

Abstract
Stimulated endothelial cells (EC) assume an activated phenotype with pro-inflammatory and prothrombotic features, requiring new gene and protein expression. New protein synthesis in activated EC is largely regulated by transcriptional events controlled by a variety of transcription factors. However, post-transcriptional control of gene expression also influences phenotype and allows the cell to alter protein expression in a faster and more direct way than is typically possible with transcriptional mechanisms. We sought to demonstrate that post-transcriptional control of gene expression occurs during EC activation. Using thrombin-activated EC and a high-throughput, microarray-based approach, we identified a number of gene products that may be regulated through post-transcriptional mechanisms, including the AP-1 transcription factor JunB. Using polysome profiling, cytoplasts and other standard cell biologic techniques, JunB is shown to be regulated at a post-transcriptional level during EC activation. In activated EC, the AP-1 transcription factor JunB, is regulated on a post-transcriptional level. Signal-dependent control of translation may regulate transcription factor expression and therefore, subsequent transcriptional events in stimulated EC.
Keywords: JunB, ENDOTHELIUM, POST-TRANSCRIPTIONAL REGULATION, POLYRIBOSOMAL PROFILING, DEEP RNA SEQUENCING
When stimulated, endothelial cells (EC) assume an activated phenotype with pro-inflammatory and prothrombotic features, some of which do not require new gene expression while many others do [Cines et al., 1998; Kraiss et al., 2005]. New protein synthesis in activated EC occurs as a result of new gene transcription controlled by a variety of transcription factors such as NF-κB and the activator protein-1 (AP-1) family [Collins et al., 1995; Minami et al., 2004]. Yet post-transcriptional gene regulation also occurs in EC, and may predominate in some conditions [Kraiss et al., 2000, 2001, 2003; Maeshima et al., 2002].
Perhaps 5–15% of a cell’s expressed gene products are regulated at translational control points [Kozak, 1991]. Translational control allows the cell to alter protein expression more rapidly, more directly, and with greater precision than is typically possible with transcriptional mechanisms [Mathews et al., 2000]. Post-transcriptional mechanisms also diversify the cell’s repertoire of gene regulatory mechanisms by allowing multiple levels of regulation to be exerted on crucial gene products and providing different patterns of control at different times. Given the rapidity, complexity, and spectrum of altered gene expression required to produce the activated endothelial phenotype, we speculated that translational mechanisms of gene regulation act in concert with transcriptional control to determine this response. We and others have shown that translational control occurs in EC [Kraiss et al., 2000, 2001; Maeshima et al., 2002]. Therefore, we sought to identify additional signal-dependent translational mechanisms and characterize important gene products regulated in this fashion in stimulated endothelium. Using a high-throughput, microarray-based approach that estimates the translation activity of large numbers of mRNA transcripts simultaneously [Zong et al., 1999], the AP-1 transcription factor JunB emerged as a particularly intriguing candidate for translational control in activated EC. Little is known about JunB function in the vasculature, and translational control of AP-1 gene expression has not previously been described in non-transformed cells. In this report, we show that JunB expression is regulated by translational control in activated EC and demonstrate that signal-dependent translation is a key mechanism of transcription factor expression in response to hemostatic and inflammatory stimuli.
METHODS
Confluent primary cultures of human umbilical vein endothelial cells were established in 0.2% gelatin-coated six-well dishes as previously described [Zimmerman et al., 1990]. Cultures were used for experiments 1 day after they reached confluence (~3–4 days after initial plating).
Translation state array analysis (TSAA), a high-throughput method of quantifying the relative distribution of mRNA transcripts among monosome and polysome subcellular fractions, was performed after the method of Zong et al. [1999] with some modifications (Supplementary Fig. 1). The Translation Index (TLI) for any given gene product is derived from TSAA results and reflects the redistribution of that gene product’s mRNA between monosome and polysome in response to a stimulus or change in conditions. A TLI ≫ 1 indicates redistribution of mRNA into polysomes (translational upregulation). A TLI ≪ 1 indicates redistribution of mRNA into monosomes (translational repression). A TLI ~1 indicates no discrete translational control as a result of the stimulus or change in condition. TSAA and the derivation of the TLI is described in more detail in the online supplement.
Additional assays including immunofluorescence, immunoblot analysis, polysome profiling, translational state array analysis, quantitative real-time RT-PCR (qRT-PCR), nuclear isolations, next-generation RNA-sequencing, and cytoplast preparations were performed according to standard techniques. A detailed Methods Section is available in the online data Supplement.
This investigation conforms with the principles outlined in the Declaration of Helsinki [1997] and the University of Utah Institutional Review Board (approval number 392).
RESULTS
HIGH-THROUGHPUT MICROARRAY ANALYSIS IDENTIFIES JunB AS A TRANSLATIONALLY REGULATED GENE PRODUCT IN ACTIVATED EC
In a systematic effort to identify translationally regulated gene products in activated EC, we stimulated human EC cultures with various agonists and performed separate TSAA for each condition. The TSAA results for an experiment using thrombin as a stimulus are presented in Figure 1A–C. Complete TSAA data have been deposited in NCBI’s Gene Expression Omnibus [GEO, http://www.ncbi.nlm.nih.gov/geo/] accession number GSE4919. The great majority (94%) of expressed genes had a TLI ~ 1 indicating absence of translational control. However, this approach identified a number of candidates (2.2%) for translational upregulation (Fig. 1C). When the TSAA results for a variety of agonists were compared (Fig. 1D and Supplementary Fig. 2), the TLI value for the AP-1 transcription factor JunB was frequently but not invariably >2.5, indicating that its mRNA had redistributed to the polysome fraction in the stimulated cells and providing further evidence for translational control under a variety of conditions. The TLI for β-actin never varied significantly from a value of 1 (Fig. 1D).
Fig. 1.
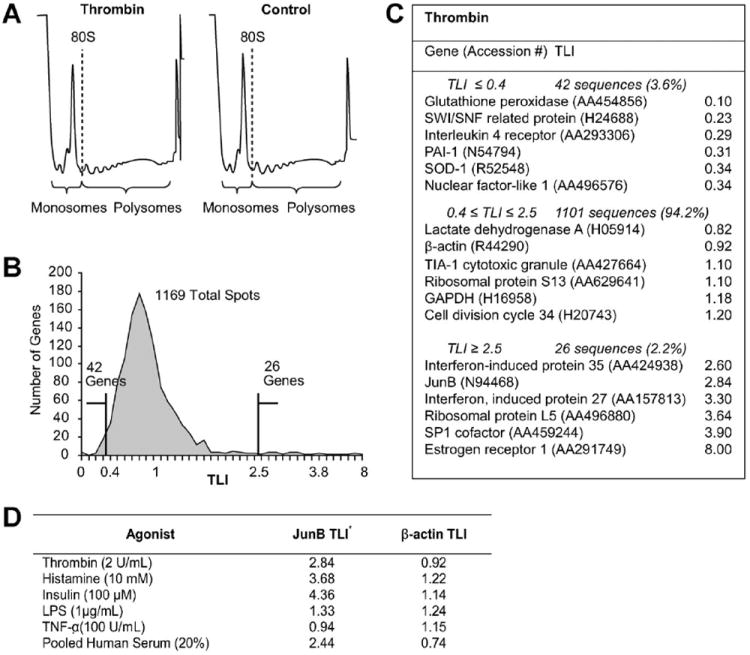
TSAA indicates that a subset of genes in thrombin-activated EC are translationally regulated. Confluent human EC were treated with thrombin (2 U/ml, 2 h) or vehicle and polyribosome preparations were fractionated and pooled into monosome or polysome samples and analyzed by microarray. TLI values for each array element were derived as described in Methods Section and the online data supplement. Panel A: Polysome profile tracings from thrombin stimulated (left panel) or control cells (right panel) illustrating the demarcation used to separate monosome and polysome fractions. Panel B: Histogram of TLI values for 1169 genes expressed above background in either control or stimulated EC. Panel C: Selected gene products whose TLI suggests translational repression (TLI ≤ 0.4), absence of translational regulation (0.4 <TLI <2.5) or translational upregulation (TLI ≥ 2.5). Note JunB TLI = 2.84. Panel D: Translationally upregulated candidate genes. HUVEC were treated with the indicated agonist or control for 2 h (except serum, 6 h) and then processed for TSAA (see on-line supplement for detailed Methods Section).
We studied JunB’s expression in response to thrombin because human EC respond extremely rapidly to this agonist and AP-1 family members are implicated in EC activation [Delerive et al., 1999; Wang et al., 2001; Bavendiek et al., 2002; Eyries et al., 2002; Hipp et al., 2002]. Further, JunB frequently appeared as a candidate for translational control in several TSAA experiments yet mechanisms regulating JunB expression in activated EC are uncharacterized and translational control of AP-1 factors in the vasculature has not been described.
POLYRIBOSOMES ARE ENRICHED WITH JunB mRNA FOLLOWING STIMULATION OF EC BY THROMBIN
In order to verify that cellular activation does indeed produce a redistribution of JunB mRNA to the polysome as predicted by TSAA, EC cultures were treated with thrombin and processed to yield ribosomal fractions that span the progression from monosomes to higher order polysomes. Amounts of β-actin and JunB mRNA in each fraction were determined by quantitative real-time polymerase chain reaction (qRT-PCR). Consistent with its role as an intracellular structural protein, β-actin mRNA was distributed throughout the polysome profile suggesting constitutive ongoing translation and did not show a significant response to thrombin (Fig. 2A). This is also consistent with TSAA results (Fig. 1C,D). In contrast, the relative abundance of JunB mRNA was markedly increased in polysomes after thrombin stimulation and was distributed across the majority of polysome fractions (Fig. 2B).
Fig. 2.
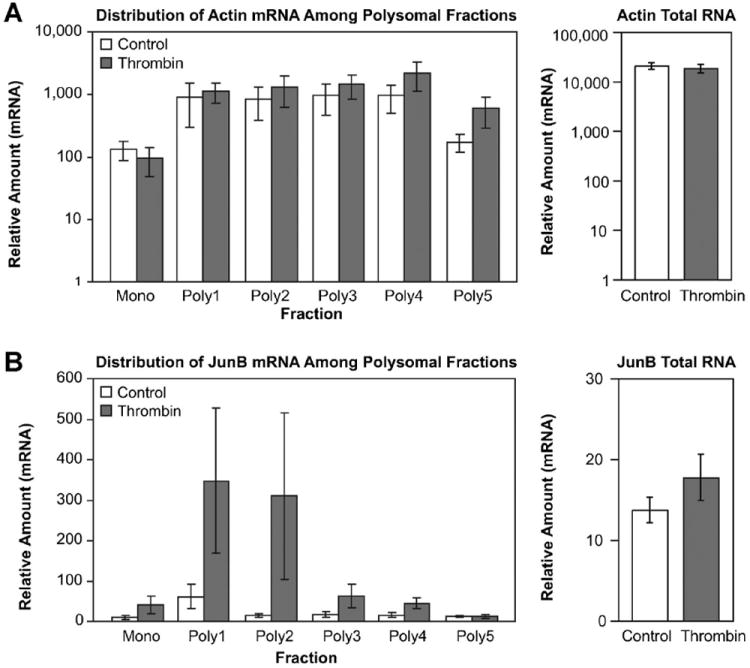
JunB mRNA is selectively enriched in polyribosomes of thrombin-stimulated EC. Ribosome samples corresponding to monosomes or polysomes were prepared from cellular lysates by centrifugation through a sucrose density gradient. Gradients were collected in eight fractions from top to bottom by upward displacement. RNA from the first-three fractions was pooled to form the Mono fraction, which consists of monosomes migrating in the least dense sucrose region. The remaining five fractions (poly1–poly 5) contain polyribosomes in the more dense sucrose regions. Relative amounts of mRNA for β-actin (panel A) and JunB (panel B) in monosome or individual polysome fractions were determined simultaneously using quantitative PCR. Values for total cellular mRNA for β-actin and JunB (right panels) were determined from aliquots of cell lysates sampled before polyribosome purification. Differences in overall JunB expression between control and thrombin stimulation were not statistically significant (P-values = 0.2–0.3). Results represent three separate experiments.
By inspection of the qRT-PCR results, thrombin-treated EC contained an overall increase in polyribosome-associated JunB mRNA. However, thrombin stimulation did not produce an overall change in steady state mRNA levels at 2 h for either β-actin or JunB mRNA (right panels, Fig. 2A,B; see also Fig. 3A). We surmized that the apparent increase in JunB mRNA present in the thrombin-treated polyribosomal samples represents mobilization of JunB mRNA to the translational apparatus from storage ribonucleoprotein complexes [Zong et al., 1999; Anderson and Kedersha, 2006].
Fig. 3.
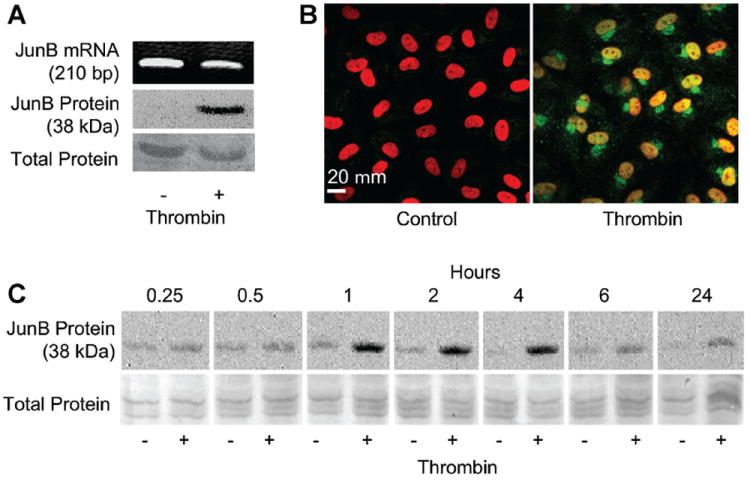
Endothelial JunB protein expression and nuclear localization are rapidly induced by thrombin without an increase in overall JunB mRNA. Panel A: Thrombin induces JunB protein expression in the nucleus without a corresponding change in overall JunB mRNA. Human EC were stimulated with thrombin (2 U/ml, 2 h). Separate samples were prepared for total RNA analysis by PCR and Western blotting of nuclear protein lysates. Bottom panel represents total protein staining to assure equal loading. Results are representative of multiple (>3) experiments. Panel B: Thrombin induces appearance of JunB protein in the nucleus and perinuclear cytoplasm. Human EC were stimulated with thrombin (2 U/ml, 2 h) and then incubated with anti-JunB antibody (green) and propidium iodide to stain nuclei (red) as described in Methods Section. Controls included non-immune IgG, which revealed no staining (not shown). Results represent three independent experiments. Panel C: Increased JunB protein expression by EC is rapidly apparent following thrombin stimulation. Human EC were stimulated with thrombin (2 U/ml, 2 h) and assayed for JunB by Western blotting at the indicated times. Bottom panel represents total protein staining to assure equal loading. The blots are representative of four separate experiments.
THROMBIN-TREATED EC RAPIDLY EXPRESS JunB PROTEIN IN THE NUCLEUS
Immunodetection techniques were then used to determine if enrichment of polyribosomes with JunB mRNA in thrombin-stimulated EC produced an increase in JunB protein. EC were treated for 2 h with thrombin or vehicle and parallel samples were prepared for nuclear isolation and total RNA analysis. Nuclear lysates were analyzed for the presence of JunB by Western blotting while total RNA was probed for JunB by semi-quantitative polymerase chain reaction (PCR; Fig. 3A). Under control conditions, JunB mRNA was readily detectable but little or no protein was present in nuclear lysates (Fig. 3A). Nonetheless, JunB protein was invariably detected in the nuclear compartment following thrombin stimulation, without a change in total mRNA levels (Fig. 3A,B), a classic pattern indicating post-transcriptional control [Mathews et al., 2000]. Since these changes in nuclear JunB protein levels might have resulted from translocation of constitutive protein from the cytoplasm, immunocytochemical studies were also performed (Fig. 3B). These studies demonstrated no detectable JunB protein in the cytoplasm of resting, unstimulated EC but abundant protein in the perinuclear region and nuclei of thrombin-stimulated EC.
One advantage of regulating gene expression at the translational level is the potential for rapid protein expression in response to appropriate signals, assuming a pre-existing reservoir of cognate mRNA [Mathews et al., 2000]. This biologic advantage is satisfied for JunB. Figure 3C illustrates that increases in JunB protein are detected in thrombin-treated EC as soon as 15–30 min after stimulation and dramatically by 1 h. Relative to transcriptionally regulated EC gene products such as E-selectin [Bevilacqua et al., 1989], this rapid appearance of protein is again consistent with signal-dependent post-transcriptional control [Mathews et al., 2000]. Thrombin receptor activating peptide-6 (TRAP-6), a specific activator of protease activated receptor 1 (PAR-1), stimulated JunB production in a similar manner (not shown), demonstrating that this response was thrombin specific and receptor dependent. Small differences in basal levels of JunB protein expression are likely due to variations in handling and experimental manipulation producing a low-level of activation in some primary EC cultures.
TRANSCRIPTION IS NOT REQUIRED FOR JunB SYNTHESIS IN EC
In order to determine whether thrombin-induced JunB protein expression occurred independent of new transcription, we stimulated EC in the presence of the transcriptional inhibitor actinomycin D. At concentrations of actinomycin D that blocked uridine incorporation (0.125 μg/ml; Fig. 4A), thrombin continued to induce JunB protein (Fig. 4B). In additional experiments we confirmed that actinomycin D inhibited thrombin-induced IL-8 expression, a protein known to transcriptionally regulated in activiated EC (Fig. 4C). Thus, continued expression of JunB despite pretreatment with actinomycin D implies a post-transcriptional mode of regulation.
Fig. 4.
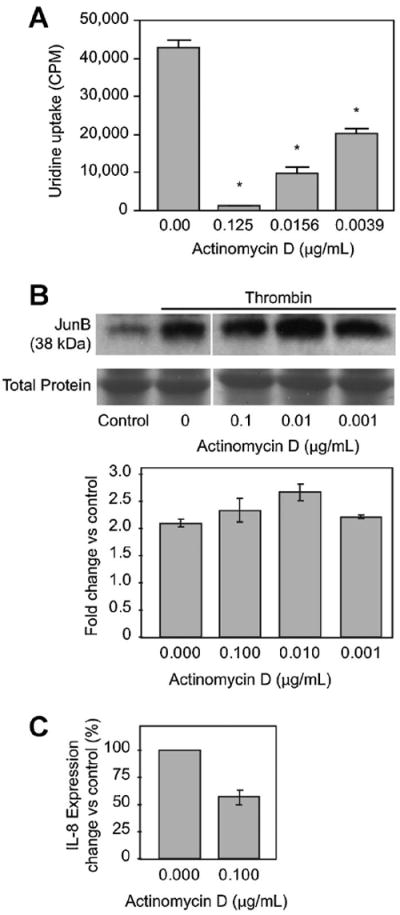
Thrombin induces JunB protein expression despite transcriptional inhibition by actinomycin D. Panel A: Endothelial uptake of 3H-uridine is inhibited by actinomycin D. Human EC were cultured in the presence of actinomycin D for 30 min before addition of 3H-uridine (1 μCi/ml) and thrombin (2 U/ml, 2 h). Samples were then processed for beta emission spectroscopy (n = 3). Statistically significant differences (P <0.05) relative to no treatment are indicated by an asterisk. Panel B: JunB protein expression is preserved in the presence of actinomycin D in thrombin-stimulated EC. EC were pre-treated with the indicated concentration of actinomycin D (or an equivalent amount of DMSO) for 30 min prior to thrombin stimulation (2 U/ml, 2 h). Cells were then processed for Western blotting. Bottom panel represents total protein staining to assure equal loading. The immunoblot is representative of three separate experiments. Densitometry, using ImageJ (http://rsb.info.nih.-gov/ij/), of the three immunoblots is graphically represented. Differences in JunB expression were not statistically significant. Panel C: IL-8 protein expression is reduced in the presence of actinomycin D in thrombin-stimulated EC. EC were pre-treated with the indicated concentration of actinomycin D (or an equivalent amount of DMSO) for 30 min prior to thrombin stimulation (2 U/ml, 2 h). Cells were then processed for ELISA. The bargraph represents the results from two independent experiments.
To further demonstrate transcriptional independence of JunB protein synthesis in activated EC, we utilized an anucleate EC system. EC were enucleated by centrifugation to produce cytoplasts incapable of transcription [Prescott et al., 1972; Veomett et al., 1976] but with intact signal transduction as evidenced by actin rearrangement and p38 MAPK phosphorylation in response to activation (unpublished observations, 2006). In situ hybridization confirmed the presence of JunB mRNA in the cytoplasm (Fig. 5A) consistent with the PCR data displayed in Figure 3A. When stimulated with thrombin, enucleated endothelial cytoplasts expressed JunB protein detected by immunohistochemical analysis (Fig. 5B, lower left panel). JunB detection in nucleated cells not stimulated with thrombin (Fig. 5B, upper left panel) results from activation of EC during the cytoplast preparation (see online Materials and Methods Section). Nevertheless, the anucleate cytoplasts clearly show an inducible staining pattern specific for JunB which depends on the activation by thrombin (Fig. 5B, left panels).
Fig. 5.
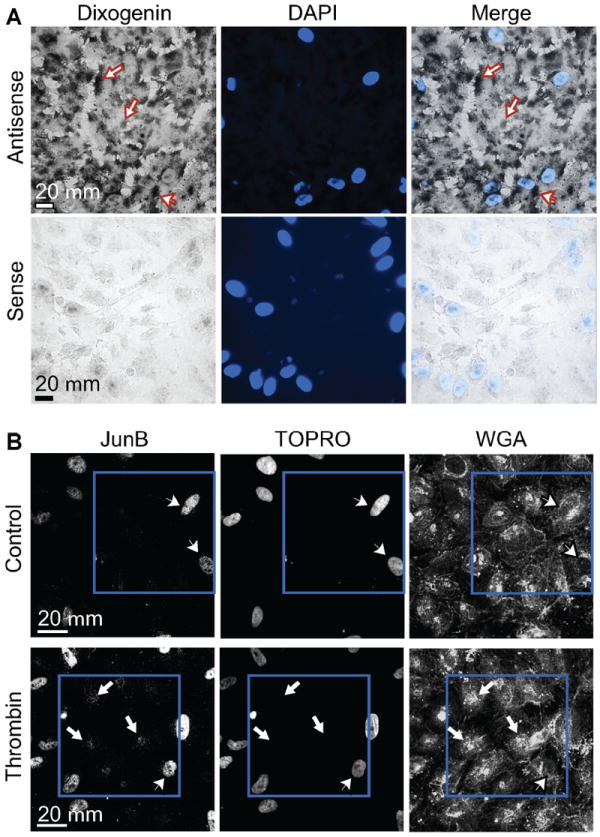
Enucleated endothelial cytoplasts retain JunB mRNA in the cytosol and express JunB protein in response to thrombin. Panel A: In situ hybridization demonstrates the presence of JunB mRNA in quiescent, unstimulated EC cytoplasts. Cytoplasts were prepared as described in Methods Section and incubated with antisense or sense digoxigenin-labeled riboprobes directed against JunB. Black reaction product in the cytoplasm of both intact EC (arrowhead) and anucleate cytoplasts labeled with antisense probes (arrows) is readily observable while minimal to no signal is present in the sense-probe labeled slides. Panel B: Thrombin stimulation induces JunB protein expression in EC cytoplasts. Cytoplasts were prepared as described and treated with either HBSS (control, top row) or thrombin for 2 h (bottom row). Immunocytochemistry was performed using Alexa 488 streptavidin to label bound anti-JunB antibody (left panels) while TOPRO-3 (middle panels) and wheat germ agglutinin (WGA, right panels) were respectively used to identify nuclei and cell membranes in the cytoplast preparations. After thrombin stimulation, signal for JunB protein was detected in enucleated cytoplasts (white arrows, lower left panel) but not in cytoplasts treated with buffer (upper left panel). Residual intact nucleated EC (arrowheads) in the cytoplast preparation also stain robustly for JunB after incubation with HBSS (due to the stress induced by cytoplast preparation) or after thrombin stimulation. The blue squares highlight a group of cytoplasts in both rows.
Taken together, the actinomycin D and cytoplast experiments (Figs. 4 and 5, respectively) indicate that JunB mRNA is basally present and can be recruited to polysomes (Fig. 2) for translation of the protein product independent of new transcription.
THROMBIN INDUCES ASSOCIATION OF JunB mRNA AND eIF4E
Translational control is most commonly exerted at its initiation [Mathews et al., 2000]. Eukaryotic initiation factor 4E (eIF4E) is a key regulator that binds to the 5′ cap of mRNAs destined for translation [Gingras et al., 1999]. According to the scanning model of translation, the initiation complex, which cannot assemble without eIF4E, binds to the 5′ cap and scans the 5′-untranslated region (UTR) until a translation start codon is encountered [Hershey and Merrick, 2000]. Messenger RNA molecules that are translationally regulated commonly have 5′-UTRs with highly complex secondary structures that present a physical barrier to unregulated scanning [Hershey and Merrick, 2000]. The eIF4E-containing multi-protein initiation complex has helicase activity, which facilitates scanning through highly structured regions by linearizing the secondary structure [Gingras et al., 1999].
We performed next-generation deep RNA sequencing [Wang et al., 2009] on EC-derived mRNA (Fig. 6A) as well as TA cloning and sequencing of the 5′-UTR for EC-derived JunB (data not shown) followed by computerized analysis of its secondary structure. The TA cloning derived sequence of the 5′-UTR has been deposited in GenBank, accession number DQ650707. Using these techniques, the JunB 5′-UTR was unambiguously detected with a length of ~300 bp and possesses a calculated free energy (ΔG) of −221.6 kcal/mol while the 5′-UTR of β-actin has a relatively simple secondary structure (ΔG = −30.3 kcal/mol; Fig. 6B). A commonly accepted threshold for regulated translation is a 5′-UTR ΔG = −50 kcal/mol [Brown and Schreiber, 1996; Hershey and Merrick, 2000]. Because the predicted structure of the JunB 5′-UTR and our experimental results to this point were consistent with translational control, we next asked if thrombin stimulation induces association of the JunB transcript with eIF4E. Anti-eIF4E immunoprecipitates from thrombin-activated EC contained JunB mRNA whereas eIF4E immunoprecipitates from control cells did not, thus demonstrating a signal-dependent association between a crucial translational control factor and the JunB transcript (Fig. 6C). In contrast, thrombin treatment did not affect the levels of β-actin mRNA associated with eIF4E. Furthermore, consistent with constitutive translation of this structural protein (see also Fig. 2A), β-actin mRNA was associated with eIF4E in control, unstimulated EC.
Fig. 6.
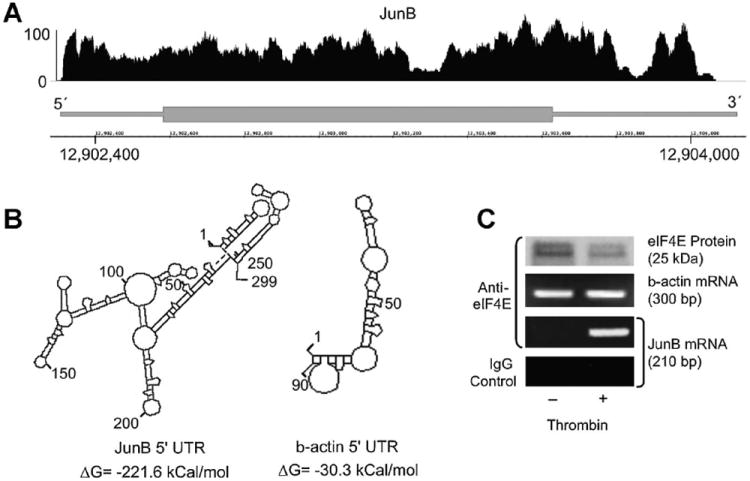
Thrombin stimulation induces association of eIF4E and JunB mRNA in human EC. Panel A: Next-generation RNA-sequencing of endothelial cell JunB transcript 5′-UTR. Polyadenylated RNA was isolated from unstimulated EC and was subsequently used for next-generation RNA-sequencing. The JunB gene (bottom portion) is represented by a thick (exon) lines, 5′- and 3′- ends of the gene are labeled accordingly. The position on the chromosome is indicated by the numbers on the x-axis. The diagram above the gene pictorial (top portion) represents EC-derived transcripts that were fragmented, sequenced, and subsequently matched and aligned to JunB using the Integrated Genome Browser (IGB). The y-axis represents the abundance of hits matched to the appropriate part of the gene. Panel B: Predicted folding structures and associated free energy values for the 5′-UTRs of JunB and b-actin (DNASIS, Hitachi Genetic Systems). The predicted secondary structures and free energies of the JunB and β-actin 5′ UTR are illustrated. A free energy value ≤50 kcal/mol suggests that a regulated event to linearize the secondary structure is necessary to efficiently translate the mRNA transcript. Panel C: JunB mRNA is detected only in eIF4E immunoprecipitates derived from thrombin-stimulated EC. Human EC were treated with thrombin (2 U/ml, 2 h). Immunoprecipitates were prepared using anti-eIF4E (or non-immune IgG) antibodies and probed for either β-actin or JunB mRNA by PCR.
DISCUSSION
TRANSLATIONAL CONTROL AS A REGULATOR OF TRANSCRIPTION FACTOR EXPRESSION AND FACILITATOR OF RAPID EC ACTIVATION
EC stimulated with thrombin assume a prothrombotic and pro-inflammatory phenotype [Cines et al., 1998; Kraiss et al., 2005]. A major mechanism contributing to this activated phenotype is induction of a new transcriptional program [Minami et al., 2004]. A variety of transcription factors regulate this program, implying the existence of thrombin-sensitive pathways that modulate transcription factor activity or expression at even earlier time points.
Mobilization of a pre-existing reservoir of mRNA to polyribosomes is a method of rapidly inducing new protein expression without the delay and energy expenditure of upstream transcription [Kozak, 1991; Mathews et al., 2000]. Our studies demonstrate that this is a major mechanism by which JunB protein expression is regulated in EC. Pre-existing JunB mRNA resides in the cytoplasm (Fig. 5A). It has a highly complex 5′-UTR with a calculated free energy consistent with translational regulation (Fig. 6A,B), and associates with eIF4E in response to an activating stimulus (Fig. 6C). JunB protein is then rapidly synthesized and accumulates in the nucleus (Fig. 3A,B).
Translational regulation also represents an additional checkpoint for regulating expression of biologically potent gene products such as transcription factors [Kozak, 1991] pointing to a complex interplay between translational and transcriptional events that regulate gene expression. We have identified other transcription factors whose expression is controlled by translational regulation responding to outside-in signals. Bcl-3, a member of the extended NF-κB family of transcription factors, is rapidly synthesized from pre-existing mRNA in activated platelets [Weyrich et al., 1998] and shear-stressed EC [Kraiss et al., 2000]. Neutrophils synthesize retinoic acid receptor-α (RAR-α) from constitutively expressed mRNA in response to cellular activation [Yost et al., 2004]. Our studies of JunB in activated EC add to this body of evidence and suggest that translational control is a common mode of regulation used by a variety of cells to determine transcription factor levels over relatively short-time frames in response to cellular activation. Translational regulatory mechanisms thereby influence subsequent transcriptional activity [Kraiss et al., 2000; Yost et al., 2004] and patterns of expressed proteins.
Discrete control of translational events by extracellular stimuli such as thrombin implies the existence of signaling pathways that directly target and regulate ribosomal function. AP-1 family activity has previously been shown to be under the control of both the ERK and p38 MAPK signaling pathways [Karin, 1995]. Thrombin activates both ERK and p38 MAPK in EC within 5 min but only blockade of the p38 MAPK pathway inhibits JunB expression in response to thrombin (Supplementary Fig. 3). These data suggest that the p38 MAPK pathway directly impinges on translational pathways as part of the mechanism by which it regulates AP-1 transcription factor activity. Interestingly, a highly specialized translational control pathway centered on mTOR, which is responsible for early synthesis of RAR-α and the urokinase plasminogen activator receptor in myeloid leukocytes [Mahoney et al., 2001; Yost et al., 2004], is not activated in ECs by thrombin and appears not to be involved in JunB expression induced by thrombin (Supplementary Fig. 3). These findings differ from those of a recent report in which the mTOR pathway was found to mediate translational control of JunB in some anaplastic large cell lymphomas [Staber et al., 2007]. Thus, regulation of JunB synthesis may vary in a cell-specific fashion in neoplastic and non-neoplastic cell types.
AP-1 TRANSCRIPTION FACTORS AND EC ACTIVATION
The biology of AP-1 is complex. The family includes at least 12 separate proteins of the Jun, Fos, or activating transcription factor (ATF) isoforms that form homo- or heterodimers which then bind to sequence-specific sites in the promoter regions of the genes they regulate [Karin et al., 1997]. AP-1 dimer composition and DNA binding activity reflect the relative amounts of AP-1 family members available for interaction [Mechta-Grigoriou et al., 2001]. AP-1 dimer biological activity is highly cell-type dependent as is dimer composition, which can also vary temporally in different conditions [Chinenov and Kerppola, 2001]. Thus, a full understanding of transcriptional regulation by AP-1 in a particular biologic context requires knowledge of the relative abundance of individual AP-1 components in the DNA-bound dimers. The relative abundance of various AP-1 family members in activated endothelial cells is largely uncharacterized. Nonetheless, this regulatory system clearly plays an important role in producing the activated endothelial phenotype in response to a variety of agonists [Shono et al., 1996; Chien et al., 1998; Delerive et al., 1999; Wang et al., 2001, 2002; Bavendiek et al., 2002; Dichtl et al., 2003; Fantozzi et al., 2003; Fisslthaler et al., 2003; Jensen and Whitehead, 2003; Rodriguez-Pascual et al., 2003; Wedgwood et al., 2003; Hsu et al., 2004].
The individual role(s) of the various AP-1 family members is better described in oncological studies, where certain generalizations have emerged [Shaulian and Karin, 2002]. Numerous stress cytokines and mitogens activate c-Jun and promote cellular proliferation. In most instances, JunB functions as a biological inhibitor of c-Jun and inhibits cell growth (and perhaps other c-Jun-dependent processes). On the other hand, we observed JunB induction in response to serum stimulation (Supplementary Table I), as have others [Mechta-Grigoriou et al., 2001], and we found that JunB antisense oligonucleotides inhibit growth in juvenile, subconfluent EC cultures (Supplementary Fig. 4). These results are consistent with observations that anti-JunB antibodies prevent cell cycle entry in other cell types [Kovary and Bravo, 1991].
The role of JunB in mature, confluent EC exposed to activating stimuli may be different than its role in subconfluent or proliferating endothelium. Perhaps induction of JunB by the same stimuli that induce c-Jun activity occurs to dampen or temper the effects that would result from unregulated transcriptional activation on the part of c-Jun. We found that thrombin triggers c-Jun phosphorylation (and presumably activation) in EC as early as 30 min after stimulation (unpublished data, 2005)—At virtually the same time that newly synthesized JunB is detectable. Therefore, one role of JunB in activated EC may be to modulate c-Jun function.
Thus, the function of JunB in EC as an individual member may be as complex and context-dependent as the entire AP-1 family, with different effects that depend on the biological conditions, including the growth state of the cell and relative abundance of other AP-1 components available for interaction [Shaulian and Karin, 2001; Eferl and Wagner, 2003].
In summary, strategies such as TSAA that examine post-transcriptional regulation have not been previously applied to EC which, conversely, have received extensive attention regarding transcriptional control. We validated TSAA results predicting that JunB is translationally regulated; prior to these studies, there was no indication that JunB expression was under discrete translational control in EC. Thus, TSAA should be a useful strategy to identify many heretofore unrecognized instances of signal-dependent translational control in EC biology. The results of such studies will expand our insight into the diversity of gene expression control mechanisms that produce the activated EC phenotype.
Supplementary Material
Acknowledgments
Diana Lim provided expert assistance in preparation of the figures.
Grant sponsor: National Institutes of Health; Grant numbers: HL 075507, HL 66277, R37HL 44525; Grant sponsor: Western States Affiliate American Heart Association Awards; Grant numbers: 0625098Y, 09BGIA2250381; Grant sponsor: Lichtenberg-Professorship.
Abbreviations used
- AP-1
activator protein-1
- eIF4E
eukaryotic initiation factor 4E
- HUVEC
human umbilical vein endothelial cells
- mTOR
mammalian target of rapamycin
- PCR
polymerase chain reaction
- qRT-PCR
quantitative real-time PCR
- TLI
translation index
- TSAA
translation state array analysis
- UTR
untranslated region
Footnotes
Additional supporting information may be found in the online version of this article.
The authors declared they have no conflicts of interest.
References
- Anderson P, Kedersha N. RNA granules. J Cell Biol. 2006;172:803–808. doi: 10.1083/jcb.200512082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bavendiek U, Libby P, Kilbride M, Reynolds R, Mackman N, Schonbeck U. Induction of tissue factor expression in human endothelial cells by CD40 ligand is mediated via activator protein 1, nuclear factor kappa B, and Egr-1. J Biol Chem. 2002;277:25032–25039. doi: 10.1074/jbc.M204003200. [DOI] [PubMed] [Google Scholar]
- Bevilacqua MP, Stengelin S, Gimbrone MA, Jr, Seed B. Endothelial leukocyte adhesion molecule 1: An inducible receptor for neutrophils related to complement regulatory proteins and lectins. Science. 1989;243:1160–1165. doi: 10.1126/science.2466335. [DOI] [PubMed] [Google Scholar]
- Brown EJ, Schreiber SL. A signaling pathway to translational control. Cell. 1996;86:517–520. doi: 10.1016/s0092-8674(00)80125-7. [DOI] [PubMed] [Google Scholar]
- Chien S, Li S, Shyy YJ. Effects of mechanical forces on signal transduction and gene expression in endothelial cells. Hypertension. 1998;31:162–169. doi: 10.1161/01.hyp.31.1.162. [DOI] [PubMed] [Google Scholar]
- Chinenov Y, Kerppola TK. Close encounters of many kinds: Fos–Jun interactions that mediate transcription regulatory specificity. Oncogene. 2001;20:2438–2452. doi: 10.1038/sj.onc.1204385. [DOI] [PubMed] [Google Scholar]
- Cines DB, Pollak ES, Buck CA, Loscalzo J, Zimmerman GA, McEver RP, Pober JS, Wick TM, Konkle BA, Schwartz BS, Barnathan ES, McCrae KR, Hug BA, Schmidt AM, Stern DM. Endothelial cells in physiology and in the pathophysiology of vascular disorders. Blood. 1998;91:3527–3561. [PubMed] [Google Scholar]
- Collins T, Read MA, Neish AS, Whitley MZ, Thanos D, Maniatis T. Transcriptional regulation of endothelial cell adhesion molecules: NF-kappa B and cytokine-inducible enhancers. FASEB J. 1995;9:899–909. [PubMed] [Google Scholar]
- World Medical Association Declaration of Helsinki. Recommendations guiding physicians in biomedical research involving human subjects. Cardiovasc Res. 1997;35:2–3. [PubMed] [Google Scholar]
- Delerive P, Martin-Nizard F, Chinetti G, Trottein F, Fruchart JC, Najib J, Duriez P, Staels B. Peroxisome proliferator-activated receptor activators inhibit thrombin-induced endothelin-1 production in human vascular endothelial cells by inhibiting the activator protein-1 signaling pathway. Circ Res. 1999;85:394–402. doi: 10.1161/01.res.85.5.394. [DOI] [PubMed] [Google Scholar]
- Dichtl W, Dulak J, Frick M, Alber HF, Schwarzacher SP, Ares MP, Nilsson J, Pachinger O, Weidinger F. HMG-CoA reductase inhibitors regulate inflammatory transcription factors in human endothelial and vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2003;23:58–63. doi: 10.1161/01.atv.0000043456.48735.20. [DOI] [PubMed] [Google Scholar]
- Eferl R, Wagner EF. AP-1: A double-edged sword in tumorigenesis. Nat Rev Cancer. 2003;3:859–868. doi: 10.1038/nrc1209. [DOI] [PubMed] [Google Scholar]
- Eyries M, Agrapart M, Alonso A, Soubrier F. Phorbol ester induction of angiotensin-converting enzyme transcription is mediated by Egr-1 and AP-1 in human endothelial cells via ERK1/2 pathway. Circ Res. 2002;91:899–906. doi: 10.1161/01.res.0000042703.39845.b4. [DOI] [PubMed] [Google Scholar]
- Fantozzi I, Zhang S, Platoshyn O, Remillard CV, Cowling RT, Yuan JX. Hypoxia increases AP-1 binding activity by enhancing capacitative Ca2+ entry in human pulmonary artery endothelial cells. Am J Physiol Lung Cell Mol Physiol. 2003;285:L1233–L12345. doi: 10.1152/ajplung.00445.2002. [DOI] [PubMed] [Google Scholar]
- Fisslthaler B, Benzing T, Busse R, Fleming I. Insulin enhances the expression of the endothelial nitric oxide synthase in native endothelial cells: A dual role for Akt and AP-1. Nitric Oxide. 2003;8:253–261. doi: 10.1016/s1089-8603(03)00042-9. [DOI] [PubMed] [Google Scholar]
- Gingras AC, Raught B, Sonenberg N. eIF4 initiation factors: Effectors of mRNA recruitment to ribosomes and regulators of translation. Annu Rev Biochem. 1999;68:913–963. doi: 10.1146/annurev.biochem.68.1.913. [DOI] [PubMed] [Google Scholar]
- Hershey J, Merrick WC. The pathway and mechanism of initiation of protein synthesis. In: Mathews MB, editor. Translational control of gene expression. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 2000. [Google Scholar]
- Hipp MS, Urbich C, Mayer P, Wischhusen J, Weller M, Kracht M, Spyridopoulos I. Proteasome inhibition leads to NF-kappaB-independent IL-8 transactivation in human endothelial cells through induction of AP-1. Eur J Immunol. 2002;32:2208–2217. doi: 10.1002/1521-4141(200208)32:8<2208::AID-IMMU2208>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Hsu YH, Chen JJ, Chang NC, Chen CH, Liu JC, Chen TH, Jeng CJ, Chao HH, Cheng TH. Role of reactive oxygen species-sensitive extracellular signal-regulated kinase pathway in angiotensin II-induced endothelin-1 gene expression in vascular endothelial cells. J Vasc Res. 2004;41:64–74. doi: 10.1159/000076247. [DOI] [PubMed] [Google Scholar]
- Jensen LE, Whitehead AS. ELAM-1/E-selectin promoter contains an inducible AP-1/CREB site and is not NF-kappa B-specific. Biotechniques. 2003;35:54–56. 58. doi: 10.2144/03351bm05. [DOI] [PubMed] [Google Scholar]
- Karin M. The regulation of AP-1 activity by mitogen-activated protein kinases. J Biol Chem. 1995;270:16483–16486. doi: 10.1074/jbc.270.28.16483. [DOI] [PubMed] [Google Scholar]
- Karin M, Liu Z, Zandi E. AP-1 function and regulation. Curr Opin Cell Biol. 1997;9:240–246. doi: 10.1016/s0955-0674(97)80068-3. [DOI] [PubMed] [Google Scholar]
- Kovary K, Bravo R. The Jun and Fos protein families are both required for cell cycle progression in fibroblasts. Mol Cell Biol. 1991;11:4466–4472. doi: 10.1128/mcb.11.9.4466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozak M. An analysis of vertebrate mRNA sequences: Intimations of translational control. J Cell Biol. 1991;115:887–903. doi: 10.1083/jcb.115.4.887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraiss LW, Weyrich AS, Alto NM, Dixon DA, Ennis TM, Modur V, McIntyre TM, Prescott SM, Zimmerman GA. Fluid flow activates a regulator of translation, p70/p85 S6 kinase, in human endothelial cells. Am J Physiol Heart Circ Physiol. 2000;278:H1537–H1544. doi: 10.1152/ajpheart.2000.278.5.H1537. [DOI] [PubMed] [Google Scholar]
- Kraiss LW, Ennis TM, Alto NM. Flow-induced DNA synthesis requires signaling to a translational control pathway. J Surg Res. 2001;97:20–26. doi: 10.1006/jsre.2001.6091. [DOI] [PubMed] [Google Scholar]
- Kraiss LW, Alto NM, Dixon DA, McIntyre TM, Weyrich AS, Zimmerman GA. Fluid flow regulates E-selectin protein levels in human endothelial cells by inhibiting translation. J Vasc Surg. 2003;37:161–168. doi: 10.1067/mva.2003.67. [DOI] [PubMed] [Google Scholar]
- Kraiss L, Martinez M, Prescott S. Endothelial function. In: Fink MP, Abraham E, Vincent J-L, editors. Textbook of critical care. 5. Philadelphia, PA: Elsevier Saunders; 2005. [Google Scholar]
- Maeshima Y, Sudhakar A, Lively JC, Ueki K, Kharbanda S, Kahn CR, Sonenberg N, Hynes RO, Kalluri R. Tumstatin, an endothelial cell-specific inhibitor of protein synthesis. Science. 2002;295:140–143. doi: 10.1126/science.1065298. [DOI] [PubMed] [Google Scholar]
- Mahoney TS, Weyrich AS, Dixon DA, McIntyre T, Prescott SM, Zimmerman GA. Cell adhesion regulates gene expression at translational checkpoints in human myeloid leukocytes. Proc Natl Acad Sci USA. 2001;98:10284–10289. doi: 10.1073/pnas.181201398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathews M, Sonenberg N, Hershey JWB. Origins and principles of translational control. In: Sonenberg N, Hershey JWB, Mathews MB, editors. Translational control of gene expression. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 2000. [Google Scholar]
- Mechta-Grigoriou F, Gerald D, Yaniv M. The mammalian Jun proteins: Redundancy and specificity. Oncogene. 2001;20:2378–2389. doi: 10.1038/sj.onc.1204381. [DOI] [PubMed] [Google Scholar]
- Minami T, Sugiyama A, Wu SQ, Abid R, Kodama T, Aird WC. Thrombin and phenotypic modulation of the endothelium. Arterioscler Thromb Vasc Biol. 2004;24:41–53. doi: 10.1161/01.ATV.0000099880.09014.7D. [DOI] [PubMed] [Google Scholar]
- Prescott DM, Myerson D, Wallace J. Enucleation of mammalian cells with cytochalasin B. Exp Cell Res. 1972;71:480–485. doi: 10.1016/0014-4827(72)90322-9. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Pascual F, Redondo-Horcajo M, Lamas S. Functional cooperation between Smad proteins and activator protein-1 regulates transforming growth factor-beta-mediated induction of endothelin-1 expression. Circ Res. 2003;92:1288–1295. doi: 10.1161/01.RES.0000078491.79697.7F. [DOI] [PubMed] [Google Scholar]
- Shaulian E, Karin M. AP-1 in cell proliferation and survival. Oncogene. 2001;20:2390–2400. doi: 10.1038/sj.onc.1204383. [DOI] [PubMed] [Google Scholar]
- Shaulian E, Karin M. AP-1 as a regulator of cell life and death. Nat Cell Biol. 2002;4:E131–E136. doi: 10.1038/ncb0502-e131. [DOI] [PubMed] [Google Scholar]
- Shono T, Ono M, Izumi H, Jimi SI, Matsushima K, Okamoto T, Kohno K, Kuwano M. Involvement of the transcription factor NF-kappaB in tubular morphogenesis of human microvascular endothelial cells by oxidative stress. Mol Cell Biol. 1996;16:4231–4239. doi: 10.1128/mcb.16.8.4231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staber PB, Vesely P, Haq N, Ott RG, Funato K, Bambach I, Fuchs C, Schauer S, Linkesch W, Hrzenjak A, Dirks WG, Sexl V, Bergler H, Kadin ME, Sternberg DW, Kenner L, Hoefler G. The oncoprotein NPM-ALK of anaplastic large-cell lymphoma induces JUNB transcription via ERK1/2 and JunB translation via mTOR signaling. Blood. 2007;110:3374–3383. doi: 10.1182/blood-2007-02-071258. [DOI] [PubMed] [Google Scholar]
- Veomett G, Shay J, Hough PV, Prescott DM. Large-scale enucleation of mammalian cells. Methods Cell Biol. 1976;13:1–6. doi: 10.1016/s0091-679x(08)61794-x. [DOI] [PubMed] [Google Scholar]
- Wang N, Verna L, Liao H, Ballard A, Zhu Y, Stemerman MB. Adenovirus-mediated overexpression of dominant-negative mutant of c-Jun prevents intercellular adhesion molecule-1 induction by LDL: A critical role for activator protein-1 in endothelial activation. Arterioscler Thromb Vasc Biol. 2001;21:1414–1420. doi: 10.1161/hq0901.095549. [DOI] [PubMed] [Google Scholar]
- Wang N, Verna L, Chen NG, Chen J, Li H, Forman BM, Stemerman MB. Constitutive activation of peroxisome proliferator-activated receptor-gamma suppresses pro-inflammatory adhesion molecules in human vascular endothelial cells. J Biol Chem. 2002;277:34176–34181. doi: 10.1074/jbc.M203436200. [DOI] [PubMed] [Google Scholar]
- Wang Z, Gerstein M, Snyder M. RNA-Seq: A revolutionary tool for transcriptomics. Nat Rev Genet. 2009;10:57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wedgwood S, Mitchell CJ, Fineman JR, Black SM. Developmental differences in the shear stress-induced expression of endothelial NO synthase: Changing role of AP-1. Am J Physiol Lung Cell Mol Physiol. 2003;284:L650–L662. doi: 10.1152/ajplung.00252.2002. [DOI] [PubMed] [Google Scholar]
- Weyrich AS, Dixon DA, Pabla R, Elstad MR, McIntyre TM, Prescott SM, Zimmerman GA. Signal-dependent translation of a regulatory protein, Bcl-3, in activated human platelets. Proc Natl Acad Sci USA. 1998;95:5556–5561. doi: 10.1073/pnas.95.10.5556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yost CC, Denis MM, Lindemann S, Rubner FJ, Marathe GK, Buerke M, McIntyre TM, Weyrich AS, Zimmerman GA. Activated polymorphonuclear leukocytes rapidly synthesize retinoic acid receptor-alpha: A mechanism for translational control of transcriptional events. J Exp Med. 2004;200:671–680. doi: 10.1084/jem.20040224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmerman GA, Whatley RE, McIntyre TM, Benson DM, Prescott SM. Endothelial cells for studies of platelet-activating factor and arachidonate metabolites. Methods Enzymol. 1990;187:520–535. doi: 10.1016/0076-6879(90)87059-c. [DOI] [PubMed] [Google Scholar]
- Zong Q, Schummer M, Hood L, Morris DR. Messenger RNA translation state: The second dimension of high-throughput expression screening. Proc Natl Acad Sci USA. 1999;96:10632–10636. doi: 10.1073/pnas.96.19.10632. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.