

Introduction
Exercise is one of the mainstay clinical interventions for the prevention and treatment of cardiovascular disease. Not only does exercise reduce cardiovascular risk factors, such as diabetes and hypertension, thereby helping prevent heart disease, it also appears to improve the functional status and outcomes in patients with existing heart disease.1-6 The cardiovascular benefits of exercise are multifactorial, and include important systemic effects (Figure 1) on skeletal muscle, the peripheral vasculature, and metabolism, as well as beneficial alterations within the myocardium itself.7, 8
Figure 1. Overview of the systemic and cardiac-specific effects of exercise.
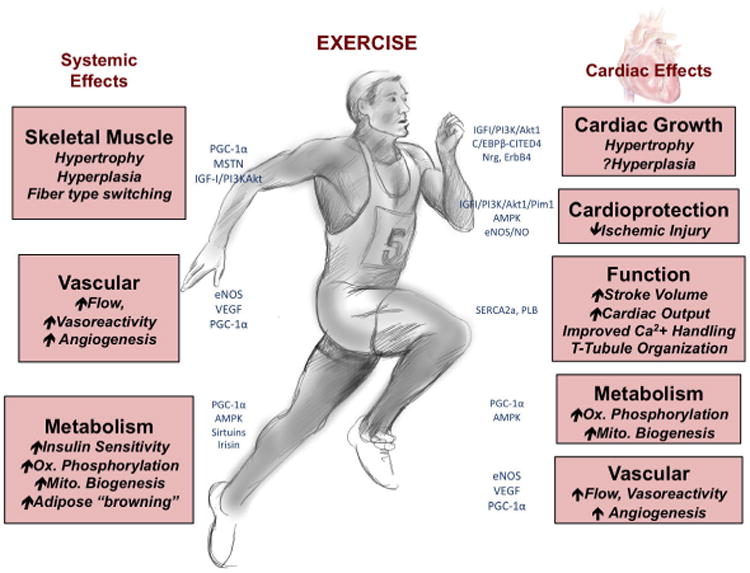
Endurance exercise has multiple systemic effects, ranging from increased skeletal muscle growth to vascular remodeling and improved energetics. Exercise also exerts direct effects on the heart itself, including increased cardiac growth, protection against ischemic damage, and modulation of cardiac function, metabolism, and vascular supply. (Illustration by AR)
Many current pharmacological treatments for cardiovascular disease are targeted towards inhibiting the adverse remodeling process associated with pathological stress. Specifically, they focus on abrogating the pathological hypertrophy, fibrosis, electrical remodeling, and cavity dilatation that accompany disease states such as longstanding hypertension and myocardial infarction.9-11 Interestingly, exercise, like many of these pathological stimuli, can also induce cardiac and cardiomyocyte hypertrophy. However, growing evidence suggests that such physiological remodeling, rather than leading to adverse sequelae, may actually be cardioprotective and that activating pathways associated with exercise can help to prevent and treat cardiovascular disease.8, 12, 13
In this review, we discuss recent advances in our understanding of the cellular and molecular mechanisms (Figure 2) that mediate the cardiac response to exercise, including cardiomyocyte hypertrophy and renewal, vascular remodeling, and alterations in calcium handling and metabolism. In addition to classical signaling mechanisms and transcriptional networks, we describe the role of secreted molecules and miRNAs. Finally, an emerging theme is that pathways that are either regulated by exercise or that mediate the heart's response to exercise often also have the potential to mitigate or even reverse cardiac disease. Thus, we suggest that understanding the effects of exercise more fully may provide useful biological insights and open the door to new therapeutic approaches aimed at restoring cardiovascular health.
Figure 2. Key signaling pathways involved in mediating exercise-induced cardiac remodeling.
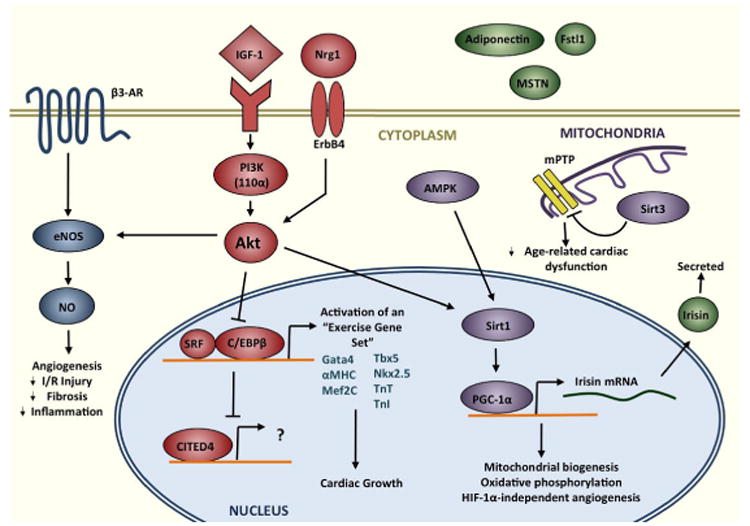
Exercise activates the IGF1-PI3K-Akt cascade. Signals then converge at the level of the nucleus, resulting in inhibition of the transcription factor C/EBPβ. Down-regulation of C/EPBβ, in turn, frees SRF to bind target gene promotors, contributing to activation of an “exercise gene set,” and, ultimately, cardiac growth. Meanwhile, activation of CITED4 may drive cardiomyocyte proliferation, as does signaling through Nrg1 and ErbB4. Akt also mediates angiogenesis and vascular remodeling via eNOS, and exerts beneficial metabolic effects through cross-talk with AMPK, Sirt1, and PGC-1α. In mitochondria, Sirt3, which is activated by exercise, works to protect against age-related cardiac dysfunction. Finally, exercise also modulates the secretion of circulating factors, primarily from skeletal muscle and adipocytes, such as adiponectin, myostatin (MSTN), irisin, and Fstl1.
Physiological Cardiac Remodeling
Exercise is perhaps one of the cheapest — and most effective — interventions for reducing the morbidity and mortality of cardiovascular disease.14 In fact, as little as 45-75 minutes of brisk walking each week appears to reduce the relative risk for adverse cardiac events.15, 16 Additionally, exercise-based cardiac rehabilitation is recommended by the American Heart Association (AHA) as one of the mainstay interventions following acute myocardial infarction (MI), with maximal benefit derived from early initiation of exercise (as early as one week post MI-hospital discharge) and from increased duration of exercise rehabilitation.1-3 Multiple studies have also demonstrated a dose-response relationship between exercise and cardiovascular benefit, but the shape of that curve, and the optimal dosage, intensity, frequency, and duration of exercise remain incompletely defined.15, 17, 18
The health benefits of exercise are multifactorial. Studies have demonstrated that physical activity is effective in reducing adipocyte mass and body mass index as well as positively affecting insulin sensitivity, glucose uptake by skeletal muscle, and cholesterol profiles.19 Physical activity — aerobic exercise, in particular — has also been associated with beneficial changes in both the systemic and coronary vasculature, including enhanced endothelial-mediated vasodilation, improved arterial compliance, and reductions in both systolic and diastolic blood pressure.20-22 Although these global effects of exercise are all implicated in improving cardiovascular health, here, we will focus primarily on the cardiac-specific effects of exercise.
Cardiac Growth
The heart has considerable plasticity9 and its capacity to hypertrophy in response to pathological stimuli, such as hypertension, aortic stenosis, or genetic mutations, is familiar to clinical cardiologists. However, a robust hypertrophic response is also seen with physiological stimuli, including exercise, pregnancy, and postnatal growth. Endurance exercise and pregnancy, for example, can induce up to a 20% increase in left ventricular (LV) mass, while, even more impressively, the hearts of Burmese pythons can grow by up to 40% following meals.23, 24
The cellular response to growth signals is often categorized as either hypertrophic — an increase in cell size — or hyperplastic — an increase in cell number. The adult heart has traditionally been viewed as capable only of hypertrophic growth; however, recent data from animal models and human studies suggest that the heart also has a limited capacity to generate new cardiomyocytes from progenitor cells and existing cardiomyocytes.25-27 In clinical practice, it is impossible, with current imaging modalities, to distinguish between these two distinct mechanisms of growth when characterizing cardiac hypertrophy. However, animal studies suggest that an increase in both cardiomyocyte size and number may contribute to heart growth in response to pathological and physiological stimuli.12, 28
Exercise-induced cardiac remodeling is the prototypical example of physiological cardiac growth, and the hypertrophic response to exercise can broadly be described as either concentric or eccentric hypertrophy, or a combination of the two. Isometric exercises — strength training activities like weight lifting — lead to transient increases in systemic vascular resistance, thereby increasing afterload and predominantly produce concentric hypertrophy, in which sarcomere fibers are added in parallel with subsequent thickening of the ventricular wall. Endurance — or isotonic — exercise, such as swimming and running, present a volume challenge to the heart and tend to result in eccentric hypertrophy, with increased preload and end-diastolic volume.29, 30 Cardiac MRI studies have suggested that isometric exercises induce minimal changes in right ventricular (RV) structure and function, while isotonic exercises lead to a balanced biventricular hypertrophy with symmetric enlargement of both the right and left ventricles.31 Cardiomyocyte hypertrophy is likely the dominant contributor to exercise-induced heart growth, and studies have reported an increase in cardiomyocyte size by up to 17-32% following exercise training.32 As noted above, however, recent work suggests that exercise also induces markers of cardiomyocyte proliferation, although the fate and contribution of these newly formed cells remains to be established.12
A recently described model for studying physiological remodeling is the Burmese python, which demonstrates an impressive increase in cardiac size — up to 40% — following meals, which regresses over the subsequent 28 days.24 Emerging data suggest that this increase in heart size is primarily a hypertrophic, rather than hyperplastic, process, that it is not associated with the characteristic changes seen in pathological cardiac growth such as fibrosis and upregulation of the fetal gene program.33 This lends support to the idea that physiological hypertrophy is primarily an adaptive and beneficial process. Interestingly, new evidence suggests that some of these postprandial cardiac growth effects are mediated by secreted lipids,33 which will be discussed in more detail below. It should be acknowledged that the clinical relevance of post-prandial changes in the Burmese python remain unclear. Interestingly, the combination of fatty acids identified in python serum also induced cardiomyocyte hypertrophy in mice.33
Altered Ca2+ Handling
In contrast to pathological cardiac remodeling, in which hypertrophy is associated with fibrosis, impaired relaxation and contractility, and potential progression to heart failure, both systolic and diastolic cardiac function are preserved, or even enhanced, in exercise-induced hypertrophy.
Exercise has been shown to improve cardiomyocyte Ca2+ sensitivity and contractility.34 In animal models, fractional shortening can increase by as much as 40-50% after endurance exercise, with concomitant improvements in contraction and relaxation.35 Up-regulation of SERCA2a, which is characteristically decreased in pathological remodeling, as well as increased phosphorylation of phospholamban (PLB), which reduces its inhibition of SERCA2a, appear to contribute to the observed benefits of exercise on Ca2+ handling.36, 37 Interestingly, studies in animal models suggest that exercise can improve contractility and normalize SERCA2a function following myocardial infarction.38-40 Structurally, physiological hypertrophy, unlike pathological hypertrophy, is associated with preservation of t-tubule density and organization, which allows for coordinated myocyte contraction.41
Vascular Remodeling
Exercise significantly increases myocardial oxygen demand, and induces changes within the macro- and microvasculature to meet these requirements. While important vascular changes occur both in the heart and the periphery, here we focus on changes within the heart itself. Specifically, exercise is associated with increased coronary blood flow and oxygen extraction, as well as improved endothelial function.42
Endurance training increases coronary blood flow in a number of different ways. Imaging studies in humans show an increase in the caliber of the large proximal coronary arteries following exercise, in proportion to the increase in LV mass.43, 44 In patients with coronary artery disease, endothelium-dependent coronary vasodilation, and subsequently myocardial perfusion, is improved, albeit not to normal levels.20 Exercise also induces angiogenesis in a VEGF-dependent manner.45 Capillary density increases following initiation of exercise, but, after sustained activity, normalizes to the extent of cardiac hypertrophy.46 This is in direct contrast to pathological cardiac remodeling: while pathological stimuli initially induce angiogenesis, some studies suggest that coordination of hypertrophy and angiogenesis is ultimately disrupted, contributing to the progression to heart failure.47, 48 Interestingly, animal studies have shown that exercise can promote angiogenesis following myocardial infarction, with significant improvements in myocardial perfusion and pump function.49 Thus some of the beneficial effects of exercise are likely related to increased angiogenesis and protective changes in the coronary vasculature. A crucial component of these vascular changes is the up-regulation of nitric oxide (NO) production by vascular endothelial cells. The precise cellular and biochemical mechanisms regulating NO production and its downstream effects will be discussed below.
Metabolism
The heart has tremendous energy requirements, both in physiological and pathological states, and a prominent feature of cardiovascular disease is myocardial metabolic dysregulation. Notably, pathological remodeling is associated with a switch from fatty acid metabolism, the primary energy source for the healthy adult human heart, to glucose utilization, which is the main energy source in fetal life.33 In contrast, energy consumption and homeostasis is preserved in physiological cardiac remodeling. An acute bout of physical activity can increase cardiac output as much as six-fold, and this significant ATP demand is met primarily by mitochondrial oxidative phosphorylation.50 Exercise training promotes efficient glucose and fatty acid handling, as well as mitochondrial biogenesis via up-regulation of the glucose sensor AMPK and its downstream target PGC-1α.50
Cardiomyocyte Renewal
As noted above, multiple studies have demonstrated that the heart does indeed have some capacity for regeneration and renewal, with data supporting both the proliferation of pre-existing cardiomyocytes and resident stem cells.25, 51-53 Adult zebrafish and neonatal mice (up to 7 days old) are able to fully regenerate cardiac muscle following apical resection, with restoration of contractile function.54, 55 Independent studies involving carbon-14 dating of genomic DNA from people who were alive during nuclear testing or iododeoxyuridine incorporation into the DNA of chemotherapy patients suggest that cardiomyocyte renewal also occurs in humans throughout life.25, 51 However, the physiological signals that regulate this process remain unclear.
Interestingly, recent collaborative work from our group suggests that the heart's regenerative potential is dynamically regulated, and that endurance exercise may stimulate cardiomyocyte proliferation in vivo.12 These studies profiled expression of all known and putative transcriptional components of the mouse genome in hearts from exercised mice, and contrasted differentially regulated genes with those altered in response to pressure overload.12 Intriguingly, a significant subset of altered transcriptional components had known functions related to cell cycle progression or proliferation in other systems, and confocal immunohistochemistry confirmed a significant increase in all examined markers of cardiomyocyte proliferation in exercised hearts.12 These results are reminiscent of the hippocampal neurogenesis well-documented to occur in response to exercise, which involves the proliferation of precursor cells.56, 57, 58 Ultimately, cardiocyte lineage tracing experiments will be needed to determine the sources and fate of new cardiomyocytes that may form in response to exercise. However, an appealing hypothesis is that exercise may provide a proliferative and potentially regenerative signal affecting multiple tissues.
Molecular Mechanisms
IGF-1-PI3K-AKT Pathway
Genetic interventions in vitro and in vivo have elucidated many of the pathways that regulate cardiac growth. Pathological hypertrophy has previously been reviewed in detail,9, 10 and is associated with activation of G-protein coupled receptors by soluble factors such as angiotensin II and endothelin, signal transduction via, among others, the calcineurin-calmodulin axis and the MAPK pathway, and, ultimately, increases in protein synthesis, cellular growth, and a switch to the fetal gene program.9, 10
Physiological hypertrophy, on the other hand, is mediated primarily by the IGF-1-PI3Kinase (PI3K)-Akt axis. IGF-1 is produced by the liver, and, to a lesser extent, the heart. Exercise induces both hepatic secretion of IGF-1 into the bloodstream, as well as cardiac expression of IGF-1.59, 60 In the heart, IGF-1 binds its tyrosine kinase receptor, IGF-1R, and activates the PI3K-Akt cascade.61 Mice with constitutive overexpression of IGF-1 develop an increase in heart size, characterized by both cardiomyocyte hypertrophy and hyperplasia, and are protected against ischemic injury and heart failure.62-66 Activation of the IGF-1 receptor also recapitulates the physiologic hypertrophic phenotype, with increased cardiomyocyte size and preserved contractile function.67
The IGF1-R activates PI3K, which consists of a family of heterodimeric kinases composed of regulatory and catalytic subunits.68 Specifically, activation of the PI3K(110α) isoform has been implicated in the development of physiological cardiac hypertrophy. Mice with constitutively active PI3K(110α) exhibit significantly increased heart weights and are protected from heart failure after pathological stress, such as aortic banding and myocardial infarction.8, 69 In contrast, mice with cardiac expression of a dominant negative PI3K(110α) hypertrophy normally in response to pressure overload, but have blunted cardiac growth in response to swimming.70 This observation provides important evidence that distinct intracellular signaling mechanisms mediate physiological and pathological cardiac hypertrophy. Further support for this model is provided by our recent genome-wide profiling of transcriptional regulators, which revealed dramatically different profiles associated with these two kinds of growth.12
Akt1 is a major downstream effector of PI3K and becomes phosphorylated (activated) in physiological cardiac hypertrophy.68, 71 The effects of Akt1 in the heart are diverse, though generally beneficial.72, 73 These include inhibiting cardiomyocyte death,72, 73 improving calcium handling,74 and modulating cardiac growth and metabolism. Interestingly, germline genetic deletion of Akt1 abrogates the cardiac growth response to exercise, but results in exacerbated hypertrophy in response to pressure overload.71 Thus Akt1 is required for physiological hypertrophy, but appears to inhibit pathological hypertrophy in a manner thought to be mediated through cross-talk with MAPK signaling.71 Most recently, Akt1 has been implicated in promoting proliferation of cardiac stem cells and cardiomyocytes, largely through nuclear activation of a Pim1-dependent pathway.75-78 Akt1 also acts downstream of Nrg1-ErbB4 signaling, which induces cardiomyocyte proliferation in vitro and in vivo.27, 79 In turn, Akt1 exerts pro-proliferative effects via repression of the transcription factor C/EBPβ and activation of CITED4.12 C/EBPβ interacts with serum response factor (SRF) contributing to regulation of a so-called “physiological or exercise gene set,” a collection of genes with known roles in cardiomyocyte hypertrophy and differentiation, including Gata4, Tbx5, and Nkx2.5, whose expression levels are altered following endurance exercise. In fact, mice heterozygous for C/EBPβ recapitulate the physiological hypertrophic phenotype, with both cardiomyocyte hypertrophy and low levels of hyperplasia, suggesting that C/EBPβ is essential in mediating the cardiac effects of exercise.12
eNOS
Nitric Oxide (NO) is a ubiquitously expressed molecule that modulates multiple cardiovascular processes, including vascular tone, platelet activation, smooth muscle cell proliferation, and cardiomyocyte contractility.80 NO is produced by the vascular endothelium as well as the myocardium, where it is generated by the enzyme endothelial nitric oxide synthase (eNOS). Multiple studies have shown that eNOS-mediated production of NO is diminished in patients with heart failure. Interestingly, forced overexpression of eNOS in animal models can reduce the extent of LV dysfunction and remodeling following pathological stress, with overall improvements in mortality.81, 82
Exercise training reduces ischemic injury in animal models, and at least part of this benefit appears mediated by upregulation of eNOS. Exercise increases circulating catecholamines, such as epinephrine and norepinephrine, which act on β3-adrenergic receptors to increase eNOS phosphorylation and activity.13 This increases the bioavailability of the NO metabolites, nitrite and nitrosothiol, which can then be utilized in times of stress to antagonize adverse remodeling processes, such as fibrosis and pathological hypertrophy.13 Of note, the cardioprotective effects of exercise in preventing adverse remodeling are not seen in mice that are deficient in eNOS,13, 83 although this is potentially confounded by a reduction in the exercise levels achieved by such mice.84 Interestingly, this pathway also appears essential in promoting the cardioprotective effects of exercise that can persist after exercise training has ceased.13
Sirtuins, AMPK, and PGC-1α
Sirtuins are a family of NAD-dependent deacetylases, seven of which, Sirt1-7, are found in mammals and regulate a variety of cellular functions, including metabolism, cell growth, apoptosis, and aging.85 Sirt1 and Sirt3 are the best studied of the sirtuins in the heart. Sirt1 is upregulated following exercise, and has pro-growth and pro-survival functions in cardiomyocytes.86 Sirt3 is a mitochondrial sirtuin, and, like Sirt1, is upregulated with exercise.87 In the heart, Sirt3 protects against oxidative stress in a Foxo3a dependent manner, and has also been shown to regulate opening of the mitochondrial permeability transition pore (mPTP) via deacetylation of cyclophilin D.88, 89 This latter effect is thought to be protective against age-related cardiac dysfunction, and, in fact, Sirt3 knock-out mice exhibit accelerated pathological hypertrophy, fibrosis, and heart failure.89
Additionally, Sirt3 regulates cardiac metabolism via activation of AMPK and PGC-1α, both of which have been shown to inhibit maladaptive cardiac remodeling.88 AMPK is a serine/threonine kinase that senses energy levels within the cell and, together with its downstream effector, PGC-1α, coordinates metabolic responses to maintain energy homeostasis. Animals deficient in AMPK exhibit increased hypertrophy, accelerated heart failure, and also have increased infarct sizes after coronary artery ligation.90 PGC-1α is a transcriptional coactivator upregulated with exercise, and a potent inducer of mitochondrial biogenesis and oxidative phosphorylation. PGC-1α-deficient mice develop early signs of heart failure due to an inability of the heart to meet energy demands, thus emphasizing the importance of metabolic and energy homeostasis in cardiac health.50, 91 PGC-1α has also been shown to regulate a HIF-1-independent pathway of angiogenesis,92 thereby providing a mechanism for coordinately regulating mitochondrial function and blood supply in exercise.
Myokines, Adipokines, and other Secreted Molecules
Exercise also exerts a number of indirect benefits on the heart, mediated, in part, by endocrine factors secreted from skeletal muscle and adipocytes, termed myokines and adipokines, respectively. Fstl1, for example, is a glycoprotein secreted by both skeletal muscle and cardiac myocytes following exercise. Fstl1 activates the PI3K-Akt pathway in cardiomyocytes and vascular endothelial cells, and exerts antiapoptotic and vasodilatory effects.93
A novel hormone and myokine under the regulation of PGC-1α, called irisin, was recently described,94 and is implicated in some systemic effects of exercise. Following exercise, up-regulation of PGC-1α results in increased skeletal muscle secretion of irisin, the proteolytic cleavage product of the type I membrane protein, FNDC5.94 Irisin plays an important role in the browning of white adipose tissue and thermogenesis, and may play a role in many of the beneficial systemic effects of muscle PGC-1α expression, such as protection against age-related obesity and diabetes.94 Whether irisin also contributes to the cardiac benefits of PGC-1α will be of great interest for future studies.
Adiponectin is a hormone secreted by adipocytes, and has anti-inflammatory and anti-hypertrophic effects on the heart. Plasma adiponectin levels are decreased in patients with obesity and insulin resistance, as well as in associated cardiovascular diseases, such coronary artery disease and hypertension.95 Interestingly, there is a paradoxical increase in plasma adiponectin levels in heart failure patients, which has been attributed to downregulation of the adiponectin receptor (AdipoR1) and subsequent skeletal muscle adiponectin resistance.96, 97 Exercise training normalizes levels of adiponectin in heart failure patients, decreases adiponectin resistance, and also reverses heart failure-associated skeletal muscle wasting, or cardiac cachexia, which is an independent risk factor for mortality.98-100
In addition to adiponectin, an important regulator of cardiac cachexia is myostatin (MSTN), a member of the TGFβ superfamily and a potent negative regulator of skeletal muscle growth.101 Notably, cardiac-specific MSTN knockout mice are protected from pressure-overload-induced cardiac cachexia, as well as aging-related cardiac fibrosis and dysfunction.102 We also found that MSTN directly regulates cardiomyocyte growth in a stimulus-specific way,103 and knockout mice are protected against aging-related cardiac fibrosis and dysfunction.104 There are also data to suggest that MSTN levels are dynamically regulated with exercise. In a rat model of heart failure, elevated skeletal and myocardial MSTN levels associated with heart failure returned to baseline following four weeks of endurance exercise.105 In human heart failure patients, exercise also resulted in a significant reduction in skeletal muscle MSTN.106 Thus, it is possible, though speculative, that a reduction in MSTN and related peptides contributes to the cardiac benefits of exercise.
In addition to proteins and peptides, studies in Burmese pythons have also implicated lipids as being potential secreted factors that mediate physiological cardiac hypertrophy. As described above, Burmese pythons exhibit significant cardiac growth following meals. It has recently been shown that an infusion of three lipids—myristic acid, palmitic acid, and palmitoleic acid—found to be elevated in the serum of Burmese pythons postprandially, can induce cardiac growth in the fasting state, thereby recapitulating the hypertrophic phenotype seen following meals.33 Importantly, these lipids can also induce hypertrophy of cultured mammalian cardiomyocytes.33 Finally, in a clinical context, the Gerszten laboratory has recently performed plasma metabolite profiling on subjects before and after exercise.107 In addition to identifying metabolite signatures of potential clinical utility, these studies also revealed a subset of exercise-induced metabolites that regulate muscle expression of Nurr77, a nuclear hormone receptor important in regulating glucose and lipid metabolism.107 These studies add to growing evidence that metabolites and other small molecules regulate diverse physiological processes, likely including the cardiac response to exercise.
miRNAs
Since their discovery a little over a decade ago, miRNAs have become increasingly recognized for their pivotal roles in the regulation of development and disease.108 Recent studies identified multiple miRNAs that are highly, and almost exclusively, expressed in the heart and skeletal muscle.109, 110 Among these are miR-208a, miR-208b, and miR-499, which comprise a family of myosin heavy chain-encoded miRNAs, collectively termed Myomirs. These Myomirs have been implicated in a wide array of cardiovascular diseases, including cardiac hypertrophy, heart failure, arrhythmias, and congenital heart disease.111
In addition, there has also been much interest in examining the role of miRNAs in regulating the physiological changes of exercise. Multiple studies in both animal models and humans suggest that miRNAs are dynamically regulated with physical activity, and, moreover, that acute and chronic bouts of exercise impart differential changes in miRNA expression.112, 113 Of note, miRNAs are also known to be secreted into the bloodstream, both at rest and following tissue injury.114 Recently, changes in levels of such circulating miRNAs, or c-miRNAs, have been described following exercise.113 Further studies will be needed, however, to identify the cellular sources of these c-miRNAs, and also to better characterize their biological roles. For instance, are c-miRNAs secreted as a byproduct of stress and tissue injury, or might they also have important endocrine and paracrine functions?
Although exercise has been shown to regulate miRNA expression, the precise role of miRNAs in regulating physiological hypertrophy and exercise-induced cardiovascular remodeling remains unclear. It has been shown that miR-1 and miR-133, two of the most abundant miRNAs in cardiac myocytes, are down-regulated in both pathological and physiological hypertrophy, suggesting that they may mediate a nonspecific “cardiomyocyte growth” pathway.115 In a more specific exercise-focused study, female Wistar Rats subjected to swimming training were found to exhibit up-regulation of miR-29c.116 Interestingly, the miR-29 family targets a number of mRNAs that encode proteins essential for fibrosis, and up-regulation of these miRNAs has been associated with repression of fibroblast collagen deposition.117 It is possible, although still speculative, that the miR-29 family might be important in actively suppressing a fibrotic response in exercise-induced cardiac hypertrophy. Of note, miRNA-29c has also been shown to suppress the PI3K-Akt pathway.118 While activation of the PI3K-Akt axis is crucial to physiological remodeling, it is also known that its effects are dependent on the timing and chronicity of Akt activity.119 In particular, chronic Akt overexpression has been linked to maladaptive remodeling and heart failure. Thus miR-29 could be important in fine-tuning the activity of the PI3K-Akt axis, thereby maintaining tissue homeostasis and healthy cardiovascular function.
It has become clear that miRNA expression is dynamically, and differentially, regulated by pathological and physiological processes. This provides important implications for the development of both diagnostic and therapeutic tools for the treatment of cardiovascular disease, as will be discussed in more detail below.
Limitations
While substantial progress has been made in the understanding of exercise physiology, particularly as it pertains to the heart, certain limitations to these studies deserve mention. One of the most widely accepted animal models of pathological stress is the use of transverse aortic constriction (TAC) to increase afterload, thereby mimicking chronic hypertension or aortic stenosis and resulting in concentric hypertrophy. However, animal models of endurance training most commonly involve the use of isotonic exercises like treadmill running or swimming, which primarily result in cardiac growth via eccentric hypertrophy.30 These different patterns of growth could potentially confound any direct comparisons made between the two models. Nonetheless, in the absence of more directly comparable animal models, current studies have still provided important insight into mechanisms regulating pathological and physiological cardiac hypertrophy.
Most pathological stimuli are chronic persistent, whereas exercise, is intermittent raising the possibility that the distinct outcomes associated with these stimuli reflect quantitative rather than qualitative differences in exposure. To address this issue, Rockman and colleagues cleverly designed a model of intermittent pressure overload in mice. Interestingly, this induced pathological changes, which – while milder than those seen with persistent pressure overload – emphasizes the importance of qualitative differences independent of exposure duration120. Of interest, while most of the cardiac changes associated with endurance exercise are thought to be cardioprotective, with beneficial adaptations to calcium handling, metabolism, and vascular remodeling, recent cardiac MRI data identified a link between lifelong, competitive endurance exercise and an increased prevalence of myocardial fibrosis with subsequent risk for arrhythmias.121 Such clinical data raise the possibility that too much exercise may have adverse effects. However, observational data on exercise are inherently limited by issues such as self-selection and the possibility of unrecognized confounding. A recent analysis of exercise studies suggests that even moderate exercise may have adverse effects on risk factors such as systolic blood pressure and HDL in some subjects (126 of 1,687 analyzed)122. Why this occurs in a subset of subjects and whether these effects result in adverse clinical outcomes despite the other benefits of exercise remain unclear. Further studies are needed to delineate the precise shape of the exercise dose-response curve and characterize the contribution(s) of exercise duration, intensity, and frequency to cardiovascular effects of exercise.
Finally, as with all experimental animal models, findings in mice may not correlate with human pathophysiology. The molecular mechanisms of exercise-induced cardiac remodeling is particularly difficult to study in human beings, due to the limited availability of tissue samples from healthy subjects. The development of novel technologies to better identify and characterize secreted peptides, circulating miRNAs, and metabolites can help further our understanding of exercise-induced changes in humans in the absence of cardiac tissue. Identification of serum components that are similarly regulated in animals and people such as the secreted molecule irisin,94 may help establish parallels between human biology and animal models of exercise.
Clinical Implications
The beneficial effects of exercise in preventing and treating cardiovascular disease have long been appreciated. Data from the Framingham Heart Study, for example, show that exercise, or lack thereof, is an independent risk factor for cardiovascular disease, even after controlling for its effects on other risk factors, such as hypertension, hyperlipidemia, and diabetes.123, 124 Additionally, meta-analyses, and, more recently, a large, prospective randomized controlled trial, HF-ACTION, suggest that exercise training has significant benefits for patients with coronary artery disease or heart failure.4-6 Of note, in HF-ACTION, the largest randomized exercise trial to date, heart failure patients randomized to exercise had improved quality of life and a trend to reduced mortality that was only significant after adjustment for differences in baseline characteristics4, 5. Nevertheless, some might argue that we already have a simple prescription for these benefits: exercise. However, many patients may be unable to exercise, and thus understanding the pathways that mediate these benefits, and learning how to manipulate them in vivo could yield novel therapeutic approaches.
Genetic interventions in mice provide evidence that manipulation of pathways to mimic the changes that occur with exercise can protect against heart failure following pathological stress. For instance, upregulation of PI3K, Akt1, eNOS, and PGC-1α, as well as repression of the transcription factor C/EBPβ, all result in preserved cardiac contractility and decreased mortality after aortic banding or ischemia-reperfusion.8, 69, 82, 125, 126 The beneficial effects of these interventions are likely multifactorial, involving a combination of pro-growth and proliferation signals, increased angiogenesis, improved calcium handling and energetics, as well as suppression of fibrosis — a constellation of changes that encompass the physiological hypertrophic phenotype. The molecular regulators of these physiological adaptations may hold promise as potential targets for intervention in the treatment of cardiovascular disease.
These genetic studies provide important proof-of-concept that benefits accrue from recapitulating some of the central molecular changes induced by exercise. However, not all the pathways implicated are ideal candidates for therapeutic targeting. Current attempts at molecular interventions targeting intracellular molecules have been limited, in part, by the difficulty of designing small molecules that activate kinases or inhibit nuclear transcription factors. Moreover, many of these molecules have multi-systemic, pleiotropic effects, requiring cardiac-specificity to be engineered elsewhere in the system. On the other hand, secreted factors such as adiponectin, myostatin, or irsin may be more promising drug targets. Inhibitors of myostatin and related peptides, in fact, are already in clinical trials for the treatment of skeletal muscle dystrophies, and could conceivably be extended to use in heart failure patients should additional research support this concept.104, 127
miRNAs are also promising targets for clinical intervention, as, unlike intracellular enzymes or signaling molecules, they are easily targetable through the use of miRNA mimics and antagonists (antiMirs). Delivery of both miRNA mimics and antiMirs have been efficacious in vitro and in vivo, and could potentially be extended to the clinical setting as well.109, 128 Some miRNAs also have the advantage of tissue specificity, and most simultaneously modulate multiple signaling cascades via the inhibition of multiple cellular targets. However, many miRNAs are still ubiquitously expressed, and the efficacy of miRNA therapy, like that of peptide or small molecule drugs, may be limited by off-target effects on other organs or pathways.
As a result, there has been much interest in the development of targeted drug delivery tools to enhance delivery of therapies specifically to the heart. For instance, miRNA mimics, antiMirs, or other small molecules could be conjugated to targeting peptides or antibodies for specific uptake by cardiomyocytes. The use of similar nanoparticle drug delivery systems for the targeted drug delivery of chemotherapeutics to prostate cancer is in clinical trials,129 and it is conceivable that this technology could be extended for the delivery of medications to the heart as well. Additionally, adenoviral delivery systems are currently being investigated as a way to enhance cardiac-specificity as well as for the continuous expression of a particular gene or miRNA of interest, and a number of clinical trials involving adeno-associated viral systems for gene delivery are already under way and have demonstrated a favorable safety profile130 and even a suggestion of efficacy131. Finally, catheter-based intracardiac delivery is also being explored as a potential drug delivery tool to increase cardiac-specificity. It seems likely that none of these “exercise mimetics” will fully reproduce all the benefits of exercise. However, the discovery of novel therapeutic targets and the development of improved drug delivery technologies may lead to improved treatments for heart disease, particularly for those patients unable to exercise.
Finally, the exercised heart provides a prototype of the healthy heart that may serve as a useful tool for gauging the response to therapy. Our initial studies of transcriptional co-activators altered in exercised hearts demonstrated a distinct pattern from those altered in response to pressure-overload.12 More extensive profiling of exercised (and diseased) hearts could ultimately lay a foundation for a systems biology132 approach to cardiac health and disease, allowing interventions to be judged not simply by their effect on one putative target but by their ability to recapitulate the healthy profile (or reverse that of disease). Our studies defined a “physiological gene set” that was characteristically altered in exercised hearts.12 C/EBPβ appeared to function as a hub in this network, and genetically reproducing the exercise-induced change in C/EBPβ was sufficient to recapitulate the changes seen in slightly more than half of this gene set, and protected against heart failure.12 As noted above, the Gerszten laboratory has recently identified plasma metabolite profiles in humans indicative of exercise performance and cardiovascular disease susceptibility.107 These signatures may provide a useful gauge of the response to interventions. Taken together, these studies suggest value in delineating such signatures as a therapeutic road map and aid to evaluating clinical intervention.
Conclusion
Current pharmacological interventions in clinical use for heart failure are focused on preventing the maladaptive changes associated with sympathetic overdrive and activation of the renin-angiotensin-aldosterone system. Emerging technological advancements and a better understanding of fundamental biological processes have provided us with deepened insight into the cardiovascular effects of exercise. Indeed, physiological cardiac growth appears to encompass not only cardiomyocyte hypertrophy, but also low levels of cardiomyocyte hyperplasia, as well as increased angiogenesis, alterations in calcium handling and metabolism, and secretion of paracrine and endocrine mediators. Overall, these changes are cardioprotective, leading to preserved, possibly even enhanced, cardiac function, and present an enticing avenue both for identifying therapeutic interventions and judging their efficacy.
Acknowledgments
Funding Sources: This work was supported in part by grants from the NIH and a Leducq Foundation Network of Research Excellence (AR). NM is a trainee the Harvard/MIT Health Sciences and Technology Program and was supported by a Howard Hughes Predoctoral Fellowship.
Footnotes
Disclosures and conflicts of interest: None.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Anderson JL, Adams CD, Antman EM, Bridges CR, Califf RM, Casey DE, Chavey WE, Fesmire FM, Hochman JS, Levin TN, Lincoff AM, Peterson ED, Theroux P, Wenger NK, Wright RS, Smith SC, Jacobs AK, Adams CD, Anderson JL, Antman EM, Halperin JL, Hunt SA, Krumholz HM, Kushner FG, Lytle BW, Nishimura R, Ornato JP, Page RL, Riegel B. Acc/aha 2007 guidelines for the management of patients with unstable angina/non–st-elevation myocardial infarction—executive summary. Journal of the American College of Cardiology. 2007;50:652–726. [Google Scholar]
- 2.Antman EM, Anbe DT, Armstrong PW, Bates ER, Green LA, Hand M, Hochman JS, Krumholz HM, Kushner FG, Lamas GA, Mullany CJ, Ornato JP, Pearle DL, Sloan MA, Smith SC, Jr, Alpert JS, Anderson JL, Faxon DP, Fuster V, Gibbons RJ, Gregoratos G, Halperin JL, Hiratzka LF, Hunt SA, Jacobs AK. Acc/aha guidelines for the management of patients with st-elevation myocardial infarction--executive summary: A report of the american college of cardiology/american heart association task force on practice guidelines (writing committee to revise the 1999 guidelines for the management of patients with acute myocardial infarction) Circulation. 2004;110:588–636. doi: 10.1161/01.CIR.0000134791.68010.FA. [DOI] [PubMed] [Google Scholar]
- 3.Haykowsky MJ, Liang Y, Pechter D, Jones LW, McAlister FA, Clark AM. A meta-analysis of the effect of exercise training on left ventricular remodeling in heart failure patients: The benefit depends on the type of training performed. Journal of the American College of Cardiology. 2007;49:2329–2336. doi: 10.1016/j.jacc.2007.02.055. [DOI] [PubMed] [Google Scholar]
- 4.Flynn KE, Pina IL, Whellan DJ, Lin L, Blumenthal JA, Ellis SJ, Fine LJ, Howlett JG, Keteyian SJ, Kitzman DW, Kraus WE, Miller NH, Schulman KA, Spertus JA, O'Connor CM, Weinfurt KP. Effects of exercise training on health status in patients with chronic heart failure: Hf-action randomized controlled trial. JAMA. 2009;301:1451–1459. doi: 10.1001/jama.2009.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.O'Connor CM, Whellan DJ, Lee KL, Keteyian SJ, Cooper LS, Ellis SJ, Leifer ES, Kraus WE, Kitzman DW, Blumenthal JA, Rendall DS, Miller NH, Fleg JL, Schulman KA, McKelvie RS, Zannad F, Pina IL. Efficacy and safety of exercise training in patients with chronic heart failure: Hf-action randomized controlled trial. JAMA. 2009;301:1439–1450. doi: 10.1001/jama.2009.454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lawler PR, Filion KB, Eisenberg MJ. Efficacy of exercise-based cardiac rehabilitation post-myocardial infarction: A systematic review and meta-analysis of randomized controlled trials. American heart journal. 2011;162:571–584. e572. doi: 10.1016/j.ahj.2011.07.017. [DOI] [PubMed] [Google Scholar]
- 7.Bowles DK, Farrar RP, Starnes JW. Exercise training improves cardiac function after ischemia in the isolated, working rat heart. The American journal of physiology. 1992;263:H804–809. doi: 10.1152/ajpheart.1992.263.3.H804. [DOI] [PubMed] [Google Scholar]
- 8.McMullen JR, Amirahmadi F, Woodcock EA, Schinke-Braun M, Bouwman RD, Hewitt KA, Mollica JP, Zhang L, Zhang Y, Shioi T, Buerger A, Izumo S, Jay PY, Jennings GL. Protective effects of exercise and phosphoinositide 3-kinase(p110alpha) signaling in dilated and hypertrophic cardiomyopathy. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:612–617. doi: 10.1073/pnas.0606663104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hill JA, Olson EN. Cardiac plasticity. N Engl J Med. 2008;358:1370–1380. doi: 10.1056/NEJMra072139. [DOI] [PubMed] [Google Scholar]
- 10.Heineke J, Molkentin JD. Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat Rev Mol Cell Biol. 2006;7:589–600. doi: 10.1038/nrm1983. [DOI] [PubMed] [Google Scholar]
- 11.Roger VL, Go AS, Lloyd-Jones DM, Adams RJ, Berry JD, Brown TM, Carnethon MR, Dai S, de Simone G, Ford ES, Fox CS, Fullerton HJ, Gillespie C, Greenlund KJ, Hailpern SM, Heit JA, Ho PM, Howard VJ, Kissela BM, Kittner SJ, Lackland DT, Lichtman JH, Lisabeth LD, Makuc DM, Marcus GM, Marelli A, Matchar DB, McDermott MM, Meigs JB, Moy CS, Mozaffarian D, Mussolino ME, Nichol G, Paynter NP, Rosamond WD, Sorlie PD, Stafford RS, Turan TN, Turner MB, Wong ND, Wylie-Rosett J. Heart disease and stroke statistics--2011 update: A report from the american heart association. Circulation. 2011;123:e18–e209. doi: 10.1161/CIR.0b013e3182009701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bostrom P, Mann N, Wu J, Quintero PA, Plovie ER, Panakova D, Gupta RK, Xiao C, MacRae CA, Rosenzweig A, Spiegelman BM. C/ebpbeta controls exercise-induced cardiac growth and protects against pathological cardiac remodeling. Cell. 2010;143:1072–1083. doi: 10.1016/j.cell.2010.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Calvert JW, Condit ME, Aragon JP, Nicholson CK, Moody BF, Hood RL, Sindler AL, Gundewar S, Seals DR, Barouch LA, Lefer DJ. Exercise protects against myocardial ischemia-reperfusion injury via stimulation of beta(3)-adrenergic receptors and increased nitric oxide signaling: Role of nitrite and nitrosothiols. Circ Res. 2011;108:1448–1458. doi: 10.1161/CIRCRESAHA.111.241117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Booth FW, Gordon SE, Carlson CJ, Hamilton MT. Waging war on modern chronic diseases: Primary prevention through exercise biology. Journal of applied physiology: respiratory, environmental and exercise physiology. 2000;88:774–787. doi: 10.1152/jappl.2000.88.2.774. [DOI] [PubMed] [Google Scholar]
- 15.Manson JE, Greenland P, LaCroix AZ, Stefanick ML, Mouton CP, Oberman A, Perri MG, Sheps DS, Pettinger MB, Siscovick DS. Walking compared with vigorous exercise for the prevention of cardiovascular events in women. N Engl J Med. 2002;347:716–725. doi: 10.1056/NEJMoa021067. [DOI] [PubMed] [Google Scholar]
- 16.Haskell WL, Lee IM, Pate RR, Powell KE, Blair SN, Franklin BA, Macera CA, Heath GW, Thompson PD, Bauman A. Physical activity and public health: Updated recommendation for adults from the american college of sports medicine and the american heart association. Medicine and science in sports and exercise. 2007;39:1423–1434. doi: 10.1249/mss.0b013e3180616b27. [DOI] [PubMed] [Google Scholar]
- 17.Tanasescu M, Leitzmann MF, Rimm EB, Willett WC, Stampfer MJ, Hu FB. Exercise type and intensity in relation to coronary heart disease in men. JAMA: the journal of the American Medical Association. 2002;288:1994–2000. doi: 10.1001/jama.288.16.1994. [DOI] [PubMed] [Google Scholar]
- 18.Lee IM, Skerrett PJ. Physical activity and all-cause mortality: What is the dose-response relation? Medicine and science in sports and exercise. 2001;33:S459–471. doi: 10.1097/00005768-200106001-00016. discussion S493-454. [DOI] [PubMed] [Google Scholar]
- 19.Thompson D, Karpe F, Lafontan M, Frayn K. Physical activity and exercise in the regulation of human adipose tissue physiology. Physiological reviews. 2012;92:157–191. doi: 10.1152/physrev.00012.2011. [DOI] [PubMed] [Google Scholar]
- 20.Hambrecht R, Wolf A, Gielen S, Linke A, Hofer J, Erbs S, Schoene N, Schuler G. Effect of exercise on coronary endothelial function in patients with coronary artery disease. N Engl J Med. 2000;342:454–460. doi: 10.1056/NEJM200002173420702. [DOI] [PubMed] [Google Scholar]
- 21.Nualnim N, Parkhurst K, Dhindsa M, Tarumi T, Vavrek J, Tanaka H. Effects of swimming training on blood pressure and vascular function in adults >50 years of age. The American journal of cardiology. 2012;109:1005–1010. doi: 10.1016/j.amjcard.2011.11.029. [DOI] [PubMed] [Google Scholar]
- 22.Pescatello LS, Franklin BA, Fagard R, Farquhar WB, Kelley GA, Ray CA. American college of sports medicine position stand. Exercise and hypertension. Medicine and science in sports and exercise. 2004;36:533–553. doi: 10.1249/01.mss.0000115224.88514.3a. [DOI] [PubMed] [Google Scholar]
- 23.DeMaria AN, Neumann A, Lee G, Fowler W, Mason DT. Alterations in ventricular mass and performance induced by exercise training in man evaluated by echocardiography. Circulation. 1978;57:237–244. doi: 10.1161/01.cir.57.2.237. [DOI] [PubMed] [Google Scholar]
- 24.Andersen RB, Rourke BC, Caiozzo VJ, Bennett AF, Hicks JW. Postprandial cardiac hypertrophy in pythons. Nature. 2005;434:37–38. doi: 10.1038/434037a. [DOI] [PubMed] [Google Scholar]
- 25.Bergmann O, Bhardwaj RD, Bernard S, Zdunek S, Barnabe-Heider F, Walsh S, Zupicich J, Alkass K, Buchholz BA, Druid H, Jovinge S, Frisen J. Evidence for cardiomyocyte renewal in humans. Science. 2009;324:98–102. doi: 10.1126/science.1164680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Beltrami AP, Barlucchi L, Torella D, Baker M, Limana F, Chimenti S, Kasahara H, Rota M, Musso E, Urbanek K, Leri A, Kajstura J, Nadal-Ginard B, Anversa P. Adult cardiac stem cells are multipotent and support myocardial regeneration. Cell. 2003;114:763–776. doi: 10.1016/s0092-8674(03)00687-1. [DOI] [PubMed] [Google Scholar]
- 27.Bersell K, Arab S, Haring B, Kuhn B. Neuregulin1/erbb4 signaling induces cardiomyocyte proliferation and repair of heart injury. Cell. 2009;138:257–270. doi: 10.1016/j.cell.2009.04.060. [DOI] [PubMed] [Google Scholar]
- 28.Hsieh PC, Segers VF, Davis ME, MacGillivray C, Gannon J, Molkentin JD, Robbins J, Lee RT. Evidence from a genetic fate-mapping study that stem cells refresh adult mammalian cardiomyocytes after injury. Nature medicine. 2007;13:970–974. doi: 10.1038/nm1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Morganroth J, Maron BJ, Henry WL, Epstein SE. Comparative left ventricular dimensions in trained athletes. Annals of internal medicine. 1975;82:521–524. doi: 10.7326/0003-4819-82-4-521. [DOI] [PubMed] [Google Scholar]
- 30.Mihl C, Dassen WRM, Kuipers H. Cardiac remodelling: Concentric versus eccentric hypertrophy in strength and endurance athletes. Neth Heart J. 2008;16:129–133. doi: 10.1007/BF03086131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Weiner RB, Baggish AL. Exercise-induced cardiac remodeling. Progress in cardiovascular diseases. 2012;54:380–386. doi: 10.1016/j.pcad.2012.01.006. [DOI] [PubMed] [Google Scholar]
- 32.Ellison GM, Waring CD, Vicinanza C, Torella D. Physiological cardiac remodelling in response to endurance exercise training: Cellular and molecular mechanisms. Heart. 2011 doi: 10.1136/heartjnl-2011-300639. [DOI] [PubMed] [Google Scholar]
- 33.Riquelme CA, Magida JA, Harrison BC, Wall CE, Marr TG, Secor SM, Leinwand LA. Fatty acids identified in the burmese python promote beneficial cardiac growth. Science. 2011;334:528–531. doi: 10.1126/science.1210558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wisloff U, Loennechen JP, Falck G, Beisvag V, Currie S, Smith G, Ellingsen O. Increased contractility and calcium sensitivity in cardiac myocytes isolated from endurance trained rats. Cardiovascular research. 2001;50:495–508. doi: 10.1016/s0008-6363(01)00210-3. [DOI] [PubMed] [Google Scholar]
- 35.Kemi OJ, Wisloff U. Mechanisms of exercise-induced improvements in the contractile apparatus of the mammalian myocardium. Acta Physiol (Oxf) 2010;199:425–439. doi: 10.1111/j.1748-1716.2010.02132.x. [DOI] [PubMed] [Google Scholar]
- 36.Kemi OJ, Ellingsen O, Ceci M, Grimaldi S, Smith GL, Condorelli G, Wisloff U. Aerobic interval training enhances cardiomyocyte contractility and ca2+ cycling by phosphorylation of camkii and thr-17 of phospholamban. Journal of molecular and cellular cardiology. 2007;43:354–361. doi: 10.1016/j.yjmcc.2007.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kemi OJ, Ceci M, Condorelli G, Smith GL, Wisloff U. Myocardial sarcoplasmic reticulum ca2+ atpase function is increased by aerobic interval training. European journal of cardiovascular prevention and rehabilitation: official journal of the European Society of Cardiology, Working Groups on Epidemiology & Prevention and Cardiac Rehabilitation and Exercise Physiology. 2008;15:145–148. doi: 10.1097/HJR.0b013e3282efd4e0. [DOI] [PubMed] [Google Scholar]
- 38.Wisloff U, Loennechen JP, Currie S, Smith GL, Ellingsen O. Aerobic exercise reduces cardiomyocyte hypertrophy and increases contractility, ca2+ sensitivity and serca-2 in rat after myocardial infarction. Cardiovascular research. 2002;54:162–174. doi: 10.1016/s0008-6363(01)00565-x. [DOI] [PubMed] [Google Scholar]
- 39.Zhang LQ, Zhang XQ, Musch TI, Moore RL, Cheung JY. Sprint training restores normal contractility in postinfarction rat myocytes. Journal of applied physiology: respiratory, environmental and exercise physiology. 2000;89:1099–1105. doi: 10.1152/jappl.2000.89.3.1099. [DOI] [PubMed] [Google Scholar]
- 40.Zhang LQ, Zhang XQ, Ng YC, Rothblum LI, Musch TI, Moore RL, Cheung JY. Sprint training normalizes ca(2+) transients and sr function in postinfarction rat myocytes. Journal of applied physiology: respiratory, environmental and exercise physiology. 2000;89:38–46. doi: 10.1152/jappl.2000.89.1.38. [DOI] [PubMed] [Google Scholar]
- 41.Kemi OJ, Hoydal MA, MacQuaide N, Haram PM, Koch LG, Britton SL, Ellingsen O, Smith GL, Wisloff U. The effect of exercise training on transverse tubules in normal, remodeled, and reverse remodeled hearts. Journal of Cellular Physiology. 2011;226:2235–2243. doi: 10.1002/jcp.22559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Duncker DJ, Bache RJ. Regulation of coronary blood flow during exercise. Physiological reviews. 2008;88:1009–1086. doi: 10.1152/physrev.00045.2006. [DOI] [PubMed] [Google Scholar]
- 43.Windecker S, Allemann Y, Billinger M, Pohl T, Hutter D, Orsucci T, Blaga L, Meier B, Seiler C. Effect of endurance training on coronary artery size and function in healthy men: An invasive followup study. American journal of physiology Heart and circulatory physiology. 2002;282:H2216–2223. doi: 10.1152/ajpheart.00977.2001. [DOI] [PubMed] [Google Scholar]
- 44.Indermuhle A, Vogel R, Meier P, Wirth S, Stoop R, Mohaupt MG, Seiler C. The relative myocardial blood volume differentiates between hypertensive heart disease and athlete's heart in humans. European heart journal. 2006;27:1571–1578. doi: 10.1093/eurheartj/ehl024. [DOI] [PubMed] [Google Scholar]
- 45.Iemitsu M, Maeda S, Jesmin S, Otsuki T, Miyauchi T. Exercise training improves aging-induced downregulation of vegf angiogenic signaling cascade in hearts. American journal of physiology Heart and circulatory physiology. 2006;291:H1290–1298. doi: 10.1152/ajpheart.00820.2005. [DOI] [PubMed] [Google Scholar]
- 46.White FC, Bloor CM, McKirnan MD, Carroll SM. Exercise training in swine promotes growth of arteriolar bed and capillary angiogenesis in heart. Journal of applied physiology: respiratory, environmental and exercise physiology. 1998;85:1160–1168. doi: 10.1152/jappl.1998.85.3.1160. [DOI] [PubMed] [Google Scholar]
- 47.Sano M, Minamino T, Toko H, Miyauchi H, Orimo M, Qin Y, Akazawa H, Tateno K, Kayama Y, Harada M, Shimizu I, Asahara T, Hamada H, Tomita S, Molkentin JD, Zou Y, Komuro I. P53-induced inhibition of hif-1 causes cardiac dysfunction during pressure overload. Nature. 2007;446:444–448. doi: 10.1038/nature05602. [DOI] [PubMed] [Google Scholar]
- 48.Shiojima I, Sato K, Izumiya Y, Schiekofer S, Ito M, Liao R, Colucci WS, Walsh K. Disruption of coordinated cardiac hypertrophy and angiogenesis contributes to the transition to heart failure. J Clin Invest. 2005;115:2108–2118. doi: 10.1172/JCI24682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Leosco D, Rengo G, Iaccarino G, Golino L, Marchese M, Fortunato F, Zincarelli C, Sanzari E, Ciccarelli M, Galasso G, Altobelli GG, Conti V, Matrone G, Cimini V, Ferrara N, Filippelli A, Koch WJ, Rengo F. Exercise promotes angiogenesis and improves beta-adrenergic receptor signalling in the post-ischaemic failing rat heart. Cardiovascular research. 2008;78:385–394. doi: 10.1093/cvr/cvm109. [DOI] [PubMed] [Google Scholar]
- 50.Arany Z, He H, Lin J, Hoyer K, Handschin C, Toka O, Ahmad F, Matsui T, Chin S, Wu PH, Rybkin II, Shelton JM, Manieri M, Cinti S, Schoen FJ, Bassel-Duby R, Rosenzweig A, Ingwall JS, Spiegelman BM. Transcriptional coactivator pgc-1 alpha controls the energy state and contractile function of cardiac muscle. Cell Metab. 2005;1:259–271. doi: 10.1016/j.cmet.2005.03.002. [DOI] [PubMed] [Google Scholar]
- 51.Kajstura J, Urbanek K, Perl S, Hosoda T, Zheng H, Ogorek B, Ferreira-Martins J, Goichberg P, Rondon-Clavo C, Sanada F, D'Amario D, Rota M, Del Monte F, Orlic D, Tisdale J, Leri A, Anversa P. Cardiomyogenesis in the adult human heart. Circ Res. 2010;107:305–315. doi: 10.1161/CIRCRESAHA.110.223024. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 52.Urbanek K, Quaini F, Tasca G, Torella D, Castaldo C, Nadal-Ginard B, Leri A, Kajstura J, Quaini E, Anversa P. Intense myocyte formation from cardiac stem cells in human cardiac hypertrophy. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:10440–10445. doi: 10.1073/pnas.1832855100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Quaini F, Urbanek K, Graiani G, Lagrasta C, Maestri R, Monica M, Boni A, Ferraro F, Delsignore R, Tasca G, Leri A, Kajstura J, Quaini E, Anversa P. The regenerative potential of the human heart. International journal of cardiology. 2004;95(Suppl 1):S26–28. doi: 10.1016/s0167-5273(04)90008-3. [DOI] [PubMed] [Google Scholar]
- 54.Poss KD, Wilson LG, Keating MT. Heart regeneration in zebrafish. Science. 2002;298:2188–2190. doi: 10.1126/science.1077857. [DOI] [PubMed] [Google Scholar]
- 55.Porrello ER, Mahmoud AI, Simpson E, Hill JA, Richardson JA, Olson EN, Sadek HA. Transient regenerative potential of the neonatal mouse heart. Science. 2011;331:1078–1080. doi: 10.1126/science.1200708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kuhn HG, Dickinson-Anson H, Gage FH. Neurogenesis in the dentate gyrus of the adult rat: Age-related decrease of neuronal progenitor proliferation. The Journal of neuroscience: the official journal of the Society for Neuroscience. 1996;16:2027–2033. doi: 10.1523/JNEUROSCI.16-06-02027.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.van Praag H, Shubert T, Zhao C, Gage FH. Exercise enhances learning and hippocampal neurogenesis in aged mice. J Neurosci. 2005;25:8680–8685. doi: 10.1523/JNEUROSCI.1731-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Erickson KI, Voss MW, Prakash RS, Basak C, Szabo A, Chaddock L, Kim JS, Heo S, Alves H, White SM, Wojcicki TR, Mailey E, Vieira VJ, Martin SA, Pence BD, Woods JA, McAuley E, Kramer AF. Exercise training increases size of hippocampus and improves memory. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:3017–3022. doi: 10.1073/pnas.1015950108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Frystyk J. Exercise and the growth hormone-insulin-like growth factor axis. Medicine and science in sports and exercise. 2010;42:58–66. doi: 10.1249/MSS.0b013e3181b07d2d. [DOI] [PubMed] [Google Scholar]
- 60.Neri Serneri GG, Boddi M, Modesti PA, Cecioni I, Coppo M, Padeletti L, Michelucci A, Colella A, Galanti G. Increased cardiac sympathetic activity and insulin-like growth factor-i formation are associated with physiological hypertrophy in athletes. Circulation Research. 2001;89:977–982. doi: 10.1161/hh2301.100982. [DOI] [PubMed] [Google Scholar]
- 61.Chao W, Matsui T, Novikov MS, Tao J, Li L, Liu H, Ahn Y, Rosenzweig A. Strategic advantages of insulin-like growth factor-i expression for cardioprotection. J Gene Med. 2003;5:277–286. doi: 10.1002/jgm.347. [DOI] [PubMed] [Google Scholar]
- 62.Li B, Setoguchi M, Wang X, Andreoli AM, Leri A, Malhotra A, Kajstura J, Anversa P. Insulin-like growth factor-1 attenuates the detrimental impact of nonocclusive coronary artery constriction on the heart. Circ Res. 1999;84:1007–1019. doi: 10.1161/01.res.84.9.1007. [DOI] [PubMed] [Google Scholar]
- 63.Welch S, Plank D, Witt S, Glascock B, Schaefer E, Chimenti S, Andreoli AM, Limana F, Leri A, Kajstura J, Anversa P, Sussman MA. Cardiac-specific igf-1 expression attenuates dilated cardiomyopathy in tropomodulin-overexpressing transgenic mice. Circ Res. 2002;90:641–648. doi: 10.1161/01.res.0000013780.77774.75. [DOI] [PubMed] [Google Scholar]
- 64.Yamashita K, Kajstura J, Discher DJ, Wasserlauf BJ, Bishopric NH, Anversa P, Webster KA. Reperfusion-activated akt kinase prevents apoptosis in transgenic mouse hearts overexpressing insulin-like growth factor-1. Circ Res. 2001;88:609–614. doi: 10.1161/01.res.88.6.609. [DOI] [PubMed] [Google Scholar]
- 65.Kajstura J, Fiordaliso F, Andreoli AM, Li B, Chimenti S, Medow MS, Limana F, Nadal-Ginard B, Leri A, Anversa P. Igf-1 overexpression inhibits the development of diabetic cardiomyopathy and angiotensin ii-mediated oxidative stress. Diabetes. 2001;50:1414–1424. doi: 10.2337/diabetes.50.6.1414. [DOI] [PubMed] [Google Scholar]
- 66.Reiss K, Cheng W, Ferber A, Kajstura J, Li P, Li B, Olivetti G, Homcy CJ, Baserga R, Anversa P. Overexpression of insulin-like growth factor-1 in the heart is coupled with myocyte proliferation in transgenic mice. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:8630–8635. doi: 10.1073/pnas.93.16.8630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.McMullen JR, Shioi T, Huang WY, Zhang L, Tarnavski O, Bisping E, Schinke M, Kong S, Sherwood MC, Brown J, Riggi L, Kang PM, Izumo S. The insulin-like growth factor 1 receptor induces physiological heart growth via the phosphoinositide 3-kinase(p110alpha) pathway. The Journal of biological chemistry. 2004;279:4782–4793. doi: 10.1074/jbc.M310405200. [DOI] [PubMed] [Google Scholar]
- 68.Matsui T, Nagoshi T, Rosenzweig A. Akt and pi 3-kinase signaling in cardiomyocyte hypertrophy and survival. Cell Cycle. 2003;2:220–223. [PubMed] [Google Scholar]
- 69.Lin RC, Weeks KL, Gao XM, Williams RB, Bernardo BC, Kiriazis H, Matthews VB, Woodcock EA, Bouwman RD, Mollica JP, Speirs HJ, Dawes IW, Daly RJ, Shioi T, Izumo S, Febbraio MA, Du XJ, McMullen JR. Pi3k(p110 alpha) protects against myocardial infarction-induced heart failure: Identification of pi3k-regulated mirna and mrna. Arteriosclerosis, thrombosis, and vascular biology. 2010;30:724–732. doi: 10.1161/ATVBAHA.109.201988. [DOI] [PubMed] [Google Scholar]
- 70.McMullen JR, Shioi T, Zhang L, Tarnavski O, Sherwood MC, Kang PM, Izumo S. Phosphoinositide 3-kinase(p110alpha) plays a critical role for the induction of physiological, but not pathological, cardiac hypertrophy. Proc Natl Acad Sci U S A. 2003;100:12355–12360. doi: 10.1073/pnas.1934654100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.DeBosch B, Treskov I, Lupu TS, Weinheimer C, Kovacs A, Courtois M, Muslin AJ. Akt1 is required for physiological cardiac growth. Circulation. 2006;113:2097–2104. doi: 10.1161/CIRCULATIONAHA.105.595231. [DOI] [PubMed] [Google Scholar]
- 72.Matsui T, Tao J, del Monte F, Lee KH, Li L, Picard M, Force TL, Franke TF, Hajjar RJ, Rosenzweig A. Akt activation preserves cardiac function and prevents injury after transient cardiac ischemia in vivo. Circulation. 2001;104:330–335. doi: 10.1161/01.cir.104.3.330. [DOI] [PubMed] [Google Scholar]
- 73.Matsui T, Li L, del Monte F, Fukui Y, Franke T, Hajjar R, Rosenzweig A. Adenoviral gene transfer of activated pi 3-kinase and akt inhibits apoptosis of hypoxic cardiomyocytes in vitro. Circulation. 1999;100:2373–2379. doi: 10.1161/01.cir.100.23.2373. [DOI] [PubMed] [Google Scholar]
- 74.Kim YK, Kim SJ, Yatani A, Huang Y, Castelli G, Vatner DE, Liu J, Zhang Q, Diaz G, Zieba R, Thaisz J, Drusco A, Croce C, Sadoshima J, Condorelli G, Vatner SF. Mechanism of enhanced cardiac function in mice with hypertrophy induced by overexpressed akt. J Biol Chem. 2003;278:47622–47628. doi: 10.1074/jbc.M305909200. [DOI] [PubMed] [Google Scholar]
- 75.Shiraishi I, Melendez J, Ahn Y, Skavdahl M, Murphy E, Welch S, Schaefer E, Walsh K, Rosenzweig A, Torella D, Nurzynska D, Kajstura J, Leri A, Anversa P, Sussman MA. Nuclear targeting of akt enhances kinase activity and survival of cardiomyocytes. Circ Res. 2004;94:884–891. doi: 10.1161/01.RES.0000124394.01180.BE. [DOI] [PubMed] [Google Scholar]
- 76.Rota M, Boni A, Urbanek K, Padin-Iruegas ME, Kajstura TJ, Fiore G, Kubo H, Sonnenblick EH, Musso E, Houser SR, Leri A, Sussman MA, Anversa P. Nuclear targeting of akt enhances ventricular function and myocyte contractility. Circ Res. 2005;97:1332–1341. doi: 10.1161/01.RES.0000196568.11624.ae. [DOI] [PubMed] [Google Scholar]
- 77.Gude N, Muraski J, Rubio M, Kajstura J, Schaefer E, Anversa P, Sussman MA. Akt promotes increased cardiomyocyte cycling and expansion of the cardiac progenitor cell population. Circ Res. 2006;99:381–388. doi: 10.1161/01.RES.0000236754.21499.1c. [DOI] [PubMed] [Google Scholar]
- 78.Muraski JA, Rota M, Misao Y, Fransioli J, Cottage C, Gude N, Esposito G, Delucchi F, Arcarese M, Alvarez R, Siddiqi S, Emmanuel GN, Wu W, Fischer K, Martindale JJ, Glembotski CC, Leri A, Kajstura J, Magnuson N, Berns A, Beretta RM, Houser SR, Schaefer EM, Anversa P, Sussman MA. Pim-1 regulates cardiomyocyte survival downstream of akt. Nat Med. 2007;13:1467–1475. doi: 10.1038/nm1671. [DOI] [PubMed] [Google Scholar]
- 79.Bersell K, Arab S, Haring B, Kühn B. Neuregulin1/erbb4 signaling induces cardiomyocyte proliferation and repair of heart injury. Cell. 2009;138:257–270. doi: 10.1016/j.cell.2009.04.060. [DOI] [PubMed] [Google Scholar]
- 80.Loscalzo J, Welch G. Nitric oxide and its role in the cardiovascular system. Progress in cardiovascular diseases. 1995;38:87–104. doi: 10.1016/s0033-0620(05)80001-5. [DOI] [PubMed] [Google Scholar]
- 81.Katz SD, Khan T, Zeballos GA, Mathew L, Potharlanka P, Knecht M, Whelan J. Decreased activity of the l-arginine-nitric oxide metabolic pathway in patients with congestive heart failure. Circulation. 1999;99:2113–2117. doi: 10.1161/01.cir.99.16.2113. [DOI] [PubMed] [Google Scholar]
- 82.Jones SP, Greer JJ, van Haperen R, Duncker DJ, de Crom R, Lefer DJ. Endothelial nitric oxide synthase overexpression attenuates congestive heart failure in mice. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:4891–4896. doi: 10.1073/pnas.0837428100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.de Waard MC, van Haperen R, Soullie T, Tempel D, de Crom R, Duncker DJ. Beneficial effects of exercise training after myocardial infarction require full enos expression. Journal of molecular and cellular cardiology. 2010;48:1041–1049. doi: 10.1016/j.yjmcc.2010.02.005. [DOI] [PubMed] [Google Scholar]
- 84.Bezzerides V, Rosenzweig A. Saying yes to exercise and no to cardiac injury. Circ Res. 2011;108:1414–1416. doi: 10.1161/CIRCRESAHA.111.247122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Finkel T, Deng CX, Mostoslavsky R. Recent progress in the biology and physiology of sirtuins. Nature. 2009;460:587–591. doi: 10.1038/nature08197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ferrara N, Rinaldi B, Corbi G, Conti V, Stiuso P, Boccuti S, Rengo G, Rossi F, Filippelli A. Exercise training promotes sirt1 activity in aged rats. Rejuvenation research. 2008;11:139–150. doi: 10.1089/rej.2007.0576. [DOI] [PubMed] [Google Scholar]
- 87.Pillai VB, Sundaresan NR, Jeevanandam V, Gupta MP. Mitochondrial sirt3 and heart disease. Cardiovascular research. 2010;88:250–256. doi: 10.1093/cvr/cvq250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Sundaresan NR, Gupta M, Kim G, Rajamohan SB, Isbatan A, Gupta MP. Sirt3 blocks the cardiac hypertrophic response by augmenting foxo3a-dependent antioxidant defense mechanisms in mice. The Journal of clinical investigation. 2009;119:2758–2771. doi: 10.1172/JCI39162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hafner AV, Dai J, Gomes AP, Xiao CY, Palmeira CM, Rosenzweig A, Sinclair DA. Regulation of the mptp by sirt3-mediated deacetylation of cypd at lysine 166 suppresses age-related cardiac hypertrophy. Aging. 2010;2:914–923. doi: 10.18632/aging.100252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zhang P, Hu X, Xu X, Fassett J, Zhu G, Viollet B, Xu W, Wiczer B, Bernlohr DA, Bache RJ, Chen Y. Amp activated protein kinase-alpha2 deficiency exacerbates pressure-overload-induced left ventricular hypertrophy and dysfunction in mice. Hypertension. 2008;52:918–924. doi: 10.1161/HYPERTENSIONAHA.108.114702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lin J, Handschin C, Spiegelman BM. Metabolic control through the pgc-1 family of transcription coactivators. Cell Metab. 2005;1:361–370. doi: 10.1016/j.cmet.2005.05.004. [DOI] [PubMed] [Google Scholar]
- 92.Arany Z, Foo SY, Ma Y, Ruas JL, Bommi-Reddy A, Girnun G, Cooper M, Laznik D, Chinsomboon J, Rangwala SM, Baek KH, Rosenzweig A, Spiegelman BM. Hif-independent regulation of vegf and angiogenesis by the transcriptional coactivator pgc-1alpha. Nature. 2008;451:1008–1012. doi: 10.1038/nature06613. [DOI] [PubMed] [Google Scholar]
- 93.Oshima Y, Ouchi N, Sato K, Izumiya Y, Pimentel DR, Walsh K. Follistatin-like 1 is an akt-regulated cardioprotective factor that is secreted by the heart. Circulation. 2008;117:3099–3108. doi: 10.1161/CIRCULATIONAHA.108.767673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Bostrom P, Wu J, Jedrychowski MP, Korde A, Ye L, Lo JC, Rasbach KA, Bostrom EA, Choi JH, Long JZ, Kajimura S, Zangaretti MC, Vind BF, Tu H, Cinti S, Hojlund K, Gygi SP, Spiegelman BM. A pgc1-alpha-dependent myokine that drives brown-fat-like development of white fat and thermogenesis. Nature. 2012;481:6. doi: 10.1038/nature10777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Walsh K. Adipokines, myokines and cardiovascular disease. Circulation journal: official journal of the Japanese Circulation Society. 2009;73:13–18. doi: 10.1253/circj.cj-08-0961. [DOI] [PubMed] [Google Scholar]
- 96.Biolo A, Shibata R, Ouchi N, Kihara S, Sonoda M, Walsh K, Sam F. Determinants of adiponectin levels in patients with chronic systolic heart failure. The American journal of cardiology. 2010;105:1147–1152. doi: 10.1016/j.amjcard.2009.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Van Berendoncks AM, Garnier A, Beckers P, Hoymans VY, Possemiers N, Fortin D, Martinet W, Van Hoof V, Vrints CJ, Ventura-Clapier R, Conraads VM. Functional adiponectin resistance at the level of the skeletal muscle in mild to moderate chronic heart failure. Circulation Heart failure. 2010;3:185–194. doi: 10.1161/CIRCHEARTFAILURE.109.885525. [DOI] [PubMed] [Google Scholar]
- 98.Van Berendoncks AM, Beckers P, Hoymans VY, Possemiers N, Wuytss FL, Vrints CJ, Conraads VM. Exercise training reduces circulating adiponectin levels in patients with chronic heart failure. Clin Sci (Lond) 2010;118:281–289. doi: 10.1042/CS20090213. [DOI] [PubMed] [Google Scholar]
- 99.Van Berendoncks AM, Garnier A, Beckers P, Hoymans VY, Possemiers N, Fortin D, Van Hoof V, Dewilde S, Vrints CJ, Ventura-Clapier R, Conraads VM. Exercise training reverses adiponectin resistance in skeletal muscle of patients with chronic heart failure. Heart. 2011;97:1403–1409. doi: 10.1136/hrt.2011.226373. [DOI] [PubMed] [Google Scholar]
- 100.Anker SD, Ponikowski P, Varney S, Chua TP, Clark AL, Webb-Peploe KM, Harrington D, Kox WJ, Poole-Wilson PA, Coats AJ. Wasting as independent risk factor for mortality in chronic heart failure. Lancet. 1997;349:1050–1053. doi: 10.1016/S0140-6736(96)07015-8. [DOI] [PubMed] [Google Scholar]
- 101.McPherron AC, Lawler AM, Lee SJ. Regulation of skeletal muscle mass in mice by a new tgf-beta superfamily member. Nature. 1997;387:83–90. doi: 10.1038/387083a0. [DOI] [PubMed] [Google Scholar]
- 102.Heineke J, Auger-Messier M, Xu J, Sargent M, York A, Welle S, Molkentin JD. Genetic deletion of myostatin from the heart prevents skeletal muscle atrophy in heart failure. Circulation. 2010;121:419–425. doi: 10.1161/CIRCULATIONAHA.109.882068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Morissette MR, Cook SA, Foo S, McKoy G, Ashida N, Novikov M, Scherrer-Crosbie M, Li L, Matsui T, Brooks G, Rosenzweig A. Myostatin regulates cardiomyocyte growth through modulation of akt signaling. Circ Res. 2006;99:15–24. doi: 10.1161/01.RES.0000231290.45676.d4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Morissette MR, Stricker JC, Rosenberg MA, Buranasombati C, Levitan EB, Mittleman MA, Rosenzweig A. Effects of myostatin deletion in aging mice. Aging Cell. 2009;8:573–583. doi: 10.1111/j.1474-9726.2009.00508.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Lenk K, Schur R, Linke A, Erbs S, Matsumoto Y, Adams V, Schuler G. Impact of exercise training on myostatin expression in the myocardium and skeletal muscle in a chronic heart failure model. Eur J Heart Fail. 2009;11:342–348. doi: 10.1093/eurjhf/hfp020. [DOI] [PubMed] [Google Scholar]
- 106.Lenk K, Erbs S, Hollriege R, Beck E, Linke A, Gielen S, Mobius Winkler S, Sandri M, Hambrecht R, Schuler G, Adams V. Exercise training leads to a reduction of elevated myostatin levels in patients with chronic heart failure. European journal of cardiovascular prevention and rehabilitation: official journal of the European Society of Cardiology, Working Groups on Epidemiology & Prevention and Cardiac Rehabilitation and Exercise Physiology. 2011 [Google Scholar]
- 107.Lewis GD, Farrell L, Wood MJ, Martinovic M, Arany Z, Rowe GC, Souza A, Cheng S, McCabe EL, Yang E, Shi X, Deo R, Roth FP, Asnani A, Rhee EP, Systrom DM, Semigran MJ, Vasan RS, Carr SA, Wang TJ, Sabatine MS, Clish CB, Gerszten RE. Metabolic signatures of exercise in human plasma. Sci Transl Med. 2:33ra37. doi: 10.1126/scitranslmed.3001006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Bartel DP. Micrornas: Genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 109.Small EM, Olson EN. Pervasive roles of micrornas in cardiovascular biology. Nature. 2011;469:336–342. doi: 10.1038/nature09783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Catalucci D, Latronico MV, Condorelli G. Micrornas control gene expression: Importance for cardiac development and pathophysiology. Annals of the New York Academy of Sciences. 2008;1123:20–29. doi: 10.1196/annals.1420.004. [DOI] [PubMed] [Google Scholar]
- 111.van Rooij E, Liu N, Olson EN. Micrornas flex their muscles. Trends Genet. 2008;24:159–166. doi: 10.1016/j.tig.2008.01.007. [DOI] [PubMed] [Google Scholar]
- 112.Nielsen S, Scheele C, Yfanti C, Akerstrom T, Nielsen AR, Pedersen BK, Laye MJ. Muscle specific micrornas are regulated by endurance exercise in human skeletal muscle. The Journal of physiology. 2010;588:4029–4037. doi: 10.1113/jphysiol.2010.189860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Baggish AL, Hale A, Weiner RB, Lewis GD, Systrom D, Wang F, Wang TJ, Chan SY. Dynamic regulation of circulating microrna during acute exhaustive exercise and sustained aerobic exercise training. The Journal of physiology. 2011;589:3983–3994. doi: 10.1113/jphysiol.2011.213363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Laterza OF, Lim L, Garrett-Engele PW, Vlasakova K, Muniappa N, Tanaka WK, Johnson JM, Sina JF, Fare TL, Sistare FD, Glaab WE. Plasma micrornas as sensitive and specific biomarkers of tissue injury. Clinical chemistry. 2009;55:1977–1983. doi: 10.1373/clinchem.2009.131797. [DOI] [PubMed] [Google Scholar]
- 115.Care A, Catalucci D, Felicetti F, Bonci D, Addario A, Gallo P, Bang ML, Segnalini P, Gu Y, Dalton ND, Elia L, Latronico MV, Hoydal M, Autore C, Russo MA, Dorn GW, 2nd, Ellingsen O, Ruiz-Lozano P, Peterson KL, Croce CM, Peschle C, Condorelli G. Microrna-133 controls cardiac hypertrophy. Nature medicine. 2007;13:613–618. doi: 10.1038/nm1582. [DOI] [PubMed] [Google Scholar]
- 116.Soci UP, Fernandes T, Hashimoto NY, Mota GF, Amadeu MA, Rosa KT, Irigoyen MC, Phillips MI, Oliveira EM. Micrornas 29 are involved in the improvement of ventricular compliance promoted by aerobic exercise training in rats. Physiological genomics. 2011;43:665–673. doi: 10.1152/physiolgenomics.00145.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.van Rooij E, Sutherland LB, Thatcher JE, DiMaio JM, Naseem RH, Marshall WS, Hill JA, Olson EN. Dysregulation of micrornas after myocardial infarction reveals a role of mir-29 in cardiac fibrosis. Proc Natl Acad Sci U S A. 2008;105:13027–13032. doi: 10.1073/pnas.0805038105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Park SY, Lee JH, Ha M, Nam JW, Kim VN. Mir-29 mirnas activate p53 by targeting p85 alpha and cdc42. Nature structural & molecular biology. 2009;16:23–29. doi: 10.1038/nsmb.1533. [DOI] [PubMed] [Google Scholar]
- 119.Matsui T, Li L, Wu JC, Cook SA, Nagoshi T, Picard MH, Liao R, Rosenzweig A. Phenotypic spectrum caused by transgenic overexpression of activated akt in the heart. The Journal of biological chemistry. 2002;277:22896–22901. doi: 10.1074/jbc.M200347200. [DOI] [PubMed] [Google Scholar]
- 120.Perrino C, Naga Prasad SV, Mao L, Noma T, Yan Z, Kim HS, Smithies O, Rockman HA. Intermittent pressure overload triggers hypertrophy-independent cardiac dysfunction and vascular rarefaction. J Clin Invest. 2006;116:1547–1560. doi: 10.1172/JCI25397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Wilson DH, Hanlon DW, Provuncher GK, Chang L, Song L, Patel PP, Ferrell EP, Lepor H, Partin AW, Chan DW, Sokoll LJ, Cheli CD, Thiel RP, Fournier DR, Duffy DC. Fifth-generation digital immunoassay for prostate-specific antigen by single molecule array technology. Clinical chemistry. 2011;57:1712–1721. doi: 10.1373/clinchem.2011.169540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Bouchard C, Blair SN, Church TS, Earnest CP, Hagberg JM, Häkkinen K, Jenkins NT, Karavirta L, Kraus WE, Leon AS, Rao DC, Sarzynski MA, Skinner JS, Slentz CA, Rankinen T. Adverse metabolic response to regular exercise: Is it a rare or common occurrence? PLoS ONE. 2012;7:e37887. doi: 10.1371/journal.pone.0037887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Blair SN, Kampert JB, Kohl HW, 3rd, Barlow CE, Macera CA, Paffenbarger RS, Jr, Gibbons LW. Influences of cardiorespiratory fitness and other precursors on cardiovascular disease and all-cause mortality in men and women. JAMA: the journal of the American Medical Association. 1996;276:205–210. [PubMed] [Google Scholar]
- 124.D'Amore S, Mora S. Gender-specific prediction of cardiac disease: Importance of risk factors and exercise variables. Cardiology in review. 2006;14:281–285. doi: 10.1097/01.crd.0000244460.25429.c8. [DOI] [PubMed] [Google Scholar]
- 125.Fujio Y, Nguyen T, Wencker D, Kitsis RN, Walsh K. Akt promotes survival of cardiomyocytes in vitro and protects against ischemia-reperfusion injury in mouse heart. Circulation. 2000;101:660–667. doi: 10.1161/01.cir.101.6.660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Arany Z, Novikov M, Chin S, Ma Y, Rosenzweig A, Spiegelman BM. Transverse aortic constriction leads to accelerated heart failure in mice lacking ppar-gamma coactivator 1alpha. Proc Natl Acad Sci U S A. 2006;103:10086–10091. doi: 10.1073/pnas.0603615103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Wagner KR, Fleckenstein JL, Amato AA, Barohn RJ, Bushby K, Escolar DM, Flanigan KM, Pestronk A, Tawil R, Wolfe GI, Eagle M, Florence JM, King WM, Pandya S, Straub V, Juneau P, Meyers K, Csimma C, Araujo T, Allen R, Parsons SA, Wozney JM, Lavallie ER, Mendell JR. A phase i/iitrial of myo-029 in adult subjects with muscular dystrophy. Annals of neurology. 2008;63:561–571. doi: 10.1002/ana.21338. [DOI] [PubMed] [Google Scholar]
- 128.Montgomery RL, Hullinger TG, Semus HM, Dickinson BA, Seto AG, Lynch JM, Stack C, Latimer PA, Olson EN, van Rooij E. Therapeutic inhibition of mir-208a improves cardiac function and survival during heart failure. Circulation. 2011;124:1537–1547. doi: 10.1161/CIRCULATIONAHA.111.030932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Hrkach J, Von Hoff D, Ali MM, Andrianova E, Auer J, Campbell T, De Witt D, Figa M, Figueiredo M, Horhota A, Low S, McDonnell K, Peeke E, Retnarajan B, Sabnis A, Schnipper E, Song JJ, Song YH, Summa J, Tompsett D, Troiano G, Van Geen Hoven T, Wright J, Lorusso P, Kantoff PW, Bander NH, Sweeney C, Farokhzad OC, Langer R, Zale S. Preclinical development and clinical translation of a psma-targeted docetaxel nanoparticle with a differentiated pharmacological profile. Science translational medicine. 2012;4:128ra139. doi: 10.1126/scitranslmed.3003651. [DOI] [PubMed] [Google Scholar]
- 130.Montgomery RL, van Rooij E. Therapeutic advances in microrna targeting. Journal of cardiovascular pharmacology. 2011;57:1–7. doi: 10.1097/FJC.0b013e3181f603d0. [DOI] [PubMed] [Google Scholar]
- 131.Jessup M, Greenberg B, Mancini D, Cappola T, Pauly DF, Jaski B, Yaroshinsky A, Zsebo KM, Dittrich H, Hajjar RJ. Calcium upregulation by percutaneous administration of gene therapy in cardiac disease (cupid): A phase 2 trial of intracoronary gene therapy of sarcoplasmic reticulum ca2+-atpase in patients with advanced heart failure. Circulation. 2011;124:304–313. doi: 10.1161/CIRCULATIONAHA.111.022889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Loscalzo J, Kohane I, Barabasi AL. Human disease classification in the postgenomic era: A complex systems approach to human pathobiology. Mol Syst Biol. 2007;3:124. doi: 10.1038/msb4100163. [DOI] [PMC free article] [PubMed] [Google Scholar]