

SUMMARY
The hallmark of the cerebral neocortex is its organization into six layers, each containing a characteristic set of cell types and synaptic connections. The transcriptional events involved in laminar development and function still remain elusive. Here we employed deep sequencing of mRNA and small RNA species to gain insights into transcriptional differences among layers and their temporal dynamics during postnatal development of the mouse primary somatosensory neocortex. A number of novel coding and noncoding transcripts were identified with specific spatiotemporal expression and splicing patterns. We also identified signature trajectories and gene co-expression networks associated with distinct biological processes and transcriptional overlap between these processes. Finally, we provide data that allows the study of potential miRNA and mRNA interactions. Overall, this study provides an integrated view of the laminar and temporal expression dynamics of coding and noncoding transcripts in the mouse neocortex and a resource for studies of neurodevelopment and transcriptome.
INTRODUCTION
The cerebral neocortex (NCX) is stereotypically organized into six distinct layers. Both glutamatergic excitatory projection (aka pyramidal) neurons and GABAergic inhibitory neurons display laminar variations in their morphological, molecular and functional properties (DeFelipe et al., 2013; Kwan et al., 2012; Leone et al., 2008; Molyneaux et al., 2007). The proper development and function of cortical neurons depends on glial cells and the neurovascular system, whose distribution also appears to vary across layers and areas (Fonta and Imbert, 2002).
The formation of layers occurs progressively and requires the orchestrated execution of a series of developmental events. These events include the migration of young neurons into appropriate positions within the emerging NCX and development of specific neuronal dendritic arbors and axonal projections (Kwan et al., 2012; Leone et al., 2008; Molyneaux et al., 2007), generation and maturation of glial cells (Rowitch and Kriegstein, 2010), development of the neurovascular system (Tam and Watts, 2010), emergence of early spontaneous activity and experience-driven activity (Kilb et al., 2011) and synaptogenesis (West and Greenberg, 2011) and circuit refinement (Espinosa and Stryker, 2012). The formation of cortical layers occurs in an inside-out manner, with the deep layers (L) 5 and 6 (infragranular layers, IgL) being formed first, followed by L4 (granular layer due to the presence of small-sized stellate and pyramidal neurons), and finally the superficial L2/3 (supragranular layers, SgL).
Studies of transcriptional events involved in the development and function of neocortical layers have been greatly advanced with the emergence of high-throughput transcriptome profiling techniques. A number of studies have analyzed the transcriptome of different mouse neocortical layers and/or areas at specific developmental time points (Arlotta et al., 2005; Belgard et al., 2011; Chen et al., 2005; Dillman et al., 2013; Han et al., 2011; Lein et al., 2007; Lyckman et al., 2008; Rossner et al., 2006; Sugino et al., 2006). Also, these studies have largely focused on the expression of protein-coding mRNA, providing limited information on noncoding RNAs (ncRNA), which play an important role in neural development and function (McNeill and Van Vactor, 2012). In an attempt to profile the spatiotemporal transcriptome dynamics of both coding and ncRNA transcripts, we deep sequenced mRNA (mRNA-seq hereafter) and small ncRNA (smRNA-seq hereafter) transcripts from the IgL, L4 and SgL of the mouse somatosensory cortex (S1C hereafter) across multiple early postnatal time points and adult. After testing different RNA collection methods as laser capture microdissection and fluorescence-activated cell sorting (data not shown), we opted to microdissect distinct cortical layers from tissue sections of the Dcdc2a-Gfp transgenic mouse (Heintz, 2004), which expressed GFP in L4 of the S1C. This approach allowed us to distinguish IgL, L4 and SgL across different time points, sequence transcripts expressed in all neural and non-neural cells types present in these layers in vivo, and analyze transcripts present not only in cell somata but also those localized to neuronal dendrites and axons (Hengst and Jaffrey, 2007). Furthermore, this approach does not expose cells to substantial chemical and mechanical manipulations or stress, which can distort RNA integrity and transcriptional states (Okaty et al., 2011). Our initial data analysis provides novel and functionally relevant insights into transcriptome dynamics of NCX layers and their relationship to specific neurodevelopmental processes.
RESULTS
Study Design, Data Generation and Quality Assessment
In order to obtain a global and unbiased view of the transcriptional dynamics in the postnatal NCX, we analyzed the transcriptome of the IgL, L4 and SgL from S1C of mouse brain at postnatal day (P) P4, P6, P8, P10, P14 and P180 (adult). To delineate the layers we used the Dcdc2a-Gfp reporter mouse that expressed GFP selectively in L4 of the S1C starting from around P2 (Figure 1A). We developed a microdissection protocol that lasted less than 2 hours and resulted in high yield and quality of RNA (RNA integrity number >8)(Supplemental information and Table S1A).
Figure 1. Study Design and Quality Control Measures.

(A) Representative sagittal tissue section of the Dcdc2a-Gfp mouse forebrain showing Gfp expression in layer 4 (L4) of the primary somatosensory cortex (S1C). Dashed lines outline the infragranular layers (IgL), L4 and supragranular layers (SgL). HIP, hippocampus; NCX, neocortex.
(B) qPCR analysis of the expression of well-established layer-enriched genes.
(C) Gfp expression across layers
(D) Expression of laminar markers depicted in a heat map of the log ratio RPKM data.
(E) Box plots representing uniquely mapped reads for either miRNA or mRNA transcriptomes in each sample.
(F) Violin plots representing the distribution of the transcribed ratios of the genome (black) and the transcriptome (grey). Fchr, female chromosome; Mchr, male chromosome; chrM, mitochondrial chromosome.
(G) Violin plots representing percentage distribution of smRNA reads across different length of reads. See also Figure S1 and Table S1.
We extracted total RNA from laminar samples microdissected from two mouse brains (one male and one female) per time point, for a total of 12 mice and 36 samples (Table S1A). We analyzed the expression of several known layer-specific markers by quantitative real-time PCR to verify the accuracy of our laminar microdissection (Figure 1B). The mRNA-seq and smRNA-seq libraries, containing spike-in RNAs to tag samples and assess the quality of sequencing, were prepared according to manufacturer’s instructions (Table S1B). On average, we observed less than 2% of mismatches per read, indicative of high quality sequenced reads (Figure S1A). Since we used a GFP reporter mouse, we mapped the mRNA-seq reads to the Gfp sequence and confirmed that L4 samples had consistently higher RPM (reads per million sequenced reads) values (Figure 1C).In addition, a list of known and well-characterized layer-specific marker genes was used to verify the identity of the samples after mRNA-seq (Figures 1D, S1B and S1C).
We obtained 611 and 143 million of single-end reads from the mRNA-seq and smRNA-seq samples, respectively. We found that approximately 431 million reads from the mRNA-seq libraries uniquely matched to mouse reference genome, that is, an average >10 million of reads per sample, with no particular laminar bias (Figures 1E and S1D; Table S1A). On average, only 3% of each autosomal chromosome was transcribed, slightly lower in sex chromosomes, while most of the mitochondrial chromosome was transcribed (~70%, Figure 1F). As expected, all female samples had 0% hits on the reference genome for the Y chromosome. Next, we determined the number of exons of known protein-coding genes matched by uniquely mapped reads (i.e., the fraction of exons transcribed). On average, the ratio of transcribed exons in annotated protein-coding genes was ~50% for the autosomal chromosomes, ~38% for the X chromosome, ~35% for the Y chromosome, and ~70% of the mitochondrial chromosome (Figure 1F). The transcribed exons occupied the majority of mRNA-seq reads (~88.6%), and only ~4.1% of reads were within intronic regions of known protein-coding genes (Figure S1E). Therefore, we found 12,729 of protein-coding genes were reliably expressed (gene RPKM ≥ 1 in at least two samples) in any layer or time point analyzed. The remaining ~7.3% of reads aligned within intergenic regions (Figure S1E), suggesting the expression of novel transcripts including long intergenic noncoding RNAs (lincRNAs), a potentially interesting discovery in light of recent work on the possible regulatory function of lincRNA (Mercer and Mattick, 2013).
As for smRNA-seq samples, the length of the 3′-adapter clipped reads were clearly enriched for the microRNA (miRNA) length (i.e., 22 nt) (Figure 1G). Approximately 50 million high quality reads were uniquely mapped (Figure S1F), that is, an average of >1 million reads per sample (Figure 1E; Table S1A). We found that 436 miRNAs were reliably expressed (reads count ≥ 10 in at least 2 samples). Among these, >80% of reads matched the top 10 reliably expressed miRNAs, which belong to 8 families (i.e., let-7, mir-25, mir-125, mir-28, mir-151, mir-127, mir-181 and mir-486) (Figure S1G).
To assess similarities and differences among samples, we performed principal component analysis (PCA) for both mRNA-seq and smRNA-seq samples. The results demonstrate that mRNA-seq samples cluster first according to the age of the mouse (PC1, 54.98%) and second to their laminar location (PC2, 11.76%) (Figures 2A and S2A–D). In contrast, the contribution of age and laminar location to smRNA-seq samples was smaller (Figures 2B and S2E–H).
Figure 2. Spatiotemporal Dynamics of Mouse Neocortical Transcriptome.

(A and B) Principal component analyses of mRNA (A) and smRNA (B) transcriptomes.
(C and D) Venn diagrams representing the number of DEX protein-coding mRNAs (C) and miRNAs (D). See also Figure S2, S3 and Table S2.
We also performed hierarchical cluster analysis of mRNA-seq samples and found that samples from P6–P10 mice cluster together, while P4 samples cluster alone in one extreme and P14 and adult samples cluster together in the other extreme (Figure S3A). These results suggest that P14 samples are molecularly more similar to adults than to any other early postnatal samples, while the molecular signature of P4 samples appeared to be distinct from P6–P10 and P14-adult mice. We also found that transcriptome differs more prominently across time and layers than it does between males and females (Figure S2).
Spatiotemporal Expression of Coding RNAs and miRNAs
To identify differentially expressed (DEX) protein-coding genes and miRNAs we used the R package DESeq (Anders and Huber, 2010). DEX genes were split into three categories according to their differential expression across layers [spatial DEX (sDEX)], age [temporal DEX (tDEX)], or both [spatiotemporal DEX (stDEX)]. For these analyses we only considered reliably expressed protein-coding genes and miRNA (Figures S3B and S3C). Additionally, only transcripts found to have a false discovery rate (FDR) <0.01 were considered as significant DEX transcripts.
Of all reliably expressed protein-coding genes (12,729), ~10% (1,321) were tDEX, ~5% (662) were sDEX, and ~8% (1,051) were stDEX (Figure 2C; Table S2A). Our analysis of the reliably expressed miRNAs (436) revealed that most were tDEX (86, ~20%), while only ~1% were sDEX and ~3% were stDEX (Figure 2D; Table S2B). These results are in agreement with the PCA analysis, which indicated that time had more influence on miRNA expression than location within NCX.
Gene function enrichment analysis of the protein-coding genes that were strictly tDEX revealed that these genes are highly associated with categories related to the cell membrane, synapses, and cell junctions (Bonferroni-adjusted P < 0.01), and are mostly involved in ion channel activity or potassium ion transport (Bonferroni-adjusted P< 0.01) (Table S3A) (Huang et al., 2009). On the other hand, protein-coding genes that were solely spatially regulated are mostly associated with categories related to the regulation of cell and vascular development, as well as neuronal differentiation (Bonferroni-adjusted P< 0.0001) (Table S3B). Protein-coding genes that were both spatially and temporally DEX are most associated with categories related to the development of projection neurons, synapses, and cell-cell signaling (Bonferroni-adjusted P< 0.00001) (Table S3C). These analyses suggest that we can use these data to explore molecular correlates of developmental events occurring during laminar maturation.
Spatiotemporal Alternative Splicing
Alternative splicing is a process by which one precursor mRNA (pre-mRNA) can give rise to more than one distinct mature mRNA via combination of alternative exon usage (Nilsen and Graveley, 2010). We analyzed five basic splicing modalities: cassette exon(s), mutually exclusive exon(s), alternative donors or acceptors, alternative first or last exon, and intron retention (Figures 3A, S3D, S3E; 2Table S4). Similar to DEX analysis, we divided the differentially alternative splicing (DAS) events into three categories: spatial (sDAS), temporal (tDAS) and spatiotemporal (stDAS). We found that tDAS were more abundant than sDAS or stDAS events (Figure 3A). Additionally, DAS events were split into known and novel groups (Figure 3A; Table S4). It is noteworthy that the apparent enrichment of novel DAS of alternative donors/acceptors and alternative first exon modalities may be skewed by the existence of more reads matching the 3′-end of transcripts than the 5′ end, as well as by reads that cannot distinguish introns or adjacent exons.
Figure 3. Spatiotemporal Dynamics of Alternative Splicing Events.

(A) Numbers of known (dark blue) and novel (red) splicing events.
B) Reads coverage in the Dlg2 gene region correspondent to exons present in X and Y (isoforms 1 and 3) and 9 and Y (isoform 2). The yellow box highlights the temporal coverage of mRNA-seq reads mapped to exon 9. Black bars/boxes underneath the exonic read distribution indicates exon junctions. Red arrows depict location of exon-specific PCR primers.
(C and D) Exon-specific PCR of the cassette exons “X-9-Y” in the mouse IgL (C) and human S1C of equivalent developmental time points (D). Biological replicates indicated as “a” or “b”. See also Figure S3 and Table S4.
mRNA-seq can reliably identify cassette exon events. As an example, we confirmed a predicted cassette exon of Dlg2 (aka Psd-93) gene, and found that it was temporally regulated (Figures 3B and 3C). There are three mouse RefSeq RNAs registered for mouse Dlg2 (isoform 1–3). During the first week of postnatal development, we detected reads that span an exon common to Dlg2 isoforms 1 and 3 (exons 17 and 5 respectively) hereafter referred to as exon X, to exon 9 present in Dlg2 isoform 2. We also detected reads that span exon 9 to another exon common to Dlg2 isoforms 1, 2, and 3 (exons 18, 10, and 6 respectively), hereafter referred to as exon Y. These observations suggest that the exons “X-9-Y” are being co-expressed in the same transcript. Interestingly, around P6, we can also detect junctions that connect exons X and Y, which becomes the main junction present at P14 and the only junction expressed in adulthood. We confirmed this observation by PCR analysis and observed that the cassette exon “X-9-Y” and “X-Y” have inverse expression profiles with the first peaking at P6 and fading out after; and the second appearing around P6 and increasing into adulthood (Figure 3D).
In humans, the orthologous “X-9-Y” cassette exons are described in DLG2 gene isoforms 2 and 4. However, these exons are not included in the RefSeq RNA and, thus their isoform expression was not reported in previous Exon Array studies of the developing human brain (Johnson et al., 2009; Kang et al., 2011; Pletikos et al., 2014). We performed PCR analysis in human S1C of equivalent developmental periods (www.translatingtime.net, Table S1C). We observed that the human “X-9-Y” versus “X-Y” cassette exons were expressed in similar pattern to that detected in mouse tissue samples (Figure 3D), indicating that this is a conserved developmental variant.
DLG2 encodes a postsynaptic density protein known as PSD-93, and is a member of the membrane-associated guanylate kinase (MAGUK) family (Hough et al., 1997). The Havana project has predicted based on expressed sequence tags, the existence of a processed transcript containing the cassette exon “X-9-Y”. Our data provides the direct evidence to the expression of the cassette exon “X-9-Y”. Since we do not know the entire structure of the new mouse transcript containing the exon cassette “X-9-Y” and resulting changes in the protein sequence, it is difficult to predict the functional consequence of its expression. However, it is noteworthy that the cassette exon “X-9-Y” matches a genomic region of Dlg2 isoform 1 that sits between the hook and GK protein domains that are thought to be involved in defining the subcellular localization of MAGUK proteins (Hough et al., 1997), as well as their ability to bind other proteins (Brenman et al., 1998; Paarmann et al., 2002). Moreover, a recent report described a similar alternative splicing event in Dlg4 (aka Psd-95) that seems to be important for the regulation of neuronal synapse maturation (Zheng et al., 2012). Together, these findings illuminate the spatiotemporal complexity of alternative splicing and provide examples of likely functionally relevant DAS events.
Developmental and Laminar Specificity of Co-Expression Networks
Co-expressed genes share spatiotemporal localization and often participate in the same biological processes (Barabasi and Oltvai, 2004; Oldham et al., 2008; Johnson et al., 2009; Kang et al., 2011). We therefore performed weighted gene co-expression network analysis (WGCNA) to construct and visualize modules of co-expressed genes across all samples (Langfelder and Horvath, 2008). We identified 40 modules with distinct spatiotemporal patterns that are characterized by the trajectories of module’s eigengenes (Figure S4). M40 (n=14 genes) shows sex biased expression of genes (i.e. Y chromosome enriched genes) and was thus excluded from downstream analyses. Of the other 39 modules, we performed hierarchical cluster analysis and found that they can be organized into 5 clusters (Figure 4A). The first (cluster I) and fifth (cluster V) are temporal clusters, in which the expression of clustered genes are increasing and decreasing along time points, respectively, in an anti-correlated fashion (Figure 4B). The remaining clusters are mainly spatially defined (i.e., cluster II is IgL-enriched, cluster III is SgL-enriched, and cluster IV is L4-enriched).
Figure 4. Weighted Gene Co-Expression Networks.

(A) Heatmap matrix showing modular eigengenes across ages for each layer.
(B) Pairwise Pearson correlations among modules.
(C) Developmental trajectories of modules M5, M7 (top panels), and proportion of genes reported to be enriched in different neural cell types (bottom bar graphs). See also Figure S4 and Table S5.
(D) Proportion of genes reported to be enriched in different neural cell types in spatial clusters II–IV.
(E) Proportion of genes reported to be enriched in immature and mature astrocytes in spatial clusters II–IV. See also Figure S5.
Functional Annotation of Co-Expression Networks
Gene function enrichment analysis revealed that a large fraction of genes grouped in a given module are associated with a specific biological process (Table S5). Interestingly, some biological categories were significantly enriched (Bonferroni-adjusted P < 0.01) in more than one module. For example, cluster III modules M5, M7 were associated with blood vessel development (Table S5). However, in M5 we found genes involved in the initial stages of angiogenesis– Notch1, Robo4, Angpt2; while in M7 we found genes involved in later stages of angiogenesis– Tek and EphB4 (Chung and Ferrara, 2011).
Due to the presence of multiple cell types in our tissue samples, we wanted to investigate whether the genes clustered in our 40 modules could be associated with the developmental program of a specific neural cell type (i.e., neurons, astrocytes and oligodendrocytes). For this analysis we intersected each module with lists of genes enriched in each neural cell type (Cahoy et al., 2008). We found that each module tends to be enriched in a defined neural cell type (Figure S5). Interestingly, modules M5 and M7 were enriched in astrocyte-enriched genes (Binominal test P = 6.4e-51, P = 4.7e-5, and P = 6.8e-6, respectively), which is in agreement with known interaction between astrocyte and blood vessel development (Zerlin and Goldman, 1997) (Figure 4C). Additionally, we found that cluster II (IgL-enriched) and cluster IV (L4-enriched) had a higher percentage of oligondendrocyte- and neuron-enriched genes than astrocyte-enriched genes (Chi-squared test P = 1e-35 and P = 4.4e-24, respectively), which is in contrast to cluster III (SgL-enriched) (Figure 4D). Interestingly, the ratio of immature/mature astrocytes was significantly higher in cluster III compared to clusters II and IV (Chi-squared test P= 9.2e-7, Figure 4E). Together, these observations are consistent with the inside-outside progressive maturation of neural cell types in developing neocortical layers.
We also analyzed the degree of connectivity among the top 10 hub genes within each module (M1–M39) and among different modules within each cluster (I–V) (Figures 5 and S6). The hub genes were selected based on gene expression profile proximity to that of the respective eigengene, and the degree of connectivity was assessed based on the co-expression correlation co-efficiency (Table S5). We found that modules enriched in genes highly expressed in a specific neural cell type were more interconnected than they were connected with modules of genes enriched in a different neural cell type (Figure 5A). This observation suggests that some modules are associated with the development of a distinct neural cell type.
Figure 5. Spatial Cluster II Inter- and Intra- Modular Connectivity.

(A) Developmental trajectories (left column) and neural cell type enrichment (right column) of cluster II modules.
(B) Connectivity of cluster II inter- and intra-modular hub genes. See also Figure S6.
Interestingly, we also observed that inter-modular connections could happen between different cell types. For example, in spatial cluster II, M13 genes were highly connected to four hub genes in M27 (Slc25A22, Tmem163, Trbj2-3, Sema5A) and two hub genes in M31 (Prkce and EphA7) (Figure 5B). M13 showed functional enrichment in genes associated with behavior and cell morphogenesis in neuronal differentiation, while M27 shows functional enrichment in genes associated with oligodendrocyte development (Table S5). Of the four genes in M27 interconnected with the M13 genes, Sema5A was enriched in the IgL and previously reported to be expressed in early postnatal oligodendrocytes (Cahoy et al. 2008). However, consistent with our finding of association of Sema5a with both infragranular oligodendrocytic and neuronal modules (M27 and M13, respectively), examination of its expression pattern in early postnatal mice in the Allen Brain Atlas (http://developingmouse.brain-map.org) revealed that Sema5a could be expressed by both oligodendroctyes and neurons in the IgL. Furthermore, analyses of whole body and retinal oligodendrocyte conditional Sema5A knock-out mice indicated a key role of this semaphorin in retinal axon outgrowth (Goldberg et al., 2004; Matsuoka et al., 2011). Interestingly, in humans, SEMA5A is an autism risk gene, and epigenetic studies show that it is highly methylated in non-neuronal cells of autistic patients (Shulha et al., 2012; Weiss et al., 2009). Thus, Sema5a may play an important role in linking different gene modules and alterations in its function may have pleiotropic effects in the developing NCX. Furthermore, these and other related findings suggest that there is considerable overlap in transcriptional programs and possible cross-talk between developing neural cell types undergoing distinct biological processes.
miRNA-mRNA Relationships
Protein-coding transcript levels can be post-transcriptionally regulated by the activity of miRNA. These small RNA molecules of 20 – 24 nucleotides are known to silence mRNA translation through sequence-specific targeting (Bartel, 2009). This prompted us to analyze miRNA-mRNA targeting relationships in the context of spatiotemporal dynamics across neocortical layers and development ages.
We first used TargetScan to find putative miRNA targets based on sequence-complementarity to mRNA 3′UTR of reliably expressed protein-coding genes (Figure S7A) (Grimson et al., 2007). Next, we compared the expression profile between miRNA-mRNA pairs and selected the top anti-correlated pairs with rank scores ≥ 80, as these most likely represent down-regulation of the target mRNAs (Table S6) (Lu et al., 2011). To determine whether one miRNA could have enhanced activity towards a particular spatial or temporal cluster or certain neural cell type, we looked for the distribution of the targeted mRNAs throughout the spatiotemporal clusters previously analyzed, and filtered them for neural cell type enrichment.
Overall there were no significant differences in enrichment of targeted mRNAs clustered in temporal, spatial or spatiotemporal modules (Figure S7B). However, we found that 32 miRNAs preferentially targeted mRNAs that cluster in predominantly temporal modules (i.e., clusters I and V), and 8 miRNAs targeted preferentially mRNAs that cluster in predominantly spatial modules (i.e., clusters II–IV) (Figure S7C). We focused our attention on the putative targets of the most reliably expressed miRNAs to increase our confidence in any possible miRNA-mRNA regulatory interaction. One of these miRNAs is miR-92b, whose 119 anti-correlated mRNA targets were enriched in the IgL-cluster (Binominal test FDR < 0.01) (Figures 6A and S7C), and in neurons and oligodendrocytes (Binominal test P = 0.01 and P = 0.002, respectively, Figure 6C). There was a strong anti-correlation between the normalized expression trajectory of miR-92b and the expression profile of its putative target mRNAs (Pearson correlation R = −0.74, Figure 6A). One of the mRNAs predicted as targets of miR-92b is Foxp2, which is enriched in IgL projection neurons (Figures 1D, and 6B; Table S5). In addition, we also predict that Foxp2 is targeted by other miRNAs (Figure 6C), suggesting that the regulation of Foxp2 levels in neocortical cells occurs through multiple miRNAs.
Figure 6. miRNA-mRNA Regulation Prediction.
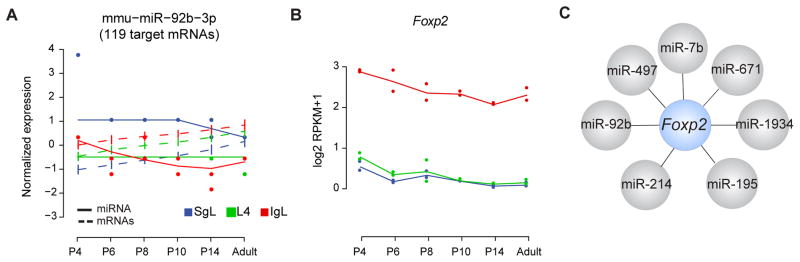
(A) Normalized expression trajectory of miR-92b to the expression profile of its putative target mRNAs (n=119). Data are expressed as mean ± 95% confident intervals for target mRNAs.
(B) Developmental trajectories of Foxp2 expression in different layers.
(C) miRNAs predicted to regulate Foxp2 mRNA. See also Figure S7 and Table S6.
By combining the temporal and spatial expression profiles of putative targeted mRNAs, we observed patterns of miRNA expression that revealed spatiotemporal aspects of gene regulation that were previously obscure. In addition, analysis of miRNAs that target mRNAs enriched in specific neural cell types may help to predict the mechanism of action of particular miRNAs.
Transcriptional correlates of neurodevelopmental events
To gain insights into progression of major neurodevelopmental processes across different layers and time points, we explore expression trajectories of genes associated with development of neuronal morphology (i.e., axonogenesis and dendritogenesis), synaptogenesis, and neural activity (i.e., excitatory and inhibitory neurotransmissions, and experience-driven activity) (Figure 7A and Table S7). As expected, the expression patterns of genes associated with axonogenesis were generally down regulated across time, while genes involved in synaptogenesis and neural activity were up regulated. Genes previously associated with dendritogenesis did not seem to have a signature trajectory (Figure 7A). This might be due to the multiple roles that these molecules have in development. Interestingly, the expression of the majority of genes associated with experience-driven activity (i.e., immediate early genes - IEG) increased dramatically after P8 first in L4, followed closely in IgL, and more robustly after P10 in SgL (Figure 7A, grey shade). This increment in expression of IEGs coincides with the emergence of exploratory whisking around P11-P14 (Takatoh et al., 2013). The fact that not all layers respond at the same time to sensorial input is reflective of the sequential maturation of the neocortical layers in an inside first – outside last fashion. Indeed, the same trend was consistently observed with the other neurodevelopmental processes.
Figure 7. Transcriptional Correlates of Neurodevelopmental Events.

(A) Developmental trajectories of genes associated with select major neurodevelopmental processes in different laminar compartment (SgL, blue; L4, green; IgL, red).
(B) Schematic of changes in dendritic morphology of L4 spiny stellate cells (SSCs; dark green) and the formation of L4 barrels (light green)
(C) Expression trajectories of genes correlated with developmental changes in dendritic morphology of L4 (green) SSCs.
We also used our dataset to identify genes that may be involved in developmental sculpting of dendritic morphology of L4 spiny stellate cells (SSCs) in S1C. These L4 excitatory neurons aggregate to form barrel-like structures that surround clusters of thalamocortical afferents (TCAs) relaying information from individual facial whiskers (Li and Crair, 2011; Li et al., 2013). After arriving to the cortex, two morphological changes occur to immature L4 excitatory neurons: they cease to have a distinct apical dendrite, growing dendrites of roughly equal size; and they direct their dendrites towards one specific TCA bundle (Figure 7B). These changes seem to be concluded by P5 and for which thalamic activity may play an important role (Callaway and Borrell, 2011; Li et al., 2013). However, the molecular mechanisms underlying these changes are unknown.
We hypothesized that genes involved in SSC dendritic development should be enriched in L4 between P4–P6, after which their expression may decrease. Expression profiles of four genes matched this expression pattern – Fat3, Lhfp, Ptgfrn, and Tgfbr1 (Figure 7C). Interestingly, all four genes encode transmembrane proteins involved in signal transduction that lead to a plethora of physiological (e.g., cell differentiation) and pathological (e.g., schizophrenia) processes. Analysis of Fat3 expression in the Allen Brain Atlas (http://developingmouse.brain-map.org; Lein et al., 2007) confirms its enrichment in S1C at P4 but not afterwards. Additionally, the atypical cadherin FAT3 has recently been shown to control the dendritic morphology of retinal neurons (Deans et al., 2011). Thus, Fat3 is a plausible candidate for regulating SSC dendritic development.
DISCUSSION
Here we profiled the neocortical developmental transcriptome by deep sequencing both mRNAs and smRNAs in the S1C, across different layers and multiple developmental time points. All generated data are publicly available as Mouse NCX Transcriptome database (http://medicine.yale.edu/lab/sestan/resources) providing the basis for a variety of future investigations and comparisons with other transcriptome related datasets.
Our analysis revealed that time, more than space (layers) or sex, defines the dynamics of coding and non-coding transcripts in the mouse S1C. In particular, P4 neocortical transcriptome was substantially different from the one at P6, P8 and P10, which were similar to each other; and P14 was more similar to adult than to P6 – P10 or P4. This segregation likely reflects the progression of major neurodevelopmental processes. Neuronal migration of the last neurons to the SgL is believed to cease around P3–P4 (Kwan et al., 2012). Interestingly, Mdga1, which has been specifically implicated in the migration of SgL neurons (Takeuchi and O’Leary, 2006), shows maximal expression at P4 in the SgL and L4, with subsequent decline (Figure 1D). After P4 there is a period of approximately one and a half weeks of intensive elaboration of axons and dendrites, development of the vascular system; and overproduction of synapses and spines (Ashby and Isaac, 2011; Yu et al., 1994 and Figure 7A). Around P14 mice open their eyes and begin to exhibit exploratory behavior and coordinated movement of their whiskers, more indicative of independent adult behaviors.
In addition to general temporal transcriptional change, there is also a spatial gradient in maturation among layers due to the inside first-outside last nature of laminar differentiation. Accordingly, at the level of protein-coding genes our samples also segregated according to their laminar identities (i.e., IgL, L4 and SgL). Gene function enrichment analysis of the sDEX genes as well as the analysis of the signature trajectory of neocortical developmental events reflected the inside-out gradient of laminar maturation (Table S3D). Genes enriched in the IgL (inside) were associated with the development of neuronal morphology as these layers are the first to go through the elaboration of dendritic trees and long reaching axons, while those enriched in L4 were genes associated with signal transduction (e.g., G protein-coupled receptors), synapse and channel activity, consistent with the specific function of this layer as main recipient of thalamic afferents and its dependence on thalamocortical neurotransmission (Li et al. 2013). On the other hand, the genes enriched in SgL (outside) were those associated with cell adhesion, suggestive of the ongoing processes of cell migration and blood vessel development, and there was no enrichment in genes associated with neuronal development, a hint at its less mature state.
Both WCGNA and hypothesis-driven analysis reinforced their value in identifying genes and networks associated with distinct biological processes. We were able to uncover transcriptional overlaps between known phenotypic interactions (e.g., blood vessel development and astrogliogenesis) and provide new mechanistic insights into related biological processes (e.g., neuronal activity and gene expression). Furthermore, we also identified gene candidates for regulating neurodevelopmental processes specific to a layer or area, such as the developmental sculpting of dendritic morphology of L4 SSCs. Finally, we also demonstrated that one can embrace the cellular and molecular complexity of distinct layers for integrated data analysis by combining our data with available data on specific neural cell types.
In addition, this dataset is also helpful in analyzing the laminar and temporal expressions of genes linked to psychiatric and neurological disorders. Our initial analysis revealed that some genes linked to schizophrenia (i.e., Zfp804a) or autism (i.e., Fezf2, Sema5a, Sox5, and Tbr1) are enriched in IgL during development (Figure 1D). Together with similar findings from a recent study on the expression of autism related genes in the human NCX (Willsey et al., 2013), the development of IgL projection neurons and their circuits might be affected in these disorders.
The mRNA-seq dataset further allows the study of the expression profile of new gene isoforms (e.g., Dlg2), which should prove especially valuable for the study of gene expression and alternative splicing across development and species (Keren et al., 2010). Additionally, the smRNA-seq dataset enables the identification of distinct spatiotemporal profiles of miRNAs and potential miRNA-mRNA interactions (e.g., miR-92b and Foxp2). The data presented here and other neural cell-type-specific miRNA datasets (see Jovicic et al., 2013) should provide valuable resources for interpreting these putative interactions.
EXPERIMENTAL PROCEDURES
Laminar Microdissection
Dcdc2a-Gfp reporter mice were acutely sacrificed and the brain was submerged in ice-cold oxygenated artificial cerebrospinal fluid (ACSF) for 5 min. Using a vibratome we prepared live sagittal brain slices (250 μm), kept in ice-cold and oxygenated ACSF at all times. Layers were dissected under a fluorescence stereoscope and collected into separate safe-lock microcentrifuge tubes with 30 μL of RNAlater®.
RNA Extraction and Library Preparation
Tissue homogenization was performed by adding stainless steel beads (Next Advance) and 2 volumes of lysis buffer to the tube with tissue and homogenized in the Bullet Blender (Next Advance), 1 min, and speed 6. Total RNA was extracted using RNeasy Plus Mini Kit (Qiagen). cDNA libraries were prepared using the TruSeq mRNA and TruSeq SmallSample Prep Kits (Illumina), as per the manufacturer’s instructions with some modifications (see Supplemental information).
Read Filtering, Processing and Alignment
The mRNA-seq reads were aligned to mouse reference genome (NCBI37/mm9) using Tophat (v1.0.13) (Trapnell et al., 2009) with up to 2 mismatches. The uniquely mapped reads were used to calculate the expression level of genes annotated in Ensembl (NCBI37/mm9, released version 63) using RSEQTools (Habegger et al., 2011). The smRNA-seq reads were clipped and aligned to miRNA and pre-miRNA retrieved from miRBase (released version 18) using miRanalyzer (released version 0.2) with up to 1 mismatch (Hackenberg et al., 2011; Kozomara and Griffiths-Jones, 2011). The uniquely aligned to either library were taken to calculate the reads count for miRNA. Reads aligned to pre-miRNA but not miRNA were attributed to divergence from the consensus sequence, and then were assigned to miRNA based on its aligned locus. See supplemental information for more details.
Data Quality Assessment of mRNA-seq and smRNA-seq
The spike-in RNAs were added to the libraries to tag samples. The percentage of mismatches within sequenced spike-in RNA reads was considered sequencing errors. The sequencing quality score of smRNA-seq reads was evaluated by FastQC. For each chromosome, the genome transcription ratio and exon transcribed ratio were quantified respectively, so as to overview the mRNA-seq sequencing coverage. To investigate the relationship between samples, we used R package prcomp to perform PCA for mRNA-seq and smRNA-seq samples. In addition, we used WGCNA to perform average-linkage hierarchical clustering for mRNA-seq samples. See supplemental information for more details.
Spatiotemporal DEX Analysis
We used R package DESeq to identify DEX genes and miRNAs between different layers and between different ages (Anders and Huber, 2010). FDR < 0.01, was chosen to detect statistically significant DEX transcripts. See supplemental information for more details.
Alternative Splicing and Intron Retention
We used JuncBASE to identify alternative splicing (AS) events (Brooks et al., 2011). We performed pairwise comparisons between different layers and between different ages. For the AS events expressed in both variables we used a threshold of FDR < 0.01. For AS events expressed only in one variable we used the supported reads count > 25 as the threshold. We used RSEQtools to build intron annotations and calculated the RPKM and reads count (Habegger et al., 2011). The combination of RPKM ≥ 0.5 and raw reads count ≥ 10 were used as the threshold. We used RPKM fold change > 2 to detect spatial and temporal different intron retention events. See supplemental information for more details.
qRT-PCR and Exon-Specific PCR
One μg of total RNA of each sample was used for cDNA synthesis using SuperScript III First-strand synthesis Supermix (Invitrogen). TaqMan Gene Expression Assay was used for each gene of interest along with TaqMan Universal Master Mix (Applied Biosystems). Exon-specific high-melting temperature primers were designed using NCBI/Primer-BLAST (http://www.ncbi.nlm.nih.gov/tools/primer-blast/) (see Supplemental information). PCR was performed using Phusion High-Fidelity DNA Polymerase (NEB), as indicated by manufacture.
Weighted Gene Co-Expression Network Analysis
We used R package WGCNA to perform weighted gene co-expression network analysis (Langfelder and Horvath, 2008). In each module, the top 10 genes highly correlated with modular eigengene were selected as modular hub genes to build and visualize inter- and intra-module connectivity using Cytoscape (Smoot et al., 2011). See supplemental information for more details.
miRNA-mRNA Regulation Prediction
We retrieved conserved miRNA-mRNA regulation pairs from TargetScan database (released version 6.2). We then used lasso_mir.R to predict the regulation between mRNA and miRNA expression levels (Lu et al., 2011) with rank score ≥ 80. Finally, we used TargetScan database and Lasso prediction to calculate the proportion of targeted mRNAs. We used FDR < 0.01 to detect reliable cluster-enriched miRNAs. See supplemental information for more details.
Supplementary Material
Highlights.
RNA-seq analysis of layers in the developing mouse neocortex
Identification of coding and noncoding RNAs and putative miRNA-mRNA interactions
Identification of spatiotemporal expression patterns and transcript variants
Prediction of transcriptional overlap between distinct biological processes
Acknowledgments
We thank A. Sousa and M. Pletikos for helping with human tissue processing and data analyses, and the members of the Sestan laboratory for valuable comments. Human postmortem tissue was obtained from sources listed in Supplemental Information. This work was supported by grants from the NIH (MH081896, MH062639, NS051869), the Kavli Foundation (M.C. and N.S.), and by a James S. McDonnell Foundation Scholar Award (N.S.).
Footnotes
ACCESSION NUMBERS
The raw sequencing reads have been deposited in NCBI Sequence Read Archive (SRA) database under the accession number SRP031888.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11:R106. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arlotta P, Molyneaux BJ, Chen J, Inoue J, Kominami R, Macklis JD. Neuronal subtype-specific genes that control corticospinal motor neuron development in vivo. Neuron. 2005;45:207–221. doi: 10.1016/j.neuron.2004.12.036. [DOI] [PubMed] [Google Scholar]
- Ashby MC, Isaac JT. Maturation of a recurrent excitatory neocortical circuit by experience-dependent unsilencing of newly formed dendritic spines. Neuron. 2011;70:510–521. doi: 10.1016/j.neuron.2011.02.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barabasi AL, Oltvai ZN. Network biology: understanding the cell’s functional organization. Nat Rev Genet. 2004;5:101–113. doi: 10.1038/nrg1272. [DOI] [PubMed] [Google Scholar]
- Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belgard TG, Marques AC, Oliver PL, Abaan HO, Sirey TM, Hoerder-Suabedissen A, Garcia-Moreno F, Molnar Z, Margulies EH, Ponting CP. A transcriptomic atlas of mouse neocortical layers. Neuron. 2011;71:605–616. doi: 10.1016/j.neuron.2011.06.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenman JE, Topinka JR, Cooper EC, McGee AW, Rosen J, Milroy T, Ralston HJ, Bredt DS. Localization of postsynaptic density-93 to dendritic microtubules and interaction with microtubule-associated protein 1A. J Neurosci. 1998;18:8805–8813. doi: 10.1523/JNEUROSCI.18-21-08805.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks AN, Yang L, Duff MO, Hansen KD, Park JW, Dudoit S, Brenner SE, Graveley BR. Conservation of an RNA regulatory map between Drosophila and mammals. Genome Res. 2011;21:193–202. doi: 10.1101/gr.108662.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahoy JD, Emery B, Kaushal A, Foo LC, Zamanian JL, Christopherson KS, Xing Y, Lubischer JL, Krieg PA, Krupenko SA, et al. A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J Neurosci. 2008;28:264–278. doi: 10.1523/JNEUROSCI.4178-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callaway EM, Borrell V. Developmental sculpting of dendritic morphology of layer 4 neurons in visual cortex: influence of retinal input. J Neurosci. 2011;31:7456–7470. doi: 10.1523/JNEUROSCI.5222-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JG, Rasin MR, Kwan KY, Sestan N. Zfp312 is required for subcortical axonal projections and dendritic morphology of deep-layer pyramidal neurons of the cerebral cortex. Proc Natl Acad Sci U S A. 2005;102:17792–17797. doi: 10.1073/pnas.0509032102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung AS, Ferrara N. Developmental and pathological angiogenesis. Annu Rev Cell Dev Biol. 2011;27:563–584. doi: 10.1146/annurev-cellbio-092910-154002. [DOI] [PubMed] [Google Scholar]
- DeFelipe J, Lopez-Cruz PL, Benavides-Piccione R, Bielza C, Larranaga P, Anderson S, Burkhalter A, Cauli B, Fairen A, Feldmeyer D, et al. New insights into the classification and nomenclature of cortical GABAergic interneurons. Nat Rev Neurosci. 2013;14:202–216. doi: 10.1038/nrn3444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deans MR, Krol A, Abraira VE, Copley CO, Tucker AF, Goodrich LV. Control of neuronal morphology by the atypical cadherin Fat3. Neuron. 2011;71:820–832. doi: 10.1016/j.neuron.2011.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillman AA, Hauser DN, Gibbs JR, Nalls MA, McCoy MK, Rudenko IN, Galter D, Cookson MR. mRNA expression, splicing and editing in the embryonic and adult mouse cerebral cortex. Nat Neurosci. 2013;16:499–506. doi: 10.1038/nn.3332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espinosa JS, Stryker MP. Development and plasticity of the primary visual cortex. Neuron. 2012;75:230–249. doi: 10.1016/j.neuron.2012.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonta C, Imbert M. Vascularization in the primate visual cortex during development. Cereb Cortex. 2002;12:199–211. doi: 10.1093/cercor/12.2.199. [DOI] [PubMed] [Google Scholar]
- Goldberg JL, Vargas ME, Wang JT, Mandemakers W, Oster SF, Sretavan DW, Barres BA. An oligodendrocyte lineage-specific semaphorin, Sema5A, inhibits axon growth by retinal ganglion cells. J Neurosci. 2004;24:4989–4999. doi: 10.1523/JNEUROSCI.4390-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimson A, Farh KK, Johnston WK, Garrett-Engele P, Lim LP, Bartel DP. MicroRNA targeting specificity in mammals: determinants beyond seed pairing. Mol Cell. 2007;27:91–105. doi: 10.1016/j.molcel.2007.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habegger L, Sboner A, Gianoulis TA, Rozowsky J, Agarwal A, Snyder M, Gerstein M. RSEQtools: a modular framework to analyze RNA-Seq data using compact, anonymized data summaries. Bioinformatics. 2011;27:281–283. doi: 10.1093/bioinformatics/btq643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackenberg M, Rodriguez-Ezpeleta N, Aransay AM. miRanalyzer: an update on the detection and analysis of microRNAs in high-throughput sequencing experiments. Nucleic Acids Res. 2011;39:W132–138. doi: 10.1093/nar/gkr247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han W, Kwan KY, Shim S, Lam MM, Shin Y, Xu X, Zhu Y, Li M, Sestan N. TBR1 directly represses Fezf2 to control the laminar origin and development of the corticospinal tract. Proc Natl Acad Sci U S A. 2011;108:3041–3046. doi: 10.1073/pnas.1016723108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heintz N. Gene expression nervous system atlas (GENSAT) Nat Neurosci. 2004;7:483. doi: 10.1038/nn0504-483. [DOI] [PubMed] [Google Scholar]
- Hengst U, Jaffrey SR. Function and translational regulation of mRNA in developing axons. Semin Cell Dev Biol. 2007;18:209–215. doi: 10.1016/j.semcdb.2007.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hough CD, Woods DF, Park S, Bryant PJ. Organizing a functional junctional complex requires specific domains of the Drosophila MAGUK Discs large. Genes Dev. 1997;11:3242–3253. doi: 10.1101/gad.11.23.3242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Johnson MB, Kawasawa YI, Mason CE, Krsnik Z, Coppola G, Bogdanovic D, Geschwind DH, Mane SM, State MW, Sestan N. Functional and evolutionary insights into human brain development through global transcriptome analysis. Neuron. 2009;62:494–509. doi: 10.1016/j.neuron.2009.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jovicic A, Roshan R, Moisoi N, Pradervand S, Moser R, Pillai B, Luthi-Carter R. Comprehensive expression analyses of neural cell-type-specific miRNAs identify new determinants of the specification and maintenance of neuronal phenotypes. J Neurosci. 2013;33:5127–5137. doi: 10.1523/JNEUROSCI.0600-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang HJ, Kawasawa YI, Cheng F, Zhu Y, Xu X, Li M, Sousa AM, Pletikos M, Meyer KA, Sedmak G, et al. Spatio-temporal transcriptome of the human brain. Nature. 2011;478:483–489. doi: 10.1038/nature10523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keren H, Lev-Maor G, Ast G. Alternative splicing and evolution: diversification, exon definition and function. Nat Rev Genet. 2010;11:345–355. doi: 10.1038/nrg2776. [DOI] [PubMed] [Google Scholar]
- Kilb W, Kirischuk S, Luhmann HJ. Electrical activity patterns and the functional maturation of the neocortex. Eur J Neurosci. 2011;34:1677–1686. doi: 10.1111/j.1460-9568.2011.07878.x. [DOI] [PubMed] [Google Scholar]
- Kozomara A, Griffiths-Jones S. miRBase: integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res. 2011;39:D152–157. doi: 10.1093/nar/gkq1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwan KY, Sestan N, Anton ES. Transcriptional co-regulation of neuronal migration and laminar identity in the neocortex. Development. 2012;139:1535–1546. doi: 10.1242/dev.069963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lein ES, Hawrylycz MJ, Ao N, Ayres M, Bensinger A, Bernard A, Boe AF, Boguski MS, Brockway KS, Byrnes EJ, et al. Genome-wide atlas of gene expression in the adult mouse brain. Nature. 2007;445:168–176. doi: 10.1038/nature05453. [DOI] [PubMed] [Google Scholar]
- Leone DP, Srinivasan K, Chen B, Alcamo E, McConnell SK. The determination of projection neuron identity in the developing cerebral cortex. Curr Opin Neurobiol. 2008;18:28–35. doi: 10.1016/j.conb.2008.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Crair MC. How do barrels form in somatosensory cortex? Ann N Y Acad Sci. 2011;1225:119–129. doi: 10.1111/j.1749-6632.2011.06024.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Fertuzinhos S, Mohns E, Hnasko T, Verhage M, Edwards R, Sestan N, Crair MC. Laminar and columnar development of barrel cortex relies on thalamocortical neurotransmission. Neuron. 2013;79:970–986. doi: 10.1016/j.neuron.2013.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Zhou Y, Qu W, Deng M, Zhang C. A Lasso regression model for the construction of microRNA-target regulatory networks. Bioinformatics. 2011;27:2406–2413. doi: 10.1093/bioinformatics/btr410. [DOI] [PubMed] [Google Scholar]
- Lyckman AW, Horng S, Leamey CA, Tropea D, Watakabe A, Van Wart A, McCurry C, Yamamori T, Sur M. Gene expression patterns in visual cortex during the critical period: synaptic stabilization and reversal by visual deprivation. Proc Natl Acad Sci U S A. 2008;105:9409–9414. doi: 10.1073/pnas.0710172105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuoka RL, Chivatakarn O, Badea TC, Samuels IS, Cahill H, Katayama K, Kumar SR, Suto F, Chedotal A, Peachey NS, et al. Class 5 transmembrane semaphorins control selective mammalian retinal lamination and function. Neuron. 2011;71:460–473. doi: 10.1016/j.neuron.2011.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNeill E, Van Vactor D. MicroRNAs shape the neuronal landscape. Neuron. 2012;75:363–379. doi: 10.1016/j.neuron.2012.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercer TR, Mattick JS. Structure and function of long noncoding RNAs in epigenetic regulation. Nat Struct Mol Biol. 2013;20:300–307. doi: 10.1038/nsmb.2480. [DOI] [PubMed] [Google Scholar]
- Molyneaux BJ, Arlotta P, Menezes JRL, Macklis JD. Neuronal subtype specification in the cerebral cortex. Nat Rev Neurosci. 2007;8:427–437. doi: 10.1038/nrn2151. [DOI] [PubMed] [Google Scholar]
- Nilsen TW, Graveley BR. Expansion of the eukaryotic proteome by alternative splicing. Nature. 2010;463:457–463. doi: 10.1038/nature08909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okaty BW, Sugino K, Nelson SB. A quantitative comparison of cell-type-specific microarray gene expression profiling methods in the mouse brain. PLoS One. 2011;6:e16493. doi: 10.1371/journal.pone.0016493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldham MC, Konopka G, Iwamoto K, Langfelder P, Kato T, Horvath S, Geschwind DH. Functional organization of the transcriptome in human brain. Nat Neurosci. 2008;11:1271–1282. doi: 10.1038/nn.2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paarmann I, Spangenberg O, Lavie A, Konrad M. Formation of complexes between Ca2+. calmodulin and the synapse-associated protein SAP97 requires the SH3 domain-guanylate kinase domain-connecting HOOK region. J Biol Chem. 2002;277:40832–40838. doi: 10.1074/jbc.M205618200. [DOI] [PubMed] [Google Scholar]
- Pletikos M, Sousa AMM, Sedmak G, Meyer KA, Zhu Y, Cheng F, Li M, Kawasawa YI, Sestan N. Temporal specification and bilaterality of human neocortical topographic gene expression. Neuron. 2014 doi: 10.1016/j.neuron.2013.11.018. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossner MJ, Hirrlinger J, Wichert SP, Boehm C, Newrzella D, Hiemisch H, Eisenhardt G, Stuenkel C, von Ahsen O, Nave KA. Global transcriptome analysis of genetically identified neurons in the adult cortex. J Neurosci. 2006;26:9956–9966. doi: 10.1523/JNEUROSCI.0468-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowitch DH, Kriegstein AR. Developmental genetics of vertebrate glial-cell specification. Nature. 2010;468:214–222. doi: 10.1038/nature09611. [DOI] [PubMed] [Google Scholar]
- Shulha HP, Cheung I, Whittle C, Wang J, Virgil D, Lin CL, Guo Y, Lessard A, Akbarian S, Weng Z. Epigenetic signatures of autism: trimethylated H3K4 landscapes in prefrontal neurons. Arch Gen Psychiatry. 2012;69:314–324. doi: 10.1001/archgenpsychiatry.2011.151. [DOI] [PubMed] [Google Scholar]
- Smoot ME, Ono K, Ruscheinski J, Wang PL, Ideker T. Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics. 2011;27:431–432. doi: 10.1093/bioinformatics/btq675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugino K, Hempel CM, Miller MN, Hattox AM, Shapiro P, Wu C, Huang ZJ, Nelson SB. Molecular taxonomy of major neuronal classes in the adult mouse forebrain. Nat Neurosci. 2006;9:99–107. doi: 10.1038/nn1618. [DOI] [PubMed] [Google Scholar]
- Takeuchi A, O’Leary DD. Radial migration of superficial layer cortical neurons controlled by novel Ig cell adhesion molecule MDGA1. J Neurosci. 2006;26:4460–4464. doi: 10.1523/JNEUROSCI.4935-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam SJ, Watts RJ. Connecting vascular and nervous system development: angiogenesis and the blood-brain barrier. Annu Rev Neurosci. 2010;33:379–408. doi: 10.1146/annurev-neuro-060909-152829. [DOI] [PubMed] [Google Scholar]
- Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takatoh J, Nelson A, Zhou X, Bolton MM, Ehlers MD, Arenkiel BR, Mooney R, Wang F. New modules are added to vibrissal premotor circuitry with the emergence of exploratory whisking. Neuron. 2013;77:346–360. doi: 10.1016/j.neuron.2012.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss LA, Arking DE, Daly MJ, Chakravarti A Gene Discovery Project of Johns Hopkins & the Autism Consortium. A genome-wide linkage and association scan reveals novel loci for autism. Nature. 2009;461:802–808. doi: 10.1038/nature08490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AE, Greenberg ME. Neuronal activity-regulated gene transcription in synapse development and cognitive function. Cold Spring Harb Perspect Biol. 2011;3:a005744. doi: 10.1101/cshperspect.a005744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willsey JA, Sanders SJ, Li M, Dong S, Tebbenkamp AT, Muhle RA, Reilly SK, Lin L, Fertuzinhos S, Miller JA, et al. Co-expression networks implicate human mid-fetal deep cortical projection neurons in the pathogenesis of autism. Cell. 2013;155:997–1007. doi: 10.1016/j.cell.2013.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu BP, Yu CC, Robertson RT. Patterns of capillaries in developing cerebral and cerebellar cortices of rats. Acta Anat. 1994;149:128–133. doi: 10.1159/000147567. [DOI] [PubMed] [Google Scholar]
- Zheng S, Gray EE, Chawla G, Porse BT, O’Dell TJ, Black DL. PSD-95 is post-transcriptionally repressed during early neural development by PTBP1 and PTBP2. Nat Neurosci. 2012;15:381–388. doi: 10.1038/nn.3026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerlin M, Goldman JE. Interactions between glial progenitors and blood vessels during early postnatal corticogenesis: blood vessel contact represents an early stage of astrocyte differentiation. J Comp Neurol. 1997;387:537–546. doi: 10.1002/(sici)1096-9861(19971103)387:4<537::aid-cne5>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.