

Abstract
Repair and regeneration of bone requires mesenchymal stem cells that by self-renewal, are able to generate a critical mass of cells with the ability to differentiate into osteoblasts that can produce bone protein matrix (osteoid) and enable its mineralization. The number of human mesenchymal stem cells (hMSCs) diminishes with age and ex vivo replication of hMSCs has limited potential. While propagating hMSCs under hypoxic conditions may maintain their ability to self-renew, the strategy of using human telomerase reverse transcriptase (hTERT) to allow for hMSCs to prolong their replicative lifespan is an attractive means of ensuring a critical mass of cells with the potential to differentiate into various mesodermal structural tissues including bone. However, this strategy must be tempered by the oncogenic potential of TERT-transformed cells, or their ability to enhance already established cancers, the unknown differentiating potential of high population doubling hMSCs and the source of hMSCs (e.g., bone marrow, adipose-derived, muscle-derived, umbilical cord blood, etc.) that may provide peculiarities to self-renewal, differentiation, and physiologic function that may differ from non-transformed native cells. Tissue engineering approaches to use hMSCs to repair bone defects utilize the growth of hMSCs on three-dimensional scaffolds that can either be a base on which hMSCs can attach and grow or as a means of sequestering growth factors to assist in the chemoattraction and differentiation of native hMSCs. The use of whole native extracellular matrix (ECM) produced by hMSCs, rather than individual ECM components, appear to be advantageous in not only being utilized as a three-dimensional attachment base but also in appropriate orientation of cells and their differentiation through the growth factors that native ECM harbor or in simulating growth factor motifs. The origin of native ECM, whether from hMSCs from young or old individuals is a critical factor in “rejuvenating” hMSCs from older individuals grown on ECM from younger individuals.
Keywords: Mesenchymal stem cell, Telomerase reverse transcriptase, Extracellular matrix, Osteogenesis, Regenerative medicine, Tissue engineering, Proliferation, Differentiation
Core tip: When human telomerase reverse transcriptase (hTERT) transformed human mesenchymal stem cells (hMSCs) are used to prolong replicative potential and osteogenic differentiation, consideration should be given to using lower population doubling hTERT-transformed hMSCs to avoid potential oncogenesis. An inducible hTERT system may also avoid oncogenic transformation. Demonstration of in vivo bone forming capacity of hTERT-transformed cells should be used as standard in determining osteogenic differentiation of such cells rather than in vitro culture mineralization; the CD146 marker may be a suggested surface marker for hTERT-transformed hMSCs that may have the capacity to form bone in vivo. Native ECM from early population doubling hMSCs or hMSCs from a younger source may be best when seeking to extend the proliferative and differentiating potential of hMSCs from either young or older sources.
INTRODUCTION
The regeneration of mesodermal and neural crest-derived structural or connective tissues such as bone, cartilage, muscle and tendon continues to be a widely pursued for the reason that such structural tissues are generally homogeneous with either a predominantly single cell type or limited number of cells that contribute to the make-up of the tissue and that precursors to the mature cell types can be found in adult tissues. These precursor cells are generally multipotent, in that they can differentiate into a variety of connective tissue phenotypes. These precursor cells are generally referred to as adult mesenchymal stem cells (MSCs) or bone marrow stromal cells and can be found in the bone marrow but also as similar multipotent cells in specific tissues as well as circulating cells in blood.
Tissue engineering seeks to replace tissues that are either lost by traumatic events or by disease through the use of specific cell types that can recapitulate the lost or diseased tissue, and generally used in combination with a three-dimensional structural scaffold, and in many instances in combination with various growth factors, cytokines, and hormones or other biological molecules to assist in either the creation of a critical mass of needed cells or to assist in differentiating these cells to the required tissue type.
Because generating a critical mass of cells used in the regenerative process is a key to successful tissue engineering followed by differentiating those cells into the specific cell type comprising the tissue, stem cells have been the preferred starting cell type in many tissue engineering trials. This minireview will focus only on human adult bone marrow MSCs (herein assumed to be synonymous with bone marrow stromal cells) as much as possible and the telomerase strategy of inducing self-renewal of these cells to create a critical cell mass. Secondly, the minireview will examine the strategy of using extracellular matrix as a native scaffold upon which mesenchymal stem cells can self-renew and differentiate into bone.
MESENCHYMAL STEM CELL SELF-RENEWAL
The ability to self-renew is a hallmark of any stem cell[1]. Self-renewal is simply defined as the ability of the resulting daughter cells, after mitotic division of the original mother cell, to retain the ability to generate a variety of differentiated cell types identical to that of the ability of the mother cell to differentiate in to those same cell types, and for a daughter cell to be able to generate daughter cells that also maintain the ability to differentiate into the same variety of cell types as the original “grandmother” and mother cells[2]. The maintenance of self-renewal and pluripotency of stem cells occurs in the stem cell niche, where stem cells are able to receive cues from the stroma and other cell types either by direct contact or by secreted soluble factors within this microenvironmental niche[3,4].
Adult MSCs also share the ability to self-renew. This potential to self-replicate and to differentiate into connective tissue phenotypes has led to the exploration to utilize MSCs in the repair of injured tissues[5,6]. While the bone marrow has been a common site to harvest MSCs, other cell types similar to bone marrow-derived MSCs can also be found in other sites. Adipose-derived stem cells, satellite cells in muscle, and pericytes around blood vessels and umbilical cord blood cells also may share multipotent characteristics for differentiation into connective tissue phenotypes under specific conditions which include selective differentiation media and growth factors[7-10]. In a comparison of MSCs from bone marrow, adipose tissue, and cord blood, Rebelatto et al[11] (2008) reported that isolation rate of MSCs from umbilical cord blood was only a third that of bone marrow-derived and adipose-derived MSCs. The initial growth rate of bone marrow-derived and adipose-derived MSCs was much higher than that of umbilical cord blood MSCs. However, others have shown that the proliferation of umbilical cord tissue-derived MSCs show higher population doublings and shorter doubling times compared to adipose-derived MSCs although adipose-derived MSC had higher numbers of colony-forming units compared to MSCs from umbilical cord tissue[12]. Surface marker expression of CD34 (cluster of differentiation molecule in family of sialomucin proteins) was significantly higher in adipose-derived MSCs compared to that of bone marrow-derived MSCs. Interestingly, CD117 (tyrosine-protein kinase Kit) was found to be positive in about 98% of adipose-derived MSCs but positive in only 52% and 39% of bone marrow-derived and umbilical cord blood-derived MSCs. Additionally, while osteogenic and chondrogenic differentiation was similar in MSCs from all three sources, umbilical cord blood-derived MSCs showed a lesser propensity for adipogenic differentiation. Others have also noted differences in marker expression between bone marrow-derived and adipose-derived MSCs. For instance, CD106 (vascular cell adhesion molecule-1) is expressed in bone marrow-derived MSCs but its expression in adipose-derived MSCs is either low or non-existent while CD49d (integrin α4 subunit) is expressed in adipose-derived MSCs but not in bone-marrow-derived MSCs[13]. Culture conditions such as the use of fetal bovine serum, human serum, or serum-free medium have been shown to influence not only the expression of surface markers for adipose-derived MSCs [e.g., CD117, CD166 (activated leucocyte cell adhesion molecule)] and bone marrow-derived MSCs but also in differentiation potential of such MSCs. As an example, fetal bovine serum has a stronger influence on osteogenic differentiation of adipose-derived MSCs than it does on adipogenic differentiation while allogeneic human serum and serum-free conditions have greater propensity to drive adipose-derived MSCs towards adipogenic differentiation than towards either osteogenic or chondrogenic lineages[14]. Thus while adipose tissue and perhaps umbilical cord tissue sources may provide ample sources for MSCs compared to that of bone marrow and umbilical cord blood, differences in some specific surface markers for MSCs, proliferative potential, and differentiation potential in vitro occur based on the source of starting material to isolate MSCs, tissue culture supplements and conditions, and even human individual heterogeneity. Whether non-bone marrow-derived MSCs favor differentiation into specific connective tissue types or even non-mesodermal cell types as in the case of umbilical cord blood MSCs and adipose-derived MSCs in an in vivo environment is still a ripe area of investigation[13-15].
Age of the organism is a determinant of the number of bone marrow MSCs present as well as in vitro tissue culture conditions that are critical for MSCs to retain their ability to self-renew yet demonstrate plasticity in their ability to differentiate into various mesodermal tissues[16]. The number of cells from human bone marrow that are MSCs as determined by colony forming unit-fibroblastic (CFU-f) assay are less than 0.1% of total bone marrow mononuclear cells, thus demonstrating a minimal number of hMSCs that can be used in bone regeneration[17]. The numbers of CFU-f and the capacity of CFU-fs that can differentiate into osteoblasts further decrease as a function of age of the bone marrow donor up to age 40; after age 40, there does not appear to be any further diminishing of CFU-fs that can differentiate into osteoblasts[18]. It was suggested that hMSCs have decreased proliferative capacity as a function of age[19]. Thus hMSCs from young individuals ages 18-29 years achieved an average population doubling level of 41 whereas hMSCs from older individuals ages 66-81 years achieved an average population doubling level of 24 with about a 55% lower population doubling rate than in hMSCs from the younger individuals. However, no difference in in vivo bone formation was noted as a function of donor age with early passage cells from either age group. Thus, once placed in primary culture, hMSCs have a limited lifespan (average 20 to 40 population doublings, but the number of population doublings may differ depending on growth medium or any added growth factors)[19-21] under environmental conditions normally used for in vitro cell culture (humidified 5% CO2 and 95% air (21% O2) and when grown on tissue culture plastic. hMSCs grown in such conditions attain the Hayflick limit where cell division ceases, and the usual hMSC size becomes larger and the usual spindle shape of normal hMSCs becomes more polygonal or with a variety of shapes and sizes, at times with multinucleation, and overall with less cell density per culture than cells undergoing cell division[22]. As the number of population doublings for such cells is limited practically in primary culture, slower cell division and finally lack of cell division ensues and the above morphological changes are noted, and the expression of senescence-associated β-galactosidase, and p16, markers of cellular senescence, are increased[23]. However, it has been shown that if environmental conditions simulate the MSC niche in the bone marrow, specifically low oxygen tension, that self-renewal of hMSCs can be prolonged. D’Ippolito et al[24] (2004) developed a multilineage inducible MSC model from human cadaveric vertebral body marrow (MIAMI cells) and propagated them in 3% O2/5% CO2/92% N2. They reported that more than 50 cell doublings beyond the Hayflick limit for primary cells could be achieved from hMSCs from at least 3 of 12 donors and at least 30 population doublings could be achieved from all of their donors. In a follow-up communication, they reported that MIAMI cells grown in 3% O2 doubled more quickly than those grown at 21% O2 and maintained the embryonic transcription factors OCT-4, REX-1, and hTERT and had suppressed osteoblastic differentiation when exposed to osteogenic differentiation medium. At higher O2 concentrations of 21%, these embryonic transcription factors were lost and osteogenic differentiation was enhanced[25]. The mechanism by which hypoxia regulates stem cell self-renewal appears to be via hypoxia inducible factor-1α (HIF-1α). Low oxygen concentrations stabilize HIF-1α by inhibiting its degradation by the proteasome. Mazumdar et al[26] (2010) reported that hypoxia induced canonical Wnt/β-catenin signaling and increased transcription of Lef/Tcf genes which have hypoxia response elements in their promoter regions that bind HIF-1α. Canonical Wnt/β-catenin signaling thus can induce increased cell proliferation.
HTERT TRANSFORMATION OF HMSCS-THE “IN’S” FOR SELF-RENEWAL
In lieu of special resources needed to grow hMSCs in a hypoxic environment to maintain a proliferative state, a self-renewal strategy, engineering of hMSCs to over express telomerase has been an alternative means to maintain a longer proliferative lifespan of such cells. Telomerase, which is a multi-subunit ribonucleoprotein found in the cell nucleus and perhaps closely associated with nucleoli, allows for the addition of non-coding telomere DNA at the 3’ end of linear chromosomes[27-29]. Maintenance of telomere length by the addition of TTAGGG repeats onto the ends of telomeres allows for cells to continue to divide[30]. Telomerase is expressed in human embryonic cells and in fetal, newborn, and adult testes and ovaries but not in mature spermatozoa or oocytes. Moreover, expression of telomerase disappears in human somatic cells in the neonatal period and later in life[31]. Thus lacking telomerase, telomeres shorten with each cell division leading to replicative senescence once cells reach a critical shortened telomere length. Specifically, with respect to MSCs, a number of laboratories have reported that hMSCs from bone marrow do not express telomerase activity or have activity below detectable levels by telomeric repeat amplification protocol (TRAP) assay when hMSCs are asynchronously dividing[20,32-34]. However, human telomerase reverse transcriptase (hTERT) expression and telomerase activity could be detected when cells were synchronized to S-phase[34]. Others have found that telomere length in hMSCs is short upon initial isolation and tend to further shorten with cell passage in vitro and appear to correlate with low to undetectable levels of hTERT[35]. Thus theoretically, maintaining telomerase expression should prevent replicative senescence. Additionally, the decrease in telomere length correlates with CFU-f numbers suggesting that telomere length and telomerase activity could also be related to the ability of hMSCs to differentiate along various cell lineages including the osteogenic lineage[35]. Gronthos et al[36] (2003) reported that expression of hTERT in human bone marrow-derived MSCs not only increased proliferative capacity by up-regulating G1 to S phase transition cell cycle genes but also increased the expression of osteogenic genes for cbfa-1, osterix, and osteocalcin and induced bone formation earlier and to a much larger degree in an in vivo ectopic bone formation assay of hTERT-transformed hMSCs. Saeed et al[37] (2011) demonstrated that in telomerase-deficient mice (Terc-/-), there was delayed ossification in occipital bone, sternum, vertebrae, and metatarsals. Overall bone volume was decreased compared to wild type controls, and trabecular bone parameters showed decreased trabecular thickness and increased trabecular spacing[37]. Additionally, bone formation rate was decreased which correlated with decreased osteoblast surface per bone surface, and osteoclast surface per bone surface was increased. The proliferative ability of bone marrow-derived MSCs from Terc-/- mice was diminished compared to wild type mice, and there was increased β-galactosidase staining of Terc-/- cells suggesting a more senescent phenotype of MSCs. There was up-regulation of pro-inflammatory genes (e.g., IL-1 receptor type 2, toll-like receptor 6, leukotriene B4 receptor 1, tumor necrosis factor, etc.) indicative of osteoclastic activity as well as a decrease of osteoblast-specific bone markers. Thus both decreased bone formation and increased bone resorption as a result of an inflammatory microenvironment were found in this telomerase deficient model.
The critical components of human telomerase include the hTERT catalytic subunit and the RNA subunit, telomerase RNA (hTR), that provides a template for the synthesis of the DNA repeats at the ends of chromosomes. However, generally only hTERT is sufficient to maintain telomere length when transfected into various cell types although integration of ectopic hTERT alone to extend cell replicative ability may be dependent on integration site, availability of other proteins associated with telomeres, or cell specificity[32]. Thus a number of studies have demonstrated the feasibility of using hTERT in hMSCs to allow for prolonged replicative lifespan as well as capability of differentiating hTERT-transformed hMSCs towards the osteogenic lineage[38-42]. The strategy used to transform hMSCs to over express the hTERT gene is generally a retroviral vector approach that uses green fluorescent protein expression as a positive selection marker to enable sorting of positively transformed cells by fluorescence activated cell sorting[41]. An alternative approach to select transformed cells is an antibiotic resistance strategy[42]. A technique to control hTERT expression in transfected hMSCs on demand utilizes the tetracycline inducible approach (Tet-On) so that proliferative and differentiation ability can be assessed at selected population doublings although “leakiness” of hTERT even in the Tet-off state could be a limitation[40]. hTERT-transformed hMSCs have been reported to undergo at least 70 population doubling levels[42] but upwards of 120 to 400 population doubling levels have been reported depending on the length of time in culture, plating density of cells, and subcultured clonal populations[32,39-41]. The interesting aspect of hTERT-transformed hMSCs is that they are able to maintain their proliferative ability while being induced to differentiate along osteogenic, but also adipogenic, and chondrogenic lineages. Thus hTERT-transformed cells are different from non-transformed hMSCs and mesenchymal (stromal) cells from other species that are able to differentiate into osteoblasts where it is observed that as osteogenic differentiation proceeds, the proliferative ability of the cells diminishes[43,44].
Three important criteria must be met when hMSCs are transformed by hTERT expression to achieve a critical mass of cells via self-renewal that would be necessary to populate fabricated scaffolds for tissue engineering. Firstly, markers of hMSCs should be maintained after hTERT transformation that would suggest maintenance of multipotency of the cells to undergo differentiation into various mesenchymal cell lineages. Secondly, it is important that hTERT transformation of hMSCs does not lead to malignant transformation either in the pluripotent state or in differentiated cell types. Thirdly, it is critical that hTERT expressing hMSCs will be able to specifically differentiate along the osteogenic lineage and to form bone which is the tissue of interest in this minireivew.
Surface markers have been traditionally used to identify hMSCs. The International Society for Cellular Therapy set minimal criteria for positive markers to define hMSCs which are > 95% expression of CD105 (endoglin), CD73 (ecto-5’-nucleotidase), CD90 (Thy-1) and < 2% expression of hematopoietic stem cell markers, CD45 (protein tyrosine phosphatase, receptor type, C), CD34 (sialomucin family adhesion factor), CD14 (monocyte differentiation antigen/lipoglycan receptor) or CD11b (integrin alpha M), CD79α (immunoglobulin associated alpha) or CD19 (B-lymphocyte antigen), and HLA-DR[45]. Other markers used to identify hMSCs include STRO-1, CD146 (melanoma cell adhesion molecule/MUC18), CD49a (integrin alpha subunit), CD271 (low-affinity nerve growth factor receptor), CD63 (lysosome-associated membrane protein-3), found on only on marrow-derived hMSCs and CD166 (activated leucocyte cell adhesion molecule)[6,16,46-49]. Interestingly, stage-specific embryonic antigen-4 (SSEA-4), found on human embryonic stem cells, was identified as a marker for both mouse and human bone marrow-derived MSCs that had the ability to differentiate into both adipogenic and osteogenic lineages[50]. Most recently CD44 was identified as a negative marker in freshly isolated although acquisition of the CD44 marker may be a function of in vitro cell culture of hMSCs[51].
Telomerase expression and activity has been found in a majority of human tumors thus suggesting that hTERT expression in human cells could potentially lead to uncontrolled cell proliferation[52]. However, it has also been suggested that the immortalization induced by hTERT may only in part be due to maintaining telomere length and stabilization and that non-canonical functions of hTERT such as the up-regulation of NF-κB transcription by TERT binding to the p65 subunit of NF-κB as well as activating the Wnt/β-catenin pathway and its target genes, MYC and CCND1 (Cyclin D1), which are regulators of oncogenic targets, and the ability of NF-κB to inhibit apoptosis, may be more important in promoting tumorigenesis[53]. The loss of expression of p16INK4a, the protein transcript of the CDKN2A gene, in addition to loss of p53 tumor suppressor function, and resistance to growth inhibition by transforming growth factor-β (TGF-β), are among other observations found in the acquisition of oncogenic potential in TERT transformed cells[54].
Specifically in hMSCs that are transformed with hTERT, there is still the potential of such cells to express tumorigenic properties. Yamaoka et al[55] (2011), constructed hTERT transformed bone marrow hMSCs and found that teratocarcinoma formation could occur when such transformed cells were implanted in immune deficient mice. However, the cells that these investigators transformed with hTERT had first been selected due to their ability to be maintain a proliferative state in the presence of fibroblast growth factor-2 (FGF-2) (> 100 population doubling levels) compared to hMSCs not cultured with FGF-2 that could proliferate to only 20 population doubling levels. As telomerase activity was absent in these FGF-2 maintained clones but had maintained long telomere length, an alternative lengthening of telomeres (ALT) pathway induced by FGF-2 in combination with TERT immortalization could have accounted for the malignant transformation. Serakinci et al[56] (2004) also reported that hMSCs transformed with hTERT could exhibit neoplastic characteristics as shown by loss of contact inhibition and development of mesenchymal tumors after implantation of cells in immunodeficient mice. Loss of p16INK4a and hypermethylation of DBCCR1 (deleted in bladder cancer chromosomal region candidate 1), a cell-cycle associated gene, were observed. Interestingly, tumors were generated only in high population doubling level hTERT-transformed hMSCs and not in relatively lower population doubling level hTERT-transformed hMSCs. Similarly, Abdallah et al[39] (2005) reported that mesodermal type tumors formed from hTERT transformed hMSCs that had a short population doubling time and accelerated growth, but no tumors developed in hTERT transformed hMSC clones with longer population doubling times that were slower growing. Thus the potential for neoplastic change may be associated with loss of proliferative control as evidenced by cell cycle gene alterations with continued proliferation.
Nevertheless, others have reported that hTERT-transformed hMSCs did not exhibit changes associated with neoplasia even at higher population doubling levels (up to 275)[32,41,57]. However, whether or not potential oncogenic development occurs in hTERT-transformed hMSCs, functional changes in hMSC parameters need to be considered. Baumer et al[58] (2011) reported that hTERT-transformed human coronary artery endothelial cells demonstrated changes in an in vitro co-culture angiogenesis assay where TERT-transformed human coronary artery endothelial cells co-cultured with human fibroblasts and treated with vascular endothelial growth factor (VEGF) did not form tubular networks indicative of angiogenesis; non-TERT-transformed endothelial cells in co-culture with fibroblasts and treated with VEGF were able to form tubular networks. Moreover, hTERT-transformed endothelial cells responded differently to exogenous tumor necrosis factor-α (TNF-α) compared to non-hTERT transformed cells where vascular cell adhesion molecule-1 (VCAM-1) expression was lower, and endothelial barrier function as measured by transepithelial resistance was lost in hTERT-transformed cells. Since hMSCs are immunomodulatory cells that can affect the function of immune hematopoietically derived cells (lymphocytes, monocytes, etc.) in an inflammatory environment, there needs to be further investigation if hTERT transformation of hMSCs do not affect these immunomodulating properties of normal hMSCs or have altered function in differentiation or on angiogenesis when interacting with other cell types in a microenvironmental setting.
Perhaps the most prudent approach to ensure that hTERT transformed hMSCs would be useful for bone repair after induction of osteogenic differentiation would be to use inducible vectors for hTERT expression that can then be regulated both temporally and spatially to avert problems with continuous cell proliferation that could result in oncogenic transformation of hTERT-transformed hMSCs[40].
One other caveat involving the potential enhancement of carcinogenesis may be specific to adipose-derived stem cells (stromal cells) and endothelial cells from white adipose tissue that is independent of hTERT transformation. Zhang et al[59] (2009) reported that the stromal/vascular fraction of white adipose tissue that have proliferative and multipotent differentiative capacity as well as pericyte-like characteristics can home to human breast and prostate carcinoma cell lines, Kaposi’s sarcoma endothelial cell line, and a mouse lung carcinoma cell line implanted in xenograft and allograft mouse models. These stromal/vascular cells engrafted into the tumors and enhanced cancer progression in part through stimulating angiogenesis in the tumors but also perhaps though immunosuppressive effects of the adipose-derived mesenchymal cells found in the stromal/vascular fraction. In follow-up studies, these investigators showed that the increase in the number of adipose-derived stromal (mesenchymal) cells found in obesity could be recruited to mouse and human breast cancer and mouse ovarian cancer models and stimulate tumor growth by increasing tumor vascularity and by differentiating into adipocytes and stimulating proliferation of tumor cells[60]. In human studies, it was reported that there was increased frequency of mesenchymal stromal (CD34brightCD45-CD31-) cells (also harboring the pericyte marker, NG2) and CD34bright leucocytes (CD45brightCD34bright) in obese patients (BMI > 30) with colorectal cancer compared to obese non-cancerous subjects[61,62]. Lean patients with colorectal cancer also had a higher frequency of mesenchymal stromal cells and CD34bright leucocytes compared to lean, non-cancerous controls. However, there was a significant increase in MSCs in obese colorectal cancer patients compared to lean colorectal cancer patients. Thus mobilization of MSCs and CD34bright leucocytes may potentially be markers of colorectal cancer but that there may be a higher frequency of CD34+ MSCs (adipose stromal cells) released into circulation even in non-cancerous obese patients suggesting that adipose tissue contributes to MSC mobilization.
OSTEOGENIC DIFFERENTIATION OF HTERT-TRANSFORMED HMSCS
Differentiation of hMSCs along the osteogenic lineage has been demonstrated using both in vitro and in vivo techniques. Induction of in vitro osteogenic differentiation in hMSCs include addition of dexamethasone, ascorbate, and a source of phosphate, mainly β-glycerophosphate to a culture medium base (generally Dulbecco’s modified Eagle’s medium) containing 10% bovine serum. However, recently it was reported that hMSCs from bone marrow may not require the addition of dexamethasone and ascorbate to form bone in vivo although bone marrow-derived hMSCs respond to dexamethasone and ascorbate with increased proliferation in vitro[63]. Osteogenic marker expression by mRNA and protein is usually assessed over the course of in vitro cell culture. Early markers of osteogenesis include core binding factor 1 [cbfa1 or runx2 (Runt-related transcription factor 2)] which is found in chondro-osseous precursor cells, osterix which appears in committed osteogenic cells, and collagen type I. Intermediate markers of osteogenesis include alkaline phosphatase and osteopontin and bone sialoprotein and osteocalcin (usually induced in hMSCs by 1.25 dihydroxyvitamin D3) are generally used as later markers of terminally differentiated osteoblasts. Determination of mineralization of culture in vitro is also critical in assessing terminal differentiation along the osteogenic lineage. This is usually accomplished by staining cell cultures using alizarin red or von Kossa stains which bind to calcium and/or eluting these stains for semi-quantitation of calcium spectrophotometrially. It is also suggested that to distinguish amorphous calcium-phosphate precipitation in cultures from hydroxyapatite [Ca10P8(OH)2], X-ray diffraction, nuclear magnetic resonance, or other technique be used to compare the calcium-phosphate complexes in in vitro cell cultures with standard hydroxyapatite patterns by these techniques. Additionally, negative markers for other mesodermal cell types that can be differentiated from hMSCs should be assessed. These are usually markers for the adipogenic lineage [adipsin, peroxisome proliferator-activated receptor gamma (PPAR-γ), adiponectin], the chondrogenic lineage (sox9, collagen type II, collagen type X, aggrecan), tenogenic lineage (scleraxis)[64], and myogenic lineage Pax3, Pax7 (myogenic precursors), MyoD and myogenin (skeletal muscle), α-smooth muscle actin, vascular endothelial (VE) cadherin (smooth muscle). Essentially, similar techniques to demonstrate osteogenic differentiation have been used for hTERT-transformed hMSCs.
In vivo osteogenesis of hMSCs, whether or not transformed with hTERT, is usually accomplished by ectopic bone ossicle formation assay. In this assay, hMSCs are usually mixed with hydroxyapatite and/or treated with various bone morphogenetic proteins (BMPs) and are implanted into subcutaneous pockets in either immunocompromised rodents (e.g., nude mice; NOD/SCID mice)[32,39,65,66] or into immune competent rodents[41]. Assessment for bone formation is done by microCT and/or histology to identify trabecular bone formation and the expression of the above bone marker genes and proteins in tissue sections. hMSCs have been shown to create a locally immunosuppressive microenvironment and are able to avoid allo-recognition[67] perhaps in rodent species although it is unknown if the same holds true for transplantation of hMSCs into human recipients or if there are any consequences of immunogenicity of hMSCs once they are differentiated into specific lineages in a human recipient[68].
It is highly important that the both in vitro and in vivo confirmation of hydroxyapatite or bone formation be done especially in hTERT-transformed hMSCs. It is possible that not all hTERT-transformed hMSCs will be able to form bone in vivo. Larsen et al[69] (2010) established subclones from hTERT transformed hMSCs at a relatively early population doubling level (PDL 77) and from a later PDL 233. They found that both subclones retained surface markers for hMSCs (CD63, CD73, CD105, and CD166) as well as expressed osteoblast markers, alkaline phosphatase, collagen type I, and osteocalcin upon induction with osteogenic medium. Both clones also formed mineralized matrix in vitro as assessed by alizarin red staining. However, the PDL 77 clone was able to form bone in an in vivo ectopic bone formation assay while the PDL 233 clone did not form bone. Interestingly, these investigators reported that CD146 was highly expressed in the hTERT-transformed hMSC clone that could form bone in vivo while CD146 was minimally expressed in the hTERT-transformed clone that did not form bone in vivo. Thus the criteria for in vivo bone formation and expression of CD146 should be helpful in assessing hTERT-transformed hMSCs that may be useful for potential bone repair or regenerative therapy, and sole dependence on osteogenic markers and in vitro, two-dimensional cell culture mineralization assays may be insufficient. Also observed in additional hTERT-transformed hMSC clones that formed bone in vivo was the increased number of extracellular matrix genes expressed as well as the increased number of Sp3 binding sites in the promoter regions of these expressed genes compared to that of hTERT-transformed hMSC clones that did not form bone in vivo. Sp3 is a transcription factor necessary for bone development and ossification.
In attempts to seed hTERT-transformed hMSCs in areas requiring their presence for tissue repair, strategies such as intracardiac or intravenous injection of hMSCs expressing a fluorescent marker (e.g., green fluorescent protein) have been used to identify sites where such injected hMSCs populate as well as to assess the longevity of transplanted hMSCs in the desired regions. Bentzon et al[70] (2005) reported that hTERT-transformed hMSCs injected intracardiac or intravenously into NOD/SCID mice were trapped mainly in microvasculature of the lungs, kidneys and heart. It was also found that only a small fraction of the injected telomerized hMSCs survived or were retained possibly due to protracted trans-endothelial migration. Thus direct engraftment of hTERT-transformed hMSCs may be a better approach to healing bone.
In addition to cells, such as MSCs, that have the potential to self-replicate and differentiate into the cell type of choice, tissue engineering in regenerative medicine strategies generally combine the cellular component with various growth and differentiation factors that can promote differentiation of undifferentiated precursor cells and with the employment of a structural framework on which either such cells and/or growth and differentiation factors can be assembled. The use of three-dimensional culture platforms may simulate the natural three-dimensional in vivo tissue architecture and provide advantages over that of assessing hMSC growth and differentiation on tissue culture plastic in a two-dimensional format[71,72]. Two dimensional cultures may only yield woven type bone (random orientation of collagen fibrils) and not allow for the formation of lamellar bone, the final desired bone product, and microenvironments that may develop in a three-dimensional framework that could affect cell-cell and cell-matrix interactions cannot fully develop in a two dimensional culture system.
For in vivo uses, three-dimensional platforms or scaffolds need to be biocompatible, potentially biodegradable, have sufficient porosity to allow great surface area for cell attachment, and in general be non-immunogenic. The more rigid platforms or scaffolds composed of material such as hydroxyapatite or other calcium-phosphate bases which are osteoinductive and can induce ectopic bone formation. Titanium has been used to grow hMSCs that can then be differentiated along the osteogenic lineage with or without BMP stimulation prior to direct surgical implantation into bone defects in translational models of bone repair[73-75]. Biological scaffolds that are composed of polymer blends such as poly(l-lactide-co-glycolide) (PLGA) are biocompatible and can be degraded by the body have also been used as a base on which hMSCs can be grown and differentiated[76]. Polymer blends have also been used in combination with inorganic hydroxyapatite crystals or naturally occurring proteins such as collagen to construct composite scaffolds that improve mechanical and osteoinductive properties of the scaffolds have also been designed[77]. Hydrogels have also been used as scaffold material due to biocompatibility; natural hydrogels are derived from collagen or gelatin, while synthetic hydrogels can be made from poly(ethylene glycol). While natural hydrogels are excellent for cell adhesion and biodegradation, immunogenic reactions may be a concern if the hydrogels are derived from animal-derived extracellular matrix (ECM) protein. Synthetic hydrogels have the advantage of creating scaffolds in situ using photopolymerization and also are non-immunogenic[78]. Hydrogels as well as polymer blends with or without ceramic material (e.g., hydroxyapatite )have also been useful in serving as reservoirs for bioactive molecules such as growth factors[77-79]. Thus scaffolds impregnated with various growth factors or composed in part of ECM-derived short peptides, modified heparin, chondroitin sulfate or hyaluronic acid to tether growth factors such as the BMPs, epidermal growth factor (EGF), platelet-derived growth factor (PDGF), TGF-β, FGF-2 have been useful in the differentiation of transplanted hMSCs and/or the chemotaxis of native MSCs useful in bone repair[75,77-82].
Stromal-derived factor 1 (SDF-1), a chemokine, has also been impregnated in scaffolds to serve as a chemotactic factor for bone marrow-derived MSCs[83-85]. It has also been shown that human cord blood-derived MSCs as well as human adipose tissue-derived MSCs (the stromal/vascular fraction of adipose tissue) express CXC receptor 4 (CXCR4), the receptor for SDF-1, and are induced to migrate in response to SDF-1[86,87]. Human bone marrow-derived MSCs have also have been shown to migrate to bone marrow stroma in a CXCR4-dependent manner[88]. Bone-marrow-derived MSCs can also express SDF-1 and serve to maintain hematopoietic stem cells in a quiescent state in the bone marrow[89,90]. However, under conditions of inflammation with the release of pro-inflammatory cytokines such as TNF-α and IL-1β and hypoxia that can be found in tissue injury and the early phases of wound repair, bone marrow-derived MSCs as well as MSCs from adipose tissue or other local sources could migrate to the wound location via the SDF-1/CXCR4 axis to participate in the repair or regeneration of mesenchymal tissues (e.g., bone)[91-94]. Potential sources of SDF-1 that could potentially be involved in local MSC migration and homing to disrupted bone may be endothelium, local osteoblasts, platelets involved in initial wound hemostasis, and periosteal cells[87,95-97]. VEGF has been used to stimulate angiogenesis that would allow for improved blood supply to repairing tissues; use of VEGF incorporated into natural hydrogels or injected directly into scaffolds and in combination with BMPs and MSCs attached to scaffolds have been tested to improve bone healing[81,98]. In the absence of seeding MSCs onto scaffolds, delivery of SDF-1 via implantable infusion pump to poly-€-caprolactone scaffolds preceded by delivery of VEGF to the scaffolds and followed by BMP-6 to induce osteogenic differentiation was able to induce mature mineralized bone formation[99]. Tasso et al[100] (2009) also reported that in a mouse model of ectopic bone formation, donor murine bone marrow MSCs loaded onto hydroxyapatite scaffolds were needed in the early development of ectopic bone (up to one week after implantation) to recruit host osteoprogenitor cells, but native (host) osteoprogenitor cells actually contributed the most to the bone formation via endochondral ossification. Thus native MSCs can be induced to populate scaffolds using SDF-1 and osteogenically differentiate to form vascularized bone. MSCs harboring viral vectors (adeno-associated virus or lentivirus) to over express growth factors and chemoattractants and attached to various types of scaffolds have been used as an alternative strategy to increase local concentrations of bioactive molecules such as BMP-2, BMP-7, VEGF, and CXCR4, the transmembrane G-protein coupled receptor for SDF-1-induced chemoattraction, to enhance osteogenic marker expression[101-106]. Finally, other chemokines may also play roles in migration of MSCs. Chemokines of the α family (CXC chemokines) as well as the β family (CC chemokines) have been reported to stimulate migration of MSC from both bone marrow and omental adipose tissue[92,107,108]. Interestingly, under pro-inflammatory conditions as is found in the initial phase of wound healing, priming with TNF-α enhances the expression of these chemokines such as CXCL8 (interleukin 8), CCL5/RANTES (regulated on activation, normal T cell expressed and secreted), CCL22 (macrophage-derived chemokine) which are then able to stimulate MSC migration[94,95]. Additionally, CXC chemokines with the glu-leu-arg motif in the N-terminus of CXC chemokines are also angiogenic and thus may play a role in new blood vessel formation during wound repair during bone regeneration[109].
Three dimensional spheroid cultures consisting of high density cell aggregates in agarose or alginate have also been used to traditionally differentiate chondrocytes from hMSCs[110,111]. Burns et al[112] (2010) used a variation of this method by using caroxymethylcellulose in their high density cell preparation to form spheroids of hTERT transformed hMSC cells. When combined with hydroxyapatite/β-tricalcium phosphate scaffolds, induced with osteogenic medium, and implanted into immunodeficient mice in an in vivo ectopic bone formation assay, lamellar bone formation was observed in scaffold concavities in addition to the expression of usual osteoblastic markers of cbfa1, alkaline phosphatase, osteonectin, osteopontin, collagen type I and osteocalcin. CD146 expression which had been high in hMSCs was lost as osteogenic differentiation proceeded. Interestingly, transcriptional co-activator with PDZ binding motif (TAZ)[113], a cbfa1 binding transcription co-activator that allows for commitment to the osteogenic lineage while inhibiting adipogenic differentiation of hMSCs was also induced in the hTERT transformed hMSC spheroids. Stimulated expression of other extracellular matrix proteins such as biglycan, lumican, elastin, periostin, microfibrillar-associated proteins (MFAP2 and MFAP5), tetranectin and decorin also occurred suggesting correlation between these extracellular matrix protein and osteogenesis.
EXTRACELLULAR MATRIX AND HMSCS-THE “OUTS” FOR OSTEOGENESIS
The use of extracellular matrix (ECM) components to enhance either rigid type scaffolds or hydrogel scaffolds or to serve as scaffolds themselves has become more popular in tissue engineering. For instance, collagen type I in the form of gels or sponges or as a protein coating of hydroxyapatite platforms has been useful in providing an attachment for cells in addition to being able to deliver growth factors such as TGF-β, BMPs, or VEGF[114]. ECM contains proteoglycans which are comprised in part of heparin sulfate that can bind many types of growth factors such as FGFs and VEGF and degradation of ECM by matrix metalloproteases can release these growth factors to subsequently bind to their receptors on specific cells[115]. Other ECM proteins such as laminin and tenascin have epidermal growth factor (EGF)-like motifs that could potentially bind to EGF receptors on cells and then initiate an EGF signaling cascade through tyrosine kinase activation resulting in cell proliferation and/or differentiation[116]. The binding of cells to naturally occurring proteins such as collagen occur via integrins, comprised of α and β subunits and binding cell membranes to ECM proteins with the arginine-glycine-aspartic acid (RGD) or leucine-valine-aspartic acid (LVD) (consensus sequence L/I (isoleucine)-D/E (glutamic acid)-V (valine)/S(serine)/T(threonine)-P (proline)/S) domains[117,118]. The short cytoplasmic domains of integrins interact with various cytoskeletal elements such as talin and kindlin to initiate inside-out signaling through integrin-linked kinase that is involved in activating integrins to bind to ECM components[119,120]. Outside-in signaling occurs with the interaction of specific sequences of ECM proteins and activated integrins to activate focal adhesion kinase to allow in part for functions such as cell spreading and migration but also activating other signaling pathways enabling cell proliferation, and survival[121].
Thus scaffolds composed of native ECM proteins such as collagen have been applied as one strategy to expand hMSCs ex vivo and to promote osteogenic differentiation and to enhance bone repair[17,122,123]. Bone marrow-derived hMSCs, express various integrins such as α1β1, α2β1, α5β1, α6β1, αvβ3, and αvβ5; however, the β1 integrin subunit was found to be most responsible for hMSCs to adhere to collagen, laminin and fibronectin and be involved in proliferation of hMSCs and for their differentiation into osteoblasts[124]. However, pre-coating scaffolds with a specific protein such as collagen type 1 or MatrigelTM (BD Biosciences) composed of collagen type IV, entactin, and laminin, may not yield the natural three dimensional environment, nor account for all appropriate ECM proteins that interact with hMSCs in vivo, nor retain the natural elasticity or stiffness required for proper self-renewal or tissue-specific differentiation. Degree of stiffness or elasticity of support structures or ECM has been shown to be important in part to be a determinant of stem cell differentiation. In reference to MSCs, softer substrates favor adipocyte or chondrogenic differentiation while stiffer substrates direct osteogenic differentiation. Intermediate stiffness can assist in directing myogenesis from MSCs[125-128]. ECM or bioengineered support structure stiffness or elasticity can be sensed by cells through the organization of stress fibers composed of actin microfilaments and myosin. Specifically, non-muscle myosin II isoforms, IIA, IIB, and IIC appear to be involved in the MSC’s ability to sense matrix stiffness through their interaction with cortical actin that is linked to focal adhesions. Increased matrix stiffness is associated with increased activity of non-muscle myosin II. The increased non-muscle myosin II also correlates with specific lineage determination of MSCs[129]. Interestingly, ECM stiffness that can set the stage for specific lineage differentiation via expression or repression of specific genes is transduced to nuclear chromatin via lamin-A[130]. Cytoskeletal stresses and tension increase with increasing ECM stiffness and the degree of lamin-A expression and phosphorylation is inversely related to ECM stiffness. Thus osteogenic differentiation of MSCs is correlated with increased lamin-A levels and decreased lamin-A phosphorylation when MSCs are grown on a stiff ECM. It would follow that lamin-A would act in a manner to maintain nuclear rigidity or stiffness which could translate into epigenetic regulation of chromatin thus enabling transcription of osteogenic genes and repression of genes specific with other mesenchymal lineages through lamin A-associated domains which contain repressive heterochromatin.
Thus the use of cell-free preparations of secreted ECM proteins produced by MSCs or cells of the desired differentiated type (i.e., osteoblasts) may perhaps allow for better osteogenic differentiation of MSCs in a native three dimensional microenvironment similar to the MSC niche found in bone marrow. Chen et al[131] (2007) prepared ECM from mouse MSCs that supported self-renewal of mouse MSCs when cultured on this native ECM and the proliferative ability of the MSCs grown on native ECM was greater than MSCs grown on fibronectin or collagen type I individually. Differentiation of mouse MSCs into both adipogenic (in response to rosiglitazone) and osteogenic lineages (in the presence of exogenous BMP-2) was also enhanced in cells cultured on native ECM compared to tissue culture plastic or culture plastic coated with fibronectin alone or with collagen type I alone. However, mouse MSCs had a delay in osteogenic differentiation when grown on native ECM in the absence of exogenous BMP-2, and it was suggested that the native ECM components such as collagen and biglycan bind BMP-2, making it less available to MSCs to allow for earlier osteogenic differentiation.
hMSCs can also be used to generate native ECM that supports self-replication of hMSCs, and the degree of enhanced proliferation of hMSCs was found to be greater than that of hMSCs grown on tissue culture plastic, or fibronectin or collagen type I independently[132]. It was also found that SSEA-4, a marker for bone marrow-derived hMSCs, was maintained at a high level throughout the culture period on native ECM and interestingly, telomerase activity was stable and reactive oxygen species was low on ECM-grown hMSCs compared to hMSCs grown on plastic, fibronectin, or collagen type I. In vivo bone formation was also significantly higher in hMSCs grown on native ECM compared to those grown on plastic. Thus native ECM from hMSCs can better support self-renewal and osteogenic differentiation compared to single ECM components or a two dimensional culture platform (plastic).
It has been shown that ECM from human foreskin young fibroblasts (< 20-30 population doublings) supported the proliferation of old fibroblasts (> 68 population doublings) so that the proliferative rate of the old fibroblasts approached that of young cells grown on ECM from young cells[133]. Additionally, telomere length was restored in old fibroblasts grown on ECM from young cells by a telomerase independent mechanism and reduced reactive oxygen species similar to young cells was also found. Interestingly, SIRT 1, a gene for the NAD-dependent histone deacetylase, sirtuin 1, which was downregulated during senescence was increased when old fibroblasts were grown on ECM from young cells. This suggests that epigenetic mechanism(s) may play a role the mechanism of how young ECM can restore the proliferative ability of old fibroblasts. SIRT 1 can be directly activated by lamin A[134], which is critically involved in the process of information flow from ECM to the nucleus to perhaps determine chromatin configuration and thus confer epigenetic regulation on gene expression or repression. Thus the potential role of epigenetics in ECM rejuvenation of old fibroblast cells is an area of interesting investigation.
With regards to MSCs, the composition of ECM from young (low passage) adipose-derived MSCs compared to that of old (higher passage) MSCs is different. For instance, while collagen type I is increased in young MSCs, laminin, fibronectin, vimentin, keratin, and lamin A/C are decreased in old MSCs. When old MSCs are seeded onto ECM from young MSCs, the pluripotency markers of Oct4, Sox2, and Nanog are increased and growth factors such as TGFβ are also upregulated[135]. The ECM component, biglycan, has been shown to increase canonical Wnt/β-catenin signaling. Wnt signaling is a critical morphogen in osteoprogenitor development. Bone marrow MSCs from mice deficient in biglycan were less proficient in Wnt-induced mineral deposition in culture, did not respond to exogenous Wnt3a, and made significantly less trabecular bone when used in an in vivo ectopic bone formation assay[136]. Thus one could speculate that ECM from young MSCs may have more biglycan than ECM from old MSCs and thus young ECM would be able to enhance Wnt signaling to enhance both proliferation of osteoprogenitors and potentially more bone formation. However, the exact mechanism of how biglycan can regulate either canonical or non-canonical Wnt signaling is unclear.
In another interesting study, Sun et al[137] (2011) reported the differential effect of ECM from mouse bone marrow stromal cells derived from young (3 mo) versus old mice (18 mo). Replicative ability was restored in MSCs from old mice cultured on ECM from young mice, similar to that of the replicative ability of young mice grown on ECM from young mice. However, the replicative ability of MSCs from either young or old mice was significantly less when cultured on ECM from old mice. Telomerase levels were also increased in MSCs from young and old mice cultured on ECM from young mice compared to that of MSCs cultured on tissue culture plastic or on ECM from old animals. Examination of bone forming ability using an in vivo assay where MSCs from young or old mice pre-cultured on ECM from young or old mice demonstrated that MSCs from old mice pre-cultured on ECM from young mice had increased cancellous bone formation compared to MSCs from young or old mice pre-cultured on tissue culture plastic. Culture of MSCs from either young or old mice on ECM from old mice demonstrated less bone formation. In trying to dissect the differential effect of ECM from old versus young mice, these investigators founds that ECM from old mice contained more mineral phosphate and less collagen although the total amount of ECM produced by young or old cells was the same. Furthermore, reactive oxygen species levels were higher in MSCs grown on ECM from old mice but were reduced in MSCs grown on ECM from young mice; there was also an inverse correlation of the number of colony forming units-osteoblast and the level of reactive oxygen species. How ECM from old mice is incapable of handling reactive oxygen species and how this may relate to changes in ECM composition (lower collagen and proteoglycans) remains unknown.
In a recent communication, Prewitz et al[138] (2013) used early passage bone marrow-derived hMSCs to generate native ECM but used either osteogenic medium to allow the hMSCs to differentiate towards the osteogenic lineage or ascorbic acid alone in the growth medium to allow the hMSCs under these conditions to generate an “enriched” ECM. These generated ECMs were then tethered to tissue culture plastic using poly(octadecene-alt-maleic anhydride). These investigators reported that ascorbic acid-stimulated native ECM contained twice as much collagen and sulfated glycosaminoglycans compared to native ECM generated using osteogenic medium although the spectrum of ECM protein were the same. Release of hepatocyte growth factor, FGF, VEGF, and interleukin-8 was also higher from ascorbic acid-stimulated ECM. Nevertheless, both types of ECM supported higher population doublings of hMSCs grown on these surfaces compared to hMSCs grown on either plasma-treated tissue culture plastic, fibronectin or MatrigelTM. Both ascorbic acid and osteogenic-induced ECM also stimulated more osteogenic differentiation as well as adipogenic differentiation although the ascorbic acid-induced ECM yielded better osteogenic and adipogenic differentiation than osteogenic-induced ECM. Finally, both ascorbic acid-induced and osteogenic-induced ECM were able to support the engraftment of hematopoietic stem and progenitor cells, similar to a hematopoietic stem cell niche. Hence, bolstering native ECM by stimulation its production from hMSCs with either ascorbic acid or osteogenic medium could potentially be a useful strategy in rejuvenating old hMSCs.
Thus whether the total or individual amounts of native ECM, the breadth of composition of native ECM, the geometry of ECM organization, or the ability of ECM to sequester growth factors, retain growth factor-like motifs (e.g., similar to the EGF-like repeats found on laminin and tenascin), or regulate other morphogens such as Wnt signaling that can potentially regulate MSC proliferation and differentiation are important factors in explaining the mechanism(s) of how young ECM can rejuvenate old MSCs are salient areas for future investigation.
CLINICAL UTILITY OF MESENCHYMAL STEM CELLS IN ORTHOPAEDIC CONDITIONS
MSCs from various sources in combination with specific growth factors and/or scaffold material potentially lend themselves to a variety of clinical orthopaedic conditions involving bone and cartilage. There are a number of clinical trials and case reports using MSCs to repair critical sized defects caused by trauma or infection as well as replacing chronically degenerated tissue such as articular cartilage and intervertebral discs. There are a number of excellent and comprehensive published reviews on the subject of orthopaedic applications for MSC therapy[139-143] and which are listed in Table 1. Two clinical trials and two other case reports using MSCs in human orthopaedic conditions are also included in Table 2. The clinical trial to treat knee osteoarthritis enrolled 25 patients. Infrapatellar fat pad-derived MSCs and platelet rich plasma were injected into knee joints after arthroscopic debridement, excision of degenerative material/osteophytes, or synovectomy[144]. Comparison was made to retrospective age- sex- and follow-up period matched controls who had received only platelet rich plasma injections with arthroscopic debridement. Various scales used in knee symptoms (visual analog pain scale, Lysholm knee scoring scale, Tegner activity level scale) showed that the initial or pre-treatment scores of the study group were significantly poorer compared to controls but by the last follow-up visit (12 mo) after MSC therapy, the study group showed significantly higher degrees of improvement from pre-treatment levels in all of the assessment scales measured compared to that of the retrospective control group. Orozco et al[145] (2011) injected autologous bone marrow-derived MSCs that were expanded under Good Manufacturing Practice conditions into the nucleus pulposus in 10 patients. These patients apparently served as their own controls and pain (visual analog scale), disability (Oswestry Disability Index), and quality of life (SF-36) were improved over the 12 mo trial. Water content of the diseased discs also improved by 12 mo after treatment. Two other communications consisting of case reports are also entered into Table 2. One report used autologous adipose-derived stem cells expanded in vitro and combined with β-tricalcium phosphate scaffolding material harboring rhBMP-2 placed in a muscle flap and used to repair a maxillary bone defect[146]. The other was a series of 5 cases using collagen sponges impregnated with rhBMP-2 with or without autologous bone marrow cells and allogeneic cancellous bone (4 cases) and one case using only rhBMP-2 adsorbed onto collagen sponges to reconstruct mandibular bone defects. Although not stated, it was presumed that bone marrow MSCs were the bone marrow cells referred to in three of the cases, two of which were successful in healing the bone defects[147].
Table 1.
Reviews of mesenchymal stem cell use in human orthopaedic conditions
| Ref. | Reviewed Orthopaedic conditions treated | MSC source | Additional repair components |
| Shenaq et al[139], Stem Cell Int, 2010 | Osteonecrosis humerus, femoral heal; Fracture non-union; Cartilage defect; Osteogenesis imperfecta; Critical size defect limbs; Calvarial defect | Autologous or allogeneic bone marrow; Fetal liver; Adipose | Ceramic scaffolds; Collagen gels |
| Rastegar et al[140], World J Stem Cells, 2010 | Critical size defect in long bones; Articular cartilage of knee; Osteogenesis imperfecta; Hypophosphatasia | Autologous bone marrow; Allogeneic bone marrow; Fetal liver | |
| Zhang et al[141], Biomaterials, 2012 | Segmental bone defects of limbs; Distraction osteogenesis; Tibial osteotomy; Posterior spinal fusion; Maxilla defects; Sinus augmentation; Osteogenesis imperfecta; Articular cartilage repair; Osteoarthritis | Autologous bone marrow; Allogeneic bone marrow; Fetal liver | Hydroxyapatite scaffolds; autologous platelet rich plasma, allogeneic bone chips or bone grafts; β-tricalcium phosphate scaffolds |
| Veronesi et al[143], Stem Cell Dev, 2013 | Osteoarthritis of knee, hip, elbow, ankle; medial femoral condyle, patellar, patella-femoral joint lesions; osteochondral lesions talar dome and femoral condyle | Autologous bone marrow | Hyaluronate; collagen type 1 sheet; platelet rich plasma; periosteal patch; collagen powder |
| Kim et al[142], Korean J Int Med, 2013 | Osteogenesis imperfecta; Cartilage defects | Allogeneic bone marrow, fetal liver |
MSC: Mesenchymal stem cell.
Table 2.
Clinical trials and case reports using mesenchymal stem cells in human orthopaedic conditions
| Orthopaedic condition | MSC source | Technique | Patients/controls | Study length | Outcome | Ref. |
| Osteoarthritis-Knee | Adipose | Autologous MSCs with platelet rich plasma | 25/25 retrospective controls | 12 mo | Study group significantly higher degrees of improvement from pre-treatment levels in pain and activity | [144] |
| Intervertebral Disc Degeneration | Bone marrow | MSC injection into nucleus pulposus | 10/self-controls - pre- and post procedure | 12 mo | Pain, disability, quality of life, disc water content improved | [145] |
| Maxillary Reconstruction | Adipose | Vascular flap with ADCs, β-tricalcium phosphate, BMP-2 | 1 case | 12 mo | Regeneration of normal bone | [146] |
| Mandibular Reconstruction | Bone marrow | BMP-2, collagen sponges + bone marrow MSCs + allogeneic bone chips | 5 cases | 22 mo | Bone regeneration in 2/4 cases using MSCs; failure overall in 2 of 5 cases | [147] |
MSC: Mesenchymal stem cell; BMP: Bone morphogenetic protein.
CLOSING THOUGHTS (A WORKING MODEL)
In summary, MSCs have promising utility in resolving orthopaedic problems although there is a need for more prospective randomized controlled trials. At this point it is still unclear if MSCs from various sources (bone marrow, adipose, cord blood, cord tissue, muscle, etc.) would all be useful in orthopaedic repair and regeneration in general and bone in particular. It does appear that MSCs from either bone marrow or adipose tissue are quite similar in their capacity to serve in bone repair and regeneration. However, work still needs to be done regarding ideal scaffolding material and whether addition of MSCs or growth factors, angiogenic factors, and/or chemotactic factors onto scaffolds alone or in combination with MSCs would be the best strategy for bone repair and regeneration in the human situation.
With specific reference to MSC self-renewal and differentiation into osteogenic tissue, addition of hTERT to MSCs would seem to assist in increasing population doublings and decreasing population doubling times to enhance a critical mass of MSCs (Figure 1). However, there is still debate over initiation of tumorigenesis associated with TERT transformation of MSCs and the potential of MSCs (TERT transformed or not) to enhance the growth of already established tumors. Differentiation of TERT-transformed MSCs into osteogenic cells appear to be kept intact although whether exceeding a certain level of population doublings can lead to a decrease or change in differentiation capacity must still be considered. The use of native ECM from young MSCs appears to enhance the proliferative and differentiative capacity of MSCs and the stiffness of the ECM appears to steer MSCs to differentiate along specific lineages, with osteogenic differentiation being assisted on a stiffer ECM (Figure 1). Thus TERT expression that can be regulated in a time and stage of differentiation manner may be an ideal strategy to both enhance a critical mass of MSCs necessary for bone repair and regeneration but to try to limit the potential of malignant transformation.
Figure 1.
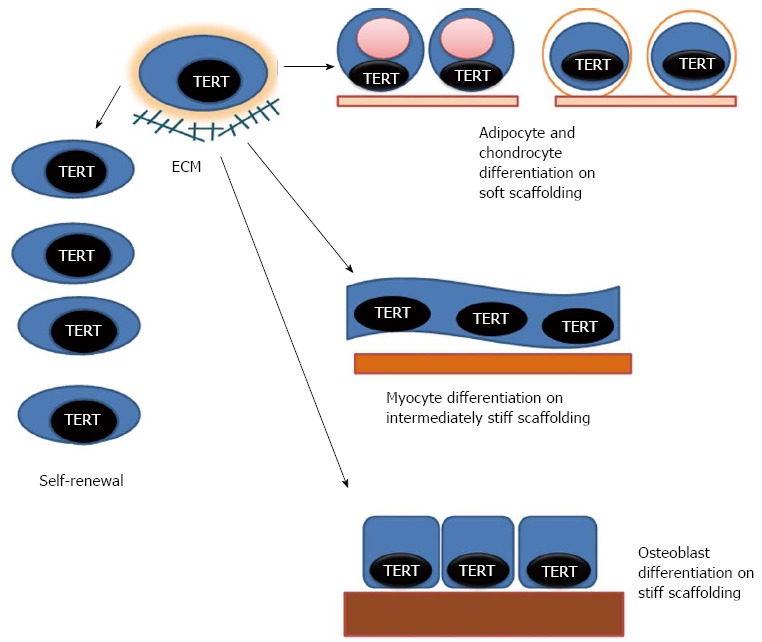
Model of human telomerase reverse transcriptase-transformed mesenchymal stem cell self-renewal and differentiation. Human telomerase reverse transcriptase (hTERT) can be expressed by transfection in human mesenchymal stem cells (hMSCs) from various sources to enhance self-renewal. hTERT transformed cells can be induced to differentiate along multiple mesenchymal lineages. Stiffness of support structures and/or extracellular matrix (ECM) upon which hMSCs are situated is important in differentiated lineage determination. Softer or less stiff support structures/ECM (lightest colored and thinnest bar under the cells) support adipogenic or chondrogenic lineages. Intermediate stiffness (medium colored and thicker bar) can direct myogenesis. Stiffer substrates (darkest colored and thickest bar) can support osteogenic differentiation. Native ECM made from hMSCs from younger hosts may also enhance self-renewal and the differentiative capacity of hMSCs from older sources, and may be superior to singular or limited number of defined ECM components in promoting self-renewal and specific lineage differentiation.
Footnotes
Supported by Veterans Administration Merit Review Award 2 I01 BX000170-05
P- Reviewers: Bonnet D, Karaoz E, Kolonin MG, Scuteri A S- Editor: Wen LL L- Editor: A E- Editor: Zhang DN
References
- 1.Fuchs E, Chen T. A matter of life and death: self-renewal in stem cells. EMBO Rep. 2013;14:39–48. doi: 10.1038/embor.2012.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rando TA. Stem cells, ageing and the quest for immortality. Nature. 2006;441:1080–1086. doi: 10.1038/nature04958. [DOI] [PubMed] [Google Scholar]
- 3.Jones DL, Wagers AJ. No place like home: anatomy and function of the stem cell niche. Nat Rev Mol Cell Biol. 2008;9:11–21. doi: 10.1038/nrm2319. [DOI] [PubMed] [Google Scholar]
- 4.Bianco P. Minireview: The stem cell next door: skeletal and hematopoietic stem cell “niches” in bone. Endocrinology. 2011;152:2957–2962. doi: 10.1210/en.2011-0217. [DOI] [PubMed] [Google Scholar]
- 5.Uccelli A, Moretta L, Pistoia V. Mesenchymal stem cells in health and disease. Nat Rev Immunol. 2008;8:726–736. doi: 10.1038/nri2395. [DOI] [PubMed] [Google Scholar]
- 6.Bianco P, Cao X, Frenette PS, Mao JJ, Robey PG, Simmons PJ, Wang CY. The meaning, the sense and the significance: translating the science of mesenchymal stem cells into medicine. Nat Med. 2013;19:35–42. doi: 10.1038/nm.3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tapp H, Hanley EN, Patt JC, Gruber HE. Adipose-derived stem cells: characterization and current application in orthopaedic tissue repair. Exp Biol Med (Maywood) 2009;234:1–9. doi: 10.3181/0805/MR-170. [DOI] [PubMed] [Google Scholar]
- 8.Fukada S, Uezumi A, Ikemoto M, Masuda S, Segawa M, Tanimura N, Yamamoto H, Miyagoe-Suzuki Y, Takeda S. Molecular signature of quiescent satellite cells in adult skeletal muscle. Stem Cells. 2007;25:2448–2459. doi: 10.1634/stemcells.2007-0019. [DOI] [PubMed] [Google Scholar]
- 9.Crisan M, Yap S, Casteilla L, Chen CW, Corselli M, Park TS, Andriolo G, Sun B, Zheng B, Zhang L, et al. A perivascular origin for mesenchymal stem cells in multiple human organs. Cell Stem Cell. 2008;3:301–313. doi: 10.1016/j.stem.2008.07.003. [DOI] [PubMed] [Google Scholar]
- 10.Ali H, Al-Mulla F. Defining umbilical cord blood stem cells. Stem Cell Discov. 2012;2:15–23. [Google Scholar]
- 11.Rebelatto CK, Aguiar AM, Moretão MP, Senegaglia AC, Hansen P, Barchiki F, Oliveira J, Martins J, Kuligovski C, Mansur F, et al. Dissimilar differentiation of mesenchymal stem cells from bone marrow, umbilical cord blood, and adipose tissue. Exp Biol Med (Maywood) 2008;233:901–913. doi: 10.3181/0712-RM-356. [DOI] [PubMed] [Google Scholar]
- 12.Choudhery MS, Badowski M, Muise A, Harris DT. Comparison of human mesenchymal stem cells derived from adipose and cord tissue. Cytotherapy. 2013;15:330–343. doi: 10.1016/j.jcyt.2012.11.010. [DOI] [PubMed] [Google Scholar]
- 13.Bassi G, Pacelli L, Carusone R, Zanoncello J, Krampera M. Adipose-derived stromal cells (ASCs) Transfus Apher Sci. 2012;47:193–198. doi: 10.1016/j.transci.2012.06.004. [DOI] [PubMed] [Google Scholar]
- 14.Lindroos B, Suuronen R, Miettinen S. The potential of adipose stem cells in regenerative medicine. Stem Cell Rev. 2011;7:269–291. doi: 10.1007/s12015-010-9193-7. [DOI] [PubMed] [Google Scholar]
- 15.Harris DT. Non-haematological uses of cord blood stem cells. Br J Haematol. 2009;147:177–184. doi: 10.1111/j.1365-2141.2009.07767.x. [DOI] [PubMed] [Google Scholar]
- 16.Delorme B, Chateauvieux S, Charbord P. The concept of mesenchymal stem cells. Regen Med. 2006;1:497–509. doi: 10.2217/17460751.1.4.497. [DOI] [PubMed] [Google Scholar]
- 17.Mauney JR, Kirker-Head C, Abrahamson L, Gronowicz G, Volloch V, Kaplan DL. Matrix-mediated retention of in vitro osteogenic differentiation potential and in vivo bone-forming capacity by human adult bone marrow-derived mesenchymal stem cells during ex vivo expansion. J Biomed Mater Res A. 2006;79:464–475. doi: 10.1002/jbm.a.30876. [DOI] [PubMed] [Google Scholar]
- 18.D’Ippolito G, Schiller PC, Ricordi C, Roos BA, Howard GA. Age-related osteogenic potential of mesenchymal stromal stem cells from human vertebral bone marrow. J Bone Miner Res. 1999;14:1115–1122. doi: 10.1359/jbmr.1999.14.7.1115. [DOI] [PubMed] [Google Scholar]
- 19.Stenderup K, Justesen J, Clausen C, Kassem M. Aging is associated with decreased maximal life span and accelerated senescence of bone marrow stromal cells. Bone. 2003;33:919–926. doi: 10.1016/j.bone.2003.07.005. [DOI] [PubMed] [Google Scholar]
- 20.Bernardo ME, Zaffaroni N, Novara F, Cometa AM, Avanzini MA, Moretta A, Montagna D, Maccario R, Villa R, Daidone MG, et al. Human bone marrow derived mesenchymal stem cells do not undergo transformation after long-term in vitro culture and do not exhibit telomere maintenance mechanisms. Cancer Res. 2007;67:9142–9149. doi: 10.1158/0008-5472.CAN-06-4690. [DOI] [PubMed] [Google Scholar]
- 21.Yu KR, Kang KS. Aging-related genes in mesenchymal stem cells: a mini-review. Gerontology. 2013;59:557–563. doi: 10.1159/000353857. [DOI] [PubMed] [Google Scholar]
- 22.Shay JW, Wright WE. Hayflick, his limit, and cellular ageing. Nat Rev Mol Cell Biol. 2000;1:72–76. doi: 10.1038/35036093. [DOI] [PubMed] [Google Scholar]
- 23.Campisi J, d’Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol. 2007;8:729–740. doi: 10.1038/nrm2233. [DOI] [PubMed] [Google Scholar]
- 24.D’Ippolito G, Diabira S, Howard GA, Menei P, Roos BA, Schiller PC. Marrow-isolated adult multilineage inducible (MIAMI) cells, a unique population of postnatal young and old human cells with extensive expansion and differentiation potential. J Cell Sci. 2004;117:2971–2981. doi: 10.1242/jcs.01103. [DOI] [PubMed] [Google Scholar]
- 25.D’Ippolito G, Diabira S, Howard GA, Roos BA, Schiller PC. Low oxygen tension inhibits osteogenic differentiation and enhances stemness of human MIAMI cells. Bone. 2006;39:513–522. doi: 10.1016/j.bone.2006.02.061. [DOI] [PubMed] [Google Scholar]
- 26.Mazumdar J, O’Brien WT, Johnson RS, LaManna JC, Chavez JC, Klein PS, Simon MC. O2 regulates stem cells through Wnt/β-catenin signalling. Nat Cell Biol. 2010;12:1007–1013. doi: 10.1038/ncb2102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bachand F, Autexier C. Functional regions of human telomerase reverse transcriptase and human telomerase RNA required for telomerase activity and RNA-protein interactions. Mol Cell Biol. 2001;21:1888–1897. doi: 10.1128/MCB.21.5.1888-1897.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nguyen BN, Elmore LW, Holt SE. Mechanism of dominant-negative telomerase function. Cell Cycle. 2009;8:3227–3233. doi: 10.4161/cc.8.19.9788. [DOI] [PubMed] [Google Scholar]
- 29.Zhang Q, Kim NK, Feigon J. Architecture of human telomerase RNA. Proc Natl Acad Sci USA. 2011;108:20325–20332. doi: 10.1073/pnas.1100279108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shay JW, Wright WE. Hallmarks of telomeres in ageing research. J Pathol. 2007;211:114–123. doi: 10.1002/path.2090. [DOI] [PubMed] [Google Scholar]
- 31.Wright WE, Piatyszek MA, Rainey WE, Byrd W, Shay JW. Telomerase activity in human germline and embryonic tissues and cells. Dev Genet. 1996;18:173–179. doi: 10.1002/(SICI)1520-6408(1996)18:2<173::AID-DVG10>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 32.Simonsen JL, Rosada C, Serakinci N, Justesen J, Stenderup K, Rattan SI, Jensen TG, Kassem M. Telomerase expression extends the proliferative life-span and maintains the osteogenic potential of human bone marrow stromal cells. Nat Biotechnol. 2002;20:592–596. doi: 10.1038/nbt0602-592. [DOI] [PubMed] [Google Scholar]
- 33.Zimmermann S, Voss M, Kaiser S, Kapp U, Waller CF, Martens UM. Lack of telomerase activity in human mesenchymal stem cells. Leukemia. 2003;17:1146–1149. doi: 10.1038/sj.leu.2402962. [DOI] [PubMed] [Google Scholar]
- 34.Zhao YM, Li JY, Lan JP, Lai XY, Luo Y, Sun J, Yu J, Zhu YY, Zeng FF, Zhou Q, et al. Cell cycle dependent telomere regulation by telomerase in human bone marrow mesenchymal stem cells. Biochem Biophys Res Commun. 2008;369:1114–1119. doi: 10.1016/j.bbrc.2008.03.011. [DOI] [PubMed] [Google Scholar]
- 35.Samsonraj RM, Raghunath M, Hui JH, Ling L, Nurcombe V, Cool SM. Telomere length analysis of human mesenchymal stem cells by quantitative PCR. Gene. 2013;519:348–355. doi: 10.1016/j.gene.2013.01.039. [DOI] [PubMed] [Google Scholar]
- 36.Gronthos S, Chen S, Wang CY, Robey PG, Shi S. Telomerase accelerates osteogenesis of bone marrow stromal stem cells by upregulation of CBFA1, osterix, and osteocalcin. J Bone Miner Res. 2003;18:716–722. doi: 10.1359/jbmr.2003.18.4.716. [DOI] [PubMed] [Google Scholar]
- 37.Saeed H, Abdallah BM, Ditzel N, Catala-Lehnen P, Qiu W, Amling M, Kassem M. Telomerase-deficient mice exhibit bone loss owing to defects in osteoblasts and increased osteoclastogenesis by inflammatory microenvironment. J Bone Miner Res. 2011;26:1494–1505. doi: 10.1002/jbmr.349. [DOI] [PubMed] [Google Scholar]
- 38.Kassem M, Abdallah BM, Yu Z, Ditzel N, Burns JS. The use of hTERT-immortalized cells in tissue engineering. Cytotechnology. 2004;45:39–46. doi: 10.1007/s10616-004-5124-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Abdallah BM, Haack-Sørensen M, Burns JS, Elsnab B, Jakob F, Hokland P, Kassem M. Maintenance of differentiation potential of human bone marrow mesenchymal stem cells immortalized by human telomerase reverse transcriptase gene despite [corrected] extensive proliferation. Biochem Biophys Res Commun. 2005;326:527–538. doi: 10.1016/j.bbrc.2004.11.059. [DOI] [PubMed] [Google Scholar]
- 40.Piper SL, Wang M, Yamamoto A, Malek F, Luu A, Kuo AC, Kim HT. Inducible immortality in hTERT-human mesenchymal stem cells. J Orthop Res. 2012;30:1879–1885. doi: 10.1002/jor.22162. [DOI] [PubMed] [Google Scholar]
- 41.Nakahara H, Misawa H, Hayashi T, Kondo E, Yuasa T, Kubota Y, Seita M, Kawamoto H, Hassan WA, Hassan RA, et al. Bone repair by transplantation of hTERT-immortalized human mesenchymal stem cells in mice. Transplantation. 2009;88:346–353. doi: 10.1097/TP.0b013e3181ae5ba2. [DOI] [PubMed] [Google Scholar]
- 42.Bischoff DS, Makhijani NS, Yamaguchi DT. Constitutive expression of human telomerase enhances the proliferation potential of human mesenchymal stem cells. Biores Open Access. 2012;1:273–279. doi: 10.1089/biores.2012.0252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bellows CG, Heersche JN, Aubin JE. Determination of the capacity for proliferation and differentiation of osteoprogenitor cells in the presence and absence of dexamethasone. Dev Biol. 1990;140:132–138. doi: 10.1016/0012-1606(90)90060-v. [DOI] [PubMed] [Google Scholar]
- 44.Malaval L, Liu F, Roche P, Aubin JE. Kinetics of osteoprogenitor proliferation and osteoblast differentiation in vitro. J Cell Biochem. 1999;74:616–627. [PubMed] [Google Scholar]
- 45.Dominici M, Le Blanc K, Mueller I, Slaper-Cortenbach I, Marini F, Krause D, Deans R, Keating A, Prockop Dj, Horwitz E. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy. 2006;8:315–317. doi: 10.1080/14653240600855905. [DOI] [PubMed] [Google Scholar]
- 46.Gronthos S, Graves SE, Ohta S, Simmons PJ. The STRO-1+ fraction of adult human bone marrow contains the osteogenic precursors. Blood. 1994;84:4164–4173. [PubMed] [Google Scholar]
- 47.Pilz GA, Braun J, Ulrich C, Felka T, Warstat K, Ruh M, Schewe B, Abele H, Larbi A, Aicher WK. Human mesenchymal stromal cells express CD14 cross-reactive epitopes. Cytometry A. 2011;79:635–645. doi: 10.1002/cyto.a.21073. [DOI] [PubMed] [Google Scholar]
- 48.Niehage C, Steenblock C, Pursche T, Bornhäuser M, Corbeil D, Hoflack B. The cell surface proteome of human mesenchymal stromal cells. PLoS One. 2011;6:e20399. doi: 10.1371/journal.pone.0020399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Deans RJ, Moseley AB. Mesenchymal stem cells: biology and potential clinical uses. Exp Hematol. 2000;28:875–884. doi: 10.1016/s0301-472x(00)00482-3. [DOI] [PubMed] [Google Scholar]
- 50.Gang EJ, Bosnakovski D, Figueiredo CA, Visser JW, Perlingeiro RC. SSEA-4 identifies mesenchymal stem cells from bone marrow. Blood. 2007;109:1743–1751. doi: 10.1182/blood-2005-11-010504. [DOI] [PubMed] [Google Scholar]
- 51.Qian H, Le Blanc K, Sigvardsson M. Primary mesenchymal stem and progenitor cells from bone marrow lack expression of CD44 protein. J Biol Chem. 2012;287:25795–25807. doi: 10.1074/jbc.M112.339622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mu J, Wei LX. Telomere and telomerase in oncology. Cell Res. 2002;12:1–7. doi: 10.1038/sj.cr.7290104. [DOI] [PubMed] [Google Scholar]
- 53.Low KC, Tergaonkar V. Telomerase: central regulator of all of the hallmarks of cancer. Trends Biochem Sci. 2013;38:426–434. doi: 10.1016/j.tibs.2013.07.001. [DOI] [PubMed] [Google Scholar]
- 54.Belgiovine C, Chiodi I, Mondello C. Telomerase: cellular immortalization and neoplastic transformation. Multiple functions of a multifaceted complex. Cytogenet Genome Res. 2008;122:255–262. doi: 10.1159/000167811. [DOI] [PubMed] [Google Scholar]
- 55.Yamaoka E, Hiyama E, Sotomaru Y, Onitake Y, Fukuba I, Sudo T, Sueda T, Hiyama K. Neoplastic transformation by TERT in FGF-2-expanded human mesenchymal stem cells. Int J Oncol. 2011;39:5–11. doi: 10.3892/ijo.2011.1029. [DOI] [PubMed] [Google Scholar]
- 56.Serakinci N, Guldberg P, Burns JS, Abdallah B, Schrødder H, Jensen T, Kassem M. Adult human mesenchymal stem cell as a target for neoplastic transformation. Oncogene. 2004;23:5095–5098. doi: 10.1038/sj.onc.1207651. [DOI] [PubMed] [Google Scholar]
- 57.Huang G, Zheng Q, Sun J, Guo C, Yang J, Chen R, Xu Y, Wang G, Shen D, Pan Z, et al. Stabilization of cellular properties and differentiation mutilpotential of human mesenchymal stem cells transduced with hTERT gene in a long-term culture. J Cell Biochem. 2008;103:1256–1269. doi: 10.1002/jcb.21502. [DOI] [PubMed] [Google Scholar]
- 58.Baumer Y, Scholz B, Ivanov S, Schlosshauer B. Telomerase-based immortalization modifies the angiogenic/inflammatory responses of human coronary artery endothelial cells. Exp Biol Med (Maywood) 2011;236:692–700. doi: 10.1258/ebm.2011.010300. [DOI] [PubMed] [Google Scholar]
- 59.Zhang Y, Daquinag A, Traktuev DO, Amaya-Manzanares F, Simmons PJ, March KL, Pasqualini R, Arap W, Kolonin MG. White adipose tissue cells are recruited by experimental tumors and promote cancer progression in mouse models. Cancer Res. 2009;69:5259–5266. doi: 10.1158/0008-5472.CAN-08-3444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhang Y, Daquinag AC, Amaya-Manzanares F, Sirin O, Tseng C, Kolonin MG. Stromal progenitor cells from endogenous adipose tissue contribute to pericytes and adipocytes that populate the tumor microenvironment. Cancer Res. 2012;72:5198–5208. doi: 10.1158/0008-5472.CAN-12-0294. [DOI] [PubMed] [Google Scholar]
- 61.Bellows CF, Zhang Y, Chen J, Frazier ML, Kolonin MG. Circulation of progenitor cells in obese and lean colorectal cancer patients. Cancer Epidemiol Biomarkers Prev. 2011;20:2461–2468. doi: 10.1158/1055-9965.EPI-11-0556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bellows CF, Zhang Y, Simmons PJ, Khalsa AS, Kolonin MG. Influence of BMI on level of circulating progenitor cells. Obesity (Silver Spring) 2011;19:1722–1726. doi: 10.1038/oby.2010.347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kuznetsov SA, Mankani MH, Robey PG. In vivo formation of bone and haematopoietic territories by transplanted human bone marrow stromal cells generated in medium with and without osteogenic supplements. J Tissue Eng Regen Med. 2013;7:226–235. doi: 10.1002/term.515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lui PP, Rui YF, Ni M, Chan KM. Tenogenic differentiation of stem cells for tendon repair-what is the current evidence? J Tissue Eng Regen Med. 2011;5:e144–e163. doi: 10.1002/term.424. [DOI] [PubMed] [Google Scholar]
- 65.Dayoub H, Dumont RJ, Li JZ, Dumont AS, Hankins GR, Kallmes DF, Helm GA. Human mesenchymal stem cells transduced with recombinant bone morphogenetic protein-9 adenovirus promote osteogenesis in rodents. Tissue Eng. 2003;9:347–356. doi: 10.1089/107632703764664819. [DOI] [PubMed] [Google Scholar]
- 66.Janicki P, Boeuf S, Steck E, Egermann M, Kasten P, Richter W. Prediction of in vivo bone forming potency of bone marrow-derived human mesenchymal stem cells. Eur Cell Mater. 2011;21:488–507. doi: 10.22203/ecm.v021a37. [DOI] [PubMed] [Google Scholar]
- 67.Ryan JM, Barry FP, Murphy JM, Mahon BP. Mesenchymal stem cells avoid allogeneic rejection. J Inflamm (Lond) 2005;2:8. doi: 10.1186/1476-9255-2-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Griffin MD, Ryan AE, Alagesan S, Lohan P, Treacy O, Ritter T. Anti-donor immune responses elicited by allogeneic mesenchymal stem cells: what have we learned so far? Immunol Cell Biol. 2013;91:40–51. doi: 10.1038/icb.2012.67. [DOI] [PubMed] [Google Scholar]
- 69.Larsen KH, Frederiksen CM, Burns JS, Abdallah BM, Kassem M. Identifying a molecular phenotype for bone marrow stromal cells with in vivo bone-forming capacity. J Bone Miner Res. 2010;25:796–808. doi: 10.1359/jbmr.091018. [DOI] [PubMed] [Google Scholar]
- 70.Bentzon JF, Stenderup K, Hansen FD, Schroder HD, Abdallah BM, Jensen TG, Kassem M. Tissue distribution and engraftment of human mesenchymal stem cells immortalized by human telomerase reverse transcriptase gene. Biochem Biophys Res Commun. 2005;330:633–640. doi: 10.1016/j.bbrc.2005.03.072. [DOI] [PubMed] [Google Scholar]
- 71.Kale S, Biermann S, Edwards C, Tarnowski C, Morris M, Long MW. Three-dimensional cellular development is essential for ex vivo formation of human bone. Nat Biotechnol. 2000;18:954–958. doi: 10.1038/79439. [DOI] [PubMed] [Google Scholar]
- 72.Fisher MB, Mauck RL. Tissue engineering and regenerative medicine: recent innovations and the transition to translation. Tissue Eng Part B Rev. 2013;19:1–13. doi: 10.1089/ten.teb.2012.0723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Billström GH, Blom AW, Larsson S, Beswick AD. Application of scaffolds for bone regeneration strategies: current trends and future directions. Injury. 2013;44 Suppl 1:S28–S33. doi: 10.1016/S0020-1383(13)70007-X. [DOI] [PubMed] [Google Scholar]
- 74.Mankani MH, Kuznetsov SA, Marshall GW, Robey PG. Creation of new bone by the percutaneous injection of human bone marrow stromal cell and HA/TCP suspensions. Tissue Eng Part A. 2008;14:1949–1958. doi: 10.1089/ten.tea.2007.0348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.van der Stok J, Wang H, Amin Yavari S, Siebelt M, Sandker M, Waarsing JH, Verhaar JA, Jahr H, Zadpoor AA, Leeuwenburgh SC, et al. Enhanced bone regeneration of cortical segmental bone defects using porous titanium scaffolds incorporated with colloidal gelatin gels for time- and dose-controlled delivery of dual growth factors. Tissue Eng Part A. 2013;19:2605–2614. doi: 10.1089/ten.TEA.2013.0181. [DOI] [PubMed] [Google Scholar]
- 76.Kruger EA, Im DD, Bischoff DS, Pereira CT, Huang W, Rudkin GH, Yamaguchi DT, Miller TA. In vitro mineralization of human mesenchymal stem cells on three-dimensional type I collagen versus PLGA scaffolds: a comparative analysis. Plast Reconstr Surg. 2011;127:2301–2311. doi: 10.1097/PRS.0b013e318213a004. [DOI] [PubMed] [Google Scholar]
- 77.Amini AR, Laurencin CT, Nukavarapu SP. Bone tissue engineering: recent advances and challenges. Crit Rev Biomed Eng. 2012;40:363–408. doi: 10.1615/critrevbiomedeng.v40.i5.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhu J. Bioactive modification of poly(ethylene glycol) hydrogels for tissue engineering. Biomaterials. 2010;31:4639–4656. doi: 10.1016/j.biomaterials.2010.02.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ohba S, Hojo H, Chung UI. Bioactive factors for tissue regeneration: state of the art. Muscles Ligaments Tendons J. 2012;2:193–203. [PMC free article] [PubMed] [Google Scholar]
- 80.Lee J, Yoo JJ, Atala A, Lee SJ. The effect of controlled release of PDGF-BB from heparin-conjugated electrospun PCL/gelatin scaffolds on cellular bioactivity and infiltration. Biomaterials. 2012;33:6709–6720. doi: 10.1016/j.biomaterials.2012.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Patel ZS, Young S, Tabata Y, Jansen JA, Wong ME, Mikos AG. Dual delivery of an angiogenic and an osteogenic growth factor for bone regeneration in a critical size defect model. Bone. 2008;43:931–940. doi: 10.1016/j.bone.2008.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Duan B, Wang M. Customized Ca-P/PHBV nanocomposite scaffolds for bone tissue engineering: design, fabrication, surface modification and sustained release of growth factor. J R Soc Interface. 2010;7 Suppl 5:S615–S629. doi: 10.1098/rsif.2010.0127.focus. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kucia M, Wojakowski W, Reca R, Machalinski B, Gozdzik J, Majka M, Baran J, Ratajczak J, Ratajczak MZ. The migration of bone marrow-derived non-hematopoietic tissue-committed stem cells is regulated in an SDF-1-, HGF-, and LIF-dependent manner. Arch Immunol Ther Exp (Warsz) 2006;54:121–135. doi: 10.1007/s00005-006-0015-1. [DOI] [PubMed] [Google Scholar]
- 84.He X, Ma J, Jabbari E. Migration of marrow stromal cells in response to sustained release of stromal-derived factor-1alpha from poly(lactide ethylene oxide fumarate) hydrogels. Int J Pharm. 2010;390:107–116. doi: 10.1016/j.ijpharm.2009.12.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lau TT, Wang DA. Stromal cell-derived factor-1 (SDF-1): homing factor for engineered regenerative medicine. Expert Opin Biol Ther. 2011;11:189–197. doi: 10.1517/14712598.2011.546338. [DOI] [PubMed] [Google Scholar]
- 86.Son BR, Marquez-Curtis LA, Kucia M, Wysoczynski M, Turner AR, Ratajczak J, Ratajczak MZ, Janowska-Wieczorek A. Migration of bone marrow and cord blood mesenchymal stem cells in vitro is regulated by stromal-derived factor-1-CXCR4 and hepatocyte growth factor-c-met axes and involves matrix metalloproteinases. Stem Cells. 2006;24:1254–1264. doi: 10.1634/stemcells.2005-0271. [DOI] [PubMed] [Google Scholar]
- 87.Sengenès C, Miranville A, Maumus M, de Barros S, Busse R, Bouloumié A. Chemotaxis and differentiation of human adipose tissue CD34+/CD31- progenitor cells: role of stromal derived factor-1 released by adipose tissue capillary endothelial cells. Stem Cells. 2007;25:2269–2276. doi: 10.1634/stemcells.2007-0180. [DOI] [PubMed] [Google Scholar]
- 88.Wynn RF, Hart CA, Corradi-Perini C, O’Neill L, Evans CA, Wraith JE, Fairbairn LJ, Bellantuono I. A small proportion of mesenchymal stem cells strongly expresses functionally active CXCR4 receptor capable of promoting migration to bone marrow. Blood. 2004;104:2643–2645. doi: 10.1182/blood-2004-02-0526. [DOI] [PubMed] [Google Scholar]
- 89.Rankin SM. Chemokines and adult bone marrow stem cells. Immunol Lett. 2012;145:47–54. doi: 10.1016/j.imlet.2012.04.009. [DOI] [PubMed] [Google Scholar]
- 90.Shi C. Recent progress toward understanding the physiological function of bone marrow mesenchymal stem cells. Immunology. 2012;136:133–138. doi: 10.1111/j.1365-2567.2012.03567.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Jung Y, Wang J, Schneider A, Sun YX, Koh-Paige AJ, Osman NI, McCauley LK, Taichman RS. Regulation of SDF-1 (CXCL12) production by osteoblasts; a possible mechanism for stem cell homing. Bone. 2006;38:497–508. doi: 10.1016/j.bone.2005.10.003. [DOI] [PubMed] [Google Scholar]
- 92.Ponte AL, Marais E, Gallay N, Langonné A, Delorme B, Hérault O, Charbord P, Domenech J. The in vitro migration capacity of human bone marrow mesenchymal stem cells: comparison of chemokine and growth factor chemotactic activities. Stem Cells. 2007;25:1737–1745. doi: 10.1634/stemcells.2007-0054. [DOI] [PubMed] [Google Scholar]
- 93.Glass GE, Chan JK, Freidin A, Feldmann M, Horwood NJ, Nanchahal J. TNF-alpha promotes fracture repair by augmenting the recruitment and differentiation of muscle-derived stromal cells. Proc Natl Acad Sci U S A. 2011;108:1585–1590. doi: 10.1073/pnas.1018501108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Maijenburg MW, van der Schoot CE, Voermans C. Mesenchymal stromal cell migration: possibilities to improve cellular therapy. Stem Cells Dev. 2012;21:19–29. doi: 10.1089/scd.2011.0270. [DOI] [PubMed] [Google Scholar]
- 95.Rafii DC, Psaila B, Butler J, Jin DK, Lyden D. Regulation of vasculogenesis by platelet-mediated recruitment of bone marrow-derived cells. Arterioscler Thromb Vasc Biol. 2008;28:217–222. doi: 10.1161/ATVBAHA.107.151159. [DOI] [PubMed] [Google Scholar]
- 96.Piva R, Manferdini C, Lambertini E, Torreggiani E, Penolazzi L, Gambari R, Pastore A, Pelucchi S, Gabusi E, Piacentini A, et al. Slug contributes to the regulation of CXCL12 expression in human osteoblasts. Exp Cell Res. 2011;317:1159–1168. doi: 10.1016/j.yexcr.2010.12.011. [DOI] [PubMed] [Google Scholar]
- 97.Kitaori T, Ito H, Schwarz EM, Tsutsumi R, Yoshitomi H, Oishi S, Nakano M, Fujii N, Nagasawa T, Nakamura T. Stromal cell-derived factor 1/CXCR4 signaling is critical for the recruitment of mesenchymal stem cells to the fracture site during skeletal repair in a mouse model. Arthritis Rheum. 2009;60:813–823. doi: 10.1002/art.24330. [DOI] [PubMed] [Google Scholar]
- 98.Young S, Patel ZS, Kretlow JD, Murphy MB, Mountziaris PM, Baggett LS, Ueda H, Tabata Y, Jansen JA, Wong M, et al. Dose effect of dual delivery of vascular endothelial growth factor and bone morphogenetic protein-2 on bone regeneration in a rat critical-size defect model. Tissue Eng Part A. 2009;15:2347–2362. doi: 10.1089/ten.tea.2008.0510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Schantz JT, Chim H, Whiteman M. Cell guidance in tissue engineering: SDF-1 mediates site-directed homing of mesenchymal stem cells within three-dimensional polycaprolactone scaffolds. Tissue Eng. 2007;13:2615–2624. doi: 10.1089/ten.2006.0438. [DOI] [PubMed] [Google Scholar]
- 100.Tasso R, Augello A, Boccardo S, Salvi S, Caridà M, Postiglione F, Fais F, Truini M, Cancedda R, Pennesi G. Recruitment of a host’s osteoprogenitor cells using exogenous mesenchymal stem cells seeded on porous ceramic. Tissue Eng Part A. 2009;15:2203–2212. doi: 10.1089/ten.tea.2008.0269. [DOI] [PubMed] [Google Scholar]
- 101.Kumar S, Chanda D, Ponnazhagan S. Therapeutic potential of genetically modified mesenchymal stem cells. Gene Ther. 2008;15:711–715. doi: 10.1038/gt.2008.35. [DOI] [PubMed] [Google Scholar]
- 102.Evans C. Gene therapy for the regeneration of bone. Injury. 2011;42:599–604. doi: 10.1016/j.injury.2011.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Evans CH. Gene delivery to bone. Adv Drug Deliv Rev. 2012;64:1331–1340. doi: 10.1016/j.addr.2012.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Huang YC, Kaigler D, Rice KG, Krebsbach PH, Mooney DJ. Combined angiogenic and osteogenic factor delivery enhances bone marrow stromal cell-driven bone regeneration. J Bone Miner Res. 2005;20:848–857. doi: 10.1359/JBMR.041226. [DOI] [PubMed] [Google Scholar]
- 105.Meinel L, Hofmann S, Betz O, Fajardo R, Merkle HP, Langer R, Evans CH, Vunjak-Novakovic G, Kaplan DL. Osteogenesis by human mesenchymal stem cells cultured on silk biomaterials: comparison of adenovirus mediated gene transfer and protein delivery of BMP-2. Biomaterials. 2006;27:4993–5002. doi: 10.1016/j.biomaterials.2006.05.021. [DOI] [PubMed] [Google Scholar]
- 106.Thieme S, Ryser M, Gentsch M, Navratiel K, Brenner S, Stiehler M, Rölfing J, Gelinsky M, Rösen-Wolff A. Stromal cell-derived factor-1alpha-directed chemoattraction of transiently CXCR4-overexpressing bone marrow stromal cells into functionalized three-dimensional biomimetic scaffolds. Tissue Eng Part C Methods. 2009;15:687–696. doi: 10.1089/ten.TEC.2008.0556. [DOI] [PubMed] [Google Scholar]
- 107.Zhang Y, Bellows CF, Kolonin MG. Adipose tissue-derived progenitor cells and cancer. World J Stem Cells. 2010;2:103–113. doi: 10.4252/wjsc.v2.i5.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Klopp AH, Zhang Y, Solley T, Amaya-Manzanares F, Marini F, Andreeff M, Debeb B, Woodward W, Schmandt R, Broaddus R, et al. Omental adipose tissue-derived stromal cells promote vascularization and growth of endometrial tumors. Clin Cancer Res. 2012;18:771–782. doi: 10.1158/1078-0432.CCR-11-1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Bischoff DS, Zhu JH, Makhijani NS, Kumar A, Yamaguchi DT. Angiogenic CXC chemokine expression during differentiation of human mesenchymal stem cells towards the osteoblastic lineage. J Cell Biochem. 2008;103:812–824. doi: 10.1002/jcb.21450. [DOI] [PubMed] [Google Scholar]
- 110.Arufe MC, De la Fuente A, Fuentes-Boquete I, De Toro FJ, Blanco FJ. Differentiation of synovial CD-105(+) human mesenchymal stem cells into chondrocyte-like cells through spheroid formation. J Cell Biochem. 2009;108:145–155. doi: 10.1002/jcb.22238. [DOI] [PubMed] [Google Scholar]
- 111.Pelttari K, Steck E, Richter W. The use of mesenchymal stem cells for chondrogenesis. Injury. 2008;39 Suppl 1:S58–S65. doi: 10.1016/j.injury.2008.01.038. [DOI] [PubMed] [Google Scholar]
- 112.Burns JS, Rasmussen PL, Larsen KH, Schrøder HD, Kassem M. Parameters in three-dimensional osteospheroids of telomerized human mesenchymal (stromal) stem cells grown on osteoconductive scaffolds that predict in vivo bone-forming potential. Tissue Eng Part A. 2010;16:2331–2342. doi: 10.1089/ten.TEA.2009.0735. [DOI] [PubMed] [Google Scholar]
- 113.Hong JH, Hwang ES, McManus MT, Amsterdam A, Tian Y, Kalmukova R, Mueller E, Benjamin T, Spiegelman BM, Sharp PA, et al. TAZ, a transcriptional modulator of mesenchymal stem cell differentiation. Science. 2005;309:1074–1078. doi: 10.1126/science.1110955. [DOI] [PubMed] [Google Scholar]
- 114.Pang Y, Greisler HP. Using a type 1 collagen-based system to understand cell-scaffold interactions and to deliver chimeric collagen-binding growth factors for vascular tissue engineering. J Investig Med. 2010;58:845–848. doi: 10.231/JIM.0b013e3181ee81f7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Hynes RO. The extracellular matrix: not just pretty fibrils. Science. 2009;326:1216–1219. doi: 10.1126/science.1176009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Brizzi MF, Tarone G, Defilippi P. Extracellular matrix, integrins, and growth factors as tailors of the stem cell niche. Curr Opin Cell Biol. 2012;24:645–651. doi: 10.1016/j.ceb.2012.07.001. [DOI] [PubMed] [Google Scholar]
- 117.Humphries JD, Byron A, Humphries MJ. Integrin ligands at a glance. J Cell Sci. 2006;119:3901–3903. doi: 10.1242/jcs.03098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kim C, Ye F, Ginsberg MH. Regulation of integrin activation. Annu Rev Cell Dev Biol. 2011;27:321–345. doi: 10.1146/annurev-cellbio-100109-104104. [DOI] [PubMed] [Google Scholar]
- 119.Shattil SJ, Kim C, Ginsberg MH. The final steps of integrin activation: the end game. Nat Rev Mol Cell Biol. 2010;11:288–300. doi: 10.1038/nrm2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Anthis NJ, Campbell ID. The tail of integrin activation. Trends Biochem Sci. 2011;36:191–198. doi: 10.1016/j.tibs.2010.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Shen B, Delaney MK, Du X. Inside-out, outside-in, and inside-outside-in: G protein signaling in integrin-mediated cell adhesion, spreading, and retraction. Curr Opin Cell Biol. 2012;24:600–606. doi: 10.1016/j.ceb.2012.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Shih YR, Chen CN, Tsai SW, Wang YJ, Lee OK. Growth of mesenchymal stem cells on electrospun type I collagen nanofibers. Stem Cells. 2006;24:2391–2397. doi: 10.1634/stemcells.2006-0253. [DOI] [PubMed] [Google Scholar]
- 123.Mauney JR, Kaplan DL, Volloch V. Matrix-mediated retention of osteogenic differentiation potential by human adult bone marrow stromal cells during ex vivo expansion. Biomaterials. 2004;25:3233–3243. doi: 10.1016/j.biomaterials.2003.10.005. [DOI] [PubMed] [Google Scholar]
- 124.Gronthos S, Simmons PJ, Graves SE, Robey PG. Integrin-mediated interactions between human bone marrow stromal precursor cells and the extracellular matrix. Bone. 2001;28:174–181. doi: 10.1016/s8756-3282(00)00424-5. [DOI] [PubMed] [Google Scholar]
- 125.Watt FM, Huck WT. Role of the extracellular matrix in regulating stem cell fate. Nat Rev Mol Cell Biol. 2013;14:467–473. doi: 10.1038/nrm3620. [DOI] [PubMed] [Google Scholar]
- 126.McBeath R, Pirone DM, Nelson CM, Bhadriraju K, Chen CS. Cell shape, cytoskeletal tension, and RhoA regulate stem cell lineage commitment. Dev Cell. 2004;6:483–495. doi: 10.1016/s1534-5807(04)00075-9. [DOI] [PubMed] [Google Scholar]
- 127.Mathieu PS, Loboa EG. Cytoskeletal and focal adhesion influences on mesenchymal stem cell shape, mechanical properties, and differentiation down osteogenic, adipogenic, and chondrogenic pathways. Tissue Eng Part B Rev. 2012;18:436–444. doi: 10.1089/ten.teb.2012.0014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Discher DE, Mooney DJ, Zandstra PW. Growth factors, matrices, and forces combine and control stem cells. Science. 2009;324:1673–1677. doi: 10.1126/science.1171643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Engler AJ, Sen S, Sweeney HL, Discher DE. Matrix elasticity directs stem cell lineage specification. Cell. 2006;126:677–689. doi: 10.1016/j.cell.2006.06.044. [DOI] [PubMed] [Google Scholar]
- 130.Swift J, Ivanovska IL, Buxboim A, Harada T, Dingal PC, Pinter J, Pajerowski JD, Spinler KR, Shin JW, Tewari M, et al. Nuclear lamin-A scales with tissue stiffness and enhances matrix-directed differentiation. Science. 2013;341:1240104. doi: 10.1126/science.1240104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Chen XD, Dusevich V, Feng JQ, Manolagas SC, Jilka RL. Extracellular matrix made by bone marrow cells facilitates expansion of marrow-derived mesenchymal progenitor cells and prevents their differentiation into osteoblasts. J Bone Miner Res. 2007;22:1943–1956. doi: 10.1359/jbmr.070725. [DOI] [PubMed] [Google Scholar]
- 132.Lai Y, Sun Y, Skinner CM, Son EL, Lu Z, Tuan RS, Jilka RL, Ling J, Chen XD. Reconstitution of marrow-derived extracellular matrix ex vivo: a robust culture system for expanding large-scale highly functional human mesenchymal stem cells. Stem Cells Dev. 2010;19:1095–1107. doi: 10.1089/scd.2009.0217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Choi HR, Cho KA, Kang HT, Lee JB, Kaeberlein M, Suh Y, Chung IK, Park SC. Restoration of senescent human diploid fibroblasts by modulation of the extracellular matrix. Aging Cell. 2011;10:148–157. doi: 10.1111/j.1474-9726.2010.00654.x. [DOI] [PubMed] [Google Scholar]
- 134.Liu B, Ghosh S, Yang X, Zheng H, Liu X, Wang Z, Jin G, Zheng B, Kennedy BK, Suh Y, et al. Resveratrol rescues SIRT1-dependent adult stem cell decline and alleviates progeroid features in laminopathy-based progeria. Cell Metab. 2012;16:738–750. doi: 10.1016/j.cmet.2012.11.007. [DOI] [PubMed] [Google Scholar]
- 135.Tang D, Zhu H, Wu J, Chen H, Zhang Y, Zhao X, Chen X, Du W, Wang D, Lin X. Silencing myostatin gene by RNAi in sheep embryos. J Biotechnol. 2012;158:69–74. doi: 10.1016/j.jbiotec.2012.01.008. [DOI] [PubMed] [Google Scholar]
- 136.Berendsen AD, Fisher LW, Kilts TM, Owens RT, Robey PG, Gutkind JS, Young MF. Modulation of canonical Wnt signaling by the extracellular matrix component biglycan. Proc Natl Acad Sci USA. 2011;108:17022–17027. doi: 10.1073/pnas.1110629108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Sun Y, Li W, Lu Z, Chen R, Ling J, Ran Q, Jilka RL, Chen XD. Rescuing replication and osteogenesis of aged mesenchymal stem cells by exposure to a young extracellular matrix. FASEB J. 2011;25:1474–1485. doi: 10.1096/fj.10-161497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Prewitz MC, Seib FP, von Bonin M, Friedrichs J, Stißel A, Niehage C, Müller K, Anastassiadis K, Waskow C, Hoflack B, et al. Tightly anchored tissue-mimetic matrices as instructive stem cell microenvironments. Nat Methods. 2013;10:788–794. doi: 10.1038/nmeth.2523. [DOI] [PubMed] [Google Scholar]
- 139.Shenaq DS, Rastegar F, Petkovic D, Zhang BQ, He BC, Chen L, Zuo GW, Luo Q, Shi Q, Wagner ER, et al. Mesenchymal Progenitor Cells and Their Orthopedic Applications: Forging a Path towards Clinical Trials. Stem Cells Int. 2010;2010:519028. doi: 10.4061/2010/519028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Rastegar F, Shenaq D, Huang J, Zhang W, Zhang BQ, He BC, Chen L, Zuo GW, Luo Q, Shi Q, et al. Mesenchymal stem cells: Molecular characteristics and clinical applications. World J Stem Cells. 2010;2:67–80. doi: 10.4252/wjsc.v2.i4.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Zhang ZY, Teoh SH, Hui JH, Fisk NM, Choolani M, Chan JK. The potential of human fetal mesenchymal stem cells for off-the-shelf bone tissue engineering application. Biomaterials. 2012;33:2656–2672. doi: 10.1016/j.biomaterials.2011.12.025. [DOI] [PubMed] [Google Scholar]
- 142.Kim N, Cho SG. Clinical applications of mesenchymal stem cells. Korean J Intern Med. 2013;28:387–402. doi: 10.3904/kjim.2013.28.4.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Veronesi F, Giavaresi G, Tschon M, Borsari V, Nicoli Aldini N, Fini M. Clinical use of bone marrow, bone marrow concentrate, and expanded bone marrow mesenchymal stem cells in cartilage disease. Stem Cells Dev. 2013;22:181–192. doi: 10.1089/scd.2012.0373. [DOI] [PubMed] [Google Scholar]
- 144.Koh YG, Choi YJ. Infrapatellar fat pad-derived mesenchymal stem cell therapy for knee osteoarthritis. Knee. 2012;19:902–907. doi: 10.1016/j.knee.2012.04.001. [DOI] [PubMed] [Google Scholar]
- 145.Orozco L, Soler R, Morera C, Alberca M, Sánchez A, García-Sancho J. Intervertebral disc repair by autologous mesenchymal bone marrow cells: a pilot study. Transplantation. 2011;92:822–828. doi: 10.1097/TP.0b013e3182298a15. [DOI] [PubMed] [Google Scholar]
- 146.Mesimäki K, Lindroos B, Törnwall J, Mauno J, Lindqvist C, Kontio R, Miettinen S, Suuronen R. Novel maxillary reconstruction with ectopic bone formation by GMP adipose stem cells. Int J Oral Maxillofac Surg. 2009;38:201–209. doi: 10.1016/j.ijom.2009.01.001. [DOI] [PubMed] [Google Scholar]
- 147.Carter TG, Brar PS, Tolas A, Beirne OR. Off-label use of recombinant human bone morphogenetic protein-2 (rhBMP-2) for reconstruction of mandibular bone defects in humans. J Oral Maxillofac Surg. 2008;66:1417–1425. doi: 10.1016/j.joms.2008.01.058. [DOI] [PubMed] [Google Scholar]