

Abstract
Stem cells (SCs) are the key to tissue genesis and regeneration. Given their central role in homeostasis, dysfunctions of the SC compartment play a pivotal role in the development of cancers, degenerative disorders, chronic inflammatory pathologies and organ failure. The gastrointestinal tract is constantly exposed to harsh mechanical and chemical conditions and most of the epithelial cells are replaced every 3 to 5 d. According to the so-called Unitarian hypothesis, this renewal is driven by a common intestinal stem cell (ISC) residing within the crypt base at the origin of the crypt-to-villus hierarchical migratory pattern. Celiac disease (CD) can be defined as a chronic immune-mediated disease that is triggered and maintained by dietary proteins (gluten) in genetically predisposed individuals. Many advances have been achieved over the last years in understanding of the pathogenic interactions among genetic, immunological and environmental factors in CD, with a particular emphasis on intestinal barrier and gut microbiota. Conversely, little is known about ISC modulation and deregulation in active celiac disease and upon a gluten-free diet. Nonetheless, bone marrow-derived SC transplantation has become an option for celiac patients with complicated or refractory disease. This manuscript summarizes the “state of the art” regarding CD and ISCs, their niche and potential role in the development and treatment of the disease.
Keywords: Intestinal stem cells, CD133, Lgr5, Celiac disease, Paneth cells, Gut microbiota, Gut barrier
Core tip: The intestinal epithelium has a high turnover rate since most of the epithelial cells are replaced every 3 to 5 d. This renewal is driven by intestinal stem cells residing within the crypt base at the origin of the crypt-to-villus hierarchical migratory pattern. Many aspects of the pathogenesis of celiac disease have been elucidated over the last years regarding the interactions among genetic and immunological factors, intestinal barrier and gut microbiota. Conversely, little is known about intestinal stem cell modulation and deregulation in celiac disease. The current knowledge regarding celiac disease and intestinal stem cells, and the potential role of stem cells in the development and treatment of the disease are summarized.
“Enthusiasm is that temper of the mind in which the imagination has got the better of the judgment” - William Warburton.
STEM CELLS AND THEIR POTENTIAL
Stemness can be defined as the capability of extensive self-maintenance and differentiation[1,2]. Stem cells (SCs) are undifferentiated cells able to give rise to diverse mature progenies and to self-renew through the alternation of symmetrical and asymmetrical divisions. SCs play a central role in tissue genesis, regeneration and homeostasis by providing differentiated cells that can increase tissue mass during pre- and post-natal growth and replace cell loss due to senescence or damage[3-6].
SCs possess a hierarchy of potentialities: from the totipotency of the zygote and its immediate progeny, to the pluripotency of embryonic stem cells (ESCs), up to the multi/unipotency of adult SCs (ASCs)[7].
ESCs are pluripotent cells derived from the inner cell mass of the blastocyst that can generate any differentiated phenotype of the three primary germ layers (endoderm, mesoderm and ectoderm), as well as germ cells. ESCs might constitute an easily available source to obtain a large number of transplantable cells for regenerative treatments. Nevertheless, ethical concerns and the possibility of immune rejection and teratoma/teratocarcinoma formation are major obstacles to the feasibility and safety of ESC clinical applications[8].
Pluripotent stem-like cells could also derive from non-pluripotent cells-typically an adult somatic cell-by inducing a “forced” expression of specific genes. These induced pluripotent stem-like cells (iPS cells) are similar to ESCs in many aspects, such as the expression of certain SC genes, potency and differentiability, formation of embryoid bodies, teratomas and viable chimeras, even if the full extent of their relationship to natural pluripotent SCs is not fully elucidated; as a consequence, they cannot be currently considered a reliable and feasible source of SCs[9,10].
Another population of SCs with high differentiation potential is represented by cells established from placental/cord tissues, which do not tend to form teratomas/teratocarcinomas and have a higher proliferation and differentiation potential than ASCs. In particular, the plasticity and accessibility of umbilical cord blood SCs (CBSCs) have given the rationale for the creation of CBSC unit banks where these cells can be collected and stored for future use[7].
The least differentiation potential is possessed by ASCs, which persist indefinitely in the tissue of origin, allowing for local tissue regeneration and renewal[11]. Despite the paradigm of unidirectional cell determination, recent studies have shown that ASCs are endowed with an unexpected plasticity as circulating adult progenitor cells can differentiate into mature cells of other tissue types[5]. A particularly high degree of plasticity is shown by hematopoietic stem cells (HSCs) and mesenchymal stem cells (MSCs).
HSCs are responsible for the renewal of blood cells[12]. Commonly used markers for HSCs identification and isolation include two membrane phosphoglycoproteins: CD34 and AC133 (CD133, or “prominin1” in rodents)[13]. It is generally accepted that the most primitive and long-term human HSCs are characterized by the expression of CD133, Thy1 (CD90) and VEGFR2 and by a variable expression of CD34 and CD38[14,15]. Bone marrow (BM) resident HSCs can be mobilized into the peripheral blood under specific stimuli such as tissue injury or administration of mobilizing agents[1]. In vitro culture and in vivo transplantation assays have demonstrated that HSCs are able to give rise to a wide array of phenotypes, including blood, cartilage, fat, tendon, lung, liver, muscle, brain, heart and kidney cells[1]. Moreover, it has been demonstrated that the number of circulating HSCs expressing early markers for muscle, nerve and hepatic differentiation increases following treatment with mobilizing agents. This phenomenon has led to speculation about the existence of BM-derived circulating pluripotent SCs which could migrate from the peripheral blood into every tissue and contribute to normal turnover and repair following injury[16].
MSCs, also called “stromal stem cells”, “stromal precursors”, “mesenchymal progenitors” and “colony-forming unit-fibroblast cells”, are highly proliferating, adherent cells which reside in a perivascular niche within the BM and also in the wall of blood vessels within most organs[17]. MSCs can differentiate into a variety of mesodermal cell lineages, including osteoblasts, chondroblasts, adipocytes, myocytes and cardiomyocytes, as well as non-mesodermal cells, such as hepatocytes and neurons[18]. In addition to BM, MSCs have been isolated from various adult tissues, including muscle, adipose tissue, connective tissue, trabecular bone, synovial fluid and from perinatal tissues (umbilical cord, amniotic fluid and placenta). The presence of MSCs in peripheral blood is still being debated as some authors identified a circulating fibroblast-like population, whereas others failed[19].
SCs colocalize with supporting cells in a physiologically limited and specialized microenvironment or niche that varies in nature and location depending upon the tissue type[20]. The reciprocal interactions between SCs and their microenvironment, through cell-cell and cell-matrix connections as well as the secretion of soluble factors, influence SC behavior, regulating the balance between quiescence and dividing state under specific pathological or physiological conditions[5]. Understanding the molecular signals which regulate SC behavior is critical for their therapeutic applications. In fact, the exogenous stimulation with specific growth factors or cytokines may be used to activate SCs in vivo and in vitro.
DEVELOPMENT AND TURNOVER OF THE INTESTINAL EPITHELIUM
The gastrointestinal tract surface derives from the endoderm. The embryonic stratified endodermal epithelium is subsequently converted into a monolayer overlying nascent villi while dividing cells segregate to the intervillous region. Intestinal crypts develop during the early postnatal period, becoming the niche for gastrointestinal SCs[21]. Once completely structured, the epithelium along the gut is characterized by a heterogeneous cell population, in terms of morpho-functional properties and proliferation kinetics, reflecting the various functions of the different gastrointestinal components[7]. The adult mammalian gut can be broadly segregated into two functionally distinct parts: the small intestine and the colon, which present with marked architectural differences, reflecting their different functions. In particular, in the small intestine, the crypts of Lieberkuhn are associated with the intestinal villi that maximize surface area, endowing the small intestine with an excellent capacity to absorb dietary nutrients from the lumen. In contrast, the absence of villi within the colonic epithelium translates to a flatter morphology, highlighting its predominant role in stool compaction[22].
As a consequence of its role in digestion, nutrient absorption and waste excretion, the gastrointestinal tract is constantly exposed to harsh mechanical and chemical conditions. Therefore, the intestinal tract has evolved mechanisms to cope with these assaults via a highly regulated process of self-renewal[23]. Mucosal proliferation plays a fundamental role in the maintenance of the gut integrity. Most of the epithelial cells are replaced every 3 to 5 d which is a high proliferation rate, second only to the hematopoietic system[7]. According to the so-called “Unitarian hypothesis”, first proposed by Cheng and Leblond in 1974[24], this epithelial renewal is driven by a common intestinal stem cell (ISC) residing within the crypt base at the origin of the well established crypt-to-villus hierarchical migratory pattern[25,26]. From their niche, ISCs give rise to transit-amplifying (TA) cells that migrate upwards and progressively lose their proliferative capability and maturate to become fully-differentiated villous epithelial cells (absorptive enterocytes or secretory cells which include goblet cells, enteroendocrine cells, Paneth cells and Tuft cells). Each adult crypt harbors approximately 5 to 15 ISCs that are responsible for the daily production of about 300 cells; up to 10 crypts are necessary to replenish the epithelium of a single villus[23]. Crypt-derived epithelial cells generally reach the villus tip after 3-5 d when they die and are exfoliated into the lumen[27], except for Paneth cells (PCs) that evade this upward migration program, instead forcing their way to the base of the crypt[28]. PCs are confined to the small intestine where they can live for up to 8 wk[29]. PCs are also unique in that they appear after birth during crypt emergence[30]. PCs secrete defensins, lysozyme and phospholipase A2 and play a central role in host defense against enteric pathogens; moreover, the antimicrobial peptides secreted by PCs shape the composition of gut microbiota and protect from bacterial translocation[29]. In addition, crypts supply less common cell types such as the M cells and cup cells, although their lineages are poorly understood[23].
ISCS AND THEIR NICHE
ISC hierarchy
Since the 1970s, several studies have supported the concept of ISCs. The ability of SCs to regenerate gut epithelium has been investigated in various animal models of intestinal injury. Such studies have led to the hypothesis of an ISC hierarchy organized in three main compartments and progressively recruited at various degrees of damage in order to ensure an effective crypt regeneration[7].
The initial location for the ISCs was deemed to be the fourth cell position from the bottom of the crypt (+4) where slowly cycling cells that show label-retention of BrdU (the so-called “+4 label retaining cells”, LRCs) were described by Potten et al[31] in 1974.
A second theory regarding the location of the ISCs was formulated in the same year by Cheng and Leblond[24]. In a series of electron microscopy studies on the small intestinal crypts, these authors described slender, immature, cycling cells wedged between PCs at the positions 1-4 of the crypt base. Upon 3H-thymidine treatment, these “crypt base columnar” (CBC) cells were able to phagocytose close damaged cells; subsequently, phagosome-labeled cells were found in all intestinal epithelial lineages, suggesting the role of CBCs as ISCs.
In 2007, a Wnt-target gene encoding a leucine-rich orphan G protein-coupled receptor named Lgr5 was identified to specifically label CBCs in the mouse small intestine[27]. Through a lineage tracing approach, Sato et al[32] demonstrated that CBCs are able to give rise to all intestinal epithelial lineages and are a self-renewing population of multipotent SCs. Further proof that Lgr5+ cells are ISCs derived from ex vivo culture assays, where single Lgr5+ cells were able to form self-renewing epithelial organoids highly reminiscent of crypt/villus epithelial units in vivo, while cells that expressed low or no Lgr5 were unable to form such structures. Unlike LRCs, Lgr5+ CBCs are resistant to radiation and are rapidly proliferating, thus challenging the previously held belief that all ASCs are quiescent or slowly cycling. In 2009, lineage tracing studies also showed that some Lgr5+ cells co-express prominin-1 (or CD133) and these CD133+ cells can generate the entire intestinal epithelium[33,34].
In addition to Lgr5 and CD133, other potential ISC markers have been identified in the last years, including musashi1 (MSI1), expressed by both LRCs and CBCs; olfactomedin 4 (OLFM4), expressed by Lgr5+ cells; PTEN, AKT1, mTERT and BMI1, predominantly expressed in LRCs (for extensive revision on this topic, see[23,35]) (Figure 1).
Figure 1.
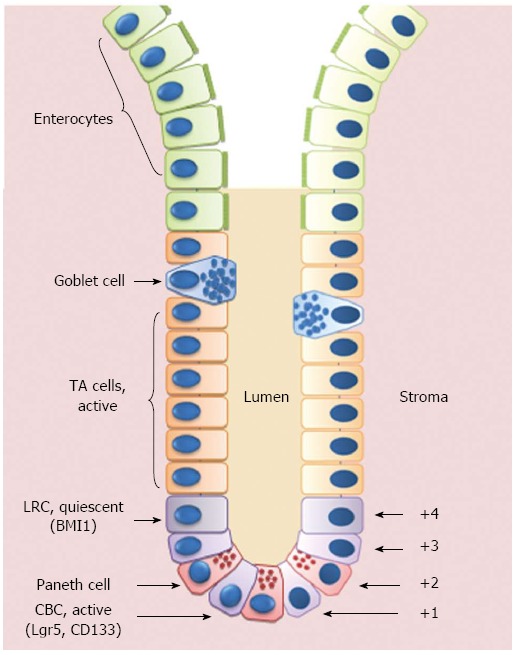
Schematic representation of the crypt/villus axis. Putative intestinal stem cells (ISCs) reside either at the crypt base, between Paneth cells, as Crypt Base Columnar Cells (CBCs), or in position +4 from the bottom of the crypt, as Label Retaining Cells (LRCs). ISCs give rise to Transit Amplifying (TA) cells that are able to migrate upwards and progressively maturate losing their proliferative capability to become fully-differentiated villous epithelial cells.
In 2008, Scoville et al[36] proposed the coexistence of two types of ISCs: the LRCs at the +4 location that are a “reserve pool” in a prolonged quiescent state and the actively cycling CBCs able to respond to stimulating signals from their microenvironment and to provide progenitor cells on an everyday basis. To support this hypothesis, Sangiorgi et al[37] found that Bmi1+ cells corresponding to 4+ LRCs can self-renew, proliferate, expand and like CBCs give rise to all the differentiated lineages of the small intestine epithelium. The authors concluded that +4 LRCs and CBCs are ISCs in different niches, able to migrate from one to the other[37]. Recently, two independent groups showed a dynamic interplay between both cell populations: Tian et al[38] demonstrated that Lgr5+ cells are dispensable for gut homeostasis and that BMI1+ cells are able to replenish the Lgr5+ cell compartment after its experimental ablation; Takeda et al[39] suggested a more complicated bidirectional relationship between Lgr5+ cells and +4 LRCs, the latter being able to either originate from or give rise to Lgr5+ cells. Whether the +4 LRCs and the CBCs truly are two distinct ISC populations and whether this is an intrinsic quality or the result of the different location within the ISC niche is still a matter of debate[40].
A third potential source of ISCs is represented by circulating multipotent SCs of BM origin that can colonize the intestinal epithelium and contribute to its turnover and regeneration[41-43]. BM stem cells may participate in gut repair by giving rise to ISCs through direct differentiation and also by providing supporting elements within the ISC niche, as demonstrated in different experimental models[44-47]. However, the reduced levels of engraftment and the low rate of differentiation into intestinal cells reported in most of these studies discouraged the practical application of these cells in a clinical setting. Recently, efforts have been made to develop strategies to enhance the levels of engraftment. Zhang et al[48] demonstrated that transplantation with BM SCs genetically modified to express CXCR-4 resulted in levels of engraftment able to ameliorate radiation enteritis. Colletti et al[49] identified a marker (EphB2) for isolating and culturing an expandable subpopulation of human BM-derived SCs with enhanced intestinal homing and contribution to ISC region.
ISC niche
ISCs reside in a physiologically limited and specialized niche that dictates the mechanisms of tissue turnover and regeneration through cell-cell interactions and molecular signals[5,50].
Traditionally, the underlying stromal cells (pericryptal myofibroblasts, enteric neurons, endothelial cells and intraepithelial lymphocytes) have been considered to constitute the niche for ISCs. Recently, it has been suggested that PCs are an essential component of the Lgr5+ ISC niche[51]. Much evidence sustains this hypothesis. In vivo, the absence of PCs compromises the recovery ability, resulting in complete loss of the intestinal epithelial integrity[52]. In vitro, the presence of PCs significantly increases the generation of epithelial organoids by Lgr5+ cells[51]. PCs produce many growth factors involved in ISC maintenance and activation, including epidermal growth factor, Wnt3 and transforming growth factor-alpha[23]. The intimate relationship between PCs and ISCs seems to be involved in the response to nutritional status of the organism. Indeed, PCs can act as a “sensor” for nutritional status and enhance ISC function in response to caloric restriction[53]. Finally, PCs seem essential to regulate ISC self-renewal by neutral competition between symmetrically dividing ISCs and a limited PC-defined niche within the crypt base[54]. Thus, PCs serve as multifunctional guardians of ISCs by secreting bactericidal products and by providing essential niche signals. As a consequence, despite the fact that SC niches are typically portrayed as pre-existing sites to which SCs migrate[55], ISCs are unique since they also receive niche support from their own specialized progeny of PCs.
The main molecular pathways involved in ISC regulation are Wnt, Notch, Hedgehog, Bmp and PTEN-PI3K-Akt.
Wnt signaling: Wnt signaling is based on the autocrine and paracrine interaction of secreted cysteine-rich Wnt-glycoproteins with a transmembrane Frizzled receptor (Fz). Binding of Wnt to its receptor activates the canonical pathway with stabilization and nuclear translocation of beta-catenin or the non-canonical pathway that encompasses the planar cell polarity and the Wnt/Ca2+ pathway. The canonical pathway is the best characterized and most relevant in SC signaling: the binding of secreted Wnt-proteins to Fz induces nuclear translocation of beta-catenin that triggers Wnt-target gene transcription. Many studies have shown the importance of this pathway in the proliferation and differentiation of the gastrointestinal epithelium (revised in[40,56]). Wnt signaling has different effects in different cell types, also depending on its localization along the crypt/villus axis.
Direct evidence of Wnt-activity in ISCs is their unique expression of Lgr5, a Wnt-target gene[27]. Other Wnt-target genes associated with proliferation of TA-cells include c-myc and cyclin D1[57,58]. R-spondins, glycoproteins likely secreted by enteroendocrine cells, amplify Wnt signaling, induce a proliferative response in human intestinal epithelium, and are responsible for the expansion of organoid cultures[59-61].
Wnt signaling is necessary for ISC proliferation and maintenance of the ISC phenotype. The previously reported PC-dependence of single Lgr5+ cells in plating efficiency can be overcome by the addition of Wnt-3 in culture[51]. Conversely, a decreased Wnt signaling results in the loss of the proliferative compartment. Over-expression of Kruppel-like factor 4 (Klf4, a negative regulator of Wnt signals) induces cell cycle arrest, while its deletion leads to increased proliferation[40,62,63].
Wnt signaling plays a pivotal role in cell differentiation: an overactive Wnt signaling impedes ISC differentiation and induces mislocalization of PCs, impaired goblet cell and enterocyte maturation; on the other hand, an underactive Wnt signaling induces depletion of progenitor cells, leading to the absence of properly differentiated cells[40,62]. The development of PCs is also directly dependent on Wnt signaling[64].
Wnt signaling is indispensable for intestinal morphogenesis and normal cell migration. Indeed, beta-catenin ensures the correct positioning of epithelial cells along the crypt/villus axis by regulating the expression of members of the Ephrin and Ephrin receptor (Eph) families[65]. Ephrins and Eph, both membrane-bound proteins, are differentially expressed in intestinal mucosa, with Eph localized in the intestinal crypt region, while Ephrin proteins colonize the villi[66,67]. A direct influence of EphB-signaling on ISC proliferation has been shown[68] and it has been demonstrated that EphB3 is essential for PC downward migration[69]. In addition to their role in promoting cell proliferation of the intestinal epithelium, tissue repair, acceleration of wound closure and maintenance of homeostasis of the intestinal barrier in adults, Ephrin/Eph signaling has been recognized to function as tumor suppressors by controlling cell migration and inhibiting tumoral invasive growth[70-72].
Given its pivotal role within the ISC niche, it is not surprising that alterations in Wnt signaling play a pivotal role in the development of non-neoplastic gastrointestinal disorders, such as chronic inflammatory bowel disease and intestinal cancers (as reviewed elsewhere[40,73]).
Notch signaling: Notch signaling is known to control cell fate decisions in the development of many tissues. The ligands Delta or Jagged bind the Notch receptor, thereby inducing its proteolytic cleavage; NCID, a cleavage fragment of Notch, translocates to the nucleus where it acts as a transcription factor, thus inducing the activation of molecular pathways involved in the control of proliferation and differentiation[74]. Manipulations of the Notch signaling in experimental models revealed its role in intestinal epithelial differentiation. Hes1, the major Notch-target gene, colocalizes with Msi1 in both the CBCs and the +4 LRCs[75,76].
Notch signaling plays a central role in preserving self-renewal in the intestinal progenitor cells by suppressing Atoh1[40]. Notch signaling seems to trigger proliferation of crypt progenitor cells in TA-cells and a regulated reduction of notch signaling in cooperation with activation of specific transcription factors (such as Atoh1 and neuroD) induces specific differentiation into the intestinal epithelial lineages[40,56].
Hedgehog and BMP pathways: The morphogens Sonic Hedgehog (Shh) and Indian Hedgehog (Ihh) are secreted by epithelial cells, while their receptor, Patched (PTCH), is expressed by subepithelial myofibroblasts. In the intestinal epithelium, Ihh is mainly expressed at the base of the villi[77]. Given the importance of the stromal-epithelial interactions in the regulation of the epithelial cell fate, hh signaling is indirectly involved in the ISC fate through its modulation of the maturation and localization of the underlying stromal cells that in turn generate signal molecules responsible for the maintenance of the ISC niche[78]. Ihh down-regulates expression of TCF4 and beta-catenin, restricting Wnt signaling to the crypt base[79]. Hh also promotes maturation of the tolerogenic immune cells in the small intestine and is critical to the ability of the gut to respond to pro-inflammatory stimuli: disruption in the hh pathway may contribute to the pathogenesis of autoimmune diseases[40,80].
Disturbed hh signaling results in severe developmental defects, enhancement of Wnt signaling, increased proliferation and structural abnormalities of crypts and villi. Such effects are mainly due to the reduced expression of bone morphogenetic proteins (BMPs) by stromal cells, which is normally triggered by hh[81].
BMPs regulate differentiation, apoptosis and cell growth depending upon the specific cellular context. BMPs bind to BMP receptors, leading to phosphorylation of SMADs, which upon heterodimerization translocate to the nucleus and act as transcriptional factors[82]. BMP pathway participates in the control of ISC numbers and self-renewal: active BMP signaling is found predominantly in differentiated intestinal epithelial cells, while its inhibition seems to confer intestinal stemness properties[81]. Physiological inhibitors of BMP signaling, Noggin and Gremlin, induce Wnt signaling activation and are produced by myofibroblasts at the crypt base, ensuring a “BMP-free” ISC niche[83]. Mesenchymal cells are the main target of BMP signaling which in turn down-regulates epithelial proliferation[40]. Of note, the BMP pathway has a direct role in the differentiation of the intestinal epithelium toward secretory lineages (especially enteroendocrine cells), while it does not affect the absorptive phenotype[84].
Mutations involving BMP signaling are associated with juvenile polyposis[81]. BMPs stabilize PTEN, thereby leading to reduced Akt activity and subsequent reduction of nuclear beta-catenin accumulation[56].
PTEN-PI3K-Akt pathway: PI3k activation leads to phosphorylation and subsequent activation of the kinase Akt, which induces cell survival, growth and proliferation programs. PTEN is a negative regulator of this pathway, thereby inhibiting Akt function[85].
This pathway is activated in many human tumors, mainly as a consequence of PTEN inactivation[86]. PTEN inherited mutations are responsible for hamartomatous polyps (Cowden syndrome)[81].
As for the role of the PI3K pathway in ISC regulation, it has been demonstrated that it enhances ISC self-renewal, probably because p-Akt can increase the transcriptional activity of beta-catenin, the main effector of the canonical Wnt pathway[87]. Moreover, PTEN might be involved in the restriction of the strong Wnt signaling to the crypt base[88].
Overall, ISC fate is regulated by a complex balance among signals controlling stem cell maintenance, proliferation and differentiation[40,56,89]. Wnt and Notch are mainly involved in ISC self-renewal and expansion. Moreover, Notch is involved in ISC differentiation, independently from Wnt. Notch inhibition leads to differentiation to a secretory phenotype, while Notch activation leads either to self-renewal within the ISC compartment or to differentiation towards an absorptive phenotype. The Wnt pathway is also implicated in PC differentiation and regulates cell migration along the crypt-villus axis, via Eph/Ephrin signaling. Hh effects on the ISCs are mainly indirect and occur through regulation of the BMP pathway. BMP signaling inhibits proliferation of ISCs, antagonizing the Wnt pathway; this suggests a homeostatic function of BMP in keeping self-renewal within the ISC niche. Most likely, this interaction is mediated by PTEN inhibition of Akt, which in turn inhibits Wnt signaling. BMPs also support the differentiation of secretory cell lineages (especially of enteroendocrine cells) (Figure 2).
Figure 2.
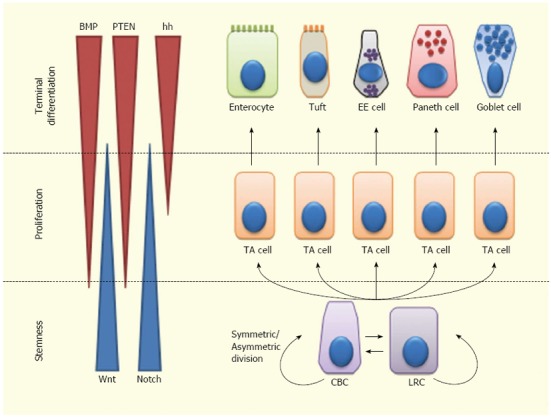
Lineage specification of intestinal stem cells. Intestinal stem cells (ISCs)-Crypt Columnar Cells (CBCs) and Label Retaining Cells (LRCs)-can divide asymmetrically or symmetrically to maintain the stem cell compartment. ISCs give rise to Transit Amplifying (TA) cells which actively proliferate and can further differentiate into enterocytes, tuft cells, enteroendocrine (EE) cells or goblet cells. Wnt signaling maintains the stem-like phenotype of ISCs, while Notch signaling maintains the proliferation of progenitor cells. In the upper crypt region, hedgehog (hh) triggers BMP expression in stromal cells which activates PTEN expression; all these factors inhibit Wnt signaling in the ISC niche.
ISCs in GI diseases
Observations that mutations in the pathways involved in ISC maintenance occur in most colon cancers have led the majority of the research on ISC biology in humans. Alterations of ISC pathways have also been reported in inflammatory bowel diseases. In particular, decreased expression of TCF4 (Wnt target gene, correlated with defensin production) has been described in ileal Crohn’s disease[90]; increased activation of Notch and PC dysfunction have been reported in both ulcerative colitis and Crohn’s disease[29,40].
A better knowledge of ISC function and dysregulation in gastrointestinal diseases will help to understand the pathophysiology of such disorders and might also offer new insight into the development of SC-based therapies.
Theoretically, ISCs would be the best source for intestinal regeneration. Although ISCs can be expanded for multiple passages in the form of organoids, most of the culture conditions provide little control over their self-renewal and differentiation. As a consequence, the inability to efficiently expand Lgr5+ SCs has so far considerably limited the translation to therapies as well as the study of intestinal epithelial biology. However, very recently Yin and co-workers identified small molecules (CHIR99021 and valproic acid) that target Wnt, Notch and BMP pathways to maintain the self-renewal of Lgr5+ ISCs, resulting in nearly homogeneous cultures with high colony-forming efficiency and preservation of the multilineage differentiation ability[91]. This might be a promising SC source for regenerative medicine, tissue engineering and drug screening.
So far, the only SCs that have “left the bench and reached the bedside” in gastroenterology are BM-derived SCs. BM SC transplantation has become an option for the treatment of selected cases of inflammatory bowel disorders (IBD). Experimental and clinical studies have suggested that both allogeneic and autologous BM SC transplants may be effective in inducing IBD remission[92-94]. The mechanisms underlying this beneficial effect are still under investigation; they might include local immune-modulation and a direct contribution to tissue repair[95,96].
BM SCs might also be used to cure other gastrointestinal pathologies, such as gastric ulcers or motility disorders, like gastroparesis, achalasia and chronic constipation[97,98].
Finally, a promising application for SC-based therapy is celiac disease (CD). The following chapters will attempt to summarize the body of knowledge regarding CD physiopathology and clinical manifestations, as well as the status of the ISC compartment during the course of the disease and the possible SC-based treatments.
CELIAC DISEASE: FROM PATHOGENESIS TO CURRENT TREATMENT
CD likely first developed after the last ice age in the fertile crescent of the Middle East with cultivation of grains[99]. The major breakthrough for the modern understanding of CD was the observation that bread shortages during World War II resulted in a dramatic decrease in death rate from celiac disease[100]. Also known as “nontropical sprue”, “celiac sprue” and “gluten-sensitive enteropathy”, CD can be defined as a chronic immune-mediated disease that is triggered and maintained by dietary proteins (gluten) in genetically predisposed individuals. Patients affected by the disease display a specific autoantibody response, various degrees of intestinal inflammation and a broad range of clinical symptoms[101,102].
Once considered a rare small bowel disease of childhood, CD is now recognized as a relatively common, systemic disease that may manifest at any age. CD affects 0.6%-1% of the population worldwide. The prevalence is up to 3-fold higher in women than in men; moreover, first-degree relatives of CD patients (10%-15%), individuals affected by autoimmune diseases, particularly type 1 diabetes (3%-16%) and Hashimoto’s thyroiditis (5%), IgA deficiency (9%), Down’s syndrome (5%) and Turner’s syndrome (3%) are at increased risk of developing the disease. The disease is less common in Hispanic Americans and it is thought to be rare in central Africa and east Asia; the frequency of CD is increasing in many developing countries because of many factors, such as increased awareness of the disease, changes in wheat production and preparation and westernization of the diet. Interestingly, serological screening studies have shown that only a small proportion of cases of CD (up to 20%) are clinically recognized[103].
Genetic background plays a pivotal role in the predisposition to CD: results from genetic linkage studies showed that CD is strongly associated with HLA-DQ genes (COELIAC1 locus, on chromosome 6p21). In particular, up to 90% of CD patients carry a variant of DQ2 (haplotype DQA1*0501/DQB1*0201), while about 5% of CD patients carry a variant of DQ8 (haplotype DQA1*0301/DQB1*0302); almost all of the remaining 5% of celiac patients have at least one of the two genes encoding DQ2[101]. DQ2 and DQ8 haplotypes are necessary for the development of CD: DQ2 and DQ8, expressed on the surface of antigen-presenting cells, can bind activated (deaminated) gluten peptides, triggering an abnormal immune response. However, DQ2 is carried by approximately a third of the general population, thus suggesting that HLA is only partly the cause of the condition.
So far, more than 30 genes, mostly involved in inflammatory and immune response, have been linked to a CD predisposition[104]. Non-HLA genes associated with CD include COELIAC2 (5q31-33) that contains cytokine gene clusters, COELIAC3 (2q33), encoding for the negative costimulatory molecule CTLA4, and COELIAC4 (19p13.1) that harbors an unconventional myosin able to alter cytoskeleton remodeling[105,106].
Almost all patients with CD develop immunoglobulin IgA antoantibodies to the enzyme tissue transglutaminase 2 (TG), which is expressed by many cell types and is associated with the extracellular matrix (endomysium or reticulin fibers). TG targets certain glutamine residues in some extracellular and intracellular proteins, usually tethering them to a lysine residue of a second protein that results in cross-linking of both proteins. Alternatively, TG merely deaminates glutamines to negatively charged glutamine acid residues. Gluten proteins are preferred substrates for TG and once deaminated, they bind more strongly to HLA-DQ2 or DQ8 on the surface of antigen presenting cells[105].
Serological tests are fundamental for CD screening. In patients with positive serology, a biopsy of the small intestine showing typical CD characteristics (increased number of intra-epithelial lymphocytes (IELs), elongation of the crypts and villous atrophy) is required to confirm the diagnosis. However, according to the most recent European guidelines, the confirmation biopsy is no longer required in children with predisposing HLA-genotypes, typical symptoms and a higher titer of anti-TG (>10 times the upper limit of normal range)[107].
CD is a “unique” model of autoimmune disease in that the key genetic components (HLA DQ2 and/or DQ8) are present in almost all patients, the autoantigen (TG) has been identified and the environmental trigger (gluten) is known. The central role of gluten in this cascade of events explains how the cornerstone of therapy for CD is a “gluten-free” diet (GFD).
Gluten is a protein complex composed of gliadins and glutenins that is responsible for the baking properties of wheat. Analysis of gliadin has identified more than one hundred components that can be grouped into four main types (omega5-, omega1, 2-, alpha/beta- and gamma-gliadins). The immunogenicity and toxicity of several gliadin epitopes has been established; although several gluten epitopes are immunostimulatory, an immunodominant peptide of 33 amino acids identified from the alpha-gliadin fraction has functional properties attributable to many proline and glutamine residues. Proline gives increased resistance to gastrointestinal proteolysis and causes a left-handed helical conformation which strengthens binding with DQ2 and DQ8 molecules on antigen-presenting cells. Additionally, glutamine residues are a preferred substrate for tissue transglutaminase-mediated deamination, which confers an enhanced immunogenicity. Storage proteins (prolamines), with similar amino acid composition to the gliadin fraction of wheat, have been identified in barley (hordeins) and rye (secalins) and show a close correlation to the taxonomy and toxic properties of wheat cereal[102].
Gluten peptides can be transported across the intestinal epithelium either paracellularly, especially in presence of an impaired gut barrier, or via transcytosis or retrotranscytosis of secretory IgA through the transferrin receptor. Gluten can elicit an innate immune response in professional antigen-presenting cells (monocytes, macrophages and dendritic cells) that activates both IELs and intestinal epithelial cells. This immediate reaction might favor the development of adaptive immunity to gluten in HLA-DQ2 or DQ8 carriers[108]. Innate immune activation of IELs by gluten induces expression of the non-classic class I molecule (MICA) on intestinal epithelium, which can in turn activate natural killer-like IELs, gamma-delta T cells and a subset of CD4+ and CD8+ T cells[109]. Epithelial MICA and production of IL-15 by epithelial cells, macrophages and dendritic cells lead to enhanced proliferation of IELs and cytokine secretion in CD patients; moreover, IL-21, produced by CD4+ Th1 cells, acts in concert with IL-15 as an additional driving force of innate immunity in CD pathogenesis[110].
Deamination or cross-linking of gluten by TG enhances the binding to HLA-DQ2 or DQ8 expressed by antigen presenting cells, leading to a more rigorous gluten-specific CD4+ Th1 T-cell activation[105]. Activated gluten-reactive CD4+ T cells produce high levels of pro-inflammatory cytokines, thus inducing a Th1-pattern dominated by interferon (IFN)-γ. Th-1 cytokines promote extracellular matrix degradation and increase cytotoxicity of IELs and NK cells. Additionally, IFN-alpha released by dendritic cells perpetuates the inflammatory reaction by inducing CD4+ T cells to produce IFN-γ. Finally, the production of Th2 cytokines by activated CD4 T cells drives the clonal expansion of B cells and subsequent production of antigliadin and anti-TG antibodies that can form deposits in the basement membrane region of the mucosal layer, leading to cytoskeleton remodeling and subsequent epithelial damage[102].
Clinical presentations of CD are extremely variable, reflecting the systemic nature of the disease. CD can be divided into 5 clinical subcategories: major (or classic), minor (or atypical), asymptomatic (or silent), latent and potential[102,111].
Major CD has three distinctive features: malabsorption (diarrhea, weight loss, vitamin and nutrient deficiencies), positive serology and pathological findings of villous atrophy. A rare life-threatening manifestation of CD is the so-called “celiac crisis”, mostly observed in children that manifests with profuse diarrhea, hypoproteinemia, metabolic and electrolyte imbalances.
Minor CD may present with only trivial, transient and apparently unrelated symptoms (fatigue, anemia, abdominal discomfort, dyspepsia, altered bowel habits, cryptic hypertransaminasemia, osteoporosis, infertility, peripheral and central neurological disorders, short stature, dental enamel defects, dermatitis herpetiformis) or isolated symptoms of associated autoimmune diseases. Most of these patients are biopsied after a positive search for anti-TG and/or anti-endomysial antibodies.
Asymptomatic CD is recognized on biopsy specimens of patients with positive serology but without symptoms of disease.
Potential CD includes subjects with positive serology but normal small bowel mucosa on a gluten-containing diet in whom CD may develop later in life. Finally, the term “latent” has been attributed to a “preclinical state” of CD, usually recognized retrospectively, or to patients with an earlier presentation of CD who recover on a GFD and later remain silent when gluten is reintroduced into the diet. Potency and latency might be transient and these patients should be followed clinically since some degree of villous atrophy with variable symptoms may develop in the future in about 80% of cases[102,111].
The only current treatment for CD involves a strict and life-long adherence to a GFD. With maintenance of a GFD, symptoms and serum celiac antibodies gradually disappear and healing of the intestinal damage typically occurs within 6 to 24 mo after initiation of the diet.
Refractory CD is diagnosed when there are persistent or recurrent malabsorptive symptoms and signs with villous atrophy detected on biopsy despite the maintenance of a strict GFD for more than 12 mo. Complications associated with untreated and/or refractory CD include ulcerative jejunoileitis, splenic hypofunction, enteropathy-associated T cell lymphoma and adenocarcinoma of the jejunum[101].
GLUTEN EFFECTS ON EPITHELIAL BARRIER AND INTESTINAL HOMEOSTASIS
The presence of gluten in the mucosa is a prerequisite for the activation of gluten-reactive T-cells and the ensuing inflammation. However, gluten also affects the intestinal mucosa by non-immune mediated mechanisms.
It has been demonstrated that gliadin-derived cytotoxic peptides can induce oxidative stress, rearrangement of actin cytoskeleton, impairment of epithelial tight junction assembly and deregulation of the epithelial homeostasis in experiments on cultured epithelial cells and celiac mucosa[112,113].
The oxidative stress induced by gliadin in epithelial cells might be responsible for the increased nuclear factor (NF)-κB activity and subsequent interleukin (IL)-15 transcription that is present in the small intestinal mucosa of celiac patients[114]. Epithelial NF-κB activation in healthy hosts is normally suppressed by anti-inflammatory cytokines produced by underlying T lymphocytes, such as transforming growth factor (TGF)-β and IL-10. In active CD, the status of chronic inflammation and the direct toxic effects of gluten worsen the epithelial layer damage, thus causing activation of NF-κB which leads to a vicious cycle of aberrant immune response, mucosal inflammation, increased mucosal permeability and impairment of the regenerative potential of the intestinal epithelium.
As for the alterations of the epithelial barrier, it is speculated that dysregulation of zonulin in many diseases may involve loss of cell junction integrity[115]. The endogenous zonulin, which is functionally and immunologically related to zonula occludins toxin from Vibrio cholera, has been found to disassemble intercellular tight junctions via interaction with cell membrane receptors. Serum zonulin is up-regulated in active CD and decreases following GFD, suggesting a role for a “leaky gut” in the development of autoimmunity[116]. Some gliadin peptides have been shown to bind to the chemokine receptor CXCR3 on the surface of epithelial cells and induce tight junction permeability and zonulin release[117].
Gliadin peptides can interfere with endocytic vesicle maturation and promote cell proliferation by prolonging epidermal growth factor receptor (EGFR) activation, which may correlate with the histological finding of crypt hyperplasia in CD[118]. Interestingly, p31-43 gluten peptide stimulation on proliferation of epithelial cells in vitro is dependent on IL-15 activity[108,118].
Furthermore, some toxic gliadin peptides have been reported to induce enterocyte apoptosis via the Fas-Fas ligand (FasL) pathway[119]. IL-15 has also been shown to induce enterocyte MICA expression in CD patients and to trigger the anti-apoptotic pathway in human IELs, which can kill intestinal epithelial cells[120].
ENVIRONMENTAL COFACTORS IN CELIAC DISEASE: GUT MICROBIOTA
Environmental cofactors that participate in the development and maintenance of CD include: intestinal pathogens that could enhance gluten immunogenicity and toxicity (i.e., rotavirus infections[121]); altered gut microbiota composition[122,123]; infant-feeding practices (with a reported 50% lower risk among infants who are still being breast-fed at the time of gluten introduction[124]); and some immune-modulatory drugs (i.e., IFN-alpha[125]) (Figure 3).
Figure 3.
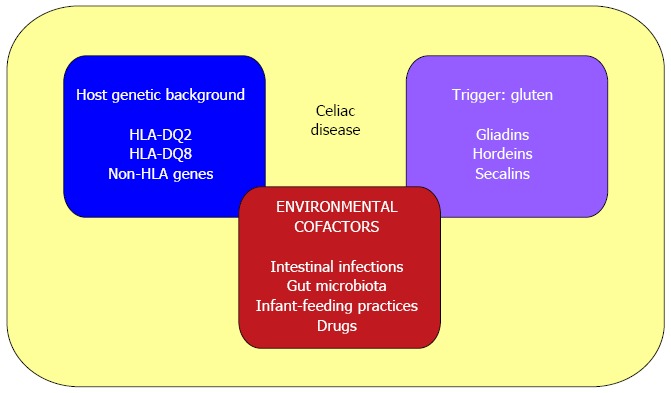
Causative factors in celiac disease. The pathogenesis of celiac disease involves: (1) host genetic background (HLA-DQ2 or DQ8 and other non-HLA genes); (2) an external trigger (gluten); and (3) environmental cofactors (such as intestinal pathogens, altered gut microbiota composition, infant-feeding practices and some immune-modulatory drugs).
In CD, the homeostatic mechanisms that allow coexistence of the host organism and the commensal microbiota are disrupted. Several studies have shown that celiac patients are characterized by a different composition of the gut microbiota when compared to healthy individuals. Rod shaped bacteria adhering to the small intestinal mucosa were frequently seen in patients with CD during the “Swedish CD epidemic”[126]. Nadal et al[127] demonstrated a higher proportion of total and gram-negative bacteria, also including potentially pro-inflammatory bacteria (Bacteroides-Prevotella and E. coli), in active CD children vs symptom-free patients and controls. Schippa et al[128] found a distinctive “microbial signature” in celiac patients, irrespective of the disease status. The duodenal mucosa of CD patients showed a higher diversity of associated bacteria population; Bacteroides vulgatus and E. coli were detected more often in celiacs than in controls[128].
The changes detected in gut microbiota of CD patients could be either a consequence or a cause of the disease. In the first scenario, the damaged mucosa covered by immature enterocytes would facilitate gram-negative bacterial colonization to the detriment of gram-positive bacteria. In the second case, the predominant colonization of gram-negative bacteria in genetically predisposed individuals would contribute to the loss of tolerance to gluten. Indeed, changes in resident microbiota composition seem to precede the onset of the disease and as such, they might be a risk factor for the development of celiac disease in susceptible individuals. Of note, interplay has been observed between HLA genes and milk feeding practice for microbial colonization that could influence the manifestation of the disease. The PROFICEL study demonstrated that infants at high genetic risk have higher numbers of B. fragilis and Staphylococcus spp. and reduced numbers of Bifidobacterium spp.; breast-feeding promoted colonization of Bifidobacteria, while formula-feeding promoted that of Bacteroides fragilis and E. coli, among others. In breast-fed infants, the increased genetic risk was associated with increased C. leptum group numbers, while in formula-fed infants it was associated with increased Staphylococcus and B. fragilis group numbers. Finally, breast-feeding reduced the genotype-related differences in microbiota composition, which could partly explain the protective role attributed to breast milk in this disorder[129].
Through a process of “cross-talk” with the mucosal immune system, gut microbiota negotiates mutual growth, survival and inflammatory control of the intestinal ecosystem. The intestinal mucosa is equipped with transmembrane and intracytoplasmic receptors referred to as pattern/pathogen recognition receptors (PRRs) that are defined by their ability to specifically recognize and bind distinctive microbial macromolecular ligands (microbial-associated or pathogen-associated molecular patterns, MAMPs or PAMPs), such as LPS, flagellin, peptidoglycans and formylated peptides. Subsequent signaling consists of an intricate and inter-relational pathway which determines the signaling output based on the initial perception of the triggering organism. Output can be a protective response to commensal microbiota, an inflammatory response to pathogenic organisms, or a trigger for apoptosis. Intestinal epithelial cells express high levels of the Toll-like receptor (TLR) inhibitor and Toll-interacting protein (TOLLIP). Expression of TOLLIP has been shown to correlate with the in vivo luminal bacterial load and is highest in healthy colonic mucosa; this inhibitory molecule is important in maintaining microbial homeostasis.
Many studies have demonstrated that the expression of TLRs is deregulated in active CD, suggesting that microbiota-associated factors may be important in the development of the disease. Higher densities of TLR4+ cells were found in active CD patients vs controls[130]. Recently, Kalliomäki et al[131] demonstrated that expression of IL-8 mRNA (marker of intestinal inflammation) and TLR-2 mRNA significantly increased in duodenal biopsies of active celiacs compared with treated celiacs and controls, while expression of TOLLIP mRNA was down-regulated.
The CD-associated bacteria and the dysbiosis they might cause in the resident microbiota through TLR/PAMP interactions might contribute to the Th1 pro-inflammatory milieu characteristic of CD. Medina et al[132] showed that gut microbiota from both active and treated CD patients increased TNF-α and IFN-γ production and decreased IL-10 production and CD4 expression in peripheral blood mononuclear cells compared with control samples. Interestingly, probiotics (Bifidobacterium strains) suppressed this pro-inflammatory cytokine pattern and increased IL-10 production. Similar beneficial effects of B. longum were found in an animal model of gliadin-induced enteropathy[133].
ISC MODULATION IN CD
Despite the many achievements in understanding of the pathogenic interactions among genetic, immunological and environmental factors in CD, little is known about ISC modulation and deregulation during the course of the disease.
In the last years it has been observed that ISC differentiation towards PCs and goblet cells may be disturbed in active CD. This may result in a defective antimicrobial and mucus barrier which enables the intestinal bacteria to invade the mucosa and trigger the inflammation. Indeed, the expression of natural antibiotics such as defensins is limited in CD. In particular, it has been demonstrated that some beta-defensins are underrepresented among celiac patients and that their expression correlated negatively with the degree of villous atrophy and rose on GFD; this suggests that increased copy numbers could protect from CD, possibly by impeding bacterial infiltration more efficiently and preserving gut epithelial integrity[134-136].
As for PC deregulation in CD, a number of studies have reported conflicting results. The earliest reports described the disappearance of PCs in patients with refractory CD and a significant decrease in patients with untreated and treated CD. However, later studies did not confirm a numeric reduction of celiac PCs and some authors even hypothesized that PCs would be increased in active CD given the high level of α-defensins found in untreated celiac mucosa[137]. Di Sabatino et al[137] adopted a multiple histochemical approach and showed no change of PC numbers in uncomplicated treated or untreated CD vs normal controls, while they observed a significant decrease of PCs in patients with complicated CD; of note, this decrease did not correlate with the degree of mucosal damage or with the duration of GFD. The proliferative pattern of PCs was not statistically different among the various groups, while crypt enterocyte proliferation was significantly higher in uncomplicated, untreated CD in comparison with treated CD and control cases[137]. More recently, Rubio found that in active CD patients, the normal production of PCs in the crypts is replaced by lysozyme-producing mucus cells. The author speculated that in CD, ISCs are re-programmed as an antimicrobial adaptation to signals generated by pathogenic duodenal bacteria[138]. The molecular mechanisms behind the abrogation of PCs in duodenal crypts and their substitution with lysozyme-producing mucus cells in CD remain to be elucidated. Further studies are needed to clarify the exact entity of PC deregulation in CD, the underlying molecular pathways and its implications in terms of ISC fate.
The intriguing hypothesis that PC secretion might be involved in the control neoplasia, thus accounting for the low incidence of neoplasms in the small bowel, encourages further investigation of the relationship between PC deficiency and premalignant and malignant complications of CD, as well as other inflammatory bowel disorders.
Regarding the deregulation of goblet cells in CD, Cinova et al[139] observed that gliadin fragments and/or IFN-γ were able to reduce the number of PAS-positive goblet cells and increase mucin secretion in rat intestinal loops; interestingly, these changes were more pronounced in the presence of potentially pathogenic enterobacteria, while the decrease in PAS-positive goblet cells by gliadin was reversed by probiotics (B. bifidum IATA-ES2).
The molecular mechanisms underlying the deregulation of ISC differentiation in CD are still being elucidated. Capuano et al[140] assessed the miRNA-based modulation of gene expression in the celiac small intestine for genes involved in intestinal differentiation and proliferation. They found a downregulation of the Notch pathway and KLF4 signals in celiac patients, whereas more nuclear beta-catenin staining (a sign of Wnt signaling activation) and more Ki67 staining (a sign of cell proliferation) were present in crypts from celiacs than in controls. Moreover, they documented a reduction of the number of goblet cells in the small intestine of children with active CD and in those on a GFD compared to controls. The authors postulated that the Notch pathway could be constitutively altered in CD and that it could drive the increased proliferation and the decreased differentiation of ISCs towards the secretory goblet cell lineage[140].
Another reported ISC niche alteration in CD regards the mucosal vasculature in the small intestine of active celiac patients that differs considerably from normal. Indeed, in celiac mucosa the capillary tufts are totally missing and the entire vasculature is disorganized. Myrsky et al[141] reported that IgA and anti-TG from CD patients disturb several steps of angiogenesis (sprouting and migration of endothelial and vascular mesenchymal cells) and also induce disorganization of the actin cytoskeleton in vitro. This disturbance of the angiogenic process could lead in vivo to the disruption of the mucosal vasculature seen in active CD[141].
Finally, little is known about the contribution of BM-derived SCs in CD. Mastrandrea et al[142] showed an increased traffic of circulating CD34+ HSCs in active CD patients vs healthy controls, but no correlation was found with anti-TG levels or histological severity. The authors postulated that this increased traffic of HSCs was more related to a defect shared by chronic inflammatory diseases than to a gliadin-specific Th1 local reaction. They hypothesized that the prevalence of apoptotic vs survival programs leading to excessive cell death in active CD might induce the mobilization of BM multipotent SCs as a supplementary source of ISCs for intestinal repair[142].
The potential contribution of extra-intestinal SCs in gut regeneration offers new insights into the development of SC-based treatments against CD.
NOVEL THERAPIES FOR CD: A ROLE FOR STEM CELLS?
Adherence to a strict GFD might restrict social activities and limit nutritional variety. Additionally, it is costly and difficult to maintain in many countries. In the last years, alternative therapeutic strategies have been tested. These include intraluminal therapies (genetic modification of wheat and/or pretreatment of flours to reduce immunotoxicity, oral enzyme therapy, intraluminal binding of gluten peptides, neutralizing gluten antibodies), transepithelial treatments (inhibition of intestinal permeability through zonulin receptor antagonists) and subepithelial actions (TG inhibitors, gluten peptides that downregulate innate responses, HLA-DQ2 inhibitors, CCR9 and integrin antagonists, IL-15 antagonists, anti-IFN-γ antibody, anti-CD3, anti-CD4 and anti-CD25 antibodies). Such approaches have been tested in experimental models and in small clinical trials with inconclusive results overall in terms of efficacy; moreover, some of these treatments have a poor safety profile and their hypothetical use should be reserved for complicated forms of CD[102,105].
Novel treatments for CD might derive from SCs. Indeed, the advancements in SC biology have led to the concept of regenerative medicine which is based on SC potential for therapies aimed at facilitating the repair of injured tissues[143]. Such therapies require a deep knowledge of the dynamics underlying SC compartment regulation, both in physiological and pathological conditions.
A potential therapeutic avenue for CD is the discovery of epithelial mitogens that stimulates mucosa growth. Recently, R-spondin-1 has been shown to stimulate crypt cell growth, accelerate mucosal regeneration and restore intestinal architecture in experimental colitis in mice[144]. In CD, the infusion of such mitogens might help to accelerate intestinal healing.
Another potential SC-based therapy for CD is transplantation of multipotent extraintestinal SCs of BM origin that can contribute to intestinal repair. In the last two decades, BM-derived SC transplantation has become an option for patients with severe autoimmune diseases refractory to conventional treatments. Such a therapy has recently found an application in gastroenterology for the treatment of selected cases of complicated CD. Bishton et al[145] reported the efficacy of autologous HSC transplantation preceded by conditioning in patients with enteropathy-associated T cell lymphoma: 4 out of 6 patients remained in a sustained complete remission for up to 4 years. Kline et al[146] treated one celiac patient affected by acute myelogenous leukemia with allogeneic HSC transplant preceded by conditioning and achieved correction of CD despite the reintroduction of a gluten-containing diet. Similarly, Hoekstra et al[147] reported that in one patient with severe aplastic anaemia and CD, allotransplant of SCs resulted in the cure of CD even after the return to a free diet. Recently, Ciccocioppo et al[148] showed that in 2 patients affected by CD and β-thalassemia major who underwent successful myeloablative allogeneic HSC transplantation for the latter condition, the introduction of a gluten-containing diet did not cause the reappearance of clinical, serological and histological markers of CD in up to 5 years of follow-up. Al-Toma et al[149] subjected patients with refractory CD type II to autologous peripheral blood SC transplant after conditioning: 6 out of 7 patients obtained a significant reduction in aberrant T cells in duodenal biopsies, associated with clinical improvement. Similar results were obtained by Tack et al[150] in refractory type-II CD patients who showed an impressive clinical improvement upon auto-transplant of HSCs.
Despite these encouraging results, further studies and longer follow-up periods are required to confirm the efficacy of HSC infusion in refractory and complicated CD.
The molecular mechanisms underlying the beneficial effects of HSC transplantation in CD are still largely unknown. It has been postulated that the immune system ablation followed by HSC transplantation provides a reset of the host immune system imbalance; this effect is likely to be more pronounced and prolonged in cases of allogeneic transplantation when HSCs that do not carry any CD-predisposing polymorphism are infused upon conditioning. Moreover, BM cells might contribute to tissue repair by differentiating into epithelial cells and myofibroblasts and by facilitating the neoangiogenesis[92,95].
CONCLUSION
SCs are the key to tissue genesis and regeneration. Given their central role in homeostasis, SC dysfunctions are involved in the pathogenesis of virtually all diseases, from cancers to degenerative disorders to chronic inflammatory pathologies. Ten years ago, SC research was compared to a “Pandora’s vase”, the opening of which could make it possible to clarify the nature and the pathophysiology of all human disease. If SCs are the source of all pathology, they can also be the ultimate cure; this is the precondition of regenerative medicine which is based on SC potential for tissue renewal and regeneration[2,7].
Despite the efforts made during the last 30 years, the body of knowledge of the physiology of ISCs and their involvement in bowel disorders appears fragmentary, incomplete and sometimes contradictory. As for CD, the role of ISCs and their niche in the development and maintenance of the disease is far from being elucidated and the clinical applications of SC-based treatments for CD are limited to a few case reports and uncontrolled trials, with small numbers of subjects affected by complicated disease.
Nonetheless, the expectations of the general population for SC-based therapies against CD are very high. Among the families that collect and bank cord blood for private storage, potential treatment of CD is the second most common motivation (19.7%)[151]. This imposes a careful consideration: further studies are needed to clarify the complex interplay among gluten, gut microbiota, gut barrier, immune system and ISC modulation and deregulation in CD. Such knowledge should be the basis for any potential clinical application of SCs against CD in order to avoid an “excess of enthusiasm” that might get “the better of judgment”.
Footnotes
P- Reviewers: Song LT, Sun J S- Editor: Song XX L- Editor: Roemmele A E- Editor: Zhang DN
References
- 1.Mimeault M, Hauke R, Batra SK. Stem cells: a revolution in therapeutics-recent advances in stem cell biology and their therapeutic applications in regenerative medicine and cancer therapies. Clin Pharmacol Ther. 2007;82:252–264. doi: 10.1038/sj.clpt.6100301. [DOI] [PubMed] [Google Scholar]
- 2.Piscaglia AC, Di Campli C, Gasbarrini G, Gasbarrini A. Stem cells: new tools in gastroenterology and hepatology. Dig Liver Dis. 2003;35:507–514. doi: 10.1016/s1590-8658(03)00226-3. [DOI] [PubMed] [Google Scholar]
- 3.Lajtha LG. Stem cell concepts. Differentiation. 1979;14:23–34. doi: 10.1111/j.1432-0436.1979.tb01007.x. [DOI] [PubMed] [Google Scholar]
- 4.Potten CS, Loeffler M. Stem cells: attributes, cycles, spirals, pitfalls and uncertainties. Lessons for and from the crypt. Development. 1990;110:1001–1020. doi: 10.1242/dev.110.4.1001. [DOI] [PubMed] [Google Scholar]
- 5.Piscaglia AC, Shupe T, Gasbarrini A, Petersen BE. Microarray RNA/DNA in different stem cell lines. Curr Pharm Biotechnol. 2007;8:167–175. doi: 10.2174/138920107780906478. [DOI] [PubMed] [Google Scholar]
- 6.Piscaglia AC, Shupe TD, Petersen BE, Gasbarrini A. Stem cells, cancer, liver, and liver cancer stem cells: finding a way out of the labyrinth. Curr Cancer Drug Targets. 2007;7:582–590. doi: 10.2174/156800907781662293. [DOI] [PubMed] [Google Scholar]
- 7.Piscaglia AC, Novi M, Campanale M, Gasbarrini A. Stem cell-based therapy in gastroenterology and hepatology. Minim Invasive Ther Allied Technol. 2008;17:100–118. doi: 10.1080/13645700801969980. [DOI] [PubMed] [Google Scholar]
- 8.Wu DC, Boyd AS, Wood KJ. Embryonic stem cell transplantation: potential applicability in cell replacement therapy and regenerative medicine. Front Biosci. 2007;12:4525–4535. doi: 10.2741/2407. [DOI] [PubMed] [Google Scholar]
- 9.Ferreira LM, Mostajo-Radji MA. How induced pluripotent stem cells are redefining personalized medicine. Gene. 2013;520:1–6. doi: 10.1016/j.gene.2013.02.037. [DOI] [PubMed] [Google Scholar]
- 10.Song L. Gene Therapy of Some Genetic Diseases by Transferring Normal Human Genomic DNA Into Somatic Cells and Stem Cells From Patients. In: Yuan XB, editor. Non-Viral Gene Therapy. Croatia: InTech; 2011. [Google Scholar]
- 11.Tarnowski M, Sieron AL. Adult stem cells and their ability to differentiate. Med Sci Monit. 2006;12:RA154–RA163. [PubMed] [Google Scholar]
- 12.Körbling M, Anderlini P. Peripheral blood stem cell versus bone marrow allotransplantation: does the source of hematopoietic stem cells matter? Blood. 2001;98:2900–2908. doi: 10.1182/blood.v98.10.2900. [DOI] [PubMed] [Google Scholar]
- 13.Guo Y, Lübbert M, Engelhardt M. CD34- hematopoietic stem cells: current concepts and controversies. Stem Cells. 2003;21:15–20. doi: 10.1634/stemcells.21-1-15. [DOI] [PubMed] [Google Scholar]
- 14.Bryder D, Rossi DJ, Weissman IL. Hematopoietic stem cells: the paradigmatic tissue-specific stem cell. Am J Pathol. 2006;169:338–346. doi: 10.2353/ajpath.2006.060312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hüttmann A, Dührsen U, Heydarian K, Klein-Hitpass L, Boes T, Boyd AW, Li CL. Gene expression profiles in murine hematopoietic stem cells revisited: analysis of cDNA libraries reveals high levels of translational and metabolic activities. Stem Cells. 2006;24:1719–1727. doi: 10.1634/stemcells.2005-0486. [DOI] [PubMed] [Google Scholar]
- 16.Ratajczak MZ, Kucia M, Reca R, Majka M, Janowska-Wieczorek A, Ratajczak J. Stem cell plasticity revisited: CXCR4-positive cells expressing mRNA for early muscle, liver and neural cells ‘hide out’ in the bone marrow. Leukemia. 2004;18:29–40. doi: 10.1038/sj.leu.2403184. [DOI] [PubMed] [Google Scholar]
- 17.Tocci A, Forte L. Mesenchymal stem cell: use and perspectives. Hematol J. 2003;4:92–96. doi: 10.1038/sj.thj.6200232. [DOI] [PubMed] [Google Scholar]
- 18.Jiang Y, Jahagirdar BN, Reinhardt RL, Schwartz RE, Keene CD, Ortiz-Gonzalez XR, Reyes M, Lenvik T, Lund T, Blackstad M, et al. Pluripotency of mesenchymal stem cells derived from adult marrow. Nature. 2002;418:41–49. doi: 10.1038/nature00870. [DOI] [PubMed] [Google Scholar]
- 19.Puglisi MA, Tesori V, Lattanzi W, Piscaglia AC, Gasbarrini GB, D’Ugo DM, Gasbarrini A. Therapeutic implications of mesenchymal stem cells in liver injury. J Biomed Biotechnol. 2011;2011:860578. doi: 10.1155/2011/860578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li L, Xie T. Stem cell niche: structure and function. Annu Rev Cell Dev Biol. 2005;21:605–631. doi: 10.1146/annurev.cellbio.21.012704.131525. [DOI] [PubMed] [Google Scholar]
- 21.Karam SM. Lineage commitment and maturation of epithelial cells in the gut. Front Biosci. 1999;4:D286–D298. doi: 10.2741/karam. [DOI] [PubMed] [Google Scholar]
- 22.Brittan M, Wright NA. Gastrointestinal stem cells. J Pathol. 2002;197:492–509. doi: 10.1002/path.1155. [DOI] [PubMed] [Google Scholar]
- 23.Lin SA, Barker N. Gastrointestinal stem cells in self-renewal and cancer. J Gastroenterol. 2011;46:1039–1055. doi: 10.1007/s00535-011-0424-8. [DOI] [PubMed] [Google Scholar]
- 24.Cheng H, Leblond CP. Origin, differentiation and renewal of the four main epithelial cell types in the mouse small intestine. V. Unitarian Theory of the origin of the four epithelial cell types. Am J Anat. 1974;141:537–561. doi: 10.1002/aja.1001410407. [DOI] [PubMed] [Google Scholar]
- 25.Hendry JH, Potten CS. Cryptogenic cells and proliferative cells in intestinal epithelium. Int J Radiat Biol Relat Stud Phys Chem Med. 1974;25:583–588. doi: 10.1080/09553007414550771. [DOI] [PubMed] [Google Scholar]
- 26.Bjerknes M, Cheng H. Intestinal epithelial stem cells and progenitors. Methods Enzymol. 2006;419:337–383. doi: 10.1016/S0076-6879(06)19014-X. [DOI] [PubMed] [Google Scholar]
- 27.Barker N, van Es JH, Kuipers J, Kujala P, van den Born M, Cozijnsen M, Haegebarth A, Korving J, Begthel H, Peters PJ, et al. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature. 2007;449:1003–1007. doi: 10.1038/nature06196. [DOI] [PubMed] [Google Scholar]
- 28.Vries RG, Huch M, Clevers H. Stem cells and cancer of the stomach and intestine. Mol Oncol. 2010;4:373–384. doi: 10.1016/j.molonc.2010.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Clevers HC, Bevins CL. Paneth cells: maestros of the small intestinal crypts. Annu Rev Physiol. 2013;75:289–311. doi: 10.1146/annurev-physiol-030212-183744. [DOI] [PubMed] [Google Scholar]
- 30.Noah TK, Donahue B, Shroyer NF. Intestinal development and differentiation. Exp Cell Res. 2011;317:2702–2710. doi: 10.1016/j.yexcr.2011.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Potten CS, Kovacs L, Hamilton E. Continuous labelling studies on mouse skin and intestine. Cell Tissue Kinet. 1974;7:271–283. doi: 10.1111/j.1365-2184.1974.tb00907.x. [DOI] [PubMed] [Google Scholar]
- 32.Sato T, Vries RG, Snippert HJ, van de Wetering M, Barker N, Stange DE, van Es JH, Abo A, Kujala P, Peters PJ, et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature. 2009;459:262–265. doi: 10.1038/nature07935. [DOI] [PubMed] [Google Scholar]
- 33.Zhu L, Gibson P, Currle DS, Tong Y, Richardson RJ, Bayazitov IT, Poppleton H, Zakharenko S, Ellison DW, Gilbertson RJ. Prominin 1 marks intestinal stem cells that are susceptible to neoplastic transformation. Nature. 2009;457:603–607. doi: 10.1038/nature07589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Snippert HJ, van Es JH, van den Born M, Begthel H, Stange DE, Barker N, Clevers H. Prominin-1/CD133 marks stem cells and early progenitors in mouse small intestine. Gastroenterology. 2009;136:2187–2194.e1. doi: 10.1053/j.gastro.2009.03.002. [DOI] [PubMed] [Google Scholar]
- 35.Quante M, Wang TC. Stem cells in gastroenterology and hepatology. Nat Rev Gastroenterol Hepatol. 2009;6:724–737. doi: 10.1038/nrgastro.2009.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Scoville DH, Sato T, He XC, Li L. Current view: intestinal stem cells and signaling. Gastroenterology. 2008;134:849–864. doi: 10.1053/j.gastro.2008.01.079. [DOI] [PubMed] [Google Scholar]
- 37.Sangiorgi E, Capecchi MR. Bmi1 is expressed in vivo in intestinal stem cells. Nat Genet. 2008;40:915–920. doi: 10.1038/ng.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tian H, Biehs B, Warming S, Leong KG, Rangell L, Klein OD, de Sauvage FJ. A reserve stem cell population in small intestine renders Lgr5-positive cells dispensable. Nature. 2011;478:255–259. doi: 10.1038/nature10408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Takeda N, Jain R, LeBoeuf MR, Wang Q, Lu MM, Epstein JA. Interconversion between intestinal stem cell populations in distinct niches. Science. 2011;334:1420–1424. doi: 10.1126/science.1213214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vanuytsel T, Senger S, Fasano A, Shea-Donohue T. Major signaling pathways in intestinal stem cells. Biochim Biophys Acta. 2013;1830:2410–2426. doi: 10.1016/j.bbagen.2012.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Körbling M, Katz RL, Khanna A, Ruifrok AC, Rondon G, Albitar M, Champlin RE, Estrov Z. Hepatocytes and epithelial cells of donor origin in recipients of peripheral-blood stem cells. N Engl J Med. 2002;346:738–746. doi: 10.1056/NEJMoa3461002. [DOI] [PubMed] [Google Scholar]
- 42.Krause DS, Theise ND, Collector MI, Henegariu O, Hwang S, Gardner R, Neutzel S, Sharkis SJ. Multi-organ, multi-lineage engraftment by a single bone marrow-derived stem cell. Cell. 2001;105:369–377. doi: 10.1016/s0092-8674(01)00328-2. [DOI] [PubMed] [Google Scholar]
- 43.Brittan M, Wright NA. Stem cell in gastrointestinal structure and neoplastic development. Gut. 2004;53:899–910. doi: 10.1136/gut.2003.025478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brittan M, Chance V, Elia G, Poulsom R, Alison MR, MacDonald TT, Wright NA. A regenerative role for bone marrow following experimental colitis: contribution to neovasculogenesis and myofibroblasts. Gastroenterology. 2005;128:1984–1995. doi: 10.1053/j.gastro.2005.03.028. [DOI] [PubMed] [Google Scholar]
- 45.Kudo K, Liu Y, Takahashi K, Tarusawa K, Osanai M, Hu DL, Kashiwakura I, Kijima H, Nakane A. Transplantation of mesenchymal stem cells to prevent radiation-induced intestinal injury in mice. J Radiat Res. 2010;51:73–79. doi: 10.1269/jrr.09091. [DOI] [PubMed] [Google Scholar]
- 46.Yabana T, Arimura Y, Tanaka H, Goto A, Hosokawa M, Nagaishi K, Yamashita K, Yamamoto H, Adachi Y, Sasaki Y, et al. Enhancing epithelial engraftment of rat mesenchymal stem cells restores epithelial barrier integrity. J Pathol. 2009;218:350–359. doi: 10.1002/path.2535. [DOI] [PubMed] [Google Scholar]
- 47.Hayashi Y, Tsuji S, Tsujii M, Nishida T, Ishii S, Nakamura T, Eguchi H, Kawano S. The transdifferentiation of bone-marrow-derived cells in colonic mucosal regeneration after dextran-sulfate-sodium-induced colitis in mice. Pharmacology. 2007;80:193–199. doi: 10.1159/000104148. [DOI] [PubMed] [Google Scholar]
- 48.Zhang J, Gong JF, Zhang W, Zhu WM, Li JS. Effects of transplanted bone marrow mesenchymal stem cells on the irradiated intestine of mice. J Biomed Sci. 2008;15:585–594. doi: 10.1007/s11373-008-9256-9. [DOI] [PubMed] [Google Scholar]
- 49.Colletti E, El Shabrawy D, Soland M, Yamagami T, Mokhtari S, Osborne C, Schlauch K, Zanjani ED, Porada CD, Almeida-Porada G. EphB2 isolates a human marrow stromal cell subpopulation with enhanced ability to contribute to the resident intestinal cellular pool. FASEB J. 2013;27:2111–2121. doi: 10.1096/fj.12-205054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen S, Lewallen M, Xie T. Adhesion in the stem cell niche: biological roles and regulation. Development. 2013;140:255–265. doi: 10.1242/dev.083139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sato T, van Es JH, Snippert HJ, Stange DE, Vries RG, van den Born M, Barker N, Shroyer NF, van de Wetering M, Clevers H. Paneth cells constitute the niche for Lgr5 stem cells in intestinal crypts. Nature. 2011;469:415–418. doi: 10.1038/nature09637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Parry L, Young M, El Marjou F, Clarke AR. Evidence for a crucial role of paneth cells in mediating the intestinal response to injury. Stem Cells. 2013;31:776–785. doi: 10.1002/stem.1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yilmaz ÖH, Katajisto P, Lamming DW, Gültekin Y, Bauer-Rowe KE, Sengupta S, Birsoy K, Dursun A, Yilmaz VO, Selig M, et al. mTORC1 in the Paneth cell niche couples intestinal stem-cell function to calorie intake. Nature. 2012;486:490–495. doi: 10.1038/nature11163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Snippert HJ, van der Flier LG, Sato T, van Es JH, van den Born M, Kroon-Veenboer C, Barker N, Klein AM, van Rheenen J, Simons BD, et al. Intestinal crypt homeostasis results from neutral competition between symmetrically dividing Lgr5 stem cells. Cell. 2010;143:134–144. doi: 10.1016/j.cell.2010.09.016. [DOI] [PubMed] [Google Scholar]
- 55.Morrison SJ, Spradling AC. Stem cells and niches: mechanisms that promote stem cell maintenance throughout life. Cell. 2008;132:598–611. doi: 10.1016/j.cell.2008.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Brabletz S, Schmalhofer O, Brabletz T. Gastrointestinal stem cells in development and cancer. J Pathol. 2009;217:307–317. doi: 10.1002/path.2475. [DOI] [PubMed] [Google Scholar]
- 57.He TC, Sparks AB, Rago C, Hermeking H, Zawel L, da Costa LT, Morin PJ, Vogelstein B, Kinzler KW. Identification of c-MYC as a target of the APC pathway. Science. 1998;281:1509–1512. doi: 10.1126/science.281.5382.1509. [DOI] [PubMed] [Google Scholar]
- 58.Shtutman M, Zhurinsky J, Simcha I, Albanese C, D’Amico M, Pestell R, Ben-Ze’ev A. The cyclin D1 gene is a target of the beta-catenin/LEF-1 pathway. Proc Natl Acad Sci USA. 1999;96:5522–5527. doi: 10.1073/pnas.96.10.5522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.de Lau W, Barker N, Low TY, Koo BK, Li VS, Teunissen H, Kujala P, Haegebarth A, Peters PJ, van de Wetering M, et al. Lgr5 homologues associate with Wnt receptors and mediate R-spondin signalling. Nature. 2011;476:293–297. doi: 10.1038/nature10337. [DOI] [PubMed] [Google Scholar]
- 60.Carmon KS, Gong X, Lin Q, Thomas A, Liu Q. R-spondins function as ligands of the orphan receptors LGR4 and LGR5 to regulate Wnt/beta-catenin signaling. Proc Natl Acad Sci USA. 2011;108:11452–11457. doi: 10.1073/pnas.1106083108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kim KA, Kakitani M, Zhao J, Oshima T, Tang T, Binnerts M, Liu Y, Boyle B, Park E, Emtage P, et al. Mitogenic influence of human R-spondin1 on the intestinal epithelium. Science. 2005;309:1256–1259. doi: 10.1126/science.1112521. [DOI] [PubMed] [Google Scholar]
- 62.Ghaleb AM, McConnell BB, Kaestner KH, Yang VW. Altered intestinal epithelial homeostasis in mice with intestine-specific deletion of the Krüppel-like factor 4 gene. Dev Biol. 2011;349:310–320. doi: 10.1016/j.ydbio.2010.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chen X, Johns DC, Geiman DE, Marban E, Dang DT, Hamlin G, Sun R, Yang VW. Krüppel-like factor 4 (gut-enriched Krüppel-like factor) inhibits cell proliferation by blocking G1/S progression of the cell cycle. J Biol Chem. 2001;276:30423–30428. doi: 10.1074/jbc.M101194200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wang Y, Giel-Moloney M, Rindi G, Leiter AB. Enteroendocrine precursors differentiate independently of Wnt and form serotonin expressing adenomas in response to active beta-catenin. Proc Natl Acad Sci USA. 2007;104:11328–11333. doi: 10.1073/pnas.0702665104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Clevers H, Batlle E. EphB/EphrinB receptors and Wnt signaling in colorectal cancer. Cancer Res. 2006;66:2–5. doi: 10.1158/0008-5472.CAN-05-3849. [DOI] [PubMed] [Google Scholar]
- 66.Batlle E, Henderson JT, Beghtel H, van den Born MM, Sancho E, Huls G, Meeldijk J, Robertson J, van de Wetering M, Pawson T, et al. Beta-catenin and TCF mediate cell positioning in the intestinal epithelium by controlling the expression of EphB/ephrinB. Cell. 2002;111:251–263. doi: 10.1016/s0092-8674(02)01015-2. [DOI] [PubMed] [Google Scholar]
- 67.Stokowski A, Shi S, Sun T, Bartold PM, Koblar SA, Gronthos S. EphB/ephrin-B interaction mediates adult stem cell attachment, spreading, and migration: implications for dental tissue repair. Stem Cells. 2007;25:156–164. doi: 10.1634/stemcells.2006-0373. [DOI] [PubMed] [Google Scholar]
- 68.Holmberg J, Genander M, Halford MM, Annerén C, Sondell M, Chumley MJ, Silvany RE, Henkemeyer M, Frisén J. EphB receptors coordinate migration and proliferation in the intestinal stem cell niche. Cell. 2006;125:1151–1163. doi: 10.1016/j.cell.2006.04.030. [DOI] [PubMed] [Google Scholar]
- 69.van Es JH, Jay P, Gregorieff A, van Gijn ME, Jonkheer S, Hatzis P, Thiele A, van den Born M, Begthel H, Brabletz T, et al. Wnt signalling induces maturation of Paneth cells in intestinal crypts. Nat Cell Biol. 2005;7:381–386. doi: 10.1038/ncb1240. [DOI] [PubMed] [Google Scholar]
- 70.Genander M, Halford MM, Xu NJ, Eriksson M, Yu Z, Qiu Z, Martling A, Greicius G, Thakar S, Catchpole T, et al. Dissociation of EphB2 signaling pathways mediating progenitor cell proliferation and tumor suppression. Cell. 2009;139:679–692. doi: 10.1016/j.cell.2009.08.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hafner C, Meyer S, Hagen I, Becker B, Roesch A, Landthaler M, Vogt T. Ephrin-B reverse signaling induces expression of wound healing associated genes in IEC-6 intestinal epithelial cells. World J Gastroenterol. 2005;11:4511–4518. doi: 10.3748/wjg.v11.i29.4511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hafner C, Meyer S, Langmann T, Schmitz G, Bataille F, Hagen I, Becker B, Roesch A, Rogler G, Landthaler M, et al. Ephrin-B2 is differentially expressed in the intestinal epithelium in Crohn’s disease and contributes to accelerated epithelial wound healing in vitro. World J Gastroenterol. 2005;11:4024–4031. doi: 10.3748/wjg.v11.i26.4024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.de Sousa EM, Vermeulen L, Richel D, Medema JP. Targeting Wnt signaling in colon cancer stem cells. Clin Cancer Res. 2011;17:647–653. doi: 10.1158/1078-0432.CCR-10-1204. [DOI] [PubMed] [Google Scholar]
- 74.Bray SJ. Notch signalling: a simple pathway becomes complex. Nat Rev Mol Cell Biol. 2006;7:678–689. doi: 10.1038/nrm2009. [DOI] [PubMed] [Google Scholar]
- 75.Kayahara T, Sawada M, Takaishi S, Fukui H, Seno H, Fukuzawa H, Suzuki K, Hiai H, Kageyama R, Okano H, et al. Candidate markers for stem and early progenitor cells, Musashi-1 and Hes1, are expressed in crypt base columnar cells of mouse small intestine. FEBS Lett. 2003;535:131–135. doi: 10.1016/s0014-5793(02)03896-6. [DOI] [PubMed] [Google Scholar]
- 76.Potten CS, Booth C, Tudor GL, Booth D, Brady G, Hurley P, Ashton G, Clarke R, Sakakibara S, Okano H. Identification of a putative intestinal stem cell and early lineage marker; musashi-1. Differentiation. 2003;71:28–41. doi: 10.1046/j.1432-0436.2003.700603.x. [DOI] [PubMed] [Google Scholar]
- 77.Kolterud A, Grosse AS, Zacharias WJ, Walton KD, Kretovich KE, Madison BB, Waghray M, Ferris JE, Hu C, Merchant JL, et al. Paracrine Hedgehog signaling in stomach and intestine: new roles for hedgehog in gastrointestinal patterning. Gastroenterology. 2009;137:618–628. doi: 10.1053/j.gastro.2009.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Madison BB, Braunstein K, Kuizon E, Portman K, Qiao XT, Gumucio DL. Epithelial hedgehog signals pattern the intestinal crypt-villus axis. Development. 2005;132:279–289. doi: 10.1242/dev.01576. [DOI] [PubMed] [Google Scholar]
- 79.van den Brink GR, Bleuming SA, Hardwick JC, Schepman BL, Offerhaus GJ, Keller JJ, Nielsen C, Gaffield W, van Deventer SJ, Roberts DJ, et al. Indian Hedgehog is an antagonist of Wnt signaling in colonic epithelial cell differentiation. Nat Genet. 2004;36:277–282. doi: 10.1038/ng1304. [DOI] [PubMed] [Google Scholar]
- 80.Lees CW, Zacharias WJ, Tremelling M, Noble CL, Nimmo ER, Tenesa A, Cornelius J, Torkvist L, Kao J, Farrington S, et al. Analysis of germline GLI1 variation implicates hedgehog signalling in the regulation of intestinal inflammatory pathways. PLoS Med. 2008;5:e239. doi: 10.1371/journal.pmed.0050239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.He XC, Zhang J, Tong WG, Tawfik O, Ross J, Scoville DH, Tian Q, Zeng X, He X, Wiedemann LM, et al. BMP signaling inhibits intestinal stem cell self-renewal through suppression of Wnt-beta-catenin signaling. Nat Genet. 2004;36:1117–1121. doi: 10.1038/ng1430. [DOI] [PubMed] [Google Scholar]
- 82.von Bubnoff A, Cho KW. Intracellular BMP signaling regulation in vertebrates: pathway or network? Dev Biol. 2001;239:1–14. doi: 10.1006/dbio.2001.0388. [DOI] [PubMed] [Google Scholar]
- 83.Kosinski C, Li VS, Chan AS, Zhang J, Ho C, Tsui WY, Chan TL, Mifflin RC, Powell DW, Yuen ST, et al. Gene expression patterns of human colon tops and basal crypts and BMP antagonists as intestinal stem cell niche factors. Proc Natl Acad Sci USA. 2007;104:15418–15423. doi: 10.1073/pnas.0707210104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Auclair BA, Benoit YD, Rivard N, Mishina Y, Perreault N. Bone morphogenetic protein signaling is essential for terminal differentiation of the intestinal secretory cell lineage. Gastroenterology. 2007;133:887–896. doi: 10.1053/j.gastro.2007.06.066. [DOI] [PubMed] [Google Scholar]
- 85.Cully M, You H, Levine AJ, Mak TW. Beyond PTEN mutations: the PI3K pathway as an integrator of multiple inputs during tumorigenesis. Nat Rev Cancer. 2006;6:184–192. doi: 10.1038/nrc1819. [DOI] [PubMed] [Google Scholar]
- 86.Parsons DW, Wang TL, Samuels Y, Bardelli A, Cummins JM, DeLong L, Silliman N, Ptak J, Szabo S, Willson JK, et al. Colorectal cancer: mutations in a signalling pathway. Nature. 2005;436:792. doi: 10.1038/436792a. [DOI] [PubMed] [Google Scholar]
- 87.He XC, Yin T, Grindley JC, Tian Q, Sato T, Tao WA, Dirisina R, Porter-Westpfahl KS, Hembree M, Johnson T, et al. PTEN-deficient intestinal stem cells initiate intestinal polyposis. Nat Genet. 2007;39:189–198. doi: 10.1038/ng1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kim S, Domon-Dell C, Wang Q, Chung DH, Di Cristofano A, Pandolfi PP, Freund JN, Evers BM. PTEN and TNF-alpha regulation of the intestinal-specific Cdx-2 homeobox gene through a PI3K, PKB/Akt, and NF-kappaB-dependent pathway. Gastroenterology. 2002;123:1163–1178. doi: 10.1053/gast.2002.36043. [DOI] [PubMed] [Google Scholar]
- 89.Yeung TM, Chia LA, Kosinski CM, Kuo CJ. Regulation of self-renewal and differentiation by the intestinal stem cell niche. Cell Mol Life Sci. 2011;68:2513–2523. doi: 10.1007/s00018-011-0687-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wehkamp J, Wang G, Kübler I, Nuding S, Gregorieff A, Schnabel A, Kays RJ, Fellermann K, Burk O, Schwab M, et al. The Paneth cell alpha-defensin deficiency of ileal Crohn’s disease is linked to Wnt/Tcf-4. J Immunol. 2007;179:3109–3118. doi: 10.4049/jimmunol.179.5.3109. [DOI] [PubMed] [Google Scholar]
- 91.Yin X, Farin HF, van Es JH, Clevers H, Langer R, Karp JM. Niche-independent high-purity cultures of Lgr5+ intestinal stem cells and their progeny. Nat Methods. 2014;11:106–112. doi: 10.1038/nmeth.2737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Brittan M, Alison MR, Schier S, Wright NA. Bone marrow stem cell-mediated regeneration in IBD: where do we go from here? Gastroenterology. 2007;132:1171–1173. doi: 10.1053/j.gastro.2007.01.064. [DOI] [PubMed] [Google Scholar]
- 93.Almeida-Porada G, Soland M, Boura J, Porada CD. Regenerative medicine: prospects for the treatment of inflammatory bowel disease. Regen Med. 2013;8:631–644. doi: 10.2217/rme.13.52. [DOI] [PubMed] [Google Scholar]
- 94.Gersemann M, Stange EF, Wehkamp J. From intestinal stem cells to inflammatory bowel diseases. World J Gastroenterol. 2011;17:3198–3203. doi: 10.3748/wjg.v17.i27.3198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Khalil PN, Weiler V, Nelson PJ, Khalil MN, Moosmann S, Mutschler WE, Siebeck M, Huss R. Nonmyeloablative stem cell therapy enhances microcirculation and tissue regeneration in murine inflammatory bowel disease. Gastroenterology. 2007;132:944–954. doi: 10.1053/j.gastro.2006.12.029. [DOI] [PubMed] [Google Scholar]
- 96.Deng X, Szabo S, Chen L, Paunovic B, Khomenko T, Tolstanova G, Tarnawski AS, Jones MK, Sandor Z. New cell therapy using bone marrow-derived stem cells/endothelial progenitor cells to accelerate neovascularization in healing of experimental ulcerative colitis. Curr Pharm Des. 2011;17:1643–1651. doi: 10.2174/138161211796197007. [DOI] [PubMed] [Google Scholar]
- 97.Komori M, Tsuji S, Tsujii M, Murata H, Iijima H, Yasumaru M, Nishida T, Irie T, Kawano S, Hori M. Efficiency of bone marrow-derived cells in regeneration of the stomach after induction of ethanol-induced ulcers in rats. J Gastroenterol. 2005;40:591–599. doi: 10.1007/s00535-005-1593-0. [DOI] [PubMed] [Google Scholar]
- 98.Sanders KM. Interstitial cells of Cajal at the clinical and scientific interface. J Physiol. 2006;576:683–687. doi: 10.1113/jphysiol.2006.116814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Losowsky MS. A history of coeliac disease. Dig Dis. 2008;26:112–120. doi: 10.1159/000116768. [DOI] [PubMed] [Google Scholar]
- 100.Dowd B, Walker-Smith J. Samuel Gee, Aretaeus, and the coeliac affection. Br Med J. 1974;2:45–47. doi: 10.1136/bmj.2.5909.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Fasano A, Catassi C. Clinical practice. Celiac disease. N Engl J Med. 2012;367:2419–2426. doi: 10.1056/NEJMcp1113994. [DOI] [PubMed] [Google Scholar]
- 102.Di Sabatino A, Corazza GR. Coeliac disease. Lancet. 2009;373:1480–1493. doi: 10.1016/S0140-6736(09)60254-3. [DOI] [PubMed] [Google Scholar]
- 103.Mustalahti K, Catassi C, Reunanen A, Fabiani E, Heier M, McMillan S, Murray L, Metzger MH, Gasparin M, Bravi E, et al. The prevalence of celiac disease in Europe: results of a centralized, international mass screening project. Ann Med. 2010;42:587–595. doi: 10.3109/07853890.2010.505931. [DOI] [PubMed] [Google Scholar]
- 104.Trynka G, Hunt KA, Bockett NA, Romanos J, Mistry V, Szperl A, Bakker SF, Bardella MT, Bhaw-Rosun L, Castillejo G, et al. Dense genotyping identifies and localizes multiple common and rare variant association signals in celiac disease. Nat Genet. 2011;43:1193–1201. doi: 10.1038/ng.998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Schuppan D, Junker Y, Barisani D. Celiac disease: from pathogenesis to novel therapies. Gastroenterology. 2009;137:1912–1933. doi: 10.1053/j.gastro.2009.09.008. [DOI] [PubMed] [Google Scholar]
- 106.Karell K, Louka AS, Moodie SJ, Ascher H, Clot F, Greco L, Ciclitira PJ, Sollid LM, Partanen J. HLA types in celiac disease patients not carrying the DQA1*05-DQB1*02 (DQ2) heterodimer: results from the European Genetics Cluster on Celiac Disease. Hum Immunol. 2003;64:469–477. doi: 10.1016/s0198-8859(03)00027-2. [DOI] [PubMed] [Google Scholar]
- 107.Husby S, Koletzko S, Korponay-Szabó IR, Mearin ML, Phillips A, Shamir R, Troncone R, Giersiepen K, Branski D, Catassi C, et al. European Society for Pediatric Gastroenterology, Hepatology, and Nutrition guidelines for the diagnosis of coeliac disease. J Pediatr Gastroenterol Nutr. 2012;54:136–160. doi: 10.1097/MPG.0b013e31821a23d0. [DOI] [PubMed] [Google Scholar]
- 108.Maiuri L, Ciacci C, Ricciardelli I, Vacca L, Raia V, Auricchio S, Picard J, Osman M, Quaratino S, Londei M. Association between innate response to gliadin and activation of pathogenic T cells in coeliac disease. Lancet. 2003;362:30–37. doi: 10.1016/S0140-6736(03)13803-2. [DOI] [PubMed] [Google Scholar]
- 109.Hüe S, Mention JJ, Monteiro RC, Zhang S, Cellier C, Schmitz J, Verkarre V, Fodil N, Bahram S, Cerf-Bensussan N, et al. A direct role for NKG2D/MICA interaction in villous atrophy during celiac disease. Immunity. 2004;21:367–377. doi: 10.1016/j.immuni.2004.06.018. [DOI] [PubMed] [Google Scholar]
- 110.De Nitto D, Monteleone I, Franzè E, Pallone F, Monteleone G. Involvement of interleukin-15 and interleukin-21, two gamma-chain-related cytokines, in celiac disease. World J Gastroenterol. 2009;15:4609–4614. doi: 10.3748/wjg.15.4609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Harris LA, Watkins D, Williams LD, Koudelka GB. Indirect readout of DNA sequence by p22 repressor: roles of DNA and protein functional groups in modulating DNA conformation. J Mol Biol. 2013;425:133–143. doi: 10.1016/j.jmb.2012.10.008. [DOI] [PubMed] [Google Scholar]
- 112.Luciani A, Villella VR, Vasaturo A, Giardino I, Pettoello-Mantovani M, Guido S, Cexus ON, Peake N, Londei M, Quaratino S, Maiuri L. Lysosomal accumulation of gliadin p31-43 peptide induces oxidative stress and tissue transglutaminase-mediated PPARgamma downregulation in intestinal epithelial cells and coeliac mucosa. Gut. 2010;59:311–319. doi: 10.1136/gut.2009.183608. [DOI] [PubMed] [Google Scholar]
- 113.Reinke Y, Behrendt M, Schmidt S, Zimmer KP, Naim HY. Impairment of protein trafficking by direct interaction of gliadin peptides with actin. Exp Cell Res. 2011;317:2124–2135. doi: 10.1016/j.yexcr.2011.05.022. [DOI] [PubMed] [Google Scholar]
- 114.Maiuri MC, De Stefano D, Mele G, Fecarotta S, Greco L, Troncone R, Carnuccio R. Nuclear factor kappa B is activated in small intestinal mucosa of celiac patients. J Mol Med (Berl) 2003;81:373–379. doi: 10.1007/s00109-003-0440-0. [DOI] [PubMed] [Google Scholar]
- 115.Fasano A, Not T, Wang W, Uzzau S, Berti I, Tommasini A, Goldblum SE. Zonulin, a newly discovered modulator of intestinal permeability, and its expression in coeliac disease. Lancet. 2000;355:1518–1519. doi: 10.1016/S0140-6736(00)02169-3. [DOI] [PubMed] [Google Scholar]
- 116.Duerksen DR, Wilhelm-Boyles C, Veitch R, Kryszak D, Parry DM. A comparison of antibody testing, permeability testing, and zonulin levels with small-bowel biopsy in celiac disease patients on a gluten-free diet. Dig Dis Sci. 2010;55:1026–1031. doi: 10.1007/s10620-009-0813-5. [DOI] [PubMed] [Google Scholar]
- 117.Lammers KM, Lu R, Brownley J, Lu B, Gerard C, Thomas K, Rallabhandi P, Shea-Donohue T, Tamiz A, Alkan S, et al. Gliadin induces an increase in intestinal permeability and zonulin release by binding to the chemokine receptor CXCR3. Gastroenterology. 2008;135:194–204.e3. doi: 10.1053/j.gastro.2008.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Barone MV, Zanzi D, Maglio M, Nanayakkara M, Santagata S, Lania G, Miele E, Ribecco MT, Maurano F, Auricchio R, et al. Gliadin-mediated proliferation and innate immune activation in celiac disease are due to alterations in vesicular trafficking. PLoS One. 2011;6:e17039. doi: 10.1371/journal.pone.0017039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Giovannini C, Matarrese P, Scazzocchio B, Varí R, D’Archivio M, Straface E, Masella R, Malorni W, De Vincenzi M. Wheat gliadin induces apoptosis of intestinal cells via an autocrine mechanism involving Fas-Fas ligand pathway. FEBS Lett. 2003;540:117–124. doi: 10.1016/s0014-5793(03)00236-9. [DOI] [PubMed] [Google Scholar]
- 120.Malamut G, El Machhour R, Montcuquet N, Martin-Lannerée S, Dusanter-Fourt I, Verkarre V, Mention JJ, Rahmi G, Kiyono H, Butz EA, et al. IL-15 triggers an antiapoptotic pathway in human intraepithelial lymphocytes that is a potential new target in celiac disease-associated inflammation and lymphomagenesis. J Clin Invest. 2010;120:2131–2143. doi: 10.1172/JCI41344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Stene LC, Honeyman MC, Hoffenberg EJ, Haas JE, Sokol RJ, Emery L, Taki I, Norris JM, Erlich HA, Eisenbarth GS, et al. Rotavirus infection frequency and risk of celiac disease autoimmunity in early childhood: a longitudinal study. Am J Gastroenterol. 2006;101:2333–2340. doi: 10.1111/j.1572-0241.2006.00741.x. [DOI] [PubMed] [Google Scholar]
- 122.Fava F, Danese S. Intestinal microbiota in inflammatory bowel disease: friend of foe? World J Gastroenterol. 2011;17:557–566. doi: 10.3748/wjg.v17.i5.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Sellitto M, Bai G, Serena G, Fricke WF, Sturgeon C, Gajer P, White JR, Koenig SS, Sakamoto J, Boothe D, et al. Proof of concept of microbiome-metabolome analysis and delayed gluten exposure on celiac disease autoimmunity in genetically at-risk infants. PLoS One. 2012;7:e33387. doi: 10.1371/journal.pone.0033387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Szajewska H, Chmielewska A, Pieścik-Lech M, Ivarsson A, Kolacek S, Koletzko S, Mearin ML, Shamir R, Auricchio R, Troncone R. Systematic review: early infant feeding and the prevention of coeliac disease. Aliment Pharmacol Ther. 2012;36:607–618. doi: 10.1111/apt.12023. [DOI] [PubMed] [Google Scholar]
- 125.Cammarota G, Cuoco L, Cianci R, Pandolfi F, Gasbarrini G. Onset of coeliac disease during treatment with interferon for chronic hepatitis C. Lancet. 2000;356:1494–1495. doi: 10.1016/S0140-6736(00)02880-4. [DOI] [PubMed] [Google Scholar]
- 126.Forsberg G, Fahlgren A, Hörstedt P, Hammarström S, Hernell O, Hammarström ML. Presence of bacteria and innate immunity of intestinal epithelium in childhood celiac disease. Am J Gastroenterol. 2004;99:894–904. doi: 10.1111/j.1572-0241.2004.04157.x. [DOI] [PubMed] [Google Scholar]
- 127.Nadal I, Donat E, Ribes-Koninckx C, Calabuig M, Sanz Y. Imbalance in the composition of the duodenal microbiota of children with coeliac disease. J Med Microbiol. 2007;56:1669–1674. doi: 10.1099/jmm.0.47410-0. [DOI] [PubMed] [Google Scholar]
- 128.Schippa S, Iebba V, Barbato M, Di Nardo G, Totino V, Checchi MP, Longhi C, Maiella G, Cucchiara S, Conte MP. A distinctive ‘microbial signature’ in celiac pediatric patients. BMC Microbiol. 2010;10:175. doi: 10.1186/1471-2180-10-175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Palma GD, Capilla A, Nova E, Castillejo G, Varea V, Pozo T, Garrote JA, Polanco I, López A, Ribes-Koninckx C, et al. Influence of milk-feeding type and genetic risk of developing coeliac disease on intestinal microbiota of infants: the PROFICEL study. PLoS One. 2012;7:e30791. doi: 10.1371/journal.pone.0030791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Westerholm-Ormio M, Vaarala O, Tiittanen M, Savilahti E. Infiltration of Foxp3- and Toll-like receptor-4-positive cells in the intestines of children with food allergy. J Pediatr Gastroenterol Nutr. 2010;50:367–376. doi: 10.1097/MPG.0b013e3181cd2636. [DOI] [PubMed] [Google Scholar]
- 131.Kalliomäki M, Satokari R, Lähteenoja H, Vähämiko S, Grönlund J, Routi T, Salminen S. Expression of microbiota, Toll-like receptors, and their regulators in the small intestinal mucosa in celiac disease. J Pediatr Gastroenterol Nutr. 2012;54:727–732. doi: 10.1097/MPG.0b013e318241cfa8. [DOI] [PubMed] [Google Scholar]
- 132.Medina M, De Palma G, Ribes-Koninckx C, Calabuig M, Sanz Y. Bifidobacterium strains suppress in vitro the pro-inflammatory milieu triggered by the large intestinal microbiota of coeliac patients. J Inflamm (Lond) 2008;5:19. doi: 10.1186/1476-9255-5-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Laparra JM, Olivares M, Gallina O, Sanz Y. Bifidobacterium longum CECT 7347 modulates immune responses in a gliadin-induced enteropathy animal model. PLoS One. 2012;7:e30744. doi: 10.1371/journal.pone.0030744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Taha AS, Faccenda E, Angerson WJ, Balsitis M, Kelly RW. Natural antibiotic expression in celiac disease--correlation with villous atrophy and response to a gluten-free diet. Dig Dis Sci. 2005;50:791–795. doi: 10.1007/s10620-005-2575-z. [DOI] [PubMed] [Google Scholar]
- 135.Fernandez-Jimenez N, Castellanos-Rubio A, Plaza-Izurieta L, Gutierrez G, Castaño L, Vitoria JC, Bilbao JR. Analysis of beta-defensin and Toll-like receptor gene copy number variation in celiac disease. Hum Immunol. 2010;71:833–836. doi: 10.1016/j.humimm.2010.05.012. [DOI] [PubMed] [Google Scholar]
- 136.Vordenbäumen S, Pilic D, Otte JM, Schmitz F, Schmidt-Choudhury A. Defensin-mRNA expression in the upper gastrointestinal tract is modulated in children with celiac disease and Helicobacter pylori-positive gastritis. J Pediatr Gastroenterol Nutr. 2010;50:596–600. doi: 10.1097/MPG.0b013e3181cd26cd. [DOI] [PubMed] [Google Scholar]
- 137.Di Sabatino A, Miceli E, Dhaliwal W, Biancheri P, Salerno R, Cantoro L, Vanoli A, De Vincenzi M, Blanco Cdel V, MacDonald TT, et al. Distribution, proliferation, and function of Paneth cells in uncomplicated and complicated adult celiac disease. Am J Clin Pathol. 2008;130:34–42. doi: 10.1309/5ADNAR4VN11TTKQ6. [DOI] [PubMed] [Google Scholar]
- 138.Rubio CA. Lysozyme-rich mucus metaplasia in duodenal crypts supersedes Paneth cells in celiac disease. Virchows Arch. 2011;459:339–346. doi: 10.1007/s00428-011-1129-3. [DOI] [PubMed] [Google Scholar]
- 139.Cinova J, De Palma G, Stepankova R, Kofronova O, Kverka M, Sanz Y, Tuckova L. Role of intestinal bacteria in gliadin-induced changes in intestinal mucosa: study in germ-free rats. PLoS One. 2011;6:e16169. doi: 10.1371/journal.pone.0016169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Capuano M, Iaffaldano L, Tinto N, Montanaro D, Capobianco V, Izzo V, Tucci F, Troncone G, Greco L, Sacchetti L. MicroRNA-449a overexpression, reduced NOTCH1 signals and scarce goblet cells characterize the small intestine of celiac patients. PLoS One. 2011;6:e29094. doi: 10.1371/journal.pone.0029094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Myrsky E, Kaukinen K, Syrjänen M, Korponay-Szabó IR, Mäki M, Lindfors K. Coeliac disease-specific autoantibodies targeted against transglutaminase 2 disturb angiogenesis. Clin Exp Immunol. 2008;152:111–119. doi: 10.1111/j.1365-2249.2008.03600.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Mastrandrea F, Semeraro FP, Coradduzza G, Manelli M, Scarcia G, Pezzuto F, Serio G. CD34+ hemopoietic precursor and stem cells traffic in peripheral blood of celiac patients is significantly increased but not directly related to epithelial damage severity. Eur Ann Allergy Clin Immunol. 2008;40:90–103. [PubMed] [Google Scholar]
- 143.Piscaglia AC. Stem cells, a two-edged sword: risks and potentials of regenerative medicine. World J Gastroenterol. 2008;14:4273–4279. doi: 10.3748/wjg.14.4273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Zhao J, de Vera J, Narushima S, Beck EX, Palencia S, Shinkawa P, Kim KA, Liu Y, Levy MD, Berg DJ, et al. R-spondin1, a novel intestinotrophic mitogen, ameliorates experimental colitis in mice. Gastroenterology. 2007;132:1331–1343. doi: 10.1053/j.gastro.2007.02.001. [DOI] [PubMed] [Google Scholar]
- 145.Bishton MJ, Haynes AP. Combination chemotherapy followed by autologous stem cell transplant for enteropathy-associated T cell lymphoma. Br J Haematol. 2007;136:111–113. doi: 10.1111/j.1365-2141.2006.06371.x. [DOI] [PubMed] [Google Scholar]
- 146.Kline RM, Neudorf SM, Baron HI. Correction of celiac disease after allogeneic hematopoietic stem cell transplantation for acute myelogenous leukemia. Pediatrics. 2007;120:e1120–e1122. doi: 10.1542/peds.2006-3397. [DOI] [PubMed] [Google Scholar]
- 147.Hoekstra JH, Groot-Loonen JJ, van der Weij A, Hoogerbrugge PM, Kooy Y, Koning F. Successful treatment of coeliac disease by allogeneic haematopoietic stem cell transplantation. J Pediatr Gastroenterol Nutr. 2010;51:793–794. doi: 10.1097/MPG.0b013e3181edf35b. [DOI] [PubMed] [Google Scholar]
- 148.Ciccocioppo R, Bernardo ME, Russo ML, Vanoli A, Franco C, Martinetti M, Catenacci L, Giorgiani G, Zecca M, Piralla A, et al. Allogeneic hematopoietic stem cell transplantation may restore gluten tolerance in patients with celiac disease. J Pediatr Gastroenterol Nutr. 2013;56:422–427. doi: 10.1097/MPG.0b013e318276a6a7. [DOI] [PubMed] [Google Scholar]
- 149.Al-toma A, Visser OJ, van Roessel HM, von Blomberg BM, Verbeek WH, Scholten PE, Ossenkoppele GJ, Huijgens PC, Mulder CJ. Autologous hematopoietic stem cell transplantation in refractory celiac disease with aberrant T cells. Blood. 2007;109:2243–2249. doi: 10.1182/blood-2006-08-042820. [DOI] [PubMed] [Google Scholar]
- 150.Tack GJ, Wondergem MJ, Al-Toma A, Verbeek WH, Schmittel A, Machado MV, Perri F, Ossenkoppele GJ, Huijgens PC, Schreurs MW, et al. Auto-SCT in refractory celiac disease type II patients unresponsive to cladribine therapy. Bone Marrow Transplant. 2011;46:840–846. doi: 10.1038/bmt.2010.199. [DOI] [PubMed] [Google Scholar]
- 151.Parco S, Vascotto F. Autologous cord blood harvesting in North Eastern Italy: ethical questions and emerging hopes for curing diabetes and celiac disease. Int J Gen Med. 2012;5:511–516. doi: 10.2147/IJGM.S31977. [DOI] [PMC free article] [PubMed] [Google Scholar]