

Abstract
DNA methylation and histone acetylation contribute to the transcriptional regulation of genes involved in apoptosis. We have demonstrated that docosahexaenoic acid (DHA, 22:6 n-3) and butyrate enhance colonocyte apoptosis. To determine if DHA and/or butyrate elevate apoptosis through epigenetic mechanisms thereby restoring the transcription of apoptosis-related genes, we examined global methylation; gene-specific promoter methylation of 24 apoptosis-related genes; transcription levels of Cideb, Dapk1, and Tnfrsf25; and global histone acetylation in the HCT-116 colon cancer cell line. Cells were treated with combinations of (50 μM) DHA or linoleic acid (18:2 n-6), (5 mM) butyrate or an inhibitor of DNA methyltransferases, and 5-aza-2′-deoxycytidine (5-Aza-dC, 2 μM). Among highly methylated genes, the combination of DHA and butyrate significantly reduced methylation of the proapoptotic Bcl2l11, Cideb, Dapk1, Ltbr, and Tnfrsf25 genes compared to untreated control cells. DHA treatment reduced the methylation of Cideb, Dapk1, and Tnfrsf25. These data suggest that the induction of apoptosis by DHA and butyrate is mediated, in part, through changes in the methylation state of apoptosis-related genes.
Keywords: docosahexaenoic acid, butyrate, apoptosis, DNA methylation, epigenetics
Introduction
During carcinogenesis, gene transcription is aberrantly regulated via epigenetic changes. These changes include DNA methylation and histone modification and occur primarily in the promoter region of select genes.1,2 Clinical studies have demonstrated a correlation between DNA methylation and the pathological and epidemiological features of colon cancer.3–5 Recent data indicate that promoter methylation of proapoptotic genes suppresses transcription, resulting in the survival of cancer cells.6 For instance, Dapk1 is a positive mediator of apoptosis, a tumor suppressor candidate, and is known to be heavily methylated in colon and bladder cancer.7,8 Restoring transcription of tumor suppressor genes by the reversal of these epigenetic processes is considered a latent target of cancer prevention and treatment.
We have previously shown that a combination of fish oil (high in docosahexaenoic acid [DHA], an n-3 polyunsaturated fatty acid [PUFA]) and the fiber pectin (fermented to short-chain fatty acids including butyrate by the colonic microflora) is protective against colon carcinogenesis in part by up-regulating the apoptotic removal of DNA damaged cells.9 In contrast, diets highly enriched in corn oil (enriched in linoleic acid [LA], an n-6 PUFA) suppress apoptosis and promote colon cancer.10 We have demonstrated that apoptosis induction by the combination of fish oil and pectin is in part contingent upon DHA incorporation into mitochondrial phospholipids,11 while butyrate functions as a energy substrate and histone deacetylase inhibitor.12 Another potential mechanism involved in tumor suppression by fish oil/pectin is through the transcriptional regulation of key tumor suppressors or oncogenes. In a previous study, we demonstrated the effect of fish oil/pectin on global changes in gene expression profiles in carcinogen-injected Sprague–Dawley rats. One result from the pathway analyses of those data was the observation that the expression of genes that promote apoptosis was up-regulated in rats consuming the fish oil/pectin diet at the tumor endpoint.13 Moreover, we have reported that cells isolated from carcinogen-induced colon tumors contain DNA that is highly methylated in the promoter region of Bcl2, an antiapoptotic mediator, which was associated with induction of apoptosis in colonocytes from rats consuming a fish oil/pectin diet.14
However, it has not been determined if DHA and/or butyrate can directly affect the aberrant promoter methylation of cancer-promoting genes. Therefore, the aim of this study was to investigate the epigenetic regulation of apoptosis-related genes in a colon cancer cell line exposed to DHA and/or butyrate. For this purpose, we examined global DNA methylation and histone acetylation in combination with gene-specific promoter methylation of 24 apoptosis-related genes, in HCT-116 human colon cancer cells. Bender et al. demonstrated that 5-Aza-dC regulated p16 DNA methylation and gene expression in seven cancer cell lines including HCT-116 cells.15 The study by Schneider-Stock et al. showed that 5-aza-cytidine, the analog of 5-Aza-dC, induced demethylation and significant down-regulation of DNMT1 and DNMT3a gene transcription in HCT-116 cells.16 Therefore, we chose to use the well-characterized HCT-116 cell line to test the effects of DHA/butyrate on DNA promoter methylation, using 5-Aza-dC as a positive control.
Materials and methods
Cell culture
HCT-116 cells were cultured in McCoy’s 5 A media (Gibco) supplemented with 10% fetal bovine serum (Hyclone) and 2 mM GlutaMAX (Gibco) at 37°C in 5% CO2. Cells (passages 11–13) were seeded onto 100-mm cell culture dishes or six-well plates at a density of 3.0 × 104 cells and allowed to attach for 24 h.
Experiment I
Fifty μM bovine serum albumin (BSA)-complexed DHA or LA (Nu Chek Prep. Inc.) was used to treat cells for 72 h.17 Cells were co-incubated with 5 mM sodium butyrate (Sigma) during the final 12 h of the fatty acid treatment period.18 5-Aza-dC (2 μM), a potent inhibitor of DNA methylation (Santa Cruz Biotechnology) served as a positive control, and cells were treated for 48 h. Negative control cells were incubated in media without BSA-complexed fatty acids or butyrate.
Experiment II
The goal of this experiment was to develop a more thorough time course for gene expression relative to treatment with 5-Aza-dC, fatty acids, and/or butyrate. For this purpose, all cells received the assigned treatments at time 0, and gene expression was monitored in cells harvested at 48, 72, and 96 h post-treatment initiation. The treatments included 50 μM BSA–DHA or BSA–LA, 5 mM butyrate, or 2 μM 5-Aza-dC (Figure 1).
Figure 1.

Design of cell culture experiments
Global DNA methylation assay
Genomic DNA was isolated from HCT-116 cells using QIAamp DNA mini kit (Qiagen). Global methylation was quantified using a MethylFlash methylated DNA quantification kit (Epigentek). Briefly, 100 ng of genomic DNA was pipetted into multiwell plates coated with a 5-methylcytosine antibody and incubated at 37°C for 90 min. DNA was subsequently washed and a capture antibody added prior to incubation at room temperature for 30 min. At the end of the incubation period, the capture antibody solution was removed and replaced with an enhancer solution followed by another 30-min incubation at room temperature. The extent of global methylation was measured by reading absorbance at 450 nm. The relative methylation status of each sample was determined using positive and negative controls provided in the kit.
Gene specific DNA methylation
DNA methylation of apoptosis-related genes was determined using a Methyl-Profiler DNA methylation PCR array (SABiosciences, MeAH-121 C-2). Genes included on the array are listed in Table 1. Briefly, 1 μM of genomic DNA from HCT-116 cells was treated with a mock enzyme, a DNA methylation sensitive enzyme, a DNA methylation-dependent enzyme, or both the DNA methylation sensitive and dependent enzymes at 37°C overnight. The relative DNA methylation status was determined using Ct values from quantitative real time PCR (qRT-PCR) as previously described.19 Data are presented as a percentage of cellular DNA containing methylated gene promoters, which included a combination of both the intermediate level of methylation and hypermethylated DNA.
Table 1.
Gene specific DNA methylation targets included in the PCR array
| RefSeq accession number | Symbol | Description |
|---|---|---|
| NM_009684 | Apaf1 | Apoptotic peptidase activating factor 1 |
| NM_007522 | Bad | BCL2-associated agonist of cell death |
| NM_009736 | Bag1 | Bcl2-associated athanogene 1 |
| NM_007527 | Bax | Bcl2-associated X protein |
| NM_009741 | Bcl2 | B-cell leukemia/lymphoma 2 |
| NM_009754 | Bcl2l11 | BCL2-like 11 (apoptosis facilitator) |
| NM_153787 | Bclaf1 | BCL2-associated transcription factor 1 |
| NM_007544 | Bid | BH3 interacting domain death agonist |
| NM_007546 | Bik | Bcl2-interacting killer |
| NM_007465 | Birc2 | Baculoviral IAP repeat-containing 2 |
| NM_009761 | Bnip3l | BCL2/adenovirus E1B interacting protein 3-like |
| NM_009810 | Casp3 | Caspase 3 |
| NM_015733 | Casp9 | Caspase 9 |
| NM_009894 | Cideb | Cell death-inducing DNA fragmentation factor, alpha subunit-like effector B |
| NM_009950 | Cradd | CASP2 and RIPK1 domain containing adaptor with death domain |
| NM_029653 | Dapk1 | Death associated protein kinase 1 |
| NM_010044 | Dffa | DNA fragmentation factor, alpha subunit |
| NM_010175 | Fadd | Fas (TNFRSF6)-associated via death domain |
| NM_007836 | Gadd45a | Growth arrest and DNA-damage-inducible 45 alpha |
| NM_007545 | Hrk | Harakiri, BCL2 interacting protein (contains only BH3 domain) |
| NM_010736 | Ltbr | Lymphotoxin B receptor |
| NM_178589 | Tnfrsf21 | Tumor necrosis factor receptor superfamily, member 21 |
| NM_033042 | Tnfrsf25 | Tumor necrosis factor receptor superfamily, member 25 |
| NM_011640 | Trp53 | Transformation related protein 53 |
Gene expression using qRT-PCR
Total RNA was extracted from HCT-116 cells using an RNAqueous Kit (Ambion) and DNase treated. The cDNA was synthesized from 2 μg total RNA using random hexamers and oligo dT primers with SuperScript II reverse transcriptase (Invitrogen). Transcript levels were determined using Taqman gene expression assays (Applied Biosystems; Cideb Hs00205339_m1, Dapk1 Hs00234489_m1, and Tnfrsf25 Hs00980365_g1) with an ABI Prism 7900HT PCR sequence detector. Expression levels were normalized to Eukaryotic 18S rRNA expression (Hs99999901_s1). Linearity of each assay was assessed prior to analysis of the samples. Negative controls that were prepared during the reverse transcription reactions by eliminating the RT enzyme were analyzed for each gene.
Histone H3 and H4 acetylation
The acetylation levels of histone H3 and H4 were assessed using the EpiQuik total histone H3/H4 acetylation detection fast kit (Epigentek). Histone proteins were extracted as per kit instructions and the sample concentrations were measured using a BCA Protein Assay Kit (Pierce). Separate assays were performed to determine acetylation status for the H3 and H4 histones. These assays used 2 μg of nuclear proteins incubated with antiacetylated histone H3-specific or H4-specific antibodies. In each assay a detection antibody was added along with a color development reagent, and the absorbance was measured at 450 nm.
Statistical analysis
Data were analyzed using one-way ANOVA and Duncan’s multiple range tests for global methylation, transcription level of Dapk1 and Tnfrsf2, and global H3 or H4 histone acetylation. To determine if gene-specific methylation was different between each treatment group and the negative control group, the statistical analysis was carried out using a Wilcoxon rank test. Data are presented as means ± SEM, and means were considered different when the resulting P value was less than 0.05.
Results
Global methylation
In Experiment I, we determined the effects of the PUFA and/or butyrate treatments on the level of global DNA methylation in HCT-116 cells exposed to the PUFA for 72 h and to butyrate for 12 h prior to sample collection. Both DHA and butyrate independently decreased global methylation whereas the combination of DHA and butyrate did not produce a significant demethylating effect compared to the negative control (media only). In comparison, the positive control, 5-Aza-dC (2 μM), significantly suppressed global methylation, as expected (Figure 2).
Figure 2.
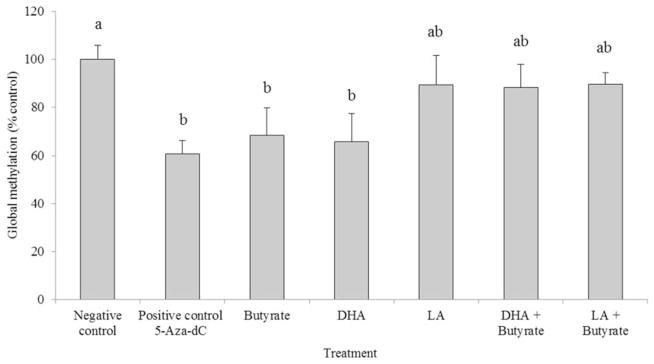
Global DNA methylation status (percent of the negative control) in HCT-116 cells cultured with 5-Aza-dC (48 h), butyrate (12 h), DHA (72 h), LA (72 h), or co-treatment (see “Experiment I” section for details of Experiment I). Values are means ± SEM. Values not sharing common letters are significantly different, P <0.05, n = 6 per treatment. Negative controls represent cells incubated in media only. DHA: docosahexaenoic acid; LA: linoleic acid
Gene-specific promoter methylation of apoptosis-related genes
We examined DNA methylation status at promoter CpG islands of 24 apoptosis-related genes using samples generated in Experiment I (Table 2). Of the 24 apoptosis-related genes on the assay card, the extent of promoter methylation in six genes (Bcl2, Bcl2l11, Cideb, Dapk1, Tnfrsf25, and Ltbr) exceeded 60% in the negative control group. The positive control treatment, 5-Aza-dC, induced demethylation only when the CpG-rich region of a gene promoter was heavily methylated (>60%).
Table 2.
Extent of apoptosis-related gene promoter methylation (%)a in HCT-116 cells cultured with 5-Aza-dC, butyrate, DHA, LA, or co-treatment
| Gene symbol | Apoptosis | Negative control | Positive control 5-Aza-dC | Butyrate | DHA | LA | DHA + butyrate | LA + butyrate |
|---|---|---|---|---|---|---|---|---|
| Highly methylated genes | ||||||||
| Bcl2 | Anti | 98.83 ± 0.27 | 80.62 ± 4.02* | 98.26 ± 0.36 | 94.55 ± 3.31 | 97.78 ± 0.25* | 97.87 ± 0.48 | 98.35 ± 0.35 |
| Bcl2l11 | Pro | 68.86 ± 1.74 | 2.10 ± 0.63* | 52.58 ± 10.24* | 53.79 ± 10.84 | 62.22 ± 2.66 | 52.11 ± 9.82* | 60.48 ± 3.02* |
| Cideb | Pro | 94.63 ± 1.52 | 77.01 ± 2.81* | 88.83 ± 2.58 | 84.69 ± 3.27* | 85.02 ± 0.94* | 86.89 ± 2.00* | 89.62 ± 2.37 |
| Dapk1 | Pro | 96.35 ± 0.28 | 69.84 ± 2.90* | 94.86 ± 0.67 | 93.45 ± 1.32* | 94.88 ± 0.16* | 94.10 ± 0.31* | 94.88 ± 0.55 |
| Ltbr | Pro | 64.55 ± 1.29 | 29.39 ± 4.87* | 61.64 ± 1.21 | 59.68 ± 2.70 | 59.99 ± 1.15* | 57.94 ± 1.54* | 61.11 ± 1.10 |
| Tnfrsf25 | Pro | 97.20 ± 0.53 | 76.79 ± 2.30* | 95.43 ± 0.90 | 92.80 ± 1.85* | 93.26 ± 0.53* | 94.07 ± 0.99* | 95.41 ± 0.93 |
| Not highly methylated genes | ||||||||
| Apaf1 | Pro | 0.76 ± 0.10 | 0.45 ± 0.13 | 0.40 ± 0.13 | 0.67 ± 0.23 | 0.53 ± 0.12 | 0.54 ± 0.22 | 0.32 ± 0.07* |
| Bad | Pro | 1.08 ± 0.09 | 2.16 ± 0.89 | 2.25 ± 0.58 | 2.32 ± 0.61 | 2.13 ± 0.52* | 1.90 ± 0.38* | 1.55 ± 0.56 |
| Bag1 | Anti | 6.81 ± 0.58 | 14.84 ± 5.43* | 15.88 ± 3.66 | 15.89 ± 3.80 | 15.67 ± 1.93* | 14.13 ± 2.93* | 12.85 ± 4.22 |
| Bax | Pro | 0.69 ± 0.07 | 0.65 ± 0.11 | 0.58 ± 0.20 | 0.81 ± 0.18 | 0.65 ± 0.14 | 0.75 ± 0.26 | 0.41 ± 0.08* |
| Bclaf1 | Pro | 0.96 ± 0.11 | 1.15 ± 0.39 | 1.40 ± 0.34 | 1.68 ± 0.38 | 1.49 ± 0.31 | 1.49 ± 0.19* | 1.30 ± 0.40 |
| Bid | Pro | 6.56 ± 0.24 | 7.52 ± 2.74 | 12.78 ± 2.89 | 11.23 ± 2.53 | 13.03 ± 1.20* | 11.60 ± 1.39* | 12.04 ± 2.4* |
| Bik | Pro | 2.84 ± 0.20 | 10.67 ± 5.25* | 9.84 ± 2.91 | 24.80 ± 9.70* | 30.42 ± 9.85* | 8.92 ± 2.35* | 7.48 ± 3.30 |
| Birc2 | Anti | 2.00 ± 0.17 | 3.67 ± 1.47 | 4.62 ± 1.06 | 4.92 ± 1.36 | 5.16 ± 1.05* | 4.16 ± 0.93* | 3.17 ± 1.15 |
| Bnip3l | Pro | 0.81 ± 0.10 | 0.69 ± 0.15 | 1.03 ± 0.39 | 1.12 ± 0.21 | 1.07 ± 0.11 | 1.03 ± 0.24 | 0.61 ± 0.19 |
| Casp3 | Pro | 1.21 ± 0.05 | 2.06 ± 0.67 | 10.74 ± 8.13* | 2.38 ± 0.61 | 2.31 ± 0.40* | 2.00 ± 0.35* | 1.71 ± 0.54 |
| Casp9 | Pro | 3.58 ± 0.34 | 5.11 ± 0.31* | 3.22 ± 0.19 | 3.44 ± 0.24 | 3.15 ± 0.26 | 3.74 ± 0.62 | 2.80 ± 0.10 |
| Cradd | Pro | 1.54 ± 0.56 | 1.33 ± 0.23 | 1.48 ± 0.32 | 1.59 ± 0.37 | 1.35 ± 0.18 | 1.69 ± 0.34 | 1.00 ± 0.20 |
| Dffa | Pro | 0.80 ± 0.05 | 0.65 ± 0.14 | 0.68 ± 0.25 | 0.77 ± 0.16 | 0.74 ± 0.19 | 0.79 ± 0.19 | 0.45 ± 0.09* |
| Fadd | Pro | 0.50 ± 0.09 | 0.50 ± 0.10 | 0.49 ± 0.19 | 0.71 ± 0.18 | 0.67 ± 0.17 | 0.59 ± 0.12 | 0.28 ± 0.08 |
| Gadd45a | Pro | 10.85 ± 1.26 | 17.73 ± 6.69 | 27.02 ± 7.71 | 32.17 ± 10.33 | 31.76 ± 5.09* | 26.42 ± 6.85 | 22.36 ± 7.30 |
| Hrk | Pro | 16.14 ± 1.23 | 32.76 ± 4.88* | 32.95 ± 6.15 | 37.31 ± 6.85* | 46.33 ± 4.10* | 33.85 ± 7.14 | 30.83 ± 6.23* |
| Tnfrsf21 | Pro | 0.67 ± 0.08 | 0.58 ± 0.19 | 0.67 ± 0.24 | 0.79 ± 0.23 | 0.48 ± 0.10 | 0.62 ± 0.22 | 0.37 ± 0.08* |
| Trp53 | Pro | 1.20 ± 0.50 | 1.75 ± 0.67 | 1.92 ± 0.47 | 2.11 ± 0.47 | 2.01 ± 0.39* | 1.88 ± 0.33 | 1.49 ± 0.54 |
Data are presented as a percentage of cellular DNA containing methylated gene promoters, which included a combination of both the intermediate level of methylation and hypermethylated DNA.
Different from the negative control group (P <0.05). Data are expressed as means ± SEM from five to six samples per treatment. The negative controls represent cells incubated in media only.
Treatment with 5-Aza-dC resulted in a 28% decrease in the level of promoter methylation in Dapk1, compared to the negative control (media only). The promoter methylation level of Bcl2l11 was 97% inhibited by 5-Aza-dC compared with the negative control. Butyrate had demethylating effects on the Bcl2l11 promoter, and DHA treatment significantly decreased the levels of promoter methylation of Cideb, Dapk1, and Tnfrsf25 compared to the negative control group. Although the combination of DHA and butyrate did not affect global methylation, it did result in gene-specific demethylation of Bcl2l11, Cideb, Dapk1, Ltbr, and Tnfrsf25.
The transcription levels of Tnfrsf25 and Dapk1
To further develop the time course of gene expression, in Experiment II HCT-116 cells were incubated with experimental agents for 48, 72, and 96 h and the transcription levels of Cideb, Dapk1, and Tnfrsf25 were quantified by qRT-PCR. These genes were chosen because of their hyper-methylated status and response to 5-Aza-dC, DHA, and the combination of DHA and butyrate noted in Experiment I. The 5-Aza-dC treatment induced the expression of Dapk1 and Tnfrsf25 after 72 and 96 h incubation (Figure 3). The transcript level of Cideb was only detectable following 5-Aza-dC treatment, suggesting that 5-Aza-dC restored the expression of this gene (data not shown). Dapk1 expression was elevated by more than 100 and 200% at 72 and 96 h of 5-Aza-dC treatment, respectively, as compared with the negative control cells. Butyrate dramatically up-regulated Tnfrsf25 and Dapk1 transcript levels in 48-h samples, but declined thereafter. Similar to the data obtained with butyrate alone, the combination of butyrate with DHA or LA resulted in a rapid elevation in transcript levels, followed by a subsequent decline in expression.
Figure 3.

Time-course analysis of Tnfrsf25 (Panel a) and Dapk1 (Panel b) transcript levels (expressed as percentage of the negative control) in HCT-116 cells cultured with 5-Aza-dC, butyrate, DHA, LA, or co-treatment for 48, 72, or 96 h (see “Experiment II” section for details of Experiment II). Values are means ± SEM. *Different from the negative control sample measured at the same time point, P <0.05, n = 3 per treatment. The negative controls were cells incubated in media only
Global histone H3/H4 acetylation
The increase in transcript levels of Tnfrsf25 and Dapk1 by butyrate treatment led us to examine whether gene expression was being influenced by its affect on histone acetylation status. Therefore, we assessed the global histone acetylation levels of H3 and H4 in HCT-116 cells from Experiment I. Cells treated with 5-Aza-dC demonstrated no significant changes in histone acetylation state. Global H3 histone acetylation levels were elevated in butyrate treated cells compared to the negative controls, but there were no interactions between butyrate and the PUFA treatments that influenced H3 acetylation beyond that obtained with butyrate alone. The global H4 histone acetylation level increased in cells treated with LA or LA combined with butyrate (Figure 4).
Figure 4.
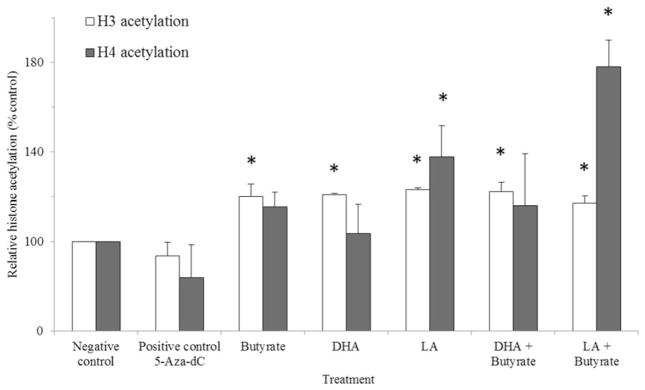
H3 and H4 histone acetylation (expressed as percentage of the negative control) in HCT-116 cells incubated with 5-Aza-dC (48 h), butyrate (12 h), DHA (72 h), LA (72 h), or co-treatment (see “Experiment I” section for details of Experiment I). Values are means ± SEM. *Different from the negative control, P <0.05, n = 3 per treatment. The negative controls were cells incubated in media only
Discussion
The transcriptional regulation of apoptosis-associated genes by DNA methylation is one of the mechanisms by which cancerous cells avoid apoptosis. The coordinated hypermethylation of proapoptotic gene promoters and hypomethylation of antiapoptotic gene promoters suppress normal regulation of apoptosis.6 Studies have been conducted on DNA methylation status of apoptosis-related genes in prostate cancer,20 glioblastoma multiforme,21 and bladder cancer.7 In general, the methylation status of apoptosis-related gene promoter regions correlates with tumor phenotype, suggesting that DNA methylation of apoptosis-related genes could be a biomarker for cancer diagnosis.
In this study, we investigated the promoter methylation status of 24 apoptosis-related genes in human colon cancer cells exposed to DHA, LA, butyrate, or a combination of PUFA and butyrate. Methylation of apoptosis-related genes was targeted because we previously demonstrated that the combination of fish oil (n-3 PUFA) and pectin (butyrate) enhanced colonocyte apoptosis, which in part, was associated with the expression of genes involved in apoptosis.13,18,22 The goal of this paper, therefore, was to determine if DHA and/or butyrate modulate the expression level of apoptosis-related genes by suppressing promoter methylation in human colon cancer cells. Hypermethylation was detected for six apoptosis-related genes, including Bcl2, Bcl2l11, Cideb, Dapk1, Ltbr, and Tnfrsf25, which are also methylated in colon cancer or other types of cancers.8,23–26 Friedrich et al. reported similar results in bladder tumor samples, wherein Dapk1 and Tnfrsf25 were hypermethylated compared with non-malignant adjacent tissue.7
Kolar et al. demonstrated that DHA or LA independently did not increase colonocyte apoptosis in HCT-116 cells.18 In the present study, DHA and LA treatments decreased methylation of Cideb, Dapk1, and Tnfrsf25 promoters, which are all linked to a proapoptotic phenotype. Cell death-inducing DFFA-like effector b (Cideb) induces apoptosis in a caspase-dependent manner via cytochrome c release from mitochondria.27 Dapk1 is a member of the serine/threonine protein kinase family and a positive regulator of apoptosis.28 TNF receptor superfamily 25 (Tnfrsf25), also known as DR3, is a membrane-bound death receptor capable of inducing apoptosis.29 The combination of LA with butyrate did not result in significant changes in the methylation status of these three genes. However, LA decreased methylation of Bcl-2, an antiapoptotic mediator.
DHA combined with butyrate also reduced promoter methylation of five proapoptotic genes Cideb, Dapk1, Tnfrsf25, Bcl2l11, and Ltbr, but did not affect methylation of the Bcl-2 promoter. We have previously demonstrated that combination chemotherapy using DHA and butyrate most effectively enhanced colonocyte apoptosis in HCT-116 cells.18 Induction of apoptosis by the combination of DHA and butyrate may therefore result from their effects on methylation, as well as other effects induced by this combination.30 We demonstrate for the first time that part of the enhanced apoptosis induced by the combination of DHA and butyrate may be attributable to the demethylation of proapoptotic genes in colon cancer cells.
Butyrate alone decreased promoter methylation of Bcl2l11 as well as global methylation. In addition, butyrate induced a fourfold increase in the transcription levels of Tnfrsf25 and Dapk1 at 48 h and increased their expression when combined with LA and DHA. These increases in gene expression may be attributed to an effect of butyrate on histone deacetylation [29]. It is known that histone acetylation mainly occurs at the promoter region of genes in the process of transcription whereas histone deacetylation occurs in the promoter region leading to gene silencing.31 The promoter region of Dapk1 and Tnfrsf25 is proximate to sites where histone modifications occur.32 Therefore, the increased histone acetylation by butyrate might contribute to the induction of gene expression of these genes. We have previously reported that butyrate enhances global histone H3 methylation [14] and this result is comparable to that of Kobori et al., who noted that 10 mM butyrate enhanced 25% more histone H3 acetylation than a control group in human colon cancer cells.33 However, it will be necessary to study site-specific histone acetylation to fully appreciate the influence of histone modifications on increases of Dapk1 and Tnfrsf25 expression. Currently, we are globally assessing histone acetylation using chromatin immunoprecipitation sequencing to fully determine the impact of diet on histone modifications in the colonic mucosa.
Bcl2l11, also known as Bim, antagonizes Bcl-2 and promotes apoptosis.34 In agreement with the increased apoptosis levels previously reported,18 promoter methylation of Bcl2l11 decreased significantly with 5-Aza-dC, butyrate, and the combination of butyrate with DHA or LA relative to the negative control group. Considering that with the exception of the positive control the demethylation of the Bcl2l11 promoter required butyrate, our findings suggest that butyrate is primarily responsible for demethylation of the Bcl2l11 promoter, and by doing so contributes to the induction of apoptosis.
With respect to the time-dependent increases in gene expression due to the 5-Aza-dC treatment, our results are comparable to those of others showing that 5-aza-cytidine, the analog of 5-Aza-dC, increased the expression level of p16 as incubation time progressed.16 Indeed, a 72-h exposure to 2 μM 5-Aza-dC suppressed Dapk1 promoter methylation by 28% and induced a 1.5-fold increase in Dapk1 transcription (Figure 3). Recently, Shu et al. reported that curcumin resulted in a similar reduction in promoter methylation of Neurog1 in prostate cancer cells and that this reduction led to the twofold increase of Neurog1 mRNA expression.35 However, the decreases in promoter methylation caused by DHA, compared to the negative control in this study were less than 50% of the decrease obtained with the positive control (5-Aza-dC, 18–27%).
The 5-Aza-dC treatment, a DNMT inhibitor, has been extensively studied and shown to inhibit DNA methylation and restores the expression level of tumor suppressor genes.36 This compound is incorporated into DNA and acts as a substrate for DNMT. However, it adheres to DNMT and remains as a covalent protein–DNA adduct, thereby suppressing DNA methylation. Dietary compounds, such as epigallocatechin-3-gallate (green tea compound) interrupt DNMT activity by blocking the active site of the enzyme.37 Therefore, it is possible that the demethylating effect of DHA might be exerted through mechanisms other than that employed by 5-Aza-dC. DHA is primarily incorporated into phosphatidylethanolamine (PE), which is converted to phosphatidylcholine through the addition of methyl groups from S-adenosyl methionine.38 Recently, Kale et al. found that when DHA was lacking, there was less PE-containing DHA, and the resulting excess in available methyl groups could be used for DNA methylation by DNMT.39 This finding suggests that DHA may be capable of suppressing DNA methylation by consuming methyl groups. Therefore, DHA may indirectly suppress DNMT activity whereas 5-Aza-dC directly inactivates DNMT. In our current study, DHA inhibited global methylation and promoter methylation of proapoptotic genes, yet the demethylating effect of DHA was not sufficient to induce gene expression. Previously, it has been reported that reduced promoter methylation by butyrate was not sufficient to allow re-expression of genes.40
In summary, we have shown that the combination of DHA and butyrate promotes apoptosis in part by suppressing promoter methylation of the proapoptotic genes Bcl2l11, Cideb, Dapk1, Ltbr, and Tnfrsf25 in colon cancer cells. We demonstrated that DHA has global and modest gene-specific demethylating activity, which may have implications for a synergistic effect with the inhibitors of DNA methylation, such as 5-Aza-dC. Although these results were obtained with a single human colon cancer cell line, they replicate, and thus further validate previous in vivo observations [14].
Acknowledgments
This study was supported by NSBRI NASA NCC 9-58 (JRL, NDT), NASA NAG-9-1523 (JRL, NDT) and NIH [CA59034 (RSC), CA129444 (RSC), CA057030 (RJC), CA168312 (RSC, NDT), and CA61750 (JRL)].
Footnotes
Author Contributions: All authors participated in the design, interpretation of the studies, and analysis of the data and review of the manuscript. YC, NDT, and LAD conducted the experiments; NDT, RSC, and JRL supplied critical input to experimental design and data interpretation; RJC provided statistical analysis and interpretation; YC and NDT were responsible for writing the manuscript, and LAD, RSC, RJC, and JRL provided editorial comments.
References
- 1.Bird A. Perceptions of epigenetics. Nature. 2007;447:396–8. doi: 10.1038/nature05913. [DOI] [PubMed] [Google Scholar]
- 2.Esteller M. Epigenetics in cancer. N Engl J Med. 2008;358:1148–59. doi: 10.1056/NEJMra072067. [DOI] [PubMed] [Google Scholar]
- 3.Issa JP. CpG island methylator phenotype in cancer. Nat Rev Cancer. 2004;4:988–93. doi: 10.1038/nrc1507. [DOI] [PubMed] [Google Scholar]
- 4.Samowitz WS, Albertsen H, Herrick J, Levin TR, Sweeney C, Murtaugh MA, Wolff RK, Slattery ML. Evaluation of a large, population-based sample supports a CpG island methylator phenotype in colon cancer. Gastroenterology. 2005;129:837–45. doi: 10.1053/j.gastro.2005.06.020. [DOI] [PubMed] [Google Scholar]
- 5.Wong JJ, Hawkins NJ, Ward RL. Colorectal cancer: a model for epigenetic tumorigenesis. Gut. 2007;56:140–8. doi: 10.1136/gut.2005.088799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gopisetty G, Ramachandran K, Singal R. DNA methylation and apoptosis. Mol Immunol. 2006;43:1729–40. doi: 10.1016/j.molimm.2005.11.010. [DOI] [PubMed] [Google Scholar]
- 7.Friedrich MG, Weisenberger DJ, Cheng JC, Chandrasoma S, Siegmund KD, Gonzalgo ML, Toma MI, Huland H, Yoo C, Tsai YC, Nichols PW, Bochner BH, Jones PA, Liang G. Detection of methylated apoptosis-associated genes in urine sediments of bladder cancer patients. Clin Cancer Res. 2004;10:7457–65. doi: 10.1158/1078-0432.CCR-04-0930. [DOI] [PubMed] [Google Scholar]
- 8.Zorko BA, Perez LB, De Blanco EJ. Effects of ILTG on DAPK1 promoter methylation in colon and leukemia cancer cell lines. Anticancer Res. 2010;30:3945–50. [PubMed] [Google Scholar]
- 9.Chang WC, Chapkin RS, Lupton JR. Predictive value of proliferation, differentiation and apoptosis as intermediate markers for colon tumorigenesis. Carcinogenesis. 1997;18:721–30. doi: 10.1093/carcin/18.4.721. [DOI] [PubMed] [Google Scholar]
- 10.Davidson LA, Nguyen DV, Hokanson RM, Callaway ES, Isett RB, Turner ND, Dougherty ER, Wang N, Lupton JR, Carroll RJ, Chapkin RS. Chemopreventive n-3 polyunsaturated fatty acids reprogram genetic signatures during colon cancer initiation and progression in the rat. Cancer Res. 2004;64:6797–804. doi: 10.1158/0008-5472.CAN-04-1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chapkin RS, Hong MY, Fan YY, Davidson LA, Sanders LM, Henderson CE, Barhoumi R, Burghardt RC, Turner ND, Lupton JR. Dietary n-3 PUFA alter colonocyte mitochondrial membrane composition and function. Lipids. 2002;37:193–9. doi: 10.1007/s11745-002-0880-8. [DOI] [PubMed] [Google Scholar]
- 12.Davie JR. Inhibition of histone deacetylase activity by butyrate. J Nutr. 2003;133:2485 S–93 S. doi: 10.1093/jn/133.7.2485S. [DOI] [PubMed] [Google Scholar]
- 13.Cho Y, Kim H, Turner ND, Mann JC, Wei J, Taddeo SS, Davidson LA, Wang N, Vannucci M, Carroll RJ, Chapkin RS, Lupton JR. A chemo-protective fish oil- and pectin-containing diet temporally alters gene expression profiles in exfoliated rat colonocytes throughout oncogenesis. J Nutr. 2011;141:1029–35. doi: 10.3945/jn.110.134973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cho Y, Turner ND, Davidson LA, Chapkin RS, Carroll RJ, Lupton JR. A chemoprotective fish oil/pectin diet enhances apoptosis via Bcl-2 promoter methylation in rat azoxymethane-induced carcinomas. Exp Biol Med (Maywood) 2012;237:1387–93. doi: 10.1258/ebm.2012.012244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bender CM, Pao MM, Jones PA. Inhibition of DNA methylation by 5-aza-2′-deoxycytidine suppresses the growth of human tumor cell lines. Cancer Res. 1998;58:95–101. [PubMed] [Google Scholar]
- 16.Schneider-Stock R, Diab-Assef M, Rohrbeck A, Foltzer-Jourdainne C, Boltze C, Hartig R, Schonfeld P, Roessner A, Gali-Muhtasib H. 5-Aza-cytidine is a potent inhibitor of DNA methyltransferase 3 a and induces apoptosis in HCT-116 colon cancer cells via Gadd45- and p53-dependent mechanisms. J Pharmacol Exp Ther. 2005;312:525–36. doi: 10.1124/jpet.104.074195. [DOI] [PubMed] [Google Scholar]
- 17.Fan YY, Zhang J, Barhoumi R, Burghardt RC, Turner ND, Lupton JR, Chapkin RS. Antagonism of CD95 signaling blocks butyrate induction of apoptosis in young adult mouse colonic cells. Am J Physiol. 1999;277:C310–9. doi: 10.1152/ajpcell.1999.277.2.C310. [DOI] [PubMed] [Google Scholar]
- 18.Kolar SS, Barhoumi R, Callaway ES, Fan YY, Wang N, Lupton JR, Chapkin RS. Synergy between docosahexaenoic acid and butyrate elicits p53-independent apoptosis via mitochondrial Ca(2+) accumulation in colonocytes. Am J Physiol Gastrointest Liver Physiol. 2007;293:G935–43. doi: 10.1152/ajpgi.00312.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ciavatta DJ, Yang J, Preston GA, Badhwar AK, Xiao H, Hewins P, Nester CM, Pendergraft WF, 3rd, Magnuson TR, Jennette JC, Falk RJ. Epigenetic basis for aberrant upregulation of autoantigen genes in humans with ANCA vasculitis. J Clin Invest. 2010;120:3209–19. doi: 10.1172/JCI40034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Suzuki M, Shigematsu H, Shivapurkar N, Reddy J, Miyajima K, Takahashi T, Gazdar AF, Frenkel EP. Methylation of apoptosis related genes in the pathogenesis and prognosis of prostate cancer. Cancer Lett. 2006;242:222–30. doi: 10.1016/j.canlet.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 21.Hervouet E, Vallette FM, Cartron PF. Impact of the DNA methyltransferases expression on the methylation status of apoptosis-associated genes in glioblastoma multiforme. Cell Death Dis. 2010;1:e8. doi: 10.1038/cddis.2009.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Turk HF, Kolar SS, Fan YY, Cozby CA, Lupton JR, Chapkin RS. Linoleic acid and butyrate synergize to increase Bcl-2 levels in colonocytes. Int J Cancer. 2011;128:63–71. doi: 10.1002/ijc.25323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yates DR, Rehman I, Abbod MF, Meuth M, Cross SS, Linkens DA, Hamdy FC, Catto JW. Promoter hypermethylation identifies progression risk in bladder cancer. Clin Cancer Res. 2007;13:2046–53. doi: 10.1158/1078-0432.CCR-06-2476. [DOI] [PubMed] [Google Scholar]
- 24.Hatada I, Fukasawa M, Kimura M, Morita S, Yamada K, Yoshikawa T, Yamanaka S, Endo C, Sakurada A, Sato M, Kondo T, Horii A, Ushijima T, Sasaki H. Genome-wide profiling of promoter methylation in human. Oncogene. 2006;25:3059–64. doi: 10.1038/sj.onc.1209331. [DOI] [PubMed] [Google Scholar]
- 25.Bell A, Bell D, Weber RS, El-Naggar AK. CpG island methylation profiling in human salivary gland adenoid cystic carcinoma. Cancer. 2011;117:2898–909. doi: 10.1002/cncr.25818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Babidge WJ, Butler LM, Burton MA, Cowled PA. Methylation of CpG sites in exon 2 of the bcl-2 gene occurs in colorectal carcinoma. Anticancer Res. 2001;21:2809–14. [PubMed] [Google Scholar]
- 27.Chen Z, Guo K, Toh SY, Zhou Z, Li P. Mitochondria localization and dimerization are required for CIDE-B to induce apoptosis. J Biol Chem. 2000;275:22619–22. doi: 10.1074/jbc.C000207200. [DOI] [PubMed] [Google Scholar]
- 28.Martoriati A, Doumont G, Alcalay M, Bellefroid E, Pelicci PG, Marine J-C. dapk1, encoding an activator of a p19ARF-p53-mediated apoptotic checkpoint, is a transcription target of p53. Oncogene. 2004;24:1461–6. doi: 10.1038/sj.onc.1208256. [DOI] [PubMed] [Google Scholar]
- 29.Mahmood Z, Shukla Y. Death receptors: Targets for cancer therapy. Exp Cell Res. 2010;316:887–99. doi: 10.1016/j.yexcr.2009.12.011. [DOI] [PubMed] [Google Scholar]
- 30.Kolar S, Barhoumi R, Jones CK, Wesley J, Lupton JR, Fan YY, Chapkin RS. Interactive effects of fatty acid and butyrate-induced mitochondrial Ca(2+) loading and apoptosis in colonocytes. Cancer. 2011;117:5294–303. doi: 10.1002/cncr.26205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Forsberg EC, Bresnick EH. Histone acetylation beyond promoters: Long-range acetylation patterns in the chromatin world. Bioessays. 2001;23:820–30. doi: 10.1002/bies.1117. [DOI] [PubMed] [Google Scholar]
- 32.Fujita PA, Rhead B, Zweig AS, Hinrichs AS, Karolchik D, Cline MS, Goldman M, Barber GP, Clawson H, Coelho A, Diekhans M, Dreszer TR, Giardine BM, Harte RA, Hillman-Jackson J, Hsu F, Kirkup V, Kuhn RM, Learned K, Li CH, Meyer LR, Pohl A, Raney BJ, Rosenbloom KR, Smith KE, Haussler D, Kent WJ. The UCSC Genome Browser database: Update 2011. Nucleic Acids Res. 2011;39:D876–82. doi: 10.1093/nar/gkq963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kobori A, Bamba S, Imaeda H, Ban H, Tsujikawa T, Saito Y, Fujiyama Y, Andoh A. Butyrate stimulates IL-32alpha expression in human intestinal epithelial cell lines. World J Gastroenterol. 2010;16:2355–61. doi: 10.3748/wjg.v16.i19.2355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.O’Connor L, Strasser A, O’Reilly LA, Hausmann G, Adams JM, Cory S, Huang DCS. Bim: A novel member of the Bcl-2 family that promotes apoptosis. EMBO J. 1998;17:384–95. doi: 10.1093/emboj/17.2.384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shu L, Khor TO, Lee JH, Boyanapalli SS, Huang Y, Wu TY, Saw CL, Cheung KL, Kong AN. Epigenetic CpG demethylation of the promoter and reactivation of the expression of Neurog1 by curcumin in prostate LNCaP cells. AAPS J. 2011;13:606–14. doi: 10.1208/s12248-011-9300-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lyko F, Brown R. DNA methyltransferase inhibitors and the development of epigenetic cancer therapies. J Natl Cancer Inst. 2005;97:1498–506. doi: 10.1093/jnci/dji311. [DOI] [PubMed] [Google Scholar]
- 37.Fang MZ, Wang Y, Ai N, Hou Z, Sun Y, Lu H, Welsh W, Yang CS. Tea polyphenol (−)-epigallocatechin-3-gallate inhibits DNA methyltransferase and reactivates methylation-silenced genes in cancer cell lines. Cancer Res. 2003;63:7563–70. [PubMed] [Google Scholar]
- 38.Stilwell W. The role of polyunsaturated lipids in membrane raft function. Scand J Food Nutr. 2006;50:107–13. [Google Scholar]
- 39.Kale A, Naphade N, Sapkale S, Kamaraju M, Pillai A, Joshi S, Mahadik S. Reduced folic acid, vitamin B12 and docosahexaenoic acid and increased homocysteine and cortisol in never-medicated schizophrenia patients: Implications for altered one-carbon metabolism. Psychiatry Res. 2010;175:47–53. doi: 10.1016/j.psychres.2009.01.013. [DOI] [PubMed] [Google Scholar]
- 40.Dronamraju SS, Coxhead JM, Kelly SB, Mathers JC. Differential anti-neoplastic effects of butyrate in cells with and without a functioning DNA mismatch repair. Nutr Cancer. 2010;62:105–15. doi: 10.1080/01635580903191486. [DOI] [PubMed] [Google Scholar]