virtually all of the deletion mutations involving coding regions were associated with the more severe clinical phenotype of Lesch-Nyhan disease, with only six reported to have the Lesch-Nyhan variant phenotype. Duplications also were only rarely associated with milder phenotypes, occurring in only three cases. Similarly, nonsense mutations leading to premature termination of protein translation were associated with classic Lesch-Nyhan disease in all cases except one. Only one Lesch-Nyhan variant case had an insertion. The majority of Lesch-Nyhan variant cases (75.4%) had missense mutations leading to single amino acid substitutions. Mutations leading to splicing errors were the second most common type of mutation in Lesch-Nyhan variants, occurring in 13 cases (9.2%, Tables 6 and 7).
Table 7.
Discrepancies in genotype-phenotype correlations
| Genotype | Type | Phenotype | Reasons for discrepancy | References |
|---|---|---|---|---|
| c.508C>T | Nonsense | HND | 10 yrs old | Burgemeister et al., 1994 |
| c.213delC | Deletion | HND | 1 yr old | Sharma et al., 2012 |
| c.-12_1del | Deletion | LNV | AUG start deleted, functional upstream GUG start | Gibbs and Caskey, 1987; Gibbs et al., 1989; Davidson et al., 1994 |
| Introns 1–2 deleted | Deletion | HND | <1 yr old | Tvrdik et al., 1998 |
| c.403_452del | Deletion | HRH | Splice junction, multiple mRNA, 9% activity in IF | Sege-Peterson et al., 1992 |
| Exon 7 deleted | Deletion | HND | 5 yrs old | Davidson et al., 1991 |
| c.648_698del | Deletion | HRH | C-terminal loss of 2 amino acids, 9% activity in LL | Igarishi et al., 1989 |
| Exons 2–3 duplicated | Duplication | HND | Gene reversion | Wilson et al., 1986; Monnat et al., 1992 |
| Exons 2–3 duplicated | Duplication | HND | Gene reversion | Yang et al., 1984, 1988; Sege-Peterson et al., 1992; Jinnah et al., 2010 |
| Exons 7–8 duplicated | Duplication | HND | Gene reversion | Marcus et al., 1993b |
| c.429_430insGCA | Insertion | HND | One amino acid added, 7.5% activity in IF | Sege-Peterson et al., 1992 |
| c.176_180delins ATAGATGTGAAA | Indels | HND | 3 yrs old | Serrano et al., 2008 |
| c.238_239delinsTT | Indels | HND | Single amino acid substitution, 3.4 yrs old | Jinnah et al., 2010 |
| c.238_239delinsTT | Indels | HND | Single amino acid substitution | New case |
| c.27+1G>A | Splice error | HND | 7 yrs old | Marcus et al., 1993a |
| c.27+1G>T | Splice error | HND | 5% activity in EL, 1% activity in LF | Jinnah et al., 2000, 2010 |
| c.27+5G>A | Splice error | HND | Multiple mRNAs | Yamada et al., 2007 |
| c.27+47C>T | Splice error | HRH | Inclusion 15 amino acids without frame shift, 7% activity in LE | Gaigl et al., 2001 |
| c.134+1G>A | Splice error | HND | Unknown | Jinnah et al., 2000 |
| c.385-1G>A | Splice error | HND | 7 yrs old | Jinnah et al., 2010 |
| c.385-2A>G | Splice error | HND | Multiple mRNAs | Jinnah et al., 2010; Torres et al., 2010 |
| c.402+1G>A | Splice error | HND | Aberrant mRNA | Jinnah et al., 2000 |
| c.402+1229A>G | Splice error | HRH | Multiple mRNAs, 50% activity in IF | Sege-Peterson et al., 1992 |
| c.485+2T>C | Splice error | HND | Unknown | Hladnik et al., 2008; de Gemmis et al., 2010 |
| c.485+5G>A | Splice error | LNV | Unknown | New case |
| c.485+5G>A | Splice error | HND | Unknown | Corrigan et al., 2011 |
| c.532+2T>C | Splice error | HND | Multiple mRNAs | Yamada et al., 2004 |
This table includes a list of cases where the reported phenotype and genotype are not consistent with the concept that mutations influence disease severity by their influence on HGprt enzyme activity.
EL = erythrocyte lysates; IF = intact fibroblasts; LL = lymphocyte lysates; LND = Lesch-Nyhan disease; LNV = Lesch-Nyhan variant; NA = not available.
These differences between Lesch-Nyhan disease and its variants suggest that mutations resulting in null enzyme function are more likely to cause the more severe Lesch-Nyhan disease phenotype, while mutations that may permit some residual enzyme function more frequently underlie the milder Lesch-Nyhan variant phenotypes (Jinnah et al., 2000). Thus deletions, insertions and duplications are uncommon in Lesch-Nyhan variants because they most often result in a structurally abnormal protein with no functional activity. Nonsense mutations are similarly uncommon in Lesch-Nyhan variants because they result in premature termination of translation and absent enzyme activity. On the other hand, missense mutations may be over-represented in Lesch-Nyhan variants, because a single amino acid substitution is more likely to permit some residual enzyme function. Nevertheless, there are several exceptions where cases with the Lesch-Nyhan variant phenotype were associated with mutations predicted to cause complete loss of enzyme activity. In addition, the high frequency of splicing errors seems unexpected since they are predicted to lead to exon skipping and often frameshifts, resulting in a dysfunctional protein. These exceptional cases are addressed in more detail below in the section on genotype–phenotype discrepancies.
Correlations with locations of genetic mutations
Taken together, the mutations have a broad but unequal distribution throughout the HPRT1 gene (Fig. 4). Because the emphasis of this review is to understand how mutations affect protein structure and enzyme activity, the following analysis focuses on missense mutations that lead to a single amino acid change. Although early studies suggested that mutations might cluster in highly conserved regions of the gene associated with the active site of the enzyme, it is now clear that many disease-causing mutations occur far from this site, where they may instead interfere with subunit interactions, conformational changes required in the catalytic cycle, or structural stability (Duan et al., 2004; Fu and Jinnah, 2012). In fact, it is possible to recognize five ‘hot clusters’ of mutations defined as 10 adjacent amino acids with a high frequency of reported cases (Fig. 4). One of these hot clusters involves amino acids 42–51, coinciding with the A–C subunit interface. The second hot cluster involves amino acids 65–74, overlapping the PPi binding loop. A third hot cluster covers positions 131–140, spanning the PRPP binding loop. The fourth cluster from 158–167 encompasses K166, which plays a key role in base recognition. The last cluster has a relatively broad range from 188–204, which contributes to the hood domain for purine base binding. No substitution mutations have been reported for 126 of 218 amino acids.
Figure 4.
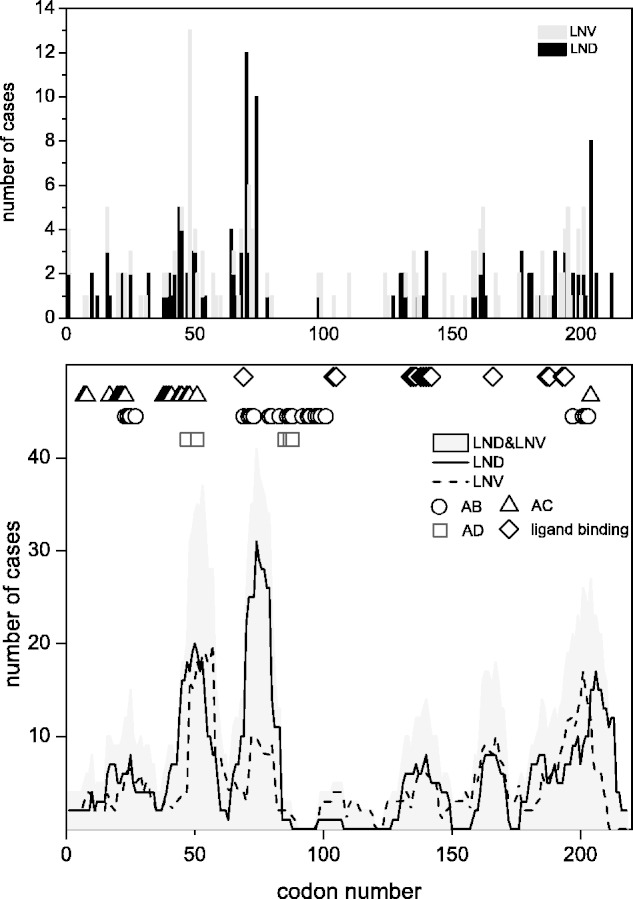
Distribution of cases with missense mutations in the HPRT1 gene. The top panel shows the numbers of cases with missense mutations reported according to codon number. Cases with Lesch-Nyhan disease (LND) are shown as black lines and Lesch-Nyhan variant (LNV) as dark grey lines. The height of each bar reflects the total numbers of independent cases reported for unrelated families. The bottom panel shows the same data as in the top panel, with a line showing a running average of 10 codons at each point. This analysis more readily permits the visualization of five hot clusters of mutations with peaks of at least 10 cases. These hot clusters show similar distributions for Lesch-Nyhan disease (solid line) and Lesch-Nyhan variant (dashed line). The total cases combined are shaded in light grey. The different symbols above the graph depict the subunit interfaces (circles for A–B; triangles for A–C; squares for A–D) and the ligand binding sites (diamonds). Cases contributing to this figure are listed individually at www.lesch-nyhan.org.
There are 11 highly conserved amino acids among HGprt protein sequences from different species that are thought to be important for the active site (Craig and Eakin, 2000). Seven of these amino acids (L68, G70, S104, D135, G190, D194 and R200) have been linked with 28 cases. Among these, 21 (75%) had Lesch-Nyhan disease and seven had attenuated phenotypes. In addition to these conserved amino acids, adjacent amino acids might be expected to disrupt important functional domains. The hot cluster of mutations from amino acids 65–74 flanks the four amino acid PPi loop. This cluster has a total of 41 cases including 31 Lesch-Nyhan disease and 10 Lesch-Nyhan variant cases. The second hot cluster occurs at amino acids 131–140, corresponds to the PRPP loop and was affected in 16 cases, including eight patients with Lesch-Nyhan disease, seven milder variants and one case for which clinical information was limited. The third hot cluster at amino acids 188–204 corresponds to the hood domain at the carboxy terminus of the HGprt and overlaps with the A–B subunit interface. This region was affected in 40 cases, with 22 Lesch-Nyhan disease cases and 18 Lesch-Nyhan variants. The only hot cluster distant from the active site of the enzyme is at the A–C interface region spanning amino acids 42-51. In this region there were 19 cases with Lesch-Nyhan disease, 18 Lesch-Nyhan variant cases and two cases with limited clinical data. Aside from the hot clusters noted above, substitution of the residues around K166 resulted in eight Lesch-Nyhan disease and 10 Lesch-Nyhan variant phenotypes, consistent with the critical role of this amino acid and its surrounding network in purine binding. There also are several cases scattered through the N-terminal region along the A–C subunit interface region from positions 7–29.
There were no obvious differences in the locations of the mutations for Lesch-Nyhan disease versus Lesch-Nyhan variants (Fig. 4). Instead, differences in phenotypic severity with mutations at the same amino acid may be explained by the fact that non-conservative mutations involving substitution of physicochemically very different amino acids cause severe phenotypes whereas more conservative mutations cause less severe phenotypes. For example, G71 is associated with Lesch-Nyhan disease with non-conservative mutations to arginine or aspartic acid, which involve replacing a small neutral amino acid with a larger charged one. In contrast, G71 is associated with milder Lesch-Nyhan variant phenotypes when more conservatively mutated to valine. Another example is D194, which is associated with the mild phenotype of HND with the conservative mutation to glutamic acid, which is physicochemically similar to aspartic acid. However, D194 is associated with the severe phenotype of Lesch-Nyhan disease when mutated to physicochemically very different amino acids histidine, tyrosine, or asparagine. Similarly, the conservative p.I136T mutation exhibited a mild clinical phenotype, whereas the less conservative p.I136K mutation showed a severe clinical phenotype.
Although many mutations may disrupt HGprt enzyme activity by falling in specific functional domains of the protein, some do not. Presumably, these mutations may affect conformational changes during the catalytic cycle from a distance, or interfere with protein folding or stability. The region between codons 80–120 has an unusually low frequency of mutations. This low frequency may reflect a less vulnerable area of the gene. Alternatively, it may reflect an area where mutations may have no significant functional consequences, and thus may never come to clinical attention. Evaluation of the HPRT1 gene in 1000 apparently normal individuals revealed only a single variant resulting in p.H60R (Fujimori et al., 1997). Whether this case should be considered a rare sequence variant or asymptomatic mutation carrier is not clear, because residual HGprt enzyme activity was 37–46% of normal and the same mutation was found for a case with the mild phenotype of HRH (Table 1).
Predicting pathogenicity of mutations
Several software algorithms have been designed to predict the pathogenicity of individual sequence variants. The most commonly employed include SIFT (http://sift/jcvi.org/) and PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/). SIFT is based on sequence comparisons from homologous or orthologous proteins and PolyPhen-2 uses both structure and sequence information (Adzhubei et al., 2010; Sim et al., 2012). Although they were designed to predict the pathogenicity of novel sequence variants of unclear functional significance, the large database of mutations in the HPRT1 gene associated with different grades of clinical severity provides an opportunity to test their performance.
SIFT correctly classified 92.8% of pathogenic missense mutations in the HPRT1 gene, but incorrectly classified 7.2% of the mutations as benign. Among those predicted to be benign, 11.8% were associated with the Lesch-Nyhan disease phenotype, and 88.2% with the Lesch-Nyhan variant phenotype. PolyPhen-2 correctly classified 83.1% of known pathogenic mutations. Among those predicted to be benign, 25% were associated with the Lesch-Nyhan disease phenotype, and 75% with the Lesch-Nyhan variant phenotype. These results demonstrate these software algorithms to have some predictive value. However, there is a significant failure rate, suggesting caution in relying on their predictions for clinical prognosis of mutations in the HPRT1 gene. These failure rates are comparable to those reported for other genes (Hicks et al., 2011).
Enzyme-phenotype correlations
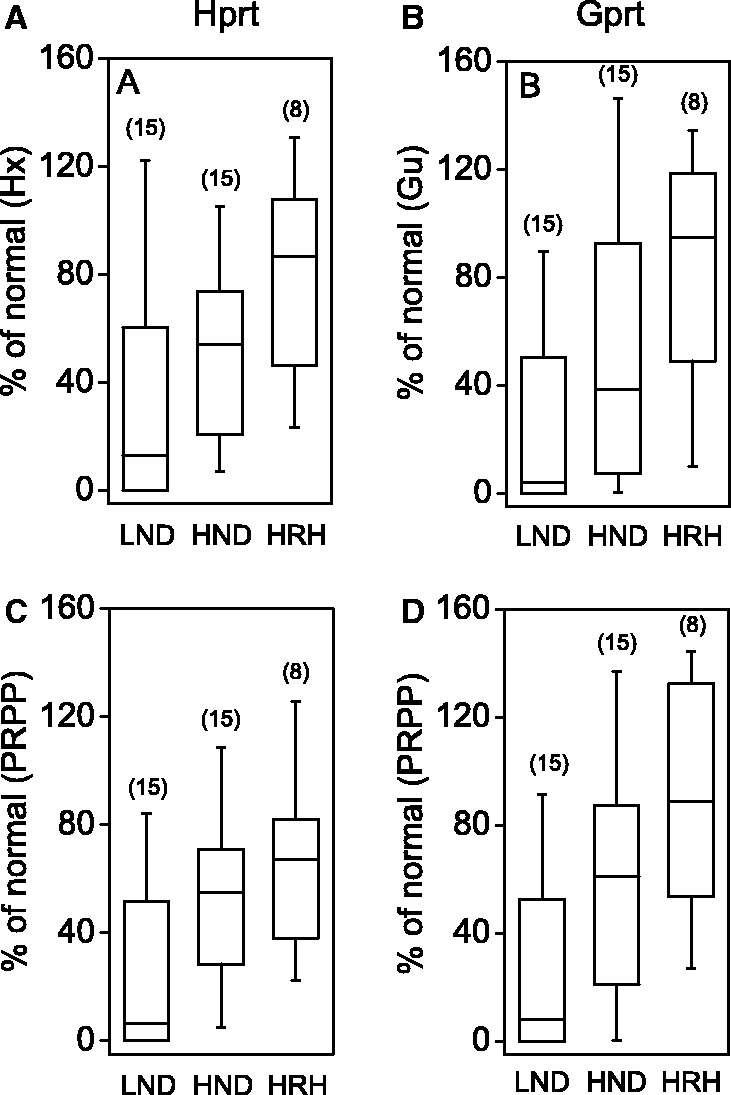
The majority of the known HPRT1 mutations are consistent with the concept that the most relevant factor for provoking disease is the effect of the mutation on residual HGprt enzyme activity. This concept is supported by several biochemical studies addressing the consequences of the mutations on enzyme function. The best results have been obtained when HGprt is studied under naturalistic conditions in living cells, rather than in artificial in vitro conditions. In one assay, skin biopsies are taken from patients to grow fibroblast cultures, which make physiological levels of PRPP (Page et al., 1981; Hersh et al., 1986; Page and Nyhan, 1989). A pulse of radiolabelled hypoxanthine or guanine is given, and the rate of incorporation into nucleotides is used as an estimate of actual activity. With this assay, patients with the most severe phenotype of Lesch-Nyhan disease typically have <1.5% of normal enzyme activity, whereas the mildest phenotype of HRH is generally associated with >10% of more activity. Patients with intermediate phenotypes of HND have activity that normally falls in between (Fig. 5).
Figure 5.
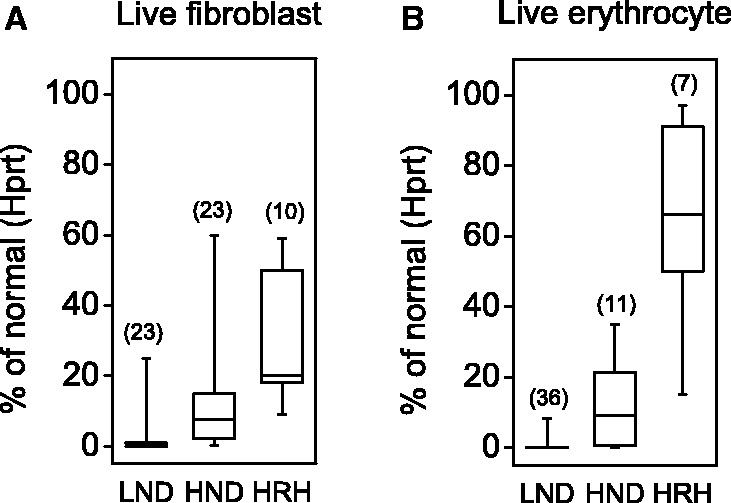
Correlations between residual enzyme activity and clinical severity in patient cells. Panel A is a box-whisker plot showing residual activities for Hprt as measured by the intact fibroblast assay for 56 patients previously reported (Itiaba et al., 1978; Bakay et al., 1979; Gottlieb et al., 1982; Warzok et al., 1982; Fattal et al., 1984; Lorentz et al., 1984; Mitchell and McInnes, 1984; Hersh et al., 1986; Tarle et al., 1991; Gilbert et al., 1992; Sege-Peterson et al., 1992; Jenkins et al., 1994; Erhard et al., 1997; Puig et al., 2001; Zoref-Shani et al., 2003, 2004; Cherian and Crompton, 2005; Cossu et al., 2006; Jinnah et al., 2010; Sarafoglou et al., 2010; Torres et al., 2010). Panel B is a box-whisker plot showing residual activities for Hprt as measured by the intact erythrocyte assay for 54 patients previously reported (Shnier et al., 1972; Ludman et al., 1992; Sege-Peterson et al., 1992; Davidson et al., 1994; Willers et al., 1999; Puig et al., 2001; Augoustides-Savvopoulou et al., 2002; Bertelli et al., 2004; Kassimatis et al., 2005; Cossu et al., 2006; Garcia et al., 2008; de Gemmis et al., 2010; Corrigan et al., 2011). Corresponding values for Gprt activity were not available. In these plots, the whiskers show the whole data range, while the boxes show the 25th and 75th percentiles of the data. The horizontal line depicts the median. These data must be interpreted with caution, because they were collected from published reports from different laboratories, each with different methods for the assay and different strategies for normalization of the results.
Another good assay is based on intact erythrocytes isolated from fresh blood samples from patients. These cells also have endogenous HGprt and PRPP, and pulsing with radiolabelled hypoxanthine or guanine can be used as an estimate of enzyme activity (Fairbanks et al., 1987). Studies using the live erythrocyte assay also provide good correlations between residual enzyme function and clinical severity (Fig. 5) (Fairbanks et al., 1987; Torres et al., 2000). However, two unusual features of erythrocytes cause artefacts in some cases. First, erythrocytes have a lifespan of ∼120 days. Second, they lack a nucleus and therefore also lack ongoing messenger RNA and protein synthesis. Thus blood samples reflect a mixture of old and new erythrocytes, so that residual HGprt activity is dependent upon the structural stability of the enzyme. The live erythrocyte assay is prone to giving artificially low activity for HGprt mutants that are structurally unstable.
Although the live cell assays may be the best choices for estimating residual HGprt enzyme activity in vivo, they are not suitable for understanding the kinetic mechanisms responsible for the loss of enzyme activity. A recent study employed an alternative approach for examining enzyme–phenotype correlations for 44 clinically relevant HGprt mutants associated with a wide spectrum of disease severity (Fu and Jinnah, 2012). The HPRT1 mutations were re-created by site-directed mutagenesis, the mutant proteins were expressed and purified in vitro, and their biochemical kinetics were studied with a sensitive spectrophotometric assay. The results confirmed a good correlation between clinical severity and residual HGprt enzyme activity (Fig. 6). There was a parallel loss of hypoxanthine and guanine recycling for most mutant enzymes, although a few mutants showed skewed loss of activity towards one or the other substrate. Clinical severity seemed to correlate best with loss of hypoxanthine recycling, although there were too few cases with skewed loss of activity to be sure. This study also pointed to the existence of several biochemical mechanisms responsible for reducing enzyme activity. These mechanisms included alterations in the affinity of the enzyme towards one or more of its substrates, altered catalytic efficiency towards one or more substrates, and reduced structural integrity of the enzyme.
Figure 6.
Correlations between residual enzyme activity and clinical severity using purified engineered mutant proteins in vitro. The two different functions of HGprt are shown separately as Hprt and Gprt. The panels show box-whisker plots for residual Hprt activity with a fixed high concentration of hypoxanthine (A) and a fixed high concentration of PRPP (C), residual Gprt activity with a fixed high concentration of guanine (B) and a fixed high concentration of PRPP (D) as previously described (Fu and Jinnah, 2012). In these plots, the whiskers show the whole data range, while the boxes show the 25th and 75th percentiles of the data. The horizontal line depicts the median.
The live cell assays and the recreation of mutant enzymes in vitro provide powerful tools for measuring residual enzyme activity and understanding mechanisms for reduced activity, but they are technically demanding and expensive. These qualities make them difficult to use for routine clinical diagnostic testing. As a result, many other studies have used simpler assays. Some of these studies have suggested the existence of rare cases that conflict with the concept that disease severity is related to residual enzyme function (Holland et al., 1976; Rijksen et al., 1981; Hersh et al., 1986; Cossu et al., 2002). These discrepancies have led some investigators to question the relationship between enzyme activity and clinical phenotype. Although it is important to acknowledge such exceptional cases, it also is important to avoid placing too much weight on unconfirmed and isolated reports, especially when they involve the use of biochemical assays that may provide inaccurate results. There are multiple reports showing that the same case may have significant differences in residual enzyme activity depending on the type of biochemical assay used (Fujimoto and Seegmiller, 1970; Holland et al., 1976; Page et al., 1984; Hersh et al., 1986; Fairbanks et al., 1987; Augoustides-Savvopoulou et al., 2002; Zoref-Shani et al., 2003; Cossu et al., 2006). These exceptional cases are addressed in detail below under the section on genotype–phenotype discrepancies.
Discrepancies in genotype–enzyme–phenotype correlations
Genotype–phenotype discrepancies
As noted above, differences in the types of mutations associated with severe and mild phenotypes suggest that those that result in null enzyme function are more likely to cause the severe Lesch-Nyhan disease phenotype, whereas mutations that may permit some residual enzyme function more frequently underlie the milder Lesch-Nyhan variant phenotypes. However, there are several exceptions that are not consistent with this rule (Table 7).
For example, there are six cases in which milder Lesch-Nyhan variant phenotypes were reported with deletion mutations affecting coding regions. In principle, deletion mutations should cause complete enzyme deficiency and the severe Lesch-Nyhan disease phenotype because they result in significant disruption of protein structure, often combined with frameshift. Additional mutations expected to cause substantial disruption to protein structure and function include three Lesch-Nyhan variant cases with partial gene duplications, three with indels, one with an insertion, and one with a nonsense mutation. There also are multiple reports describing Lesch-Nyhan variant in association with either deletions or point mutations affecting intron-exon splice junctions, which are expected to result in exon skipping often combined with frame shifts.
Detailed evaluation of each of these apparent exceptions reveals several explanations. First, several of these cases were described at a very early age (Table 7). It is conceivable that these cases might have been mislabelled as Lesch-Nyhan variants because the full phenotype had not yet developed. In fact, two of these cases (Serrano et al., 2008; Sharma et al., 2012) began to exhibit self-biting a few years after the original report (personal communication). Another explanation for the apparent exceptions involves unusual molecular mechanisms that permit residual enzyme activity. For example, one case had a 13-bp deletion, which eliminated the AUG start codon, but a small amount of residual protein with functional activity was transcribed from an upstream GUG start site (Davidson et al., 1994). Another case had a deletion at the extreme C-terminus, with loss of only two amino acids, presumably allowing residual enzyme function (Igarishi et al., 1989). Three more Lesch-Nyhan variant cases with large duplications were shown to exhibit spontaneous reversions in vitro resulting in small amounts of residual HGprt enzyme activity (Yang et al., 1988; Monnat et al., 1992; Marcus et al., 1993b). Another case had insertion of three bases, resulting in the insertion of one additional amino acid without frameshift, again presumably allowing residual enzyme activity (Sege-Peterson et al., 1992). Finally, several cases with splice site mutations were shown to have residual enzyme function. The mechanism responsible for residual enzyme activity involves variations in the fidelity of the splicing mechanism, with generation of multiple messenger RNA transcripts (Sege-Peterson et al., 1992; Yamada et al., 1992; Hunter et al., 1996). In some cases, these mutations yield a small proportion of transcripts encoding the entire normal protein (Yamada et al., 2007; Torres et al., 2010). In one example, a splice site mutation resulted in the addition of 15 amino acids to the normal HGprt protein, with 7% residual enzyme activity (Gaigl et al., 2001). Thus splicing mutations can yield unpredictable results.
In summary, there are multiple reports of mild clinical phenotypes associated with mutations that predict elimination of HGprt enzyme function. At first glance, these cases seem to disprove the concept that clinical severity is determined by the amount of residual enzyme activity. However, most of these cases can be explained by an incompletely ascertained clinical phenotype or specific molecular mechanisms permitting some residual enzyme function. As a result, these apparent exceptions do not provide strong evidence against the concept that relates clinical severity to residual enzyme function.
Enzyme–phenotype discrepancies
As noted above, there are also a small number of reported cases in which the measured HGprt enzyme activity does not correlate with the clinical phenotype. Specifically, there are a few reported cases with a severe phenotype associated with relatively high residual enzyme activity (Holland et al., 1976; Rijksen et al., 1981), and a few reported cases with attenuated phenotypes associated with non-detectable enzyme activity (Hersh et al., 1986; Cossu et al., 2002). Although uncommon, these exceptions are important to address, because they seem to provide evidence against the concept that residual HGprt enzyme activity is the primary determinant of clinical severity. Careful examination of these exceptions reveals that all involved the use of biochemical assays that may provide misleading results under certain conditions.
For example, one mechanism responsible for reduced HGprt enzyme function involves altered affinities towards hypoxanthine, guanine or PRPP (McDonald and Kelley, 1971; Benke et al., 1973; Richardson et al., 1973; Rijksen et al., 1981; Wilson et al., 1983; Snyder et al., 1984; Zanic et al., 1985; Fujimori et al., 1988; Lightfoot et al., 1994; Hikita et al., 1998; Zoref-Shani et al., 2000; Fu and Jinnah, 2012). For such mutants, in vitro assays employing artificially high substrate concentrations may yield high apparent activities, while concentrations of substrates normally available in vivo may result in little or no actual activity. An example is p.D194N, which has >200-fold increase in the Km for PRPP in association with the Lesch-Nyhan disease phenotype. This mutant had almost no activity using a live lymphoblast cell assay (<0.7%), which relies on endogenous production of physiological amounts of PRPP (Wilson et al., 1986). However, the same mutant displayed normal residual activity when PRPP was saturating, because of high PRPP concentrations used to drive the reaction in vitro (Wilson and Kelley, 1984; Wilson et al., 1986). Thus the in vitro assay incorrectly predicted a mild phenotype whereas the more naturalistic assay correctly predicted the severe phenotype.
Another mechanism leading to reduced HGprt enzyme function involves reduced structural stability of the enzyme, with rapid loss of enzyme function over time or loss of enzyme function outside of the normal cellular milieu (Richardson et al., 1973; Snyder et al., 1984; Sampat et al., 2011; Fu and Jinnah, 2012). Such mutants are likely to yield little or no activity in lysate-based assays or cell-based assays where continual messenger RNA transcription does not occur (such as erythrocytes, which lack a nucleus and ongoing messenger RNA synthesis) yet significantly higher activity in cells where ongoing messenger RNA transcription and translation will generate a constant supply of newly formed enzyme (such as live fibroblasts, where synthesis of messenger RNA and new protein are ongoing). This mechanism explains several cases where no activity was found in erythrocytes, yet significant activity was found in live fibroblasts. For example, p.R48H repeatedly has been associated with relatively mild clinical phenotypes with 20–39% residual activity in live fibroblast assays (Jinnah et al., 2010; Sampat et al., 2011). However, same mutation was reported to cause non-detectable residual activity in other assays (Jinnah et al., 2010; Sampat et al., 2011). Similar mechanisms could account for the differences between in vitro assays using purified protein and assays that rely on existing HGprt levels in live cells or lysates. There are examples of several mutants (p.D44G, p.E47G, p.F74L, and p.H204D) with no activity with assays based on lysates from human cell materials, yet high activity when the mutant enzyme is expressed in vitro and tested immediately (Davidson et al., 1988; Tarle et al., 1991; Sculley et al., 1992; Jinnah et al., 2006; Fu and Jinnah, 2012).
These observations emphasize the importance of careful attention to the biochemical assay used for the occasional reports of cases in whom residual enzyme activity does not appear to correlate with the clinical phenotype. In vitro assays are prone to give artificially high residual activity when they are conducted with non-physiological substrate concentrations or artificially low residual activity if the enzyme is structurally unstable outside of the normal cellular milieu. The live cell assays may yield results that are closer to those occurring in vivo, but erythrocyte-based assays are prone to give artificially low residual activity for mutant enzymes that are structurally unstable because of the lack of ongoing synthesis of new enzyme. The live fibroblast assay may be least susceptible to inaccurate results, although the phenomenon of somatic mosaicism may yield variable results in rare instances, such as mutations capable of reversion.
Different clinical phenotypes associated with the same mutation
If residual enzyme function is the most important factor for determining the clinical phenotype, then identical HPRT1 mutations should have the same biochemical consequences for HGprt function and thereby cause the same clinical phenotype. A total of 85 mutations were reported with at least two cases from unrelated families, for a total of 272 cases with shared mutations (Table 8). The majority of recurrent mutations (93.7%) produced a similar phenotype even in unrelated patients. However, there were a few exceptions where the same mutation caused discordant phenotypes (Table 9). For all discordant phenotypes, only one case for each mutation was discordant. Discordant phenotypes were most often observed for missense mutations, splicing errors, or duplications. There also were several reports of different phenotypes in the same family presumably carrying the same mutation (Hladnik et al., 2008; Jinnah et al., 2010; Sarafoglou et al., 2010). These cases argue that factors other than HGprt activity may influence clinical outcomes.
Table 8.
Summary of HPRT1 mutations with discordant clinical phenotypes
| Type of Mutation | Concordant | Discordant | % Discordant |
|---|---|---|---|
| Missense | 116 | 8 | 6.9 |
| Splicing errors | 48 | 5 | 10.4 |
| Nonsense | 42 | 1 | 2.4 |
| Microdeletions | 26 | 0 | 0.0 |
| Microduplications | 19 | 0 | 0.0 |
| Large duplications | 3 | 2 | 66.7 |
| Indels | 2 | 0 | 0.0 |
| Total | 256 | 16 | 6.3 |
This table summarizes the numbers of cases with the same (concordant) or different (discordant) reported phenotype associated with an identical molecular mutation.
Table 9.
Specific HPRT1 mutations with discordant clinical phenotypes
| LND | LNV | Total | |
|---|---|---|---|
| Missense | |||
| c.115C>G | 1 | 1 | 2 |
| c.125T>C | 1 | 1 | 2 |
| c.134G>A | 3 | 1 | 4 |
| c.203T>C | 2 | 1 | 3 |
| c.212G>T | 1 | 3 | 4 |
| c.293A>G | 1 | 1 | 2 |
| c.486C>G | 1 | 1 | 2 |
| Splicing errors | |||
| c.27+1G>A | 1 | 1 | 2 |
| c.27+1G>T | 2 | 1 | 3 |
| c.27+5G>A | 1 | 1 | 2 |
| c.385-1G>A | 3 | 1 | 4 |
| c.402+1G>A | 1 | 1 | 2 |
| c.485+2T>C | 1 | 1 | 2 |
| Nonsense | |||
| c.508C>T | 18 | 1 | 19 |
| Microdeletion | |||
| c.213delC | 2 | 1 | 3 |
| Duplications | |||
| Exons 2-3 | 1 | 2 | 3 |
| Exons 7-8 | 1 | 1 | 2 |
This table provides a list of the cases where different (discordant) phenotypes were reported to be associated with an identical molecular mutation.
LND = Lesch-Nyhan disease; LNV = Lesch-Nyhan variant.
Several potential mechanisms may account for these discordant cases. There are both genetic and non-genetic modifiers that can affect the amount of residual HGprt enzyme activity associated with some HPRT1 mutations. As noted above, variations in the fidelity of splicing mechanisms can result in multiple messenger RNAs including some normal messenger RNA transcripts and associated enzyme activity (O'Neill et al., 1998). Since the mechanisms responsible for splicing are inherited independently from HPRT1, the amount of normal messenger RNA encoding HGprt may vary among individuals carrying the same splicing mutation. This mechanism may explain why discordant phenotypes are commonly associated with splicing mutations (Table 8). One example involves the c.485+2T>C splicing mutation, which was reported to be associated with different phenotypes in five members of one family (Hladnik et al., 2008). Only one had self-injurious behaviour and therefore had the Lesch-Nyhan disease phenotype. Two others did not have overt self-injury, but they suffered repeated injuries to the chin from falls. This observation raises questions regarding whether these injures were entirely accidental since they were not random but limited to the chin. Two more family members were reported to have HRH because of normal intelligence, but both had abnormal gaits. The gait disorder suggests these cases may be better classified as HND. This family provides an example of phenotypic variation associated with a splicing mutation, as well as the challenges of classifying a continuously graded spectrum of clinical phenotypes into distinct categories.
A related mechanism where a single mutation may variably affect enzyme activity in different cases involves non-coding mutations. In this situation, regulatory mechanisms that control messenger RNA transcription from the mutant gene or protein translation from messenger RNA with aberrant regulatory signals may operate with varying fidelity among different cases with the same mutation. The outcome could be differences in residual HGprt enzyme activity and clinical phenotype. Here, quantitative studies of messenger RNA levels or enzyme function may be useful (Nguyen et al., 2012).
Another mechanism that may alter enzyme activity independently from the associated mutation involves structural instability of the HGprt protein (Richardson et al., 1973; Snyder et al., 1984; Sampat et al., 2011; Fu and Jinnah, 2012). For unstable proteins, enzyme function depends on the balance between elimination of damaged proteins and ongoing synthesis of new functional protein. Since mechanisms of degradation and synthesis are inherited independently from HPRT1, different individuals with the same mutation may have different steady-state levels of HGprt activity. In the case of structurally unstable enzymes, environmental influences may also play a role. For example, thermal effects associated with frequent or lengthy febrile episodes may result in an increased structural damage to HGprt proteins, especially if they occur during a critical period of early brain development. This mechanism predicts a worse clinical outcome for patients who have experienced many fevers due to infections during early childhood (Sampat et al., 2011).
A fourth mechanism could involve biochemical pathways that compensate for the loss of HGprt-mediated purine recycling. Although there are no known HGprt isoenzymes that can replace its function, the products of HGprt can be produced by an alternative pathway known as the de novo synthesis pathway. This pathway is accelerated in HGprt-deficient cells, suggesting metabolic regulatory changes in synthesis that may compensate for the lack of recycling (Nyhan et al., 1965; Rosenbloom et al., 1968; Sorensen, 1970). The ability of this alternative pathway to compensate for HGprt deficiency may vary from case to case. As the mechanisms responsible for the de novo pathway are inherited independently from HPRT1, it is feasible that the degree of compensation may vary among patients inheriting the same mutation. This mechanism for explaining discordant phenotypes with the same HPRT1 mutation is theoretically attractive, but currently there are no studies comparing the extent to which the de novo pathway is influenced by different HPRT1 mutations or across different patients. Therefore, this possibility remains speculative.
These observations suggest several molecular mechanisms by which the same HPRT1 mutation may result in different levels of residual HGprt enzyme activity and thereby discordant clinical outcomes. There also are important non-molecular explanations for discordant clinical phenotypes associated with the same HPRT1 mutation. These non-biochemical mechanisms relate to limitations in the characterization of the clinical phenotype and assignment of patients to specific disease subgroups.
Beyond biochemical genetics
Many years of research in genetics and biochemistry have led to a rich understanding of how the clinical phenotype relates to HPRT1 gene mutations and associated HGprt enzyme activity. A major unsolved question is how the loss of HGprt enzyme function affects the brain to cause the neurobehavioural syndrome in Lesch-Nyhan disease and its attenuated variants. Although HGprt is expressed throughout the brain (Jinnah et al., 1992a), many of the clinical features point to prominent dysfunction of a specific region known as the basal ganglia (Visser et al., 2000). However, autopsy studies have not revealed any consistent anatomical defects anywhere in the Lesch-Nyhan disease brain, including the basal ganglia. Notably, there is no evidence for a degenerative process (Jinnah et al., 2006). There is no evidence for major developmental malformations, and the multifocal atrophy of cerebellar Purkinje neurons reported for some cases (Del Bigio and Halliday, 2007) has not been found in other cases (unpublished observations). Structurally, there are no serious defects.
However, neurochemical studies of Lesch-Nyhan disease brains collected at autopsy have revealed 60–80% loss of dopamine, a critically important neurotransmitter in the basal ganglia (Lloyd et al., 1981; Saito et al., 1999; Saito and Takashima, 2000). Profound dysfunction of dopamine neurons also has been documented in imaging studies of patients with Lesch-Nyhan disease, which show significant loss of fluoroDOPA uptake into the basal ganglia and major reductions in the binding of WIN 35,428 to dopamine transporters in the basal ganglia (Ernst et al., 1996; Wong et al., 1996). The neurochemical and imaging studies are corroborated by studies of CSF from patients with Lesch-Nyhan disease, which show reduced monoamine neurotransmitter metabolites (Silverstein et al., 1985; Edwards et al., 1986; Manzke et al., 1986; Jankovic et al., 1988). A selective disruption of basal ganglia dopamine systems also has been documented in genetically engineered mouse models that lack HGprt (Jinnah et al., 1992b, 1994, 1999; Egami et al., 2007), and even in tissue culture models involving neuron-like cells that make dopamine as a neurotransmitter (Bitler and Howard, 1986; Yeh et al., 1998; Lewers et al., 2008). The reasons for relatively prominent disruption of dopamine neurons remain unknown, but may relate to the selective vulnerability of this neuronal population to the loss of HGprt (Egami et al., 2007; Lewers et al., 2008; Ceballos-Picot et al., 2009; Guibinga et al., 2011; Kang et al., 2011). Additional work to clarify how the loss of HGprt affects these and other neurons is still needed.
Conclusions
Lesch-Nyhan disease and its attenuated variants are associated with characteristic clinical features, including overproduction of uric acid, motor and cognitive disability, and behavioural abnormalities. Although patients are divided into three distinct subgroups based on overall severity, the reality is a more continually graded spectrum, which evolves during early development. There are many different clinically relevant mutations in the HPRT1 gene, and genotype–phenotype correlations suggest that the most relevant aspect of the mutation is its influence on residual function of HGprt enzyme activity. The mutations are distributed through all functional domains of the HGprt protein. The mutations may affect residual enzyme dysfunction by disrupting the active enzyme site, by interfering with conformational changes required during the catalytic cycle, by blocking essential subunit interactions, or by reducing protein stability. There are also examples where residual enzyme activity may be influenced by factors other than the mutation itself.
These observations have direct implications for genetic testing and counselling regarding prognosis. New cases with mutations predicted to cause null enzyme activity are likely to develop the most severe clinical phenotype of Lesch-Nyhan disease, while those with mutations that permit residual enzyme activity have a higher probability of developing a milder phenotype. New cases found to have mutations already described have a high probability of developing a phenotype similar to the previously reported case. Predicting the outcome of new cases with novel mutations is more difficult, because available predictive algorithms such as SIFT and PolyPhen-2 are not always accurate. Here, biochemical tests for residual HGprt enzyme function may be better for predicting prognoses, and it is important to recognize that the live erythrocyte or fibroblast assays are more accurate than lysate-based assays, which are prone to multiple artefacts.
The basic principles of genotype–phenotype relationships associated with mutations in the HPRT1 gene provide a valuable model for understanding genotype–phenotype relationships in many other genetic disorders. The complexities associated with clinical ascertainment of the phenotype are relevant to all genetic disorders, and the mechanisms by which different mutations can alter protein function are likely to occur with many other gene products. Although Lesch-Nyhan disease often is considered a relatively ‘simple’ disorder because it is monogenic and inherited in an X-linked recessive manner permitting evaluation of single allelic defects, the unexpected and sometimes unusual mechanisms that influence genotype–phenotype relationships provide useful principles for other more complex disorders.
Funding
This work was supported in part by the Lesch-Nyhan Syndrome Children’s Research Foundation, Lesch-Nyhan Action and Malaury Associations, the Harold A. and Madeline R. Jacobs Fund at the San Diego Foundation, the Fondo de Investigaciones Sanitarias (Health Research Fund, FIS) 11/00598 from Spain, a Gout Research Foundation Grant of Japan, and grants from the National Institutes of Health of the USA (HD053312 and DK082840).
Glossary
Abbreviations
- HGprt
hypoxanthine-guanine phosphoribosyltransferase
- HND
HGprt-related neurological dysfunction
- HRH
HGprt-related hyperuricaemia
- PRPP
phosphoribosylpyrophosphate
References
- Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–9. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson LT, Ernst M. Self-injury in Lesch-Nyhan disease. J Autism Dev Disord. 1994;24:67–81. doi: 10.1007/BF02172213. [DOI] [PubMed] [Google Scholar]
- Anderson LT, Ernst M, Davis SV. Cognitive abilities of patients with Lesch-Nyhan disease. J Autism Dev Disord. 1992;22:189–203. doi: 10.1007/BF01058150. [DOI] [PubMed] [Google Scholar]
- Aral B, de Saint B, Al-Garawi S, Kamoun P, Ceballos-Picot I. Novel nonsense mutation in the hypoxanthine guanine phosphoribosyltransferase gene and nonrandom X-inactivation causing Lesch-Nyhan syndrome in a female patient. Hum Mutat. 1996;7:52–8. doi: 10.1002/(SICI)1098-1004(1996)7:1<52::AID-HUMU7>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- Augoustides-Savvopoulou P, Papachristou F, Fairbanks LD, Dimitrakopoulos K, Marinaki AM, Simmonds HA. Partial hypoxanthine-guanine phosphoribosyltransferase deficiency as the unsuspected cause of renal disease spanning three generations: a cautionary tale. Pediatr. 2002;109:E17. doi: 10.1542/peds.109.1.e17. [DOI] [PubMed] [Google Scholar]
- Bakay B, Nissinen E, Sweetman L, Francke U, Nyhan WL. Utilization of purines by an HPRT variant in an intelligent, nonmutilative patient with features of the Lesch-Nyhan syndrome. Pediatr Res. 1979;13:1365–70. doi: 10.1203/00006450-197912000-00013. [DOI] [PubMed] [Google Scholar]
- Balendiran GK, Molina JA, Xu Y, Torres-Martinez J, Stevens R, Focia PJ, et al. Ternary complex structure of human HGPRTase, PRPP, Mg2+, and the inhibitor HPP reveals the involvement of the flexible loop in substrate binding. Protien Sci. 1999;8:1023–31. doi: 10.1110/ps.8.5.1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benke PJ, Herrick N, Hebert A. Hypoxanthine-guanine phosphoribosyltransferase variant associated with accelerated purine synthesis. J Clin Invest. 1973;52:2234–40. doi: 10.1172/JCI107409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertelli M, Randi D, Micheli V, Gallo S, Andrighetto G, Parmigiani P, et al. Molecular basis of hypoxanthine-guanine phosphoribosyltransferase deficiency in Italian Lesch-Nyhan patients: identification of nine novel mutations. J Inherit Metab Dis. 2004;27:767–73. doi: 10.1023/B:BOLI.0000045799.78633.23. [DOI] [PubMed] [Google Scholar]
- Bitler CM, Howard BD. Dopamine metabolism in hypoxanthine-guanine phosphoribosyltransferase-deficient variants of PC12 cells. J Neurochem. 1986;47:107–12. doi: 10.1111/j.1471-4159.1986.tb02837.x. [DOI] [PubMed] [Google Scholar]
- Burgemeister R, Gutensohn W, Van den Berghe G, Jaeken J. Genetic and clinical heterogeneity in hypoxanthine phosphoribosyltransferase deficiencies. Adv Exp Med Biol. 1994;370:331–5. doi: 10.1007/978-1-4615-2584-4_71. [DOI] [PubMed] [Google Scholar]
- Ceballos-Picot I, Mockel L, Potier MC, Dauphinot L, Shirley TL, Torero-Ibad R, et al. Hypoxanthine-guanine phosphoribosyl transferase regulates early developmental programming of dopamine neurons: implications for Lesch-Nyhan disease pathogenesis. Hum Mol Genet. 2009;18:2317–27. doi: 10.1093/hmg/ddp164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherian S, Crompton CH. Partial hypoxanthine-guanine phosphoribosyltransferase deficiency presenting as acute renal failure. Pediatr Nephrol. 2005;20:1811–3. doi: 10.1007/s00467-005-2065-8. [DOI] [PubMed] [Google Scholar]
- Corrigan A, Arenas M, Escuredo E, Fairbanks L, Marinaki A. HPRT deficiency: identification of twenty-four novel variants including an unusual deep intronic mutation. Nucleosides Nucleotides Nucleic Acids. 2011;30:1260–5. doi: 10.1080/15257770.2011.590172. [DOI] [PubMed] [Google Scholar]
- Cossu A, Micheli V, Jacomelli G, Carcassi A. Kelley-Seegmiller syndrome in a patient with complete hypoxanthine-guanine phosphoribosyltransferase deficiency. Clin Exp Rhematol. 2002;19:851–3. [PubMed] [Google Scholar]
- Cossu A, Orru S, Jacomelli G, Carcassi C, Contu L, Sestini S, et al. HPRT-Sardinia: a new point mutation causing HPRT deficiency without Lesch-Nyhan disease. Biochim Biophys Acta. 2006;1762:29–33. doi: 10.1016/j.bbadis.2005.03.012. [DOI] [PubMed] [Google Scholar]
- Craig SP, III, Eakin AE. Purine phosphoribosyltransferases. J Biol Chem. 2000;275:20231–4. doi: 10.1074/jbc.R000002200. [DOI] [PubMed] [Google Scholar]
- Davidson BL, Golovoy N, Roessler BJ. A 13 base pair deletion in exon 1 of HPRTIllinois forms a functional GUG initiation codon. Hum Genet. 1994;93:300–4. doi: 10.1007/BF00212027. [DOI] [PubMed] [Google Scholar]
- Davidson BL, Pashmforoush M, Kelley WN, Palella TD. Genetic basis of hypoxanthine guanine phosphoribosyltransferase deficiency in a patient with the Lesch-Nyhan syndrome (HPRTFlint) Gene. 1988;63:331–6. doi: 10.1016/0378-1119(88)90536-7. [DOI] [PubMed] [Google Scholar]
- Davidson BL, Tarle SA, Van Antwerp M, Gibbs DA, Watts RW, Kelley WN, et al. Identification of 17 independent mutations responsible for human hypoxanthine-guanine phsophoribosyltransferase (HPRT) deficiency. Am J Hum Genet. 1991;48:951–8. [PMC free article] [PubMed] [Google Scholar]
- Dawson PA, Gordon RB, Keough DT, Emmerson BT. Normal HPRT coding region in a male with gout due to HPRT deficiency. Mol Genet Metab. 2005;85:78–80. doi: 10.1016/j.ymgme.2005.01.005. [DOI] [PubMed] [Google Scholar]
- de Gemmis P, Anesi L, Lorenzetto E, Gioachini I, Fortunati E, Zandona G, et al. Analysis of the HPRT1 gene in 35 Italian Lesch-Nyhan families: 45 patients and 77 potential female carriers. Mutat Res. 2010;692:1–5. doi: 10.1016/j.mrfmmm.2010.07.003. [DOI] [PubMed] [Google Scholar]
- De Gregorio L, Jinnah HA, Nyhan WL, Trombley L, O'Neill JP. Lesch-Nyhan disease in one member of a female monozygotic twin pair heterozygous for a mutation in HPRT. Mol Genet Metab. 2005;85:70–7. doi: 10.1016/j.ymgme.2004.11.009. [DOI] [PubMed] [Google Scholar]
- De Gregorio L, Nyhan WL, Serafin E, Chanoles NA. An unexpected affected female patient in a classical Lesch-Nyhan family. Mol Genet Metab. 2000;69:263–8. doi: 10.1006/mgme.2000.2967. [DOI] [PubMed] [Google Scholar]
- Del Bigio MR, Halliday WC. Multifocal atrophy of cerebellar internal granular neurons in Lesch-Nyhan disease: case reports and review. J Neuropathol Exp Neurol. 2007;66:346–53. doi: 10.1097/nen.0b013e3180515319. [DOI] [PubMed] [Google Scholar]
- Duan J, Nilsson L, Lambert B. Structural and functional analysis of mutations at the human hypoxanthine phosporibosyl transferase (HPRT1) locus. Hum Mutat. 2004;23:599–611. doi: 10.1002/humu.20047. [DOI] [PubMed] [Google Scholar]
- Ea HK, Bardin T, Jinnah HA, Aral B, Liote F, Ceballos-Picot I. Severe gouty arthritis and mild neurological symptoms due to a new variant of the hypoxanthine-guanine phosphoriboysltransferase (F199C) Arthritis Rheum. 2009;60:2201–4. doi: 10.1002/art.24617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eads JC, Scapin G, Xu Y, Grubmeyer C, Sacchettini JC. The crystal structure of human hypoxanthine-guanine phosphoribosyltransferase with bound GMP. Cell. 1994;78:325–34. doi: 10.1016/0092-8674(94)90301-8. [DOI] [PubMed] [Google Scholar]
- Edwards NL, Silverstein FS, Johnston MV. Purine and monoamine metabolites in cerebrospinal fluid. Adv Exp Med Biol. 1986;195 (Pt B):53–6. doi: 10.1007/978-1-4684-1248-2_9. [DOI] [PubMed] [Google Scholar]
- Egami K, Yitta S, Kasim S, Lewers JC, Roberts RC, Lehar M, et al. Basal ganglia dopamine loss due to defect in purine recycling. Neurobiol Dis. 2007;26:396–407. doi: 10.1016/j.nbd.2007.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emmerson BT, Thompson L. The spectrum of hypoxanthine-guanine phosphoribosyltranferase deficiency. Quart J Med. 1973;166:423–40. [PubMed] [Google Scholar]
- Erhard U, Herkenrath P, Benz-Bohm G, Querfeld U. Lesch-Nyhan syndrome: clinical diagnosis and confirmation by biochemical and genetic methods. Pediatr Nephrol. 1997;11:124–5. [PubMed] [Google Scholar]
- Ernst M, Zametkin AJ, Matochik JA, Pascualvaca D, Jons PH, Hardy K, et al. Presynaptic dopaminergic deficits in Lesch-Nyhan disease. N Engl J Med. 1996;334:1568–72. doi: 10.1056/NEJM199606133342403. [DOI] [PubMed] [Google Scholar]
- Fairbanks LD, Simmonds HA, Webster DR. Use of intact erythrocytes in the diagnosis of inherited purine and pyrimidine disorders. J Inherit Metab Dis. 1987;10:174–86. doi: 10.1007/BF01800045. [DOI] [PubMed] [Google Scholar]
- Fattal A, Spirer Z, Zoref-Shani E, Sperling O. Lesch-Nyhan syndrome: biochemical characterization of a case with attenuated behavioral manifestation. Enzyme. 1984;31:55–60. [PubMed] [Google Scholar]
- Fox IH, Kelley WN. Phosphoribosylpyrophosphate in man: biochemical and clinical significance. Ann Int Med. 1971;74:424–33. doi: 10.7326/0003-4819-74-3-424. [DOI] [PubMed] [Google Scholar]
- Fu R, Jinnah HA. Different phenotypes among Lesch-Nyhan variants: clinical reality or limitation of ascertainment? Arch Neurol. 2011;68:270. doi: 10.1001/archneurol.2010.360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu R, Jinnah HA. Genotype-phenotype correlations in Lesch-Nyhan disease: moving beyond the gene. J Biol Chem. 2012;287:2997–3008. doi: 10.1074/jbc.M111.317701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimori S, Hidaka Y, Davidson BL, Palella TD, Kelley WN. Identification of a single nucleotide change in a mutant gene for hypoxanthine-guanine phosphoribosyltransferase (HPRTAnn Arbor) Hum Genet. 1988;79:39–43. doi: 10.1007/BF00291707. [DOI] [PubMed] [Google Scholar]
- Fujimori S, Sakuma R, Yamaoka N, Hakoda M, Yamanaka H, Kamatani N. An asymptomatic germline missense base substitution in the hypoxanthine phosphoribosyltransferase (HPRT) gene that reduces the amount of enzyme in humans. Hum Genet. 1997;99:8–10. doi: 10.1007/s004390050300. [DOI] [PubMed] [Google Scholar]
- Fujimori S, Tagaya T, Yamaoka N, Kamatani N, Akaoka I. Molecular analysis of hypoxanthine-guanine phosphoribosyltransferase deficiency in Japanese patients. Adv Exp Med Biol. 1991;309B:101–4. doi: 10.1007/978-1-4615-7703-4_22. [DOI] [PubMed] [Google Scholar]
- Fujimori S, Tagaya T, Yamaoka N, Saito H, Kamatani N, Akaoka I. Direct evidence for a hot spot of germline mutation at HPRT locus. Adv Exp Med Biol. 1994;370:679–82. doi: 10.1007/978-1-4615-2584-4_141. [DOI] [PubMed] [Google Scholar]
- Fujimoto WY, Seegmiller JE. Hypoxanthine-guanine phosphoribosyltransferase deficiency: activity in normal, mutant, and heterozygote cultured human skin fibroblasts. Proc Natl Acad Sci USA. 1970;65:577–84. doi: 10.1073/pnas.65.3.577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaigl Z, Shin YS, Gathof BS. Novel splice mutation in a patient with partial hypoxanthine-guanine phosphoribosyltransferase (HPRT) deficiency. J Inherit Metab Dis. 2001;24(Supp 1):142. [Google Scholar]
- Garcia MG, Torres RJ, Prior C, Puig JG. Normal HPRT coding region in complete and partial HPRT deficiency. Mol Genet Metab. 2008;94:167–72. doi: 10.1016/j.ymgme.2008.01.006. [DOI] [PubMed] [Google Scholar]
- Gibbs RA, Caskey CT. Identification and localization of mutations at the Lesch-Nyhan locus by ribonuclease A cleavage. Science. 1987;236:303–5. doi: 10.1126/science.3563511. [DOI] [PubMed] [Google Scholar]
- Gibbs RA, Nguyen PN, McBride LJ, Koepf SM, Caskey CT. Identification of mutations leading to the Lesch-Nyhan syndrome by automated direct DNA sequencing of in vitro amplified cDNA. Proc Natl Acad Sci USA. 1989;86:1919–23. doi: 10.1073/pnas.86.6.1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert RD, Wiggelinkhuizen J, Harley EH, Marinaki A. The Lesch-Nyhan syndrome–an under-recognised condition in South Africa? A case report. S Afr Med J. 1992;81:375–7. [PubMed] [Google Scholar]
- Gottlieb RP, Koppel MM, Nyhan WL, Bakay B, Nissinen E, Borden M, et al. Hyperuricaemia and choreoathetosis in a child without mental retardation or self-mutilation - a new HPRT variant. J Inherit Metab Dis. 1982;5:183–6. doi: 10.1007/BF02179135. [DOI] [PubMed] [Google Scholar]
- Guibinga GH, Hrustanovic G, Bouic K, Jinnah HA, Friedmann T. MicroRNA-mediated dysregulation of neural developmental genes in HPRT deficiency: clues for Lesch-Nyhan disease? Hum Mol Genet. 2011;21:609–22. doi: 10.1093/hmg/ddr495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara K, Kashiwamata S, Ogasawara N, Ohishi H, Natsume R, Yamanaka T, et al. A female case of the Lesch-Nyhan syndrome. Tohoku J Exp Med. 1982;137:275–82. doi: 10.1620/tjem.137.275. [DOI] [PubMed] [Google Scholar]
- Hersh JH, Page T, Hand ME, Seegmiller JE, Nyhan WL, Weisskopf B. Clinical correlations in partial hypoxanthine guanine phosphoribosyltransferase deficiency. Pediatr Neurol. 1986;2:302–4. doi: 10.1016/0887-8994(86)90025-1. [DOI] [PubMed] [Google Scholar]
- Hicks S, Wheeler DA, Plon SE, Kimmel M. Prediction of missense mutation functionality depends on both the algorithm and sequence alignment employed. Hum Mutat. 2011;32:661–8. doi: 10.1002/humu.21490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hikita M, Hosoya T, Ichida K, Okabe H, Saji M, Ohno I, et al. Partial deficiency of hypoxanthine-guanine-phosphoribosyltransferase manifesting as acute renal damage. Int Med. 1998;37:945–9. doi: 10.2169/internalmedicine.37.945. [DOI] [PubMed] [Google Scholar]
- Hladnik U, Nyhan WL, Bertelli M. Variable expression of HPRT deficiency in 5 members of a family with the same mutation. Arch Neurol. 2008;65:1240–3. doi: 10.1001/archneur.65.9.1240. [DOI] [PubMed] [Google Scholar]
- Holden JA, Kelley WN. Human HPRT: evidence for tetrameric structure. J Biol Chem. 1978;253:4459–64. [PubMed] [Google Scholar]
- Holland MJ, DiLorenzo AM, Dancis J, Balis ME, Yu TF, Cox RP. Hypoxanthine phosphoribosyltransferase activity in intact fibroblasts from patients with X-linked hyperuricemia. J Clin Invest. 1976;57:1600–5. doi: 10.1172/JCI108430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooft C, Van Nevel C, De Schaepdryver AF. Hyperuricosuric encephalopathy without hyperuricaemia. Arch Dis Child. 1968;43:734–7. doi: 10.1136/adc.43.232.734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter TC, Melancon SB, Dallaire L, Taft S, Skopek TR, Albertini RJ, et al. Germinal HPRT splice donor site mutation results in multiple RNA splicing products in T-lymphocyte cultures. Somat Cell Mol Genet. 1996;22:145–50. doi: 10.1007/BF02369904. [DOI] [PubMed] [Google Scholar]
- Igarishi T, Minami M, Nishida Y. Molecular analysis of hypoxanthine-guanine phosphoribosyltransferase mutation in five unrelated Japanese patients. Acta Paediatr Jpn. 1989;31:303–13. doi: 10.1111/j.1442-200x.1989.tb01306.x. [DOI] [PubMed] [Google Scholar]
- Inokuchi T, Moriwaki Y, Takahashi S, Tsutsumi Z, Ka T, Yamamoto A, et al. Identification of a new point mutation in hypoxanthine phosphoribosyl transferase responsible for hyperuricemia in a female patient. Metabolism. 2004;53:1500–2. doi: 10.1016/j.metabol.2004.04.016. [DOI] [PubMed] [Google Scholar]
- Itiaba K, Melancon SB, Dallaire L, Crawhall JC. Adenine phosphoribosyl transferase deficiency in association with sub-normal hypoxanthine phophoribosyl transferase in families of Lesch-Nyhan patients. Biochem Med. 1978;19:252–9. doi: 10.1016/0006-2944(78)90027-3. [DOI] [PubMed] [Google Scholar]
- Jankovic J, Caskey CT, Stout JT, Butler IJ. Lesch-Nyhan syndrome: a study of motor behavior and cerebrospinal fluid neurotransmitters. Ann Neurol. 1988;23:466–9. doi: 10.1002/ana.410230507. [DOI] [PubMed] [Google Scholar]
- Jenkins EA, Hallett RJ, Hull RG. Lesch-Nyhan syndrome presenting with renal insufficiency in infancy and transient neonatal hypothyroidism. Br J Rheumatol. 1994;33:392–6. doi: 10.1093/rheumatology/33.4.392. [DOI] [PubMed] [Google Scholar]
- Jinnah HA, Ceballos-Picot I, Torres RJ, Visser JE, Schretlen D, Verdu A, et al. Attenuated variants of Lesch-Nyhan disease. Brain. 2010;133:671–89. doi: 10.1093/brain/awq013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinnah HA, DeGregorio L, Harris JC, Nyhan WL, O'Neill JP. The spectrum of inherited mutations causing HPRT deficiency: 75 new cases and a review of 196 previously reported cases. Mutat Res. 2000;463:309–26. doi: 10.1016/s1383-5742(00)00052-1. [DOI] [PubMed] [Google Scholar]
- Jinnah HA, Harris JC, Nyhan WL, O'Neill JP. The spectrum of mutations causing HPRT deficiency: an update. Nucleosides Nucleotides Nucleic Acids. 2004;23:1153–60. doi: 10.1081/NCN-200027400. [DOI] [PubMed] [Google Scholar]
- Jinnah HA, Hess EJ, Wilson MC, Gage FH, Friedmann T. Localization of hypoxanthine-guanine phosphoribosyltransferase mRNA in the mouse brain by in situ hybridization. Mol Cell Neurosci. 1992a;3:64–78. doi: 10.1016/1044-7431(92)90010-y. [DOI] [PubMed] [Google Scholar]
- Jinnah HA, Jones MD, Wojcik BE, Rothstein JD, Hess EJ, Friedmann T, et al. Influence of age and strain on striatal dopamine loss in a genetic mouse model of Lesch-Nyhan disease. J Neurochem. 1999;72:225–9. doi: 10.1046/j.1471-4159.1999.0720225.x. [DOI] [PubMed] [Google Scholar]
- Jinnah HA, Langlais PJ, Friedmann T. Functional analysis of brain dopamine systems in a genetic mouse model of Lesch-Nyhan syndrome. J Pharmacol Exp Ther. 1992b;263:596–607. [PubMed] [Google Scholar]
- Jinnah HA, Visser JE, Harris JC, Verdu A, Larovere L, Ceballos-Picot I, et al. Delineation of the motor disorder of Lesch-Nyhan disease. Brain. 2006;129:1201–17. doi: 10.1093/brain/awl056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinnah HA, Wojcik BE, Hunt MA, Narang N, Lee KY, Goldstein M, et al. Dopamine deficiency in a genetic mouse model of Lesch-Nyhan disease. J Neurosci. 1994;14:1164–75. doi: 10.1523/JNEUROSCI.14-03-01164.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson GG, Eisenberg LR, Migeon BR. Human and mouse hypoxanthine-guanine phosphoribosyltransferase: dimers and tetramers. Science. 1979;203:174–6. doi: 10.1126/science.569362. [DOI] [PubMed] [Google Scholar]
- Johnson GG, Ramage AL, Littlefield JW, Kazazian HH., Jr Hypoxathine-guanine phsophoribosyltransferase in human erythroid cells: posttranslational modification. Biochemistry. 1982;21:960–6. doi: 10.1021/bi00534a022. [DOI] [PubMed] [Google Scholar]
- Jolly DJ, Okayama H, Berg P, Esty AC, Filpula D, Bohlen P, et al. Isolation and characterization of a full-length expressible cDNA for human hypoxanthine phosphoribosyltransferase. Proc Natl Acad Sci USA. 1983;80:477–81. doi: 10.1073/pnas.80.2.477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurecka A, Popowska E, Tylki-Szymanska A, Kubalska J, Ciara E, Krumina Z, et al. Hypoxanthine-guanine phosphoribosylotransferase deficiency-The spectrum of Polish mutations. J Inherit Metab Dis. 2008;31(Suppl 2):S447–51. doi: 10.1007/s10545-008-1013-8. [DOI] [PubMed] [Google Scholar]
- Kang TH, Guibinga GH, Jinnah HA, Friedmann T. HPRT deficiency coordinately dysregulates canonical Wnt and presenilin-1 signaling: a neuro-developmental regulatory role for a housekeeping gene? PLoS One. 2011;6:e16572. doi: 10.1371/journal.pone.0016572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kassimatis TI, Simmonds HA, Goudas PC, AMarinaki AM, Fairbanks LD, Diamandopoulos AA. HPRT deficiency as the cause of ESRD in a 24-year-old patient: a very rare presentaion of the disorder. J Nephrol. 2005;18:447–51. [PubMed] [Google Scholar]
- Keebaugh AC, Sullivan RT, Thomas JW. Gene duplication and inactivation in the HPRT gene family. Genomics. 2006;89:134–42. doi: 10.1016/j.ygeno.2006.07.003. [DOI] [PubMed] [Google Scholar]
- Keough DT, Brereton IM, de Jersey J, Guddat LW. The crystal structure of free human hypoxanthine-guanine phosphoribosyltransferase reveals extensive conformational plasticity throughout the catalytic cycle. J Mol Biol. 2005;351:170–81. doi: 10.1016/j.jmb.2005.05.061. [DOI] [PubMed] [Google Scholar]
- Keough DT, Emmerson BT, de Jersey J. Localization of the 5-phospho-a-D-ribosyl-1-pyrophosphate binding site of human hypoxanthine-guanine phosphoribosyltransferase. Biochim Biophys Acta. 1991;1096:95–100. doi: 10.1016/0925-4439(91)90045-b. [DOI] [PubMed] [Google Scholar]
- Kim KJ, Yamada Y, Suzumori K, Choi Y, Yang SW, Cheong HI, et al. Molecular analysis of hypoxanthine guanine phosphoribosyltransferase (HPRT) gene in five Korean families with Lesch-Nyhan syndrome. J.Korean Med Sci. 1997;12:332–9. doi: 10.3346/jkms.1997.12.4.332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SJ, Sohn YB, Park SW, Jin DK, Paik KH. Chronic peritoneal dialysis in a 4-year-old boy with Lesch-Nyhan disease. Pediatr Nephrol. 2009;24:1089–90. doi: 10.1007/s00467-008-1081-x. [DOI] [PubMed] [Google Scholar]
- Lesch M, Nyhan WL. A familial disorder of uric acid metabolism and central nervous system function. Am J Med. 1964;36:561–70. doi: 10.1016/0002-9343(64)90104-4. [DOI] [PubMed] [Google Scholar]
- Lewers JC, Ceballos-Picot I, Shirley TL, Mockel L, Egami K, Jinnah HA. Consequences of impaired purine recycling in dopaminergic neurons. Neuroscience. 2008;152:761–72. doi: 10.1016/j.neuroscience.2007.10.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lightfoot T, Lewkonia RM, Snyder FF. Sequence, expression, and characterization of HPRTMoose Jaw: a point mutation resulting in cooperativity and decreased substrate affinities. Hum Mol Genet. 1994;3:1377–81. doi: 10.1093/hmg/3.8.1377. [DOI] [PubMed] [Google Scholar]
- Lloyd KG, Hornykiewicz O, Davidson L, Shannak K, Farley I, Goldstein M, et al. Biochemical evidence of dysfunction of brain neurotransmitters in the Lesch-Nyhan syndrome. N Engl J Med. 1981;305:1106–11. doi: 10.1056/NEJM198111053051902. [DOI] [PubMed] [Google Scholar]
- Lorentz WB, Burton BK, Trillo A, Browning MC. Failure to thrive, hyperuricemia, and renal insufficiency in early infancy secondary to partial hypoxanthine-guanine phosphoribosyl transferase deficiency. J Pediatr. 1984;104:94–6. doi: 10.1016/s0022-3476(84)80600-9. [DOI] [PubMed] [Google Scholar]
- Ludman CN, Dicks-Mireaux C, Saunders AJ. Renal ultrasonographic appearances at presentation in an infant with Lesch-Nyhan syndrome. Br J Radiol. 1992;65:724–5. doi: 10.1259/0007-1285-65-776-724. [DOI] [PubMed] [Google Scholar]
- Mak BS, Shi CS, Tsai CR, Lee WJ, Lin HY. New mutations of the HPRT gene in Lesch-Nyhan syndrome. Pediatr Neurol. 2000;23:332–5. doi: 10.1016/s0887-8994(00)00199-5. [DOI] [PubMed] [Google Scholar]
- Manzke H, Gustmann H, Koke HB, Nyhan WL. Hypoxanthine and tetrahydrobiopterin treatment of a patient with features of the Lesch-Nyhan syndrome. Adv Exp Med Biol. 1986;195(Pt A):197–204. doi: 10.1007/978-1-4684-5104-7_31. [DOI] [PubMed] [Google Scholar]
- Marcus S, Christensen E, Malm G. Molecular analysis of the mutations in five unrelated patients with the Lesch Nyhan syndrome. Hum Mutat. 1993a;2:473–7. doi: 10.1002/humu.1380020608. [DOI] [PubMed] [Google Scholar]
- Marcus S, Hellgren D, Lambert B, Fallstrom SP, Wahlstrom J. Duplication in the hypoxanthine phosphoribosyl-transferase gene caused by Alu-Alu recombination in a patient with Lesch-Nyhan syndrome. Hum Genet. 1993b;90:477–82. doi: 10.1007/BF00217444. [DOI] [PubMed] [Google Scholar]
- Marmattom BV. Self-mutilation in the Lesch-Nyhan syndrome. Neurology. 2005;65:25. doi: 10.1212/01.wnl.0000184611.55051.f3. [DOI] [PubMed] [Google Scholar]
- Matthews WS, Solan A, Barabas G. Cognitive functioning in Lesch-Nyhan syndrome. Dev Med Child Neurol. 1995;37:715–22. doi: 10.1111/j.1469-8749.1995.tb15017.x. [DOI] [PubMed] [Google Scholar]
- McDonald JA, Kelley WN. Lesch-Nyhan syndrome: altered kinetic properties of mutant enzyme. Science. 1971;171:689–90. doi: 10.1126/science.171.3972.689. [DOI] [PubMed] [Google Scholar]
- Melton DW. HPRT gene organization and expression. Oxf Surv Euk Genes. 1987;4:35–75. [PubMed] [Google Scholar]
- Mitchell G, McInnes RR. Differential diagnosis of cerebral palsy: Lesch-Nyhan syndrome without self-mutilation. Can Med Assoc J. 1984;130:1323–4. [PMC free article] [PubMed] [Google Scholar]
- Mizunuma M, Yamada Y, Yamada K, Sonta S, Wakamatsu N, Kaneko K, et al. Disruption of the hypoxanthine-guanine phosphoribosyl-transferase gene caused by a translocation in a patient with Lesch-Nyhan syndrome. Nucleosides Nucleotides Nucleic Acids. 2004;23:1173–6. doi: 10.1081/NCN-200027441. [DOI] [PubMed] [Google Scholar]
- Monnat RJ, Jr, Chiaverotti TA, Hackmann AF, Maresh GA. Molecular structure and genetic stability of human hypoxanthine phosphoribosyltransferase (HPRT) gene duplications. Genomics. 1992;13:788–96. doi: 10.1016/0888-7543(92)90154-k. [DOI] [PubMed] [Google Scholar]
- Musick WD. Structural features of the phosphoribosyltransferases and their relationship to the human deficiency disorders of purine and pyrimidine metabolism. CRC Crit Rev Biochem. 1981;11:1–34. doi: 10.3109/10409238109108698. [DOI] [PubMed] [Google Scholar]
- Nguyen KV, Naviaux RK, Paik KK, Nyhan WL. Lesch-Nyhan syndrome: mRNA expression of HPRT in patients with enzyme proven deficiency of HPRT and normal HPRT coding region of DNA. Mol Genet Metab. 2012;106:498–501. doi: 10.1016/j.ymgme.2012.06.003. [DOI] [PubMed] [Google Scholar]
- Nyhan WL. Behavior in the Lesch-Nyhan syndrome. J Autism Child Schizophren. 1976;6:235–52. doi: 10.1007/BF01543464. [DOI] [PubMed] [Google Scholar]
- Nyhan WL, Oliver WJ, Lesch M. A familial disorder or uric acid metabolism and central nervous system function II. J Pediatr. 1965;67:257–63. doi: 10.1016/0002-9343(64)90104-4. [DOI] [PubMed] [Google Scholar]
- O'Neill JP, Finette BA. Transition mutations at CpG dinucleotides are the most frequent in vivo spontaneous single-base substitution mutation in the human HPRT gene. Environ Molec Mutagen. 1998;32:188–91. doi: 10.1002/(sici)1098-2280(1998)32:2<188::aid-em16>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- O'Neill JP, Rogan PK, Cariello N, Nicklas JA. Mutations that alter RNA splicing of the human HPRT gene: a review of the spectrum. Mutat Res. 1998;411:179–214. doi: 10.1016/s1383-5742(98)00013-1. [DOI] [PubMed] [Google Scholar]
- Ogasawara N, Stout JT, Goto H, Sonta SI, Matsumoto A, Caskey CT. Molecular anaylsis of a female Lesch-Nyhan patient. J Clin Invest. 1989;4:1024–7. doi: 10.1172/JCI114224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page T, Bakay B, Nissinen E, Nyhan WL. Hypoxanthine-guanine phosphoribosyltranferase variants: correlation of clinical phenotype with enzyme activity. J Inherit Metab Dis. 1981;4:203–6. doi: 10.1007/BF02263652. [DOI] [PubMed] [Google Scholar]
- Page T, Bakay B, Nyhan WL. Kinetic studies of hypoxanthine-guanine phosphoribosyltransferase in intact cells. Adv Exp Med Biol. 1984;165(Pt B):27–31. doi: 10.1007/978-1-4757-0390-0_6. [DOI] [PubMed] [Google Scholar]
- Page T, Nyhan WL. The spectrum of HPRT deficiency: an update. Adv Exp Med Biol. 1989;253A:129–133. doi: 10.1007/978-1-4684-5673-8_20. [DOI] [PubMed] [Google Scholar]
- Patel PE, Nussbaum RL, Framson PE, Ledbetter DH, Caskey CT, Chinault AC. Organization of the HPRT gene and related sequences in the human genome. Somat Cell Mol Genet. 1984;10:483–93. doi: 10.1007/BF01534853. [DOI] [PubMed] [Google Scholar]
- Puig JG, Torres RJ, Mateos FA, Ramos TH, Arcas JM, Buno AS, et al. The spectrum of hypoxanthine-guanine phosphoribosyltransferase deficiency: clinical experience based on 22 patients from 18 Spanish families. Medicine. 2001;80:102–12. doi: 10.1097/00005792-200103000-00003. [DOI] [PubMed] [Google Scholar]
- Richardson BJ, Ryckman DL, Komarnicki LM, Hamerton JL. Heterogeneity in the biochemical characteristics of red blood cell hypoxanthine-guanine phosphoribosyl transferase from two unrelated patients with the Lesch-Nyhan syndrome. Biochem Genet. 1973;9:197–202. doi: 10.1007/BF00487450. [DOI] [PubMed] [Google Scholar]
- Rijksen G, Staal GE, van der Vlist MJ, Beemer FA, Troost J, Gutensohn W, et al. Partial hypoxanthine-guanine phosphoribosyl transferase deficiency with full expression of the Lesch-Nyhan syndrome. Hum Genet. 1981;57:39–47. doi: 10.1007/BF00271165. [DOI] [PubMed] [Google Scholar]
- Rinat C, Zoref-Shani E, Ben-Neriah Z, Bromberg Y, Becker-Cohen R, Feinstein S, et al. Molecular, biochemical, and genetic characterization of a female patient with Lesch-Nyhan disease. Mol Genet Metab. 2005;87:249–52. doi: 10.1016/j.ymgme.2005.09.025. [DOI] [PubMed] [Google Scholar]
- Robey KL, Reck JF, Giacomini KD, Barabas G, Eddey GE. Modes and patterns of self-mutilation in persons with Lesch-Nyhan disease. Dev Med Child Neurol. 2003;45:167–71. doi: 10.1017/s001216220300032x. [DOI] [PubMed] [Google Scholar]
- Rosenbloom FM, Henderson JF, Caldwell IC, Kelley WN, Seegmiller JE. Biochemical bases of accelerated purine biosynthesis de novo in human fibroblasts lacking hypoxanthine-guanine phosphoribosyltransferase. J Biol Chem. 1968;243:1166–73. [PubMed] [Google Scholar]
- Saito Y, Ito M, Hanaoka S, Ohama E, Akaboshi S, Takashima S. Dopamine receptor upregulation in Lesch-Nyhan syndrome: a postmortem study. Neuropediatrics. 1999;30:66–71. doi: 10.1055/s-2007-973462. [DOI] [PubMed] [Google Scholar]
- Saito Y, Takashima S. Neurotransmitter changes in the pathophysiology of Lesch-Nyhan syndrome. Brain Dev. 2000;22(Suppl 1):S122–31. doi: 10.1016/s0387-7604(00)00143-1. [DOI] [PubMed] [Google Scholar]
- Salerno C, Giacomello A. Human hypoxanthine-guanine phosphoribosyltransferase: IMP-GMP exchange stoichiometry and steady state kinetics of the reaction. J Biol Chem. 1979;254:10232–6. [PubMed] [Google Scholar]
- Sampat R, Fu R, Larovere LE, Torres RJ, Ceballos-Picot I, Fischbach M, et al. Mechanisms for phenotypic variation in Lesch-Nyhan disease and its variants. Hum Genet. 2011;129:71–8. doi: 10.1007/s00439-010-0901-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarafoglou K, Grosse-Redlinger K, Boys CJ, Charnas L, Otten N, Broock R, et al. Lesch-Nyhan variant syndrome: variable presentation in 3 affected family members. Arch Neurol. 2010;67:761–4. doi: 10.1001/archneurol.2010.116. [DOI] [PubMed] [Google Scholar]
- Schretlen DS, Harris JC, Park KS, Jinnah HA, Ojeda del Pozo N. Neurocognitive functioning in Lesch-Nyhan disease and partial hypoxanthine-guanine phosphoribosyltransferase deficiency. J Int Neuropsychol Soc. 2001;7:805–12. doi: 10.1017/s135561770177703x. [DOI] [PubMed] [Google Scholar]
- Schretlen DS, Ward J, Meyer SM, Yun J, Puig JG, Nyhan WL, et al. Behavioral aspects of Lesch-Nyhan disease and it variants. Dev Med Child Neurol. 2005;47:673–7. doi: 10.1017/S0012162205001374. [DOI] [PubMed] [Google Scholar]
- Sculley DG, Dawson PA, Emmerson BT, Gordon RB. A review of the molecular basis of hypoxanthine-guanine phosphoribosyltransferase (HPRT) deficiency. Hum Genet. 1992;90:195–207. doi: 10.1007/BF00220062. [DOI] [PubMed] [Google Scholar]
- Sebesta I, Stibarkova B, Dvorakova L, Hrebicek M, Minks J, Stolnaja L, et al. Unusual presentation of Kelley-Seegmiller syndrome. Nucleosides Nucleotides Nucleic Acids. 2008;27:648–55. doi: 10.1080/15257770802143863. [DOI] [PubMed] [Google Scholar]
- Sege-Peterson K, Chambers J, Page T, Jones OW, Nyhan WL. Characterization of mutations in phenotypic variants of hypoxanthine phosphoribosyltransferase deficiency. Hum Mol Genet. 1992;1:427–32. doi: 10.1093/hmg/1.6.427. [DOI] [PubMed] [Google Scholar]
- Serrano M, Perez-Duenas B, Ormazabal A, Artuch R, Campistol J, Torres RJ, et al. Levodopa therapy in a Lesch-Nyhan disease patient: pathological, biochemical, neuroimaging, and therapeutic remarks. Mov Disord. 2008;23:1297–300. doi: 10.1002/mds.21786. [DOI] [PubMed] [Google Scholar]
- Sharma S, Jimenez RT, Aneja S, Garcia MG, Sethi GR. Lesch-Nyhan Syndrome in an Indian Family with Novel Mutation in the HPRT1 Gene. Indian J Pediatr. 2012;79:1520–2. doi: 10.1007/s12098-011-0657-9. [DOI] [PubMed] [Google Scholar]
- Shi W, Li CM, Tyler PC, Furneaux RH, Grubmeyer C, Schramm VL, et al. The 2.0 A structure of human hypoxanthine-guanine phosphoribosyltransferase in complex with a transition-state analog inhibitor. Nat Struct Biol. 1999;6:588–93. doi: 10.1038/9376. [DOI] [PubMed] [Google Scholar]
- Shnier MH, Sims F, Zail S. The Lesch-Nyhan syndrome: first case description in a South African family. S Afr Med J. 1972;46:947–9. [PubMed] [Google Scholar]
- Silverstein FS, Johnston MV, Hutchinson RJ, Edwards NL. Lesch-Nyhan syndrome: CSF neurotransmitter abnormalities. Neurology. 1985;35:907–11. doi: 10.1212/wnl.35.6.907. [DOI] [PubMed] [Google Scholar]
- Sim NL, Kumar P, Hu J, Henikoff S, Schneider G, Ng PC. SIFT web server: predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 2012;40:W452–7. doi: 10.1093/nar/gks539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder FF, Chudley AE, MacLeod PM, Carter RJ, Fung E, Lowe JK. Partial deficiency of hypoxanthine-guanine phosphoribosyltransferase with reduced affinity for PP-ribose-P in four related males with gout. Hum Genet. 1984;67:18–22. doi: 10.1007/BF00270552. [DOI] [PubMed] [Google Scholar]
- Solan A, Matthews W, Barabas G, Robey K. Cognition in LND: a two-year follow-up study. Dev Med Child Neurol. 1997;39:492–3. [PubMed] [Google Scholar]
- Sorensen LB. Mechanism of excessive purine biosynthesis in hypoxanthine-guanine phosphoribosyltransferase deficiency. J Clin Invest. 1970;49:968–78. doi: 10.1172/JCI106316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stout JT, Caskey CT. HPRT: gene structure, expression, and mutation. Ann Rev Genet. 1985;19:127–48. doi: 10.1146/annurev.ge.19.120185.001015. [DOI] [PubMed] [Google Scholar]
- Taniguchi A, Yamada Y, Hakoda M, Sekita C, Kawamoto M, Kaneko H, et al. Molecular characterization of a deletion in the HPRT1 gene in a patient with Lesch-Nyhan syndrome. Nucleosides Nucleotides Nucleic Acids. 2011;30:1266–71. doi: 10.1080/15257770.2011.608396. [DOI] [PubMed] [Google Scholar]
- Tarle SA, Davidson BL, Wu VC, Zidar FJ, Seegmiller JE, Kelley WN, et al. Determination of the mutations responsible for the Lesch-Nyhan syndrome in 17 subjects. Genomics. 1991;10:499–501. doi: 10.1016/0888-7543(91)90341-b. [DOI] [PubMed] [Google Scholar]
- Torres RJ, Garcia MG, Puig JG. Partial HPRT deficiency phenotype and incomplete splicing mutation. Nucleosides Nucleotides Nucleic Acids. 2010;29:295–300. doi: 10.1080/15257771003730250. [DOI] [PubMed] [Google Scholar]
- Torres RJ, Mateos FA, Molano J, Gathoff BS, O'Neill JP, Gundel RM, et al. Molecular basis of hypoxanthine-guanine phosphoribosyltransferase deficiency in thirteen Spanish families. Hum Mutat. 2000;15:383. doi: 10.1002/(SICI)1098-1004(200004)15:4<383::AID-HUMU17>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Torres RJ, Puig JG, Jinnah HA. Update on the phenotypic spectrum of Lesch-Nyhan disease and its attenuated variants. Curr Rheumatol Rep. 2011;14:189–94. doi: 10.1007/s11926-011-0231-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tvrdik T, Marcus S, Hou SM, Falt S, Noori P, Podlutskaja N, et al. Molecular characterization of two deletion events involving Alu-sequences, one novel base substitution and two tentative hotspot mutations in the hypoxanthine-phosphoribosyltransferase (HPRT) gene in five patients with Lesch-Nyhan syndrome. Hum Genet. 1998;103:311–318. doi: 10.1007/s004390050822. [DOI] [PubMed] [Google Scholar]
- Van Bogaert P, Ceballos I, Desguerre I, Telvi L, Kamoun P, Ponsot G. Lesch-Nyhan syndrome in a girl. J Inherit Metab Dis. 1992;15:790–1. doi: 10.1007/BF01800022. [DOI] [PubMed] [Google Scholar]
- Visser JE, Baer PR, Jinnah HA. Lesch-Nyhan syndrome and the basal ganglia. Brain Res Rev. 2000;32:449–75. doi: 10.1016/s0165-0173(99)00094-6. [DOI] [PubMed] [Google Scholar]
- Wang F, Shi W, Nieves E, Angeletti RH, Schramm VL, Grubmeyer C. A transition-state analogue reduces protein dynamics in hypoxanthine-guanine phosphoribosyltransferase. Biochemistry. 2001;40:8043–54. doi: 10.1021/bi010203f. [DOI] [PubMed] [Google Scholar]
- Warzok R, Schwesinger G, Knapp A, Seidlitz F. Neuropathologische befunde beim Lesch-Nyhan Syndrom. Zbl Allg Pathol U Pathol Anat. 1982;126:95–104. [PubMed] [Google Scholar]
- Watts RWE, Spellacy E, Gibbs DA, Allsop J, McKeran RO, Slavin GE. Clinical, post-mortem, biochemical and therapeutic observations on the Lesch-Nyhan syndrome with particular reference to the neurological manifestions. Q J Med. 1982;201:43–78. [PubMed] [Google Scholar]
- Willers I, Bolz H, Wehnert M, Gal A. Eighteen novel mutation in patients with Lesch-Nyhan syndrome or partial hypoxanthine phosphoribosyltransferase deficiency. J Inherit Metab Dis. 1999;22:845–6. doi: 10.1023/a:1005522527689. [DOI] [PubMed] [Google Scholar]
- Wilson JM, Kelley WN. Molecular genetics of the HPRT-deficiency syndromes. Hosp Pract. 1984;19:81–9, 93–7, 100. doi: 10.1080/21548331.1984.11702819. [DOI] [PubMed] [Google Scholar]
- Wilson JM, Landa LE, Kobayashi R, Kelley WN. Human hypoxanthine-guanine phosphoribosyltransferase: tryptic peptides and post-translational modification of the erythrocyte enzyme. J Biol Chem. 1982;257:14830–4. [PubMed] [Google Scholar]
- Wilson JM, Stout JT, Palella TD, Davidson BL, Kelley WN, Caskey CT. A molecular survey of hypoxanthine-guanine phosphoribosyltransferase deficiency in man. J Clin Invest. 1986;77:188–95. doi: 10.1172/JCI112275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson JM, Young AB, Kelley WN. Hypoxanthine-guanine phosphoribosyltransferase deficiency. The molecular basis of the clinical syndromes. N Engl J Med. 1983;309:900–10. doi: 10.1056/NEJM198310133091507. [DOI] [PubMed] [Google Scholar]
- Wong DF, Harris JC, Naidu S, Yokoi F, Marenco S, Dannals RF, et al. Dopamine transporters are markedly reduced in Lesch-Nyhan disease in vivo. Proc Natl Acad Sci USA. 1996;93:5539–43. doi: 10.1073/pnas.93.11.5539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Eads J, Sacchettini JC, Grubmeyer C. Kinetic mechanism of human hypoxanthine-guanine phosphoribosyltransferase: rapid phosphoribosyl transfer chemistry. Biochemistry. 1997;36:3700–12. doi: 10.1021/bi9616007. [DOI] [PubMed] [Google Scholar]
- Yamada Y, Goto H, Suzumori K, Adachi R, Ogasawara N. Molecular analysis of five independent Japanese mutant genes responsible for hypoxanthine guanine phosphoribosyltransferase (HPRT) deficiency. Hum Genet. 1992;90:379–84. doi: 10.1007/BF00220463. [DOI] [PubMed] [Google Scholar]
- Yamada Y, Goto H, Yukawa T, Akazawa H, Ogasawara N. Molecular mechanisms of the second female Lesch-Nyhan patient. Adv Exp Med Biol. 1994;370:337–40. doi: 10.1007/978-1-4615-2584-4_72. [DOI] [PubMed] [Google Scholar]
- Yamada Y, Nomura N, Yamada K, Wakamatsu N. Molecular analysis of HPRT deficiencies: An update of the spectrum of Asian mutations with novel mutations. Mol Genet Metab. 2007;90:70–6. doi: 10.1016/j.ymgme.2006.08.013. [DOI] [PubMed] [Google Scholar]
- Yamada Y, Wakamatsu N, Taniguchi A, Kaneko K, Fujimori S. Hypoxanthine guanine phosphoribosyltransferase (HPRT) mutations in the Asian population. Nucleosides Nucleotides Nucleic Acids. 2011a;30:1248–55. doi: 10.1080/15257770.2011.603714. [DOI] [PubMed] [Google Scholar]
- Yamada Y, Yamada K, Nomura N, Yamano A, Kimura R, Naiki M, et al. Molecular analysis of X-linked inborn errors of purine metabolism: HPRT1 and PRPS1 mutations. Nucleosides Nucleotides Nucleic Acids. 2011b;30:1272–5. doi: 10.1080/15257770.2011.597369. [DOI] [PubMed] [Google Scholar]
- Yamada Y, Yamada K, Sonta S, Wakamatsu N, Ogasawara N. Mutations in the hypoxanthine-guanine phosphoribosyltransferase gene (HPRT1) in Asian HPRT-deficient families. Nucleosides Nucleotides Nucleic Acids. 2004;23:1169–72. doi: 10.1081/NCN-200027439. [DOI] [PubMed] [Google Scholar]
- Yang TP, Patel PI, Chinault AC, Stout JT, Jackson LG, Hildebrand BM, et al. Molecular evidence for new mutation at the hprt locus in Lesch-Nyhan patients. Nature. 1984;310:412–4. doi: 10.1038/310412a0. [DOI] [PubMed] [Google Scholar]
- Yang TP, Stout JT, Konecki DS, Patel PI, Alford RL, Caskey CT. Spontaneous reversion of novel Lesch-Nyhan mutation by HPRT gene rearrangement. Somat Cell Mol Genet. 1988;14:293–303. doi: 10.1007/BF01534590. [DOI] [PubMed] [Google Scholar]
- Yeh J, Zheng S, Howard BD. Impaired differentiation of HPRT-deficient dopaminergic neurons: a possible mechanism underlying neuronal dysfunction in Lesch-Nyhan syndrome. J Neurosci Res. 1998;53:78–85. doi: 10.1002/(SICI)1097-4547(19980701)53:1<78::AID-JNR8>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- Yukawa T, Akazawa H, Miyake Y, Takahashi Y, Nagao H, Takeda E. A female patient with Lesch-Nyhan syndrome. Dev Med Child Neurol. 1992;34:543–6. doi: 10.1111/j.1469-8749.1992.tb11477.x. [DOI] [PubMed] [Google Scholar]
- Zamora A, Escarcega RO, Vazquez R, O'Neill JP. HPRT deficiency in a two-month-old child presenting acute renal failure and gout with a new deletion of two bases in exon 3 of the HPRT gene. Arch Med Res. 2007;38:460–2. doi: 10.1016/j.arcmed.2006.10.015. [DOI] [PubMed] [Google Scholar]
- Zanic T, Gamulin V, Lipovac K. A case of severe hypoxanthine-guanine phosphoribosyl transferase deficiency. J Inherit Metab Dis. 1985;8:79. doi: 10.1007/BF01801670. [DOI] [PubMed] [Google Scholar]
- Zoref-Shani E, Bromberg Y, Hirsch J, Feinstein S, Frishberg Y, Sperling O. A novel point mutation (I137T) in the conserved 5-phophorylribose-1-pyrophosphate binding motif of hypoxanthine-guanine phosphoribosyltransferase (HPRTJerusalem) in a variant of Lesch-Nyhan syndrome. Mol Genet Metab. 2003;78:158–61. doi: 10.1016/s1096-7192(03)00002-7. [DOI] [PubMed] [Google Scholar]
- Zoref-Shani E, Bromberg Y, Hirsh J, Feinstein S, Frishberg Y, Sperling O. Clinical and biochemical manifestations and molecular characterization of the mutation in HPRT Jerusalem. Nucleosides Nucleotides Nucleic Acids. 2004;23:1165–8. doi: 10.1081/NCN-200027436. [DOI] [PubMed] [Google Scholar]
- Zoref-Shani E, Feinstein S, Frishberg Y, Sperling O. Kelley-Seegmiller syndrome due to a unique variant of hypoxanthine-guanine phosphoribosyltransferase: reduced affinity for 5-phosphoribosy1-pyrophosphate manifested only at low, physiological substrate concentrations. Biochim Biophys Acta. 2000;1500:197–203. doi: 10.1016/s0925-4439(99)00103-9. [DOI] [PubMed] [Google Scholar]