

Abstract
Alzheimer’s disease (AD) is a progressive neurodegenerative disease in which patients exhibit gradual loss of memory that impairs their ability to learn or carry out daily tasks. Diagnosis of AD is difficult, particularly in early stages of the disease, and largely consists of cognitive assessments, with only one in four patients being correctly diagnosed. Development of novel therapeutics for the treatment of AD has proved to be a lengthy, costly and relatively unproductive process with attrition rates of > 90%. As a result, there are no cures for AD and few treatment options available for patients. Therefore, there is a pressing need for drug discovery platforms that can accurately and reproducibly mimic the AD phenotype and be amenable to high content screening applications. Here, we discuss the use of induced pluripotent stem cells (iPSCs), which can be derived from adult cells, as a method of recapitulation of AD phenotype in vitro. We assess their potential use in high content screening assays and the barriers that exist to realising their full potential in predictive efficacy, toxicology and disease modelling. At present, a number of limitations need to be addressed before the use of iPSC technology can be fully realised in AD therapeutic applications. However, whilst the use of AD-derived iPSCs in drug discovery remains a fledgling field, it is one with immense potential that is likely to reach fruition within the next few years.
Keywords: Human induced pluripotent stem cells, Alzheimer’s disease, Neurodegenerative diseases, High-throughput screening assays, Cholinergic neurons, Drug discovery, Stratified medicine
Core tip: Alzheimer’s disease (AD) affects 36 million people worldwide and is set to double by 2030. Progress in understanding AD has been hindered by a lack of suitable in vitro and in vivo models reflected in > 90% drug attrition rates. Induced pluripotent stem cells are an alternative source of neural cells that can be derived from patients’ somatic cells and exhibit AD pathophysiological phenotypes. These cells are amenable to HTS formats required for drug discovery applications. Harnessing this combined potential would provide an unprecedented opportunity to significantly reduce timeframes and costs associated with developing novel therapeutics, ultimately improving patient outcomes.
INTRODUCTION
Alzheimer’s disease (AD) is a progressive neurodegenerative disease in which patients exhibit gradual loss of memory that impairs their ability to learn or carry out daily tasks. The classic, post-mortem neuropathology exhibited in AD largely consists of amyloid plaques and neurofibrillary tangles[1], however, there is significant controversy within the field as to the causative mechanism(s). Worldwide nearly 36 million people have AD or related dementia, with a reported 7.7 million new dementia sufferers worldwide per year. The global cost of neurodegenerative diseases was over United States $600 billion in 2010 and affects people in all countries, with 58% living in low- and middle-income countries[2]. In the United Kingdom alone, specific neurodegenerative diseases (including AD and Parkinson’s disease), have a combined patient population in excess of 800000 and the cost for provision of care was an estimated £23bn in 2012[3].
Diagnosis of AD is difficult, particularly in early stages of the disease, and largely consists of cognitive assessments, with only one in four patients being correctly diagnosed[2]. Lack of knowledge of disease pathology is a major disadvantage in diagnosis and prescribing treatments since drug regimens are not the same for all dementias or patients. Moreover, development of a successful drug for the treatment of AD has, as yet, eluded pharmaceutical companies as current medicines treat only symptoms and not the cause(s) of AD. For example, in just over a decade there have been over 100 failed medicines for treatment of AD, including recent late stage failures of solanezumab and bapineuzumab with just five approved medications available to treat the symptoms of various stages of AD (three in United Kingdom). Therefore, a failure in pre-clinical to clinical development exists and can be attributed to several key factors; existing animal models or cellular models are inadequate, insufficient knowledge of drug action on human physiology and a lack of pharmacologically relevant biomarkers. Consequently, there is a pressing need for technologies that can provide definitive assays that can confirm disease pathology as well as predict novel or optimal drug regimens.
Since the creation of induced pluripotent stem cells (iPSCs) from human adult somatic cells in 2007[4], the potential applications of stem cells in regenerative medicine are considerable. Human pluripotent stem cells (that include iPSCs and embryonic stem cells) are self-renewing, which permits them to be grown indefinitely, and retain the potential to give rise to all cell types of the body. IPSCs are an ideal alternative cell source as they can be derived (reprogrammed) from somatic cells from any individual and are genetically identical to the donor, making them invaluable for use in cell-based models for human disease (Figure 1). Reprogramming of somatic cells is a highly inefficient and lengthy methodology and, as such, certain parameters should be considered when making disease specific iPSCs. These include; source of somatic cells (e.g., dermal fibroblast, blood cells), method of cellular reprogramming (e.g., retroviral, episomal) and the robustness of differentiation protocols for mature cell types. Here, we focus on AD-specific iPSCs and their derivatives to illustrate how they might be used in various applications in regenerative medicine. For a detailed overview of reprogramming, we refer the reader to another review[5].
Figure 1.
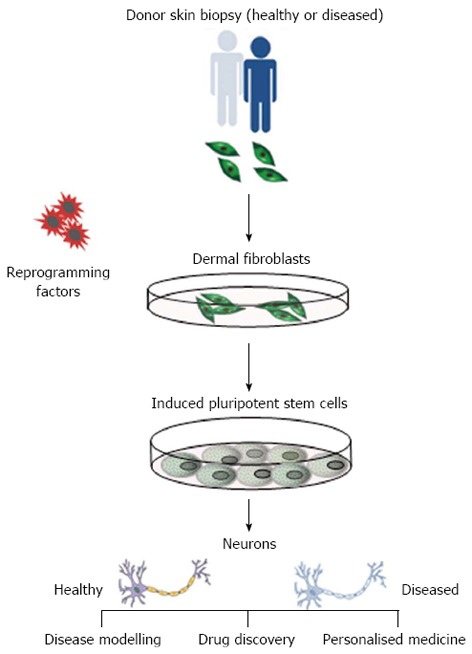
Isolation of disease specific induced pluripotent stem cells. Reprogramming of dermal fibroblasts from patients with Alzheimer’s disease into induced pluripotent stem cells provides an infinite source of cells to apply directed differentiation protocols to generate disease-specific neurons that exhibit phenotypic disease traits. This presents a unique opportunity to utilise these cells in the exploitation of drug discovery, disease modelling and personalised medicine.
Crucially, previous research demonstrates that iPSC-derived neural cells harvested from individuals suffering from a range of neurodegenerative disorders exhibit similar abnormal disease characteristics in vitro[6-9]. This observation presents an invaluable opportunity for the use of diseased cell lines in in vitro studies to further our understanding of disease modelling, early toxicity screening and in the development of novel therapeutics. Performance of a literature search using the NCBI database, PubMed, under specific search terms [disease modeling AND ips cells NOT “review” (Publication Type)] in original research publications reveals that the field of disease modelling using iPSCs has increased at a substantial rate since the creation of iPSCs in 2007 (Figure 2). A year-on-year increase in the number of publications from 2009 (n = 20) to 2011 (n = 114) is observed, however, in 2012 this trend appeared to slow. In 2013, a reduction in papers is recorded (n = 52) which could indicate that the field is maturing, whereby the initial raft of papers reflected high impact method-based publications (i.e., the production of diseased iPSCs), whereas current work is focussed on disease modelling and drug discovery, which are lengthy studies. The number of original research articles containing iPSCs for disease modelling of AD patients was very small and there are only 8 research papers that have utilised AD-derived iPSCs between 2011-2013. This demonstrates that the use of iPSCs to model AD is still in its infancy and may reflect the difficulty of isolation of these cells and identification of appropriate donor patients. This review will discuss the pathology and cellular targets of AD, how we can utilise iPSCs as a model to investigate AD, applications and limitations of these cells in high throughput analyses and future opportunities in personalised medicine.
Figure 2.
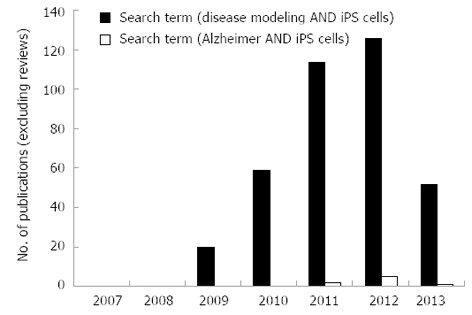
Publication statistics on original research papers using disease specific induced pluripotent stem cells between 2007 and 2013. Analysis of the search terms [disease modeling AND ips cells NOT “review” (Publication Type)] (blue bars) and (Alzheimer AND iPS cells) (red bars) for research papers published on NCBI database (PubMed) between 2007 and 2013.
DISEASE PATHOLOGY
AD can be divided into familial or sporadic genetic events with early- or late-onset. Whilst the majority of AD cases manifest as late-onset sporadic form, familial cases present a unique opportunity to investigate the inheritance of genes contributing a higher risk of AD. The familial form of AD is associated with mutations in amyloid precursor protein (APP), presenilin-1 and presenilin-2. Risk of AD is also observed to be increased where mutations in apolipoprotein E4 (APOE4) or triggering receptor expressed on myeloid cells 2 (TREM2) are present. Genes associated with the pathology of AD include APP, which results in β-amyloid plaques (Aβ), and microtubule associated protein Tau (MAPT), which results in hyperphosphorylated tau aggregates (tau tangles) within neurons of AD patients[10]. Despite tau tangles being identified as a pathological feature of AD, mutations in this gene are unusual in such patients. AD is characterized by extracellular amyloid deposition, intracellular neurofibrillary tangle formation, and neuronal loss. Below, we discuss the contribution of these genes to the pathology of AD. Other confounding factors in AD include oxidative stress, mitochondrial function, inflammation and microglia function.
Amyloid precursor protein
A significant pathological feature of AD is the presence of extracellular plaques in the brain comprised of β-amyloid (Aβ) peptides derived from the amyloid precursor protein[11,12]. APP is located on chromosome 21 in humans and is associated with dementia in Down syndrome patients, who exhibit a triplication of this chromosome (trisomy 21). Whilst APP in AD has been studied in significant detail, the events leading to Aβ deposition are less well defined and likely to involve stimulation of APP expression via the neuroinflammation-promoting cytokines IL-1 and S100B[12]. Drugs developed to target Aβ deposits for the treatment of AD have proved relatively unsuccessful. This may be due to the fact that overexpression of APP is associated with other events, such as glial activation, suggesting that the deposition of Aβ is associated with, rather than being a causal factor of, AD. As such, APP is now generally disparaged as a drug target for AD treatment with hyper-phosphorylated tau aggregates now being a major focus.
Microtubule Associated Protein Tau
The Microtubule Associated Protein Tau (MAPT; Tau) functions to assemble and stabilize microtubules within neurons, playing an important part in regulation of neuronal polarity, axonal transport and neurite outgrowth[10]. Phosphorylation of Tau allows regulation of binding and stability within neurons and aberrant phosphorylation or dephosphorylation in specific residues of the Tau protein lead to pathology, collectively known as tauopathies. The main component of the protein aggregates found in tauopathies is hyperphosphorylated tau protein within neurons. Although the exact mechanisms are unclear, the neurofibrillary tangles (NFT) associated with tauopathies may also involve conformational changes in Tau protein. Whilst tau in NFT forms the basis for pathology of tauopathies it has been suggested that tau oligomers act as a toxic species by providing a template for the misfolding of native tau and spreading from cell to cell leading to propagation of the disease[13]. Research is now focused on the targeting of Tau oligomers for drug therapies for the treatment of AD.
Apolipoprotein E4
Apolipoprotein E consists of 3 isoforms of which apoE4 is a genetic risk factor for late-onset familial and sporadic forms of AD and is also associated with dementia in DS, Huntingdon’s disease, vascular dementia and cerebrovascular disease[14]. APOE4 exhibits multifunctionality in lipid and lipoprotein transport systems, mainly in the metabolism of dietary lipids[15]. Carriers of polymorphic variants of APOE4 are between 4- and 10-times more likely to exhibit late onset AD. In the CNS, APOE4 is produced by glial cells and interacts with receptors of the low-density lipoprotein family. APOE4 binds to Aβ peptide and onset of AD is likely to reflect the inability of APOE4 to aggregate and clear Aβ in the brain, although other factors such as the effect of APOE4 on synaptic plasticity, lipid transport, neuroinflammation may also account for this[16]. Since the APOE4 isoform can be assessed prior to onset of neurodegeneration it is considered a promising target for drug therapy[17].
Presenilin-1 and -2
Presenilin-1 (PSEN1) and PSEN2 are major components of the atypical aspartyl protease complex that is required for γ-secretase complex activity and cleavage of APP. Mutations in PSEN1 are the major cause of early onset AD and also account for the most severe forms of the disease[18]. Early onset AD in PSEN1 mutation carriers can occur as early as 30 years of age, although the mean age of onset is over 58 years. More than 180 mutations have been described in PSEN1, of which the majority are missense mutations[18]. PSEN2 mutations are less common and 14 specific mutations have been associated with AD[19]. Mutations within the PSEN proteins affect APP synthesis and proteolysis leading to an increase in the ratio of Aβ42 peptide compared to Aβ40, the former a more toxic form of Aβ peptide that is more prone to oligomerisation and fibril formation[19,20]. Drug treatments have focussed on γ-secretase modulators capable of decreasing the ratio of Aβ42 to Aβ40 peptides[21].
Triggering Receptor Expressed on Myeloid Cells 2
Variants in Triggering Receptor Expressed on Myeloid Cells 2 (TREM2) have been identified that triple the risk of developing late onset AD[22]. TREM2 is a cell surface receptor, which triggers activation of the immune response in association with DAP12[23]. In the CNS, TREM2 is expressed by microglial cells and functions to activate phagocytosis in these cells and to suppress neuroinflammation and cytokine production[22]. Several functions of TREM2 include aiding clearance of Aβ and synapse remodelling. Whilst the exact mechanism of TREM2 in late onset AD is unclear it is likely that mutations in this gene contribute to disease pathogenesis via insufficient clearance of Aβ and increased localised inflammation.
AD MODELLING USING HUMAN IPSCS
The single most important factor in the utility of iPSCs in AD modelling, is that mature cell type(s) affected by the disease, e.g., neurons, exhibit phenotypic characteristics of the disease. Numerous studies have demonstrated that iPSCs can be used to model genetic diseases by showing that cells affected by the disease recapitulate these traits in vitro. iPSC AD modeling is still in its infancy and only a few studies have demonstrated successful generation and characterization of AD patient-derived neurons (Figure 2). Five out of eight publications reported isolation of iPSC-derived neurons from patients with familial AD, however a key development in the field showed that reprogramming could similarly be used to recapitulate patient specific phenotypes in vitro of sporadic forms of the disease[6,24,25]. iPSC-derived neurons generated from familial AD patients with mutation of the APP gene and sporadic AD showed, relative to non-demented controls, elevated levels of Aβ, phosphorylated tau and glycogen synthase kinase 3B[6].
A known pathology of AD progression is significant neurodegeneration in the cortical regions, with all regions of the brain registering degenerative changes as the disease progresses. Initial reports using iPSC-derived neurons from patients with familial AD utilised heterogeneous neuron populations[6,8]. Although results demonstrated an increase in Aβ42 secretion from mutant PSEN1, PSEN2 and APP iPSC-derived neurons compared to control cells both studies observed inconsistencies in Tau expression. For example, no Tau expression or tangles were observed in the Yagi et al[8] study, whereas increased levels of phosphorylated Tau were observed in both familial AD-derived neurons and one of the two sporadic AD-derived neurons compared to non-demented control neurons in the Israel et al[6] study. In addition, a recent paper reported increased levels of intracellular neuron specific amyloid aggregates in cells derived from familial (APP-E693Δ) and one of two sporadic AD derived neurons[24]. These disparities may reflect the disparate differentiation periods used in the studies and differences in the proportion of cholinergic neurons within the populations. However, it is also possible that these differences reflect inherent variability of iPSCs, which is discussed further below.
In a seminal study, iPSCs derived from patients with Downs Syndrome (a model for early onset AD) were used to generate, highly enriched populations of cholinergic neurons in significant numbers. Following differentiation times of 28-100 d following neural induction of iPSCs, analysis of these cells showed production of neuron specific Aβ secretion, amyloid aggregate formation and altered Tau protein localisation and phosphorylation[26,27]. Another key finding from this report (and others) demonstrates that early AD pathologies, such as the formation of AB42 aggregates, occur in relatively short culture periods in vitro opposed to years in vivo. Furthermore, iPS-derived neurons are able to respond functionally to various modulators highlighting their potential use in validation and identification in drug discovery[8,25].
LIMITATIONS OF IPSCS AS MODELS OF DISEASE
At present, a number of limitations need to be addressed before the full potential of iPSC technology in predictive efficacy, toxicology and disease modelling can be realised. Human iPSCs are effectively man-made cells that are similar to embryonic stem cells, which themselves only exist in vivo for a matter of days. These nuances may be reflected in the challenges faced in the differentiation of pluripotent stem cells into mature cell derivatives, despite a good understanding of the molecular mechanisms that occur during development. In order to fully exploit opportunities in disease modelling, but in particular in HTS formats, robust, efficient and cost-effective methods are fundamental. Differentiation protocols that require cocktails of growth factors are costly and are susceptible to significant batch-batch variation, however, alternative methods to acquire differentiated phenotypes are being explored, such as the use of more cost effective small molecules[28].
A significant research focus in the pluripotent stem cell field has been the development of robust differentiation protocols to enrich for specific mature cell types and populations. However, homogenous cell populations are difficult to obtain in practice and are unlikely to reflect the true pathophysiology of the disease. In addition, modelling complex, idiopathic diseases such as AD, likely requires exposing the cells to biological, chemical or environmental factors to reveal pathophysiological phenotypes. For example, Israel et al[6] demonstrated a favorably enriched neuron population (90%), however since neurons and synapses are largely dependent upon endoctyic activity they found it necessary to co-culture with astrocytes.
In addition, it has been shown by hierarchical cluster analysis that AD-derived neurons are akin to fetal neurons and, therefore, not fully mature[6]. Although, this is considered one of the major hurdles to overcome in modelling degenerative diseases, the recapitulation of a fetal phenotype presents an opportunity to isolate specific progenitors, which can be used to study developmental aberrations in congenital/developmental disorders. Conversely, for the study of late-stage onset diseases, such as sporadic AD, adult disease phenotypes might not be exhibited under standard differentiation conditions. As such, further work is necessary to identify appropriate differentiation methods for the derivation of adult neurons in vitro.
An advantage with the use of patient specific iPSCs means that each iPSC-derived cell reflects this genetic variation. Despite this being a clear advantage in the toxicological evaluation of patient populations to novel therapeutics, conclusions from studies using iPSCs from donors with different genetic backgrounds may be problematic. For example, are any phenotypic differences observed due to the mutation of interest or the genetic background of the patients? At present, parameters such as gender-, age- and ethnicity-matching are used in the selection of control donors, however, genome-wide studies show that each person has single nucleotide polymorphisms that may have disease relevance. Therefore, a fundamental feature in the use of iPSCs in regenerative applications is careful consideration of appropriate control patients. A further aspect to consider is the reprogramming event required to derive iPSCs from donors. It is well known that epigenetic variations can, and often do, occur during the reprogramming stage of iPSC derivation. Therefore, iPSC clones must be fully characterised prior to use in therapeutic analysis.
HIGH THROUGHPUT SCREENING OF NOVEL THERAPEUTICS FOR AD: IN VITRO CLINICAL TRIALS
Development of novel therapeutics for treatment of disease is a lengthy and costly process with extremely high attrition rates of > 90%, in particular, CNS therapeutics exhibit one of the lowest success rates[29]. Current practices involve evaluation of the safety and efficacy of new drugs in animal and in vitro models of relevant tissues and biological processes. Existing in vitro cell models attempt to recapitulate core pathologies or targets of AD. For example, Georgievska et al[30] recently described inhibition of Tau phosphorylation in response to AZD1080, an inhibitor of Glycogen synthase kinase-3β, using a mouse 3T3 fibroblast cell line transfected with human Tau. Stable over expression of Tau has also been achieved in the human SH-SY5Y neuroblastoma cell line[31], similarly, over expression of APP695wt in the SH-SY5Y cell line was used to determine Aβ40 secretion in response to AZD3839 in pre-clinical studies[32]. The use of animal cells, however, lacks human context and the cancer-derived SH-SY5Y cell line may not accurately reflect the cellular processes associated with AD. A recent paper highlighted the importance of the endoplasmic reticulum (ER) in protein catalysis and correlated the presence of amyloid-β plaques with age-related diminished ER function. The author went on to call for better drug discovery cell models which enable enhancement of ER function to be detected through embedding fluorescent reporter proteins within an exon of a target gene[16]. In short, these methods of target validation focus on the recapitulation of only one key feature of AD in an often-irrelevant cell line, failing to account for other components of the signalling pathway. Primary neurons offer more relevant pre-clinical cell models and are capable of synapse formation, but are costly, difficult to transfect and are typically animal derived[33]. Transgenic animal models and cell lines have undoubtedly aided our knowledge of AD mechanisms and predictive pharmacology, however, these are hindered by inter-species differences and lack of clinical relevance and genetic heterogeneity, which has resulted in poor clinical translation.
The derivation of iPSCs from patients with AD would, however, enable the applicable recapitulation of AD phenotype in a dish, since iPSCs retain the patient’s genotype. Circumventing cross species differences and negating any ethical constraints associated with the use of human embryonic stem cells would create increased translational value. Indeed, neurons derived from disease specific iPSCs have been used to validate the potency of candidate drugs in the treatment of neurological pathologies[34]. Of further importance, studies have shown treatment of AD iPSC-derived neurons with β-secretase inhibitors, but not γ-secretase inhibitors, causes significant reductions in phosphorylated Tau expression and GSK-3β levels[6,8,25]. The accessibility of iPSCs allows many compounds to be tested simultaneously, reflecting a real-life scenario of patients taking a variety of prescription and non-prescription drugs.
Harnessing this potential could provide an unprecedented opportunity to improve preclinical predictions by allowing therapeutics to be tested in multiple cell lines derived from a cohort of patients[35]. This may also allow the repositioning, reprofiling or repurposing of old, failed and existing drugs. The use of patient-derived iPSCs could be highly amenable to high throughput screening (HTS) practices using multi-well formats to enable rapid analysis of thousands of compounds. Early identification of toxic or inefficacious compounds would, thus, prevent expensive animal studies and subsequent clinical failures. Traditional HTS techniques have focussed on biochemical assays measuring enzyme activity and protein interactions using absorbance, luminescence or fluorescence based readings. For example, Haugabook et al[36] describe the use of a sandwich ELISA (in 96-well formats) to detect aggregation of amyloid plaques, a key contributor to the formation of senile plaques in AD. HTS assays have also been developed to enable detection of Aβ42 aggregation using a GFP fusion construct expressed in E. Coli, in which compound inhibition of Aβ42 aggregation resulted in the emission of a fluorescent signal[37,38]. As a result of these methods often lacking cellular context, high content screening (HCS) in whole cells has been recognised as a powerful tool for drug discovery and has been adopted largely by the pharmaceutical industry due to the large volume of multiparametric data that can be obtained[39]. HCS encompasses the automated acquisition of fluorescent images and image analysis using mathematical algorithms to extract and quantify phenotypic information, including signal shape, intensity and cellular localisation, which can be statistically analysed[40]. To increase throughput and reduce human error, additional processes such as compound storage, dosing and immunofluorescent staining can also be automated. The principle of HCS in neuronal cultures has already been demonstrated[41-43]. Neurite loss is one of the core pathologies of AD and application of HCS to quantify neuronal outgrowth has already been achieved and proven to be faster than traditional manual tracing methods[41,43]. Assessment of chemical toxicity has also been demonstrated by HCS in three neuronal cell lines, whereby proliferation was detected by BrdU incorporation (an indicator of actively proliferating cells) and cell counts were obtained with Hoechst 33342 nuclear dye in a 96-well plate format[44]. HCS has applications in additional areas of neuroscience including neurogenesis, cell signalling and inclusion formation as reviewed by Dragunow[45]. An example of HCS applications in AD therapeutics is shown in Figure 3.
Figure 3.
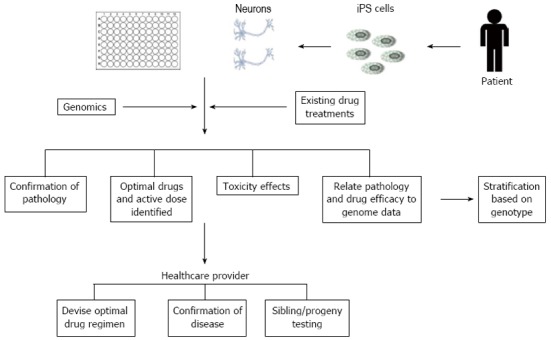
Example of some of the inputs and outputs in Alzheimer’s disease high content screening applications. iPS: Induced pluripotent stem.
Overall, powerful high-throughput and -content screening assays are in place that can be applied to multiple areas of drug discovery, but clinical success is hindered by a lack of relevant cell models in the pre-clinical stages. High throughput toxicity screening using human iPSC-derived cardiomyocytes has been reported using electrode sensors to acquire oscillating impedance measurements to detect the contraction and relaxation or beating of iPSC-derived cardiomyocytes in a 96-well plate format[46]. Arrhythmia data obtained from iPSC-derived cardiomyocytes treated with cardiac modulators was qualitatively comparable to results obtained from more traditional, low throughput microelectrode arrays in parallel experiments. Therefore, the potential use of iPSC technology in high throughput drug discovery has been demonstrated but to date has not been described in the literature for iPSC-derived neurons. The UK Government and pharmaceutical industry have recognised the potential for iPSC AD models in HCS and by late 2013 several calls for funding such technology have been announced. As a result, we expect to see significant activity in this field and the development of HCS platforms for AD.
FUTURE PERSPECTIVES: PERSONALISED MEDICINE
The potential to use patient-specific cells to generate pluripotent cells, which can be maintained indefinitely and subsequently differentiated into desired cell types, presents a real opportunity for stratified (personalised) medicine applications (Figure 4). For example, this will allow scientists and clinicians to model, in vitro, the progression of AD (or other degenerative diseases) for each individual patient, perform “customised” pharmacologic screening to determine the optimal therapeutic regimes and implement genomic testing of large cohorts of patients, representing different ethnic/genetic backgrounds in order to inform pharma of susceptible populations. There is a clear unmet drug need for the treatment of AD and the utility of iPSC technology will provide a more efficacious model to reassess (or rescue) former drug candidates that either have been withdrawn from use or aborted at a late stage of development for safety reasons. In short, the use of disease specific iPSC derived neural cells, in conjunction with high throughput/content screening methods, offer improved clinically relevant cell models that will significantly reduce timeframes and costs associated with the development of novel therapeutics, ultimately improving the number of new medicines to the market to treat patients with neurodegenerative diseases.
Figure 4.
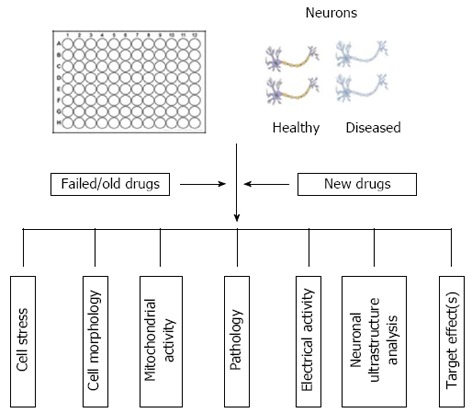
Example of how high content screening of patient-derived induced pluripotent stem cells could aid stratification of existing drug treatments and help identify genetic profiles associated with specific disease phenotypes.
Footnotes
Supported by United Kingdom Biotechnology and Biosciences Research Council, Engineering and Physical Sciences Research Council and the Technology Strategy Board
P- Reviewers: Freter R, Perron M S- Editor: Ma YJ L- Editor: A E- Editor: Zhang DN
References
- 1.Hardy J. The amyloid hypothesis for Alzheimer’s disease: a critical reappraisal. J Neurochem. 2009;110:1129–1134. doi: 10.1111/j.1471-4159.2009.06181.x. [DOI] [PubMed] [Google Scholar]
- 2.Prince M, Prina M, Guerchet M, Alzheimer’s Disease International. World Alzheimer’s Disease Report 2013, 2013. Available from: http://www.alz.co.uk/research/world-report-2013.
- 3.World Health Organisation. WHO report: Dementia: A public health priority 2012. Available from: http://www.who.int/mental_health/publications/dementia_report_2012/en/
- 4.Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 5.Cahan P, Daley GQ. Origins and implications of pluripotent stem cell variability and heterogeneity. Nat Rev Mol Cell Biol. 2013;14:357–368. doi: 10.1038/nrm3584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Israel MA, Yuan SH, Bardy C, Reyna SM, Mu Y, Herrera C, Hefferan MP, Van Gorp S, Nazor KL, Boscolo FS, et al. Probing sporadic and familial Alzheimer’s disease using induced pluripotent stem cells. Nature. 2012;482:216–220. doi: 10.1038/nature10821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Qiang L, Fujita R, Yamashita T, Angulo S, Rhinn H, Rhee D, Doege C, Chau L, Aubry L, Vanti WB, et al. Directed conversion of Alzheimer’s disease patient skin fibroblasts into functional neurons. Cell. 2011;146:359–371. doi: 10.1016/j.cell.2011.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 8.Yagi T, Ito D, Okada Y, Akamatsu W, Nihei Y, Yoshizaki T, Yamanaka S, Okano H, Suzuki N. Modeling familial Alzheimer’s disease with induced pluripotent stem cells. Hum Mol Genet. 2011;20:4530–4539. doi: 10.1093/hmg/ddr394. [DOI] [PubMed] [Google Scholar]
- 9.Cherry AB, Daley GQ. Reprogramming cellular identity for regenerative medicine. Cell. 2012;148:1110–1122. doi: 10.1016/j.cell.2012.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guzmán-Martinez L, Farías GA, Maccioni RB. Tau Oligomers as Potential Targets for Alzheimer’s Diagnosis and Novel Drugs. Front Neurol. 2013;4:167. doi: 10.3389/fneur.2013.00167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang YW, Thompson R, Zhang H, Xu H. APP processing in Alzheimer’s disease. Mol Brain. 2011;4:3. doi: 10.1186/1756-6606-4-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wilcock DM, Griffin WS. Down’s syndrome, neuroinflammation, and Alzheimer neuropathogenesis. J Neuroinflammation. 2013;10:84. doi: 10.1186/1742-2094-10-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gerson JE, Kayed R. Formation and propagation of tau oligomeric seeds. Front Neurol. 2013;4:93. doi: 10.3389/fneur.2013.00093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zlokovic BV. Cerebrovascular effects of apolipoprotein E: implications for Alzheimer disease. JAMA Neurol. 2013;70:440–444. doi: 10.1001/jamaneurol.2013.2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Frisardi V. Apolipoprotein E genotype: the innocent bystander or active bridge between metabolic syndrome and cognitive impairment? J Alzheimers Dis. 2012;30 Suppl 2:S283–S304. doi: 10.3233/JAD-2012-111568. [DOI] [PubMed] [Google Scholar]
- 16.Holtzman JL. Cellular and animal models for high-throughput screening of therapeutic agents for the treatment of the diseases of the elderly in general and Alzheimer's disease in particular(†) Front Pharmacol. 2013;4:59. doi: 10.3389/fphar.2013.00059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mahley RW, Huang Y. Small-molecule structure correctors target abnormal protein structure and function: structure corrector rescue of apolipoprotein E4-associated neuropathology. J Med Chem. 2012;55:8997–9008. doi: 10.1021/jm3008618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Alonso Vilatela ME, López-López M, Yescas-Gómez P. Genetics of Alzheimer’s disease. Arch Med Res. 2012;43:622–631. doi: 10.1016/j.arcmed.2012.10.017. [DOI] [PubMed] [Google Scholar]
- 19.Bekris LM, Yu CE, Bird TD, Tsuang DW. Genetics of Alzheimer disease. J Geriatr Psychiatry Neurol. 2010;23:213–227. doi: 10.1177/0891988710383571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu L, Rosa-Neto P, Hsiung GY, Sadovnick AD, Masellis M, Black SE, Jia J, Gauthier S. Early-onset familial Alzheimer’s disease (EOFAD) Can J Neurol Sci. 2012;39:436–445. doi: 10.1017/s0317167100013949. [DOI] [PubMed] [Google Scholar]
- 21.Kounnas MZ, Danks AM, Cheng S, Tyree C, Ackerman E, Zhang X, Ahn K, Nguyen P, Comer D, Mao L, et al. Modulation of gamma-secretase reduces beta-amyloid deposition in a transgenic mouse model of Alzheimer’s disease. Neuron. 2010;67:769–780. doi: 10.1016/j.neuron.2010.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rohn TT. The triggering receptor expressed on myeloid cells 2: “TREM-ming” the inflammatory component associated with Alzheimer’s disease. Oxid Med Cell Longev. 2013;2013:860959. doi: 10.1155/2013/860959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lanier LL, Bakker AB. The ITAM-bearing transmembrane adaptor DAP12 in lymphoid and myeloid cell function. Immunol Today. 2000;21:611–614. doi: 10.1016/s0167-5699(00)01745-x. [DOI] [PubMed] [Google Scholar]
- 24.Kondo T, Asai M, Tsukita K, Kutoku Y, Ohsawa Y, Sunada Y, Imamura K, Egawa N, Yahata N, Okita K, et al. Modeling Alzheimer’s disease with iPSCs reveals stress phenotypes associated with intracellular Aβ and differential drug responsiveness. Cell Stem Cell. 2013;12:487–496. doi: 10.1016/j.stem.2013.01.009. [DOI] [PubMed] [Google Scholar]
- 25.Koch P, Tamboli IY, Mertens J, Wunderlich P, Ladewig J, Stüber K, Esselmann H, Wiltfang J, Brüstle O, Walter J. Presenilin-1 L166P mutant human pluripotent stem cell-derived neurons exhibit partial loss of γ-secretase activity in endogenous amyloid-β generation. Am J Pathol. 2012;180:2404–2416. doi: 10.1016/j.ajpath.2012.02.012. [DOI] [PubMed] [Google Scholar]
- 26.Shi Y, Kirwan P, Livesey FJ. Directed differentiation of human pluripotent stem cells to cerebral cortex neurons and neural networks. Nat Protoc. 2012;7:1836–1846. doi: 10.1038/nprot.2012.116. [DOI] [PubMed] [Google Scholar]
- 27.Shi Y, Kirwan P, Smith J, MacLean G, Orkin SH, Livesey FJ. A human stem cell model of early Alzheimer’s disease pathology in Down syndrome. Sci Transl Med. 2012;4:124ra29. doi: 10.1126/scitranslmed.3003771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ding S, Wu TY, Brinker A, Peters EC, Hur W, Gray NS, Schultz PG. Synthetic small molecules that control stem cell fate. Proc Natl Acad Sci USA. 2003;100:7632–7637. doi: 10.1073/pnas.0732087100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kola I, Landis J. Can the pharmaceutical industry reduce attrition rates? Nat Rev Drug Discov. 2004;3:711–715. doi: 10.1038/nrd1470. [DOI] [PubMed] [Google Scholar]
- 30.Georgievska B, Sandin J, Doherty J, Mörtberg A, Neelissen J, Andersson A, Gruber S, Nilsson Y, Schött P, Arvidsson PI, et al. AZD1080, a novel GSK3 inhibitor, rescues synaptic plasticity deficits in rodent brain and exhibits peripheral target engagement in humans. J Neurochem. 2013;125:446–456. doi: 10.1111/jnc.12203. [DOI] [PubMed] [Google Scholar]
- 31.Löffler T, Flunkert S, Taub N, Schofield EL, Ward MA, Windisch M, Hutter-Paier B. Stable mutated tau441 transfected SH-SY5Y cells as screening tool for Alzheimer’s disease drug candidates. J Mol Neurosci. 2012;47:192–203. doi: 10.1007/s12031-012-9716-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jeppsson F, Eketjäll S, Janson J, Karlström S, Gustavsson S, Olsson LL, Radesäter AC, Ploeger B, Cebers G, Kolmodin K, et al. Discovery of AZD3839, a potent and selective BACE1 inhibitor clinical candidate for the treatment of Alzheimer disease. J Biol Chem. 2012;287:41245–41257. doi: 10.1074/jbc.M112.409110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Daub A, Sharma P, Finkbeiner S. High-content screening of primary neurons: ready for prime time. Curr Opin Neurobiol. 2009;19:537–543. doi: 10.1016/j.conb.2009.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bellin M, Marchetto MC, Gage FH, Mummery CL. Induced pluripotent stem cells: the new patient? Nat Rev Mol Cell Biol. 2012;13:713–726. doi: 10.1038/nrm3448. [DOI] [PubMed] [Google Scholar]
- 35.Han SS, Williams LA, Eggan KC. Constructing and deconstructing stem cell models of neurological disease. Neuron. 2011;70:626–644. doi: 10.1016/j.neuron.2011.05.003. [DOI] [PubMed] [Google Scholar]
- 36.Haugabook SJ, Yager DM, Eckman EA, Golde TE, Younkin SG, Eckman CB. High throughput screens for the identification of compounds that alter the accumulation of the Alzheimer’s amyloid beta peptide (Abeta) J Neurosci Methods. 2001;108:171–179. doi: 10.1016/s0165-0270(01)00388-0. [DOI] [PubMed] [Google Scholar]
- 37.McKoy AF, Chen J, Schupbach T, Hecht MH. A novel inhibitor of amyloid β (Aβ) peptide aggregation: from high throughput screening to efficacy in an animal model of Alzheimer disease. J Biol Chem. 2012;287:38992–39000. doi: 10.1074/jbc.M112.348037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim W, Kim Y, Min J, Kim DJ, Chang YT, Hecht MH. A high-throughput screen for compounds that inhibit aggregation of the Alzheimer’s peptide. ACS Chem Biol. 2006;1:461–469. doi: 10.1021/cb600135w. [DOI] [PubMed] [Google Scholar]
- 39.Haney SA, LaPan P, Pan J, Zhang J. High-content screening moves to the front of the line. Drug Discov Today. 2006;11:889–894. doi: 10.1016/j.drudis.2006.08.015. [DOI] [PubMed] [Google Scholar]
- 40.Zanella F, Lorens JB, Link W. High content screening: seeing is believing. Trends Biotechnol. 2010;28:237–245. doi: 10.1016/j.tibtech.2010.02.005. [DOI] [PubMed] [Google Scholar]
- 41.Ofengeim D, Shi P, Miao B, Fan J, Xia X, Fan Y, Lipinski MM, Hashimoto T, Polydoro M, Yuan J, et al. Identification of small molecule inhibitors of neurite loss induced by Aβ peptide using high content screening. J Biol Chem. 2012;287:8714–8723. doi: 10.1074/jbc.M111.290957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nguyen L, Wright S, Lee M, Ren Z, Sauer JM, Hoffman W, Zago W, Kinney GG, Bova MP. Quantifying amyloid beta (Aβ)-mediated changes in neuronal morphology in primary cultures: implications for phenotypic screening. J Biomol Screen. 2012;17:835–842. doi: 10.1177/1087057112441972. [DOI] [PubMed] [Google Scholar]
- 43.Hu M, Schurdak ME, Puttfarcken PS, El Kouhen R, Gopalakrishnan M, Li J. High content screen microscopy analysis of A beta 1-42-induced neurite outgrowth reduction in rat primary cortical neurons: neuroprotective effects of alpha 7 neuronal nicotinic acetylcholine receptor ligands. Brain Res. 2007;1151:227–235. doi: 10.1016/j.brainres.2007.03.051. [DOI] [PubMed] [Google Scholar]
- 44.Mundy WR, Radio NM, Freudenrich TM. Neuronal models for evaluation of proliferation in vitro using high content screening. Toxicology. 2010;270:121–130. doi: 10.1016/j.tox.2010.02.004. [DOI] [PubMed] [Google Scholar]
- 45.Dragunow M. High-content analysis in neuroscience. Nat Rev Neurosci. 2008;9:779–788. doi: 10.1038/nrn2492. [DOI] [PubMed] [Google Scholar]
- 46.Guo L, Abrams RM, Babiarz JE, Cohen JD, Kameoka S, Sanders MJ, Chiao E, Kolaja KL. Estimating the risk of drug-induced proarrhythmia using human induced pluripotent stem cell-derived cardiomyocytes. Toxicol Sci. 2011;123:281–289. doi: 10.1093/toxsci/kfr158. [DOI] [PubMed] [Google Scholar]