

Abstract
The TNF-related apoptosis-inducing ligand (TRAIL or Apo2L) preferentially cause apoptosis of malignant cells in vitro and in vivo without severe toxicity. Therefore, TRAIL or agonist antibodies to the TRAIL DR4 and DR5 receptors are used in cancer therapy. However, many malignant cells are intrinsically resistant or acquire resistance to TRAIL. It has been previously proposed that the multidrug transporter P-glycoprotein (Pgp) might play a role in resistance of cells to intrinsic apoptotic pathways by interfering with components of ceramide metabolism or by modulating the electrochemical gradient across the plasma membrane. In this study we investigated whether Pgp also confers resistance toward extrinsic death ligands of the TNF family. To this end we focused our study on HeLa cells carrying a tetracycline-repressible plasmid system which shuts down Pgp expression in the presence of tetracycline. Our findings demonstrate that expression of Pgp is a significant factor conferring resistance to TRAIL administration, but not to other death ligands such as TNF-α and Fas ligand. Moreover, blocking Pgp transport activity sensitizes the malignant cells toward TRAIL. Therefore, Pgp transport function is required to confer resistance to TRAIL. Although the resistance to TRAIL-induced apoptosis is Pgp specific, TRAIL itself is not a direct substrate of Pgp. Pgp expression has no effect on the level of the TRAIL receptors DR4 and DR5. These findings might have clinical implications since the combination of TRAIL therapy with administration of Pgp modulators might sensitize TRAIL resistant tumors.
Keywords: Apoptosis, P-glycoprotein, TRAIL, Resistance, Modulators
1. Introduction
In mammalian cells, apoptosis occurs through two distinct molecular pathways. The intrinsic pathway is activated by intracellular events and depends on the release of proapoptotic factors from the mitochondria [1]. Standard chemotherapy and radiotherapy for cancer predominately initiate apoptosis via the intrinsic pathway and thus may select for resistant cancer cells that can evade intrinsic apoptosis signaling [1]. However, cell death can still occur in the absence of intrinsic apoptosis via extrinsic apoptosis. The extrinsic apoptosis pathway transmits signals from extracellular death ligands through binding to proapoptotic death receptors [2]. The TNF family of proteins is among the extrinsic death ligands. This family that includes TNF, Fas ligand and TRAIL are predominantly produced by cells of the immune system [3–6] TRAIL is considered a key cytotoxic effector used by NK cells. TRAILs are expressed by NK cells and regulated by IFNγ. These ligands are important mediators of caspase-dependent target cell apoptosis via their receptors [7].
The proapoptotic member of the TNF family TRAIL binds to five identified receptors [2]. Two of these receptors are DR4 and DR5 that contain protein motifs in their cytoplasmic region, known as death domains. These death domains are essential for the ability of DR4 and DR5 to transmit apoptotic signals. TRAIL also interacts with three “decoy” receptors that are unable to transmit apoptotic signals: DcR1, DcR2, and osteoprotegerin. Interestingly, TRAIL has anti-tumorigenic properties in vitro and in vivo, causing negligible effects on normal cells when exogenously administered. This facilitated clinical studies using three pharmacological strategies including: (a) the administration of recombinant human TRAIL; (b) the use of activating humanized antibodies directed against the death receptors DR4 or DR5; or (c) the adenoviral delivery of the TRAIL coding sequence into tumor cells [8]. However, clinical trials have indicated that many human tumors demonstrate intrinsic or acquired resistance to TRAIL therapy [9–11].
P-glycoprotein (Pgp) is a membrane-bound drug-efflux transporter which belongs to the family of ATP-binding cassette (ABC) transporters. This multidrug transporter is a product of the MDR1 (ABCB1) gene [12,13]. The study of over 400 various tumors has shown a consistent association of MDR1 gene expression with several intrinsically chemotherapy resistant cancers and increased expression of the MDR1 geneinvarious cancers with acquired drug resistance [14]. The first direct evidence that the expression of this gene is associated with multidrug resistance in vivo was in the study on transgenic mice expressing the human MDR1 gene [15]. Pgp is highly expressed in many clinically resistant malignancies and, therefore, it is considered as an adverse prognostic factor in solid and hematological malignancies. In these malignancies Pgp expression has been correlated with reduced drug sensitivity and poor clinical outcome [16]. A strategy already undergoing clinical trials is the use of combination chemotherapy with chemical inhibitors of Pgp. These agents from different pharmacological classes (termed chemosensitizers or MDR-modulators) reverse cellular resistance to cytotoxic agents in experimental systems in vitro [17–19].
In this study, we examined the effect of Pgp expression and activity on extrinsic apoptosis pathways. The data supply interesting evidence that Pgp confers cellular resistance to the TRAIL death ligand. Moreover, we demonstrate that the sensitivity to TRAIL is increased by co-administration of Pgp modulators.
2. Materials and methods
2.1. The Pgp ON/OFF system
A Pgp-tetracycline-off variant of the cervical carcinoma HeLa cell line has been previously engineered [20] and used in this study. In this cell system, Pgp expression is under tetracycline control. Addition of 2 μg/ml tetracycline hydrochloride (cell culture tested, Sigma–Aldrich, St. Louis, MO, USA) turns off transcription of MDR1 mRNA, and over incubation period of 3–4 days, Pgp expression is silenced, allowing comparison of the same cells with and without Pgp in the plasma membrane.
These cells were grown as monolayer cultures in high glucose (4.5 g/l) DMEM medium supplemented with 4 mM l-glutamine (Gibco, Grand Island, NY, USA)), 100 u/ml penicillin, 100 μg/ml streptomycin and 10% heat-inactivated fetal-calf serum (all from Biological industries, Beit-Haemek, Israel), at 37 °C with 5% CO2. The Pgp-tetracycline-off cells were grown with 10% tetracycline system approved (gamma irradiated) FBS (Clontech, Mountain View, CA, USA).
2.2. RT-PCR analyses
Total RNA was purified using TRIZOL reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's protocol. cDNA was obtained from 1 μg RNA using Reverse Transcription System (Promega, Madison, WI, USA). Reverse transcriptase polymerase chain reaction (RT-PCR) reactions were performed using PCR Reddy Mix Master Mix (ABgene, Surrey, UK). To identify the presence of various MDR genes (MDR1, MRPs), we used specific primers, synthesized based on public human sequences. RT-PCR detection conditions are summarized in Supplementary Data (Table 1). PCR products were analyzed by electrophoresis in 2% agarose gel (SeaKem LE Agarose; Lonza, Basel, Switzerland). Amplification of a 600-bp region of the β-actin gene was performed as an internal control.
Table 1.
Primers and detection conditions of RT-PCR.
| Gene | Sequences of primers (5′–3′) | Amplification temperature (°C) | Product size (bp) |
|---|---|---|---|
| MDR1 | F: TTTACTGATAAAGAACTCTTA | 48 | 487 |
| R: AAACTGAAGTGAACATTTCTG | |||
| MRP1 | F: ATCGTTCTGTTTGCTGCCCT | 59 | 182 |
| R: GTCTCTGAATACTCCTTGAGCCT | |||
| MRP2 | F: ATCCAATACAGCAGACAATG | 51 | 198 |
| R: GAAAAGATCAGGATCAGGAT | |||
| MRP3 | F: GACCTCACTCCCTGCTTCCA | 61 | 298 |
| R: CATACTGTATCAGCAGGGTGGC | |||
| MXR1 | F: GATCTCTCACCCTGGGGCTTGTGG | 63 | 196 |
| R: TGTGCAACAGTGTGATGGCAAGGGA | |||
| β-Actin | F: CCAAGGCCAACCGCGAGAAGATGAC | 589 | |
| R: AGGGTACATGGTGGTGCCGCCAGAC |
2.3. Flow cytometry analyses
Immunostaining of cell surface markers was carried out with monoclonal antibodies: Anti-human P-glycoprotein monoclonal antibody MRK16 (Acris antibodies, Hiddenhausen, Germany); Anti-human DR4 (TRAIL receptor 1), PE-conjugated; Anti-human DR5 (TRAIL receptor 2), PE-conjugated; Anti-human DcR1 (TRAIL receptor 3), PE-conjugated; Anti-human DcR2 (TRAIL receptor 4), PE-conjugated; Anti-human TRAIL, PE-conjugated (all from eBioscience, San Diego, CA, USA); Anti-human TNFR1 monoclonal antibody, fluorescein isothiocyanate (FITC)-conjugated; Anti-human TNFR2 monoclonal antibody, Phycoerythrin (PE)-conjugated and various surface antigen markers (all from R&D systems, Minneapolis, MN, USA); Anti-Human CD95/Fas monoclonal antibody (DakoCytomation, Glostrup, Denmark); anti-Human TNF-α monoclonal antibody (Biosource, Camarillo, CA, USA), as recommended by the manufacturer. When unconjugated monoclonal antibodies were used for the initial step, the cells were washed and then incubated with the indicated fluorophore-conjugated F(ab′)2 fragments of polyclonal goat anti-mouse IgG (DakoCytomation, Glostrup, Denmark).
Controls used in the FACS analysis were cells that had been incubated with a relevant isotype standard or with the second antibody only.
Flow cytometry analysis of triplicate samples was performed using FACSCalibur with the CellQuest software (BD Biosciences, MD, USA). The FACS data analysis was done with the FlowJo 7.2.2 software (Ashland, OR, USA). Viable cells were defined by their forward scatter/side scatter (FSC/SSC) characteristics or propidium iodide (PI) (10 μg/ml, excitation at 535 nm and emission at 617).
2.4. Rh123 accumulation assay by FACS
The cellular uptakeof the Pgp substrate, Rh123 (Sigma–Aldrich; St. Louis, MO, USA) was measured according to a standard protocol, in the absence or presence of the Pgp-specific inhibitor, R-VRP (Sigma–Aldrich; St. Louis, MO, USA) [21]. Cells were analyzed on a FACSCalibur (Becton Dickinson) excitation at 488 nm and emission at 530 nm. Cells incubated in the absence of Rh123 and R-VRP were used as controls. Triplicate samples were measured. Assays were performed at least 3 times. Activity values are calculated as the difference between median fluorescence in the presence and absence of R-VRP, divided bymedianfluorescenceinthe absence of R-VRP.
2.5. Induction of apoptosis
Target cells were incubated with medium containing elevating concentrations of death ligands: Fas-agonistic antibody (CH-11) from MBL, Woburn, MA, USA, recombinant human TNF-α/TNFSF1A, recombinant human TRAIL/TNFSF10 (both from R&D systems; Minneapolis, MN, USA); or TNF-unrelated apoptosis inducing factors: low serum levels or camptothecin; for 24 h at 37 °C. Apoptosis by death ligands was determined in the presence or absence of Cycloheximide (CHX), which facilitates apoptosis induction by death receptors. Cells incubated with diluents alone (medium containing PBS and DMSO) were used to determine the spontaneous apoptosis of the target cells. Assays were performed in triplicate 3 times.
2.6. Annexin V binding assay to detect apoptotic cells
Translocation of phosphatidylserine (PS) from the inner to the outer leaflet of the plasma membrane, which is characteristic of early-apoptotic cells, was quantified using Annexin V-FITC apoptosis detection kit I (BD Pharmingen, Bedford, MA, USA), as recommended by the manufacturer. Adherent cells were labeled with Annexin V-FITC prior to harvesting, as previously described [22]. Early apoptotic cells were distinguished from late apoptotic/necrotic cells using PI staining, since membranes of late apoptotic/necrotic cells are permeable to PI [23]. Samples were analyzed on a FACSCalibur (Becton Dickinson), excitation at 494 nm and emission at 518 nm for FITC and excitation at 535 nm and emission at 617 for PI. Assays were performed in triplicate 3 times.
2.7. Inhibition of Pgp activity
Death-ligand induced apoptosis assays were performed in the absence or presence of the Pgp inhibitor R-VRP during the assay (20 μM, unless otherwise indicated). PBS (diluent) was used in the control experiments.
Alternatively, the irreversible combined Pgp inhibitors Cyclosporine A (CsA) from Sigma and Anti-MDR1 monoclonal antibody UIC2 (Chemicon international; Temecula, CA, USA) were used by the method described by Goda et al. [24].
2.8. Proliferation assay
Proliferation of exponentially growing target cells was determined after treatment with elevating concentrations of death ligands: Fas-agonistic antibody (CH-11), recombinant human TNF-α, recombinant human TRAIL; or other apoptosis inducing factors: low serum levels or camptothecin (Sigma–Aldrich; St. Louis, MO, USA); for 72 h at 37 °C. The growth inhibitory effect was measured using Cell-Titer 96 non-radioactive cell proliferation assay kit (Promega, Madison, WI, USA) as recommended by the manufacturer. Data are expressed as the percentage of cell proliferation inhibition compared to control cells incubated in complete medium containing relevant diluent. Assays were performed 3 times with 5 replicates.
2.9. MDR1 shift assay
Binding of the conformation-selective anti-MDR1 monoclonal antibody UIC2, which is drastically increased in the presence of Pgp substrates [25], was measured using the MDR1 Shift Assay (Chemicon international, Temecula, CA, USA), as recommended by the manufacturer. The UIC2 shift is the difference between UIC2 binding in the presence or absence of the reagents examined. This assay was used to determine whether TRAIL may serve as transport substrate of Pgp. Vinblastine (4.5 μg/ml), which is a well-known UIC2 shifting agent, and the reagent diluents (PBS for TRAIL and DMSO for vinblastine) were used as positive and negative controls, respectively.
2.10. TRAIL neutralization assays
To study the role of TRAIL receptors DR4 and DR5 on the susceptibility of target cells to TRAIL, cells were pretreated (5 μg/ ml each, unless otherwise indicated) with neutralizing anti-human DR4 monoclonal antibody, neutralizing anti-human DR5 monoclonal antibody, or both (ALEXIS, San Diego, CA, USA), 1 h before treatment with TRAIL for 24 h. Specific lysis was determined using PI (10 μg/ml). 0.5 μg/ml Mouse IgG1 (BioLegend; San Diego, CA, USA) was used in control experiments. Assays were performed in triplicate 3 times.
2.11. Statistical analysis
The Student's t-test for paired data was applied to compare the mean values for Pgp expressing and non-expressing cells using the various abovementioned assays.
3. Results
3.1. Characterization of Pgp expression and activity in PgpON/OFF cells
In this study we have utilized HeLa cell variants which express Pgp under the control of the tetracycline-promoter (PgpOFF and PgpON HeLa variants). Pgp expression in these cells is shut down in the presence of tetracycline.
Of note, the proliferation rates of both PgpOFF and PgpON HeLa variants were the same (data not shown). Moreover, the growth of the parental HeLa cells in the absence or the presence of tetracycline has shown no apparent variation in the spontaneous apoptotic rates, which were considerably low. Using the Annexin V assay, the early apoptosis rates of total cells in the absence or the presence of tetracycline were 2.3 ± 0.6% and 3.6 ± 0.8% respectively. This indicates that the tetracycline itself does not induce apoptosis.
Fig. 1 demonstrates Pgp expression and activity on the HeLa PgpOFF and HeLa PgpON variants. Four days before all assays, colchicine was removed from the growth media and cells were grown in the absence or presence of tetracycline, which shuts down Pgp expression in these cell variants. Four days after tetracycline addition, RNA levels of MDR1 were determined by RT-PCR using specific primers, as described in Section 2. PCR products were analyzed by electrophoresis in agarose gel. Fig. 1A demonstrates that the MDR1 gene is significantly downregulated in HeLa PgpOFF cells. The same level of RNA was detected for other tested ABC transporter genes. The expression of a 600-bp region of the β-actin gene is shown as a housekeeping gene internal control.
Fig. 1.
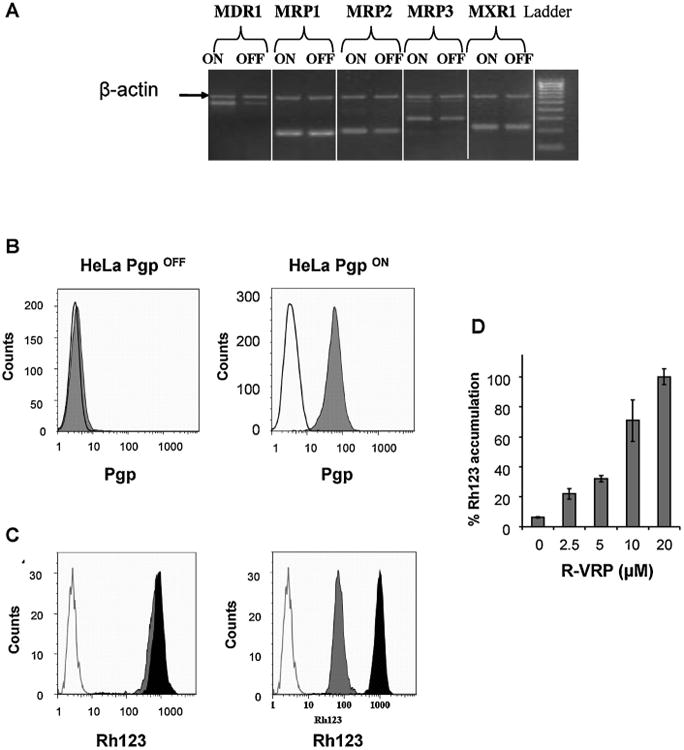
Pgp expression and activity in HeLa PgpOFF and HeLa PgpON cells. (A) Analysis of the expression of various MDR genes (MDR1 and MRPs) as determined by RT-PCR using specific primers. PCR products were analyzed by electrophoresis in agarose gel. The expression of a 600-bp region of the β-actin gene is demonstrated as an internal control of a housekeeping gene expression. (B) Analysis of cell surface expression of Pgp in the HeLa variants, as determined using Pgp specific mouse monoclonal antibody MRK16 followed by PE-conjugated goat anti-mouse antibody. Experiments were performed in triplicate 3 times. A typical experiment is shown. Isotype control (IC) was determined using staining with mouse IgG followed by PE-conjugated anti-mouse antibody, and represented by the open peak. Full gray peaks indicate surface expression of Pgp. (C) Analysis of Pgp activity as determined by accumulation of the Pgp substrate Rhodamine 123 (Rh123) in the HeLa variants. The full gray and black peaks represent Rh123 levels in the absence and presence of R-VRP, respectively. The open peak represents autofluorescence of the cells. Assays were performed in at least 3 independent experiments. (D) Dose-dependence of Rh123 accumulation in HeLa PgpON cells. Results are mean ± SD values of triplicate samples.
Pgp expression was measured using flow cytometry with Pgp-specific antibody MRK16. Pgp expression could not be detected in HeLa PgpOFF cells (Fig. 1B, left) but highly expressed on the surface of PgpON cells (Fig. 1B, right). The ratio between the median fluorescence in the HeLa PgpON cells and the IC median fluorescence was 18.7 ±2.7.
Pgp transport activity in the HeLa variants was determined by assaying the Rh123 accumulation. As demonstrated in Fig. 1C, Pgp was not active in HeLa PgpOFF cells (left), but only in HeLa PgpON cells (right). The Pgp inhibitor R-VRP blocked Pgp activity in HeLa PgpON cells, thus elevating Rh123 accumulation. Fig. 1D demonstrates that R-VRP blocks Pgp activity in HeLa PgpON cells in a dose dependent manner.
3.2. Pgp confers resistance to TRAIL-mediated apoptosis, not to FasL or TNF-α
In this study, we have investigated the direct apoptotic effects of death-ligands in relation to Pgp-expression. These death ligands included FasL, TNF-α and TRAIL. Apoptotic cell death was measured using Annexin V/PI binding assay.
Similar levels of apoptotic cells were detected in PgpOFF and PgpON cells after 24 h treatment with elevating concentrations of Fas-agonistic antibody (CH-11) or TNF-α, in the presence of cycloheximide (CHX) (Fig. 2). No apoptosis was observed without CHX (data not shown). Of note, the percentage of spontaneous lysis of PgpON/OFF cells was similar in all cases and did not exceed 10%.
Fig. 2.

Induction of apoptosis by Fas-agonistic antibody (CH-11), TNF-α and TRAIL in PgpON/OFF HeLa cells. Elevating concentrations of Fas-agonistic antibody (CH-11) (A), TNF-α (B) in the presence of CHX (500 ng/ml) and TRAIL (C and D) were added to PgpOFF (white) and PgpON (gray) HeLa cells for 24 h. Apoptotic cell death was measured using Annexin V and PI staining, as described in Section 2. No Annexin V+/PI− (early apoptotic) cells were observed after 24 h incubation with these death ligands. TRAIL effect was tested in the absence (C) or presence (D) of CHX. Results are mean ± SD values of triplicate samples from one representative experiment of 3 independent experiments (*P < 0.05, **P < 0.01).
On the other hand, Pgp expression conferred resistance to TRAIL-induced apoptosis, in a dose dependent manner. Of note, after 24 h treatment with the death ligands, no early apoptotic cells (Annexin V+/PI−) could be detected, but only late-apoptotic cells (Annexin V+/PI+).
While CHX was essential to induce apoptosis of HeLa cells by the Fas-agonistic antibody (CH-11) and TNF-α, TRAIL-apoptosis was induced in the absence of CHX. To confirm that CHX itself does not alter the resistance of Pgp-expressing cells to the death ligands, TRAIL induced apoptosis of PgpOFF and PgpON cells was also determined in the presence of CHX. Although CHX increased TRAIL-dependent apoptosis, Pgp conferred resistance to TRAIL in the presence of CHX as well (Fig. 2D).
Treating PgpOFF and PgpON HeLa cells with TRAIL (7.5 and 15 ng/ml) for 12,24, and 48 h induced significantly lower apoptosis in the Pgp-expressing cells in all time points examined (Fig. 3). Of note, after 6 h treatment with TRAIL low and similar levels of early apoptotic cells (Annexin V+/PI−) were detected in both cell variants, (5% and 7% Annexin V+/PI− in cells treated with 7.5 and 15 ng/ml TRAIL, respectively). However, late apoptotic cells (Annexin V+/PI+) were detected after treatment with TRAIL for 12 h or longer (Fig. 3A).
Fig. 3.

Effect of TRAIL, Fas-agonistic antibody (CH-11) and TNF-α on apoptosis and proliferation of PgpON/OFF HeLa cells. (A) Time dependent effects of TRAIL on cell apoptosis of PgpOFF (white) and PgpON (gray) HeLa cells, treated with 7.5 and 15 ng/ml TRAIL for 12,24 and 48 h. Apoptotic cell death was measured using Annexin V and PI staining, as described in Section 2. (B–D) Proliferation of PgpOFF (black) and PgpON (gray) HeLa cells was determined after treatment with various concentrations of TRAIL (B), Fas-agonistic antibody, CH-11 (C) and TNF-α (D), using CellTiter cell proliferation assay. Effect of Fas-agonistic antibody and TNF-α was determined in the presence of CHX. Results are expressed as the percentage of cell proliferation inhibition compared to diluent-treated control cells. Results are mean ± SD values of 5 replicates from one representative experiment of 3 independent experiments (*P < 0.05, **P < 0.01).
Furthermore, the proliferation of Pgp-expressing cells, determined using CellTiter cell proliferation assay, was less affected by treatment with TRAIL, further demonstrating that Pgp confers resistance to TRAIL (Fig. 3B). In contrast, proliferation of PgpOFF and PgpON cells in the presence of anti-Fas and TNF-α was similar (Fig. 3C and D).
3.3. Pgp expression does not influence death-receptors expression in Pgp expressing and non-expressing cells
As different expression levels of death-receptors on the surface of target cells may affect their susceptibility to killing by death-ligand pathways, we have examined using flow cytometry whether the PgpOFF and PgpON cells express different levels of the death receptors, i.e. FasL receptor (Fas), TNF-α receptors (TNFR1 and TNFR2) and TRAIL receptors (DR4, DR5 and the decoy receptors DcR1 and DcR2). Fig. 4 demonstrates that similar levels of the death receptors Fas, TNFR1, DR4 and DR5 are expressed on the surface of both PgpOFF and PgpON cells, indicating that the resistance to lysis by TRAIL that conferred by Pgp was not derived from different death receptor levels on target cells. TNFR2, DcR1 and DcR2 were not detected on the cell surface of the HeLa cells (data not shown).
Fig. 4.

Death receptor level on the surface of PgpON/OFF HeLa cells. Analysis of cell surface expression of the FasL receptor Fas (A), the TNF-α receptor TNFR1 (B) and TRAIL receptors DR4 (C) and DR5 (D) as determined by flow cytometry, using the specific monoclonal antibodies conjugated to PE. PE-conjugated mouse IgG1 was used as an isotype control. FAB = fold above background = the ratio between median fluorescence of positively labeled cells and the median fluorescence of the isotype control. Results are mean ± SD values of triplicate samples from one representative experiment of 3 independent experiments.
3.4. Pgp activity is required to confer resistance to TRAIL
To determine whether the mechanism by which Pgp confers resistance to TRAIL is structural (i.e. the presence of Pgp on the cell-surface is sufficient to confer resistance) or functional (i.e. Pgp activity is required to confer resistance), we studied the effect of the Pgp-inhibitor R-VRP on TRAIL-induced apoptosis of PgpON/OFF HeLa cells (Fig. 5A and B). Apoptosis was determined using PI staining alone, since as previously demonstrated (Fig. 3), cells were late apoptotic when incubation times were 12 h or longer.
Fig. 5.

Effect of Pgp inhibition on the sensitivity of PgpON/OFF HeLa cells to TRAIL. The sensitivity of the PgpOFF (white) and PgpON (gray) HeLa cells to 24 h treatment of 7.5 ng/ml (A) and 15 ng/ml (B) TRAIL was measured in the presence of elevating concentrations of the Pgp inhibitor R-VRP. Effect of irreversible blocking of Pgp activity on the sensitivity of PgpON/OFF HeLa cells to TRAIL. Blocking Pgp activity using the irreversible CsA + UIC2 blocking method before TRAIL addition to HeLa PgpON (gray) and PgpOFF (white) cells. 7.5 ng/ml TRAIL (C) and 15 ng/ml TRAIL (D) were applied after washing the inhibitors. Results are mean ± SD values of triplicate samples from one representative experiment of 3 independent experiments (*P < 0.05, **P < 0.01).
Blocking Pgp activity by R-VRP reversed the resistance of Pgp-expressing cells to TRAIL (7.5 and 15 ng/ml) in a dose dependent manner. Lysis of HeLa PgpOFF cells by TRAIL was unaltered by R-VRP, demonstrating the specific effect of the Pgp inhibitor. Furthermore, after blocking Pgp activity in the Pgp-expressing cells, similar levels of lysis were observed in both cell variants, further demonstrating the specific role of Pgp in conferring resistance to TRAIL. These results demonstrate that Pgp activity is required to confer resistance to TRAIL.
An additional method was developed to block Pgp activity. This method, which is a modification of the method published by Goda et al. [24], is irreversible, i.e. Pgp activity remains blocked even after washing the access of the Pgp inhibitor. To determine the optimal CsA concentration required to irreversibly block Pgp activity, adherent HeLa PgpON cells were treated with elevating concentrations of CsA for 10 min (Fig. 6). Then, 10 μg/ml of the Pgp modulating antibody UIC2 was supplemented for an additional 10 min and cells were washed allowing only bound UIC2 to remain in the system. Twenty four hours later, Rh123 accumulation assay demonstrated that 10 μM CsA followed by UIC2 binding resulted maximum inhibition of Pgp activity. Similarly, calibration curve of UIC2 antibody in the presence of 10 μM CsA demonstrated that the optimal UIC2 concentration required for blocking Pgp activity was 10 μg/ml (Fig. 6B). Of note, longer incubation times with the Pgp inhibitors resulted in higher spontaneous lysis of cells (data not shown).
Fig. 6.
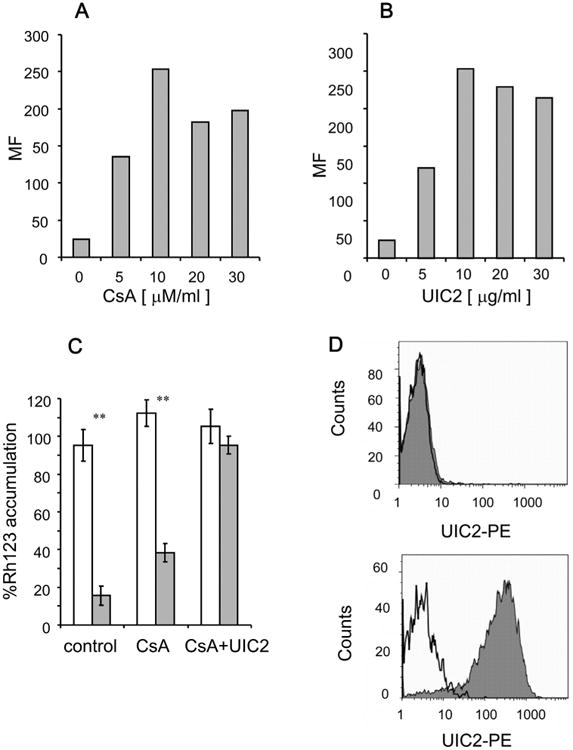
Irreversible inhibition of Pgp activity by Cyclosporine A (CsA) and the specific monoclonal antibody UIC2. Pgp activity was inhibited using a modification of the method published by Goda etal. [24]. Pgp activity was determined by accumulation of the Pgp substrate Rh123 in the HeLa cells. (A) Determination of CsA concentration required to block Pgp activity: HeLa PgpON cells were first treated with elevating concentrations of CsA for 10 min, then incubated with 10 μg/ml of the Pgp-modulating-monoclonal antibody UIC2 for an additional 10 min and then washed, allowing only bound UIC2 to remain in the system. After 24 h, Rh123 accumulation assay was performed. (B) Calibration of UIC2 antibody concentration required to block Pgp activity: HeLa PgpON cells were first treated with 10 mM CsA for 10 min, then with elevating concentrations of UIC2 antibody for an additional 10 min, and then washed. Rh123 accumulation assay was performed after 24 h. (C) Rh123 accumulation in the HeLa PgpOFF cells (white) and HeLa PgpON cells (gray) before and after treatment with 10 μM CsA and 10 μg/ml UIC2 antibody, and washing the inhibitors. Results are mean ± SD values of triplicate samples from one representative experiment of 3 independent experiments (*P < 0.05, **P < 0.01) (D) The levels of UIC2 antibody that remained bound to cells after 24 h in medium was determined using PE-conjugated goat anti mouse antibody in HeLa PgpOFF cells (upper histogram), and HeLa PgpON cells (lower histogram). Isotype control levels are represented by the open peak while gray peaks indicate surface-bound UIC2 antibody.
Blocking Pgp activity by the irreversible CsA and UIC2 blocking method before TRAIL addition reversed the resistance of Pgp-expressing cells to TRAIL even after washing of the inhibitors (Fig. 5C and D). This is consistent with results obtained when R-VRP was used during the lysis assay (Fig. 5A and B). Lysis of HeLa PgpOFF cells by TRAIL was unaltered, further demonstrating the specific effect of the Pgp inhibitor.
3.5. The resistance to TRAIL-induced apoptosis is Pgp specific, but TRAIL is not a direct substrate of Pgp
To further study whether the resistance of HeLa PgpON cells to lysis by TRAIL is Pgp-specific, we examined the sensitivity of the different cells to other apoptosis-inducing factors, which are known to lyse target cells in a Pgp-independent manner [26,27]. First, the effect of 24 h serum starvation was examined (Fig. 7). PgpON/OFF HeLa cells were lysed similarly in low concentrations of serum. Similarly, 24 h exposure to the apoptosis inducing factor camptothecin, which is not a Pgp substrate [27], resulted in similar levels of lysis in PgpOFF and PgpON HeLa cells.
Fig. 7.

Analysis of induction of apoptosis by Pgp-unrelated apoptosis inducing factors in PgpON/OFF HeLa cells and KB cells. (A and C) PgpOFF (white) and PgpON (gray) cells were treated for 24 h with medium containing low concentrations of FBS (A) or elevating concentrations of the apoptosis inducing agent camptothecin (C). Cell lysis was then evaluated using PI staining. (B and D) KB-V1 cells (gray) and KB-3-1 cells (white) sensitivity to lysis by serum starvation (B) or camptothecin (D). Results are mean ± SD values of triplicate samples from one representative experiment of 3 independent experiments d (*P < 0.05, **P < 0.01).
Since Pgp conferred resistance to lysis induced by TRAIL, we examined whether TRAIL can result a change in Pgp conformation (as other Pgp substrates do) using the MDR1 Shift Assay. This assay uses the conformation-selective anti-MDR1 antibody UIC2 which binds Pgp in higher affinity in the presence of Pgp substrates. Similar levels of UIC2 antibody were bound to Pgp-expressing cells in the absence and presence of TRAIL. Thus indicating that TRAIL did not cause a change in Pgp conformation (Fig. 8A). In the control experiment, the known Pgp substrate vinblastine induced an expected shift caused by higher binding of UIC2 to VBL-treated cells (Fig. 8B). To determine whether TRAIL can indirectly affect the conformation of Pgp in the presence of a Pgp substrate, TRAIL was supplemented in the presence of the Pgp substrate vinblastine during the MDR1 shift assay (Fig. 8C). Cells incubated with vinblastine and UIC2 demonstrated similar UIC2-shift in the absence or presence of TRAIL. Thus indicating that TRAIL has no indirect effect on Pgp conformation either.
Fig.8.

Analysis of TRAIL-induced conformational change of Pgp. (A) To determine whether TRAIL acts as a transport substrate of Pgp, the MDR1 (UIC2) Shift Assay was utilized, because the binding of the conformation-selective anti-MDR1 antibody UIC2 is drastically increased in the presence of Pgp substrates. HeLa PgpON cells were incubated at 37° C in the presence of TRAIL (100 ng/ml, full gray histogram) or control PBS (white histogram). Then control IgG2A or UIC2 was added and further incubated at 37° C. Cells were then incubated with PE-labeled anti-mouse IgG and washed. Binding of UIC2 was evaluated by flow cytometry. Cells incubated with control IgG2A (thin gray line) serves as isotype control. (B) Control MDR1 Shift Assay was performed using the known Pgp substrate vinblastine. Cells were incubated with vinblastine (22.5 μM) and UIC2 (full gray histogram) and fluorescence was compared to cells incubated with DMSO (diluent) or UIC2 (white histogram). Isotype control is depicted in the thin gray line. (C) To determine whether TRAIL can indirectly affect the conformation of Pgp in the presence of a Pgp substrate, TRAIL was supplemented to vinblastine during the MDR1 shift assay. UIC2-shift in the absence (white histogram) or presence (full gray histogram) of TRAIL is depicted. Isotype control is represented by the thin gray line. Typical analysis of 3 independent experiments is depicted.
3.6. Both DR4 and DR5 receptors play a role in TRAIL-induced lysis of Pgp expressing and non-expressing cells
To confirm the specificity of the TRAIL-induced lysis of PgpOFF and PgpON cells, we blocked each of the different TRAIL-receptors expressed in the target cells separately, by neutralizing antibodies (anti-DR4 and anti-DR5), and measured TRAIL induced lysis. Five μg/ml of each neutralizing antibody was sufficient to confer the maximal receptor neutralization for lysis induced by TRAIL (Fig. 9). Pretreatment of PgpON/OFF cells with neutralizing antibodies to DR4 or DR5 1 h before addition of TRAIL for 24 h, lowered the lysis of both cell variants, demonstrating that both receptors are active in PgpOFF and PgpON cells. Lysis of HeLa PgpOFF cells was less affected by the neutralization of TRAIL-induced lysis by anti-DR4 or anti-DR5 antibodies, but the combination of both receptors antibodies totally and synergistically prevented TRAIL-induced lysis of both PgpOFF and PgpON cells as expected. Therefore, these results confirm that Pgp effects on TRAIL-induced lysis are specific and require the binding of TRAIL to both of its receptors.
Fig. 9.

Analysis of the role of TRAIL-receptors in TRAIL-induced apoptosis of PgpON/OFF HeLa cells. (A) HeLa PgpOFF cells were pre-incubated with various concentrations of monoclonal neutralizing antibodies to TRAIL receptors DR4 (white) and DR5 (gray) 1 h prior to treatment with 15 ng/ml TRAIL for 24 h. In the control experiment, cells were pre-treated with mouse IgG1. Specific lysis was determined using PI staining. (B)PgpOFF (white) and PgpON (gray) HeLa cells were pretreated with the neutralizing antibodies anti-DR4, anti-DR5 or both (5 μg/ml each) 1 h before treatment with 15 ng/ml TRAIL for 24 h. Specific lysis was then determined using PI staining. In the control experiment, cells were pre-treated with mouse IgG1. (C) Percentage of neutralization of lysis by anti-DR4 and anti-DR5 was calculated as 100 × (1 − (%lysed cells in sample)/(%lysed cells in control). HeLa PgpOFF cells (white) cells and HeLa PgpON cells (gray) are depicted. Results are mean ± SD values of triplicate samples from one representative experiment of 3 independent experiments (*P < 0.05, **P < 0.01).
Since treatment with TRAIL may up- or down-regulate the cell surface expression levels of TRAIL receptors on the surface target cells [28,29], we analyzed the levels of DR4 and DR5 expressed in PgpON/OFF cells without or after treatment with 7.5 and 15 ng/ml TRAIL for 24 h, using flow cytometry. Fig. 10 demonstrates that TRAIL treatment slightly down regulated DR4 receptor and up regulated the DR5 receptor. However, these TRAIL-induced variations in receptor levels are similarly exhibited by both Pgp-on and Pgp-off cells. Therefore is independent of Pgp expression. The DcR1 and DcR2 receptors were not detected on the cell surface before and after treatment with TRAIL (data not shown). These results indicate that the resistance of Pgp-expressing cells to TRAIL is not related to specific variation of TRAIL-receptor expression levels in these cells.
Fig. 10.

Expression levels of DR4 and DR5 in PgpON/OFF HeLa cells before and after TRAIL-treatment. PgpOFF (white) and PgpON (gray) HeLa cells were treated with 7.5 or 15 ng/ ml TRAIL for 24 h. Cells were then stained with monoclonal anti-DR4 (A) or anti-DR5 (B) antibodies, conjugated to PE, or PE-conjugated mouse IgG1 (isotype control). After washing, cells were analyzed by flow cytometry. FAB = fold above background = the ratio between median fluorescence of positively labeled cells and the median fluorescence of the isotype control. Results are mean ± SD values of triplicate samples from one representative experiment of 3 independent experiments (*P< 0.05, **P< 0.01).
4. Discussion
It is well established that over-expression of Pgp (which is encoded by the MDR1 gene and is overexpressed in various tumors) reduces the accumulation of various drugs below therapeutic threshold, thereby causes drug resistance in malignant cells. The question whether these cells retain susceptibility toward death ligands is of extreme importance because of the possibility for targeting cancer cells via extrinsic apoptosis signaling pathway therapy [8–11]. About half of tumor cell lines are TRAIL-resistant and this resistance may vary in primary human tumor cells. Some tumor cells appear to be inherently resistant to TRAIL and resistance can also be acquired in cells that were originally found to be sensitive to TRAIL [9].
In recent years a number of laboratories have proposed a role for Pgp in regulating cell death. However, the conclusions of these studies were controversial. Some groups have correlated expression of Pgp in drug selected cell lines with either decreased or increased sensitivity to death induced by TNF and FAS ligand. These discrepancies can be explained by the fact that Pgp-overexpressed, drug selected cell lines undergo a range of different genetic mutations resulting in altered expression of many cellular proteins which could affect various biological processes including apoptosis. Therefore, it was not possible to accurately determine which effects were due to Pgp expression and which were due to increased or decreased expression of other cellular proteins [30]. To address whether Pgp activity is involved in intrinsic resistance toward death ligands, we focused our study on HeLa cells containing a tetracycline-repressible plasmid system, which shuts down Pgp expression in the presence of tetracycline [20]. This system is most specific to determine Pgp role in conferring resistance of malignant cells toward death ligands. Because in these cells, addition of tetracycline rapidly turns off transcription of MDR1 mRNA and over a period of a few days Pgp is not expressed on the cells-surface. Therefore allowing a comparison of the same cells with and without Pgp in the plasma membrane. Indeed, we detected only Pgp-specific alterations in these cells. Other ABC transporters examined remained unchanged. Moreover, the level of death receptors (Fas, TNFR1, TNFR2, DR4, DR5, DcR1 and DcR2) is similar in the PgpON/OFF system. Therefore, these receptors are not a factor involved in the resistance of Pgp-expressing cells to death ligand-mediated killing.
Pgp-expressing cells were significantly less susceptible to TRAIL-induced apoptosis, in a dose- and time-dependent manner. Lysis of these cells by the other death-ligands such as FasL and TNF-α, was found to be Pgp-unrelated. These results are of great importance since recombinant TRAIL is considered a most promising cytokine for anti-cancer therapy. Its cytotoxic activity is considered selective toward malignant cells compared to normal cells and at least six TRAIL-related drugs have already been used in humans [8–11,29,31].
Of note, it was previously proposed that Pgp might play a role in resistance of cells to intrinsic apoptosis, independent of its role in drug efflux [32–34]. It has been suggested that Pgp inhibits apoptosis by interfering with components of ceramide metabolisms or by modulating the electrochemical gradient across the plasma membrane. However, in our study Pgp expressing cells were specifically more resistant to TRAIL-induced apoptosis while other apoptosis inducing factors (FasL, TNF-α, serum starvation and camptothecin) lysed similarly both Pgp expressing and non-expressing target cells. The TRAIL apoptotic specificity in relation to Pgp expression and activity is demonstrated by the use of the specific tetracycline-repressible cellular system, which avoids developing possible evolutionary adaptations that occasionally are seen in cells subjected to drug selections over long periods of time in culture.
Blocking Pgp activity by R-VRP resulted in similar levels of TRAIL-induced apoptosis in both Pgp-expressing and non-expressing cells. These results demonstrate that Pgp transport function (and not its mere presence on the cell membrane) is required to confer resistance to TRAIL. These findings were further verified applying an inhibitor of Pgp (CsA) and the Pgp-specific UIC2 antibody [24] which specifically inhibit irreversibly Pgp on the cell surface. Thus further indicating the specific role that Pgp plays in conferring resistance to TRAIL-induced apoptosis. However, TRAIL did not induce transport-related conformational changes in Pgp, suggesting TRAIL is not a substrate transported by Pgp. Thus, it is possible that the efflux of an apoptotic mediator molecule by Pgp may be coinvolved in acquired resistance to TRAIL-induced lysis.
A previous study demonstrated that a significant fraction of Pgp resides in lipid rafts microdomains [35]. Also the death ligand receptors DR4 and DR5 are located at the same lipid rafts microdomains and the death-mediated apoptotic signaling is dependent upon lipid rafts [36]. Moreover, Pgp, DR4, and DR5 were shown to co-immunoprecipitate [28], suggesting that cross-talk between Pgp and TRAIL receptor may occur in lipid raft microdomains. Although yet speculative, this might be a mechanism by which Pgp reduces TRAIL-mediated apoptosis. However, as functional inhibition of Pgp is required to restore full TRAIL-mediated death, a functional Pgp is probably required for such Pgp-TRAIL receptors interactions. Based on these previously published studies and the current study we propose a cross-talk model between Pgp and TRAIL receptors in the extrinsic cell death pathway by TRAIL (Fig. 11). Further, extensive studies might clarify the exact interactions between TRAIL receptors and Pgp.
Fig. 11.
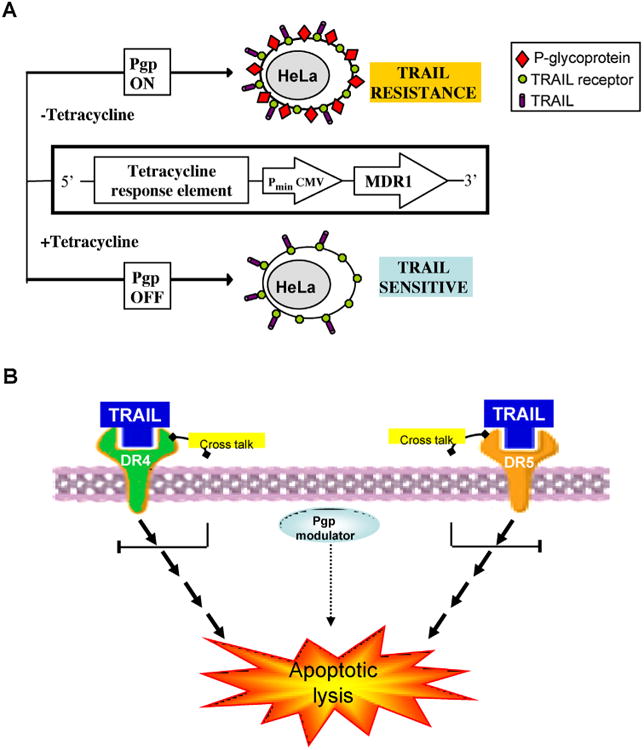
Modulation model of TRAK-extrinsic apoptosis by Pgp activity. (A) In the HeLa cellular system of tetracycline ON/OFF. The tetracycline-response-element (TRE) is located upstream of the minimal immediate early promoter of cytomegalovirus (PminCMV), which is silent in the absence of activation. In the absence of tetracycline, the tetracycline-trans-activator (tTA) binds the TRE and thereby activates transcription of MDR1 gene and its active product Pgp. In the presence of tetracycline, the (tTA) does not bind the TRE and thereby transcription of MDR1 is silenced. The Pgp-nonexpressing HeLa cells are sensitive to TRAIL-specific death. The Pgp-expressing cells are TRAIL-resistant but resensitized by Pgp-modulators, (B) the proposed model for the involvement of Pgp-activity inthe inhibition of TRAIL-dependent apoptosis. Based on the data of this study and the previous studies [35,36] that demonstrated co-localization of Pgp, TRAIL DR4 and DR5 receptors in the cell membrane lipid rafts micro-domains as well as co-immuno precipitation of both [28] – we propose a model of cross-talk between Pgp and both DR4 and DR5 receptors of TRAIL. This cross talk involves Pgp transport activity, as TRAIL-dependent death is restored by Pgp-modulators.
Since not only TRAIL itself but also monoclonal antibodies that target TRAIL receptors DR4 and DR5 are in clinical trials, we examined whether resistance conferred by Pgp to TRAIL is both DR4- and DR5-dependent. Pgp-expressing cells remained more resistant than Pgp non-expressing cells to both DR4- and DR5- mediated TRAIL induced lysis. Furthermore, TRAIL treatment had similar effects on the cell surface expression of DR4 and DR5 in both Pgp-expressing and non-expressing cells. These data suggest that TRAIL-induced apoptosis is both DR4 and DR5 dependent. Conflicting results exist on the human MCF-7 breast cancer cell line and its Pgp-expressing variant BC-19 [28]. Pgp-expressing BC-19 cells were more susceptible to TRAIL than their drug-sensitive variants, and their lysis was only DR5-dependent, while parental cells were lysed in a DR4- and DR5-dependent manner. It is possible that the other Pgp unrelated factors that were demonstrated to be changed in these cell variants (such as DR4, DR5 and DcR1 expression levels) may explain these conflicts.
In summary, we demonstrate in this research that malignant cells that express Pgp are more resistant to killing by TRAIL due to Pgp activity. Furthermore, we have shown that inhibition of Pgp activity by Pgp modulating drugs re-sensitized Pgp-expressing malignant cells to killing by TRAIL. Therefore, our findings might also have clinical implications, suggesting TRAIL-based therapy in combination with administration of Pgp-modulating agents might improve treatment outcome for Pgp expressing malignant cells.
Acknowledgments
This study has been performed as part of T. O-G Ph.D. thesis. This study supported in part by grants from The Israel Science Foundation (ISF494/04-08) and Israel Cancer Association (ICA # 20052008) granted to H.G & A.N.
Abbreviations
- ABC
ATP-binding cassette
- APC
Allophycocyanin
- BSA
bovine serum albumin
- CHX
cycloheximide
- CsA
cyclosporine A
- DMEM
Dulbecco's modified Eagle's medium
- DMSO
dimethyl sulfoxide
- D-PBS
Dulbecco's phosphate buffered saline
- FasL
Fas ligand
- FBS
foetal bovine serum
- FITC
fluorescein isothiocyanate
- HRP
horse radish peroxidase
- MDR
multidrug resistance
- PCR
polymerase chain reaction
- PE
phycoerythrin
- Pgp
P-glycoprotein
- PI
propidium iodide
- Rh123
Rhodamine 123
- R-VRP
R-verapamil
- TMB
tetramethylbenzidine
- TMD
transmembrane domains
- TNF
tumor necrosis factor
- TRAIL
TNF-related apoptosis-inducing ligand
References
- 1.Ferrín G, Linares CI, Muntané J. Mitochondrial drug targets in cell death and cancer. Curr Pharm Des. 2011;17:2002–16. doi: 10.2174/138161211796904803. [DOI] [PubMed] [Google Scholar]
- 2.Gonzalvez F, Ashkenazi A. New insights into apoptosis signaling by Apo2L/ TRAIL. Oncogene. 2010;29:4752–65. doi: 10.1038/onc.2010.221. [DOI] [PubMed] [Google Scholar]
- 3.Eissner G, Kolch W, Scheurich P. Ligands working as receptors: reverse signaling by members of the TNF superfamily enhance the plasticity of the immune system. Cytokine Growth Factor Rev. 2004;15:353–66. doi: 10.1016/j.cytogfr.2004.03.011. Review. [DOI] [PubMed] [Google Scholar]
- 4.Watts TH. TNF/TNFR family members in costimulation of T cell responses. Annu Rev Immunol. 2005;23:23–68. doi: 10.1146/annurev.immunol.23.021704.115839. Review. [DOI] [PubMed] [Google Scholar]
- 5.Strasser A, Jost PJ, Nagata S. The many roles of FAS receptor signaling in the immune system. Immunity. 2009;30:180–92. doi: 10.1016/j.immuni.2009.01.001. Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Falschlehner C, Schaefer U, Walczak H. Following TRAIL's path in the immune system. Immunology. 2009;127:145–54. doi: 10.1111/j.1365-2567.2009.03058.x. Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Smyth MJ, Cretney E, Kelly JM, Westwood JA, Street SE, Yagita H, et al. Activation of NK cell cytotoxicity. Mol Immunol. 2005;42:501–10. doi: 10.1016/j.molimm.2004.07.034. [DOI] [PubMed] [Google Scholar]
- 8.Holoch PA, Griffith TS. TNF-related apoptosis-inducing ligand (TRAIL): a new path to anti-cancer therapies. Eur J Pharmacol. 2009;625:63–72. doi: 10.1016/j.ejphar.2009.06.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Abdulghani J, El-Deiry WS. TRAIL receptor signaling and therapeutics. Expert Opin Ther Targets. 2010;14:1091–108. doi: 10.1517/14728222.2010.519701. [DOI] [PubMed] [Google Scholar]
- 10.Pavet V, Portal MM, Moulin JC, Herbrecht R, Gronemeyer H. Towards novel paradigms for cancer therapy. Oncogene. 2011;30:1–20. doi: 10.1038/onc.2010.460. [DOI] [PubMed] [Google Scholar]
- 11.Sayers TJ. Targeting the extrinsic apoptosis signaling pathway for cancer therapy. Cancer Immunol Immunother. 2011;60:1173–80. doi: 10.1007/s00262-011-1008-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ambudkar SV, Kimchi-Sarfaty C, Sauna ZE, Gottesman MM. P-glycoprotein: from genomics to mechanism. Oncogene. 2003;22:7468–85. doi: 10.1038/sj.onc.1206948. Review. [DOI] [PubMed] [Google Scholar]
- 13.Fung KL, Gottesman MM. A synonymous polymorphism in a common MDR1 (ABCB1) haplotype shapes protein function. Biochim Biophys Acta. 2009;1794:860–71. doi: 10.1016/j.bbapap.2009.02.014. Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Goldstein LJ, Galski H, Fojo A, Willingham M, Lai SL, Gazdar A, et al. Expression of a multidrug resistance gene in human cancer. J Natl Cancer Inst. 1989;81:116–24. doi: 10.1093/jnci/81.2.116. [DOI] [PubMed] [Google Scholar]
- 15.Galski H, Sullivan M, Willingham MC, Chin KV, Gottesman MM, Pastan I, et al. Expression of a human multidrug resistance cDNA (MDR1) in the bone marrow of transgenic mice: resistance to daunomycin-induced leukopenia. Mol Cell Biol. 1989;9:57–63. doi: 10.1128/mcb.9.10.4357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hirose M. Biology and modulation of multidrug resistance (MDR) in hematological malignancies. Int J Hematol. 2002;76(Suppl. 2):206–9. doi: 10.1007/BF03165119. [DOI] [PubMed] [Google Scholar]
- 17.Ford JM. Experimental reversal of P-glycoprotein mediated multidrug resistance by pharmacological chemosensitizers. Eur J Cancer. 1996;32:991–1001. doi: 10.1016/0959-8049(96)00047-0. [DOI] [PubMed] [Google Scholar]
- 18.Thomas H, Coley HM. Overcoming multidrug resistance in cancer: an update on the clinical strategy of inhibiting p-glycoprotein. Cancer Control. 2003;10:159–65. doi: 10.1177/107327480301000207. [DOI] [PubMed] [Google Scholar]
- 19.Galski H, Sivan H, Lazarovici P, Nagler A. In vitro and in vivo reversal of MDR1-mediated multidrug resistance by KT-5720: implications on hematological malignancies. Leuk Res. 2006;30:1151–8. doi: 10.1016/j.leukres.2006.02.016. [DOI] [PubMed] [Google Scholar]
- 20.Alemán C, Annereau JP, Liang XJ, Cardarelli CO, Taylor B, Yin JJ, et al. P-glycoprotein, expressed in multidrug resistant cells, is not responsible for alterations in membrane fluidity or membrane potential. Cancer Res. 2003;63:3084–91. [PubMed] [Google Scholar]
- 21.Findling-Kagan S, Sivan H, Ostrovsky O, Nagler A, Galski H. Establishment and characterization of new cellular lymphoma model expressing transgenic human MDR1. Leuk Res. 2005;29:407–14. doi: 10.1016/j.leukres.2004.09.001. [DOI] [PubMed] [Google Scholar]
- 22.van Engeland M, Ramaekers FC, Schutte B, Reutelingsperger CP. A novel assay to measure loss of plasma membrane asymmetry during apoptosis of adherent cells in culture. Cytometry. 1996;24:131–9. doi: 10.1002/(SICI)1097-0320(19960601)24:2<131::AID-CYTO5>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 23.Huerta S, Goulet EJ, Huerta-Yepez S, Livingston EH. Screening and detection of apoptosis. J Surg Res. 2007;139:143–56. doi: 10.1016/j.jss.2006.07.034. Review. [DOI] [PubMed] [Google Scholar]
- 24.Goda K, Fenyvesi F, Bacsó Z, Nagy H, Márián T, Megyeri A, et al. Complete inhibition of P-glycoprotein by simultaneous treatment with a distinct class of inhibitors and the UIC2 monoclonal antibody. J Pharmacol Exp Ther. 2007;320:81–8. doi: 10.1124/jpet.106.110155. [DOI] [PubMed] [Google Scholar]
- 25.Nagy H, Goda K, Arceci R, Cianfriglia M, Mechetner E, Szabó G., Jr P-glycoprotein conformational changes detected by antibody competition. Eur J Biochem. 2001;268:2416–20. doi: 10.1046/j.1432-1327.2001.02122.x. [DOI] [PubMed] [Google Scholar]
- 26.Notarbartolo M, Cervello M, Poma P, Dusonchet L, Meli M, D'Alessandro N. Expression of the IAPs in multidrug resistant tumor cells. Oncol Rep. 2004;11:133–6. [PubMed] [Google Scholar]
- 27.Hoki Y, Fujimori A, Pommier Y. Differential cytotoxicity of clinically important camptothecin derivatives in P-glycoprotein-overexpressing cell lines. Cancer Chemother Pharmacol. 1997;40:433–8. doi: 10.1007/s002800050682. [DOI] [PubMed] [Google Scholar]
- 28.Park SJ, Wu CH, Choi MR, Najafi F, Emami A, Safa AR. P-glycoprotein enhances TRAIL-triggered apoptosis in multidrug resistant cancer cells by interacting with the death receptor DR5. Biochem Pharmacol. 2006;72:293–307. doi: 10.1016/j.bcp.2006.04.024. [DOI] [PubMed] [Google Scholar]
- 29.Thorburn A, Behbakht K, Ford H. TRAIL receptor-targeted therapeutics: resistance mechanisms and strategies to avoid them. Drug Resist Update. 2008;2:17–24. doi: 10.1016/j.drup.2008.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Johnstone RW, Ruefli AA, Tainton KM, Smyth MJ. A role for P-glycoprotein in regulating cell death. Leuk Lymphoma. 2000;38:1–11. doi: 10.3109/10428190009060314. Review. [DOI] [PubMed] [Google Scholar]
- 31.Bellail AC, Qi L, Mulligan P, Chhabra V, Hao C. TRAIL agonists on clinical trials for cancer therapy: the promises and the challenges. Rev Recent Clin Trials. 2009;4:34–41. doi: 10.2174/157488709787047530. Review. [DOI] [PubMed] [Google Scholar]
- 32.Pallis M, Turzanski J, Higashi Y, Russell N. P-glycoprotein in acute myeloid leukaemia: therapeutic implications of its association with both a multidrug-resistant and an apoptosis-resistant phenotype. Leuk Lymphoma. 2002;43:1221–8. doi: 10.1080/10428190290026277. Review. [DOI] [PubMed] [Google Scholar]
- 33.Tainton KM, Smyth MJ, Jackson JT, Tanner JE, Cerruti L, Jane SM, et al. Mutational analysis of P-glycoprotein: suppression of caspase activation in the absence of ATP-dependent drug efflux. Cell Death Differ. 2004;11:1028–37. doi: 10.1038/sj.cdd.4401440. [DOI] [PubMed] [Google Scholar]
- 34.Mizutani T, Masuda M, Nakai E, Furumiya K, Togawa H, Nakamura Y, et al. Genuine functions of P-glycoprotein (ABCB1) Curr Drug Metab. 2008;9:167–74. doi: 10.2174/138920008783571756. Review. [DOI] [PubMed] [Google Scholar]
- 35.Bacso Z, Nagy H, Goda K, Bene L, Fenyvesi F, Matkó J, et al. Raft and cytoskeleton associations of an ABC transporter: P-glycoprotein. Cytometry A. 2004;61:105–16. doi: 10.1002/cyto.a.20081. [DOI] [PubMed] [Google Scholar]
- 36.Gajate C, Mollinedo F. Cytoskeleton-mediated death receptor and ligand concentration in lipid rafts forms apoptosis-promoting clusters in cancer chemotherapy. J Biol Chem. 2005;280:11641–47. doi: 10.1074/jbc.M411781200. [DOI] [PubMed] [Google Scholar]