

Dear Sirs
Establishing the genetic basis for complex neurological conditions with targeted gene testing is challenging, particularly in genetically heterogeneous conditions such as hereditary spastic paraplegia (HSP). An added challenge is that of precisely defining the clinical phenotype, e.g. distinguishing distal dystonia from spasticity. We report a case that illustrates the diagnostic potential of whole exome sequencing (WES) to define a genetic etiology in a complex condition, directly leading to successful treatment.
A 36 year-old female presented to a neurology clinic with progressive spastic gait. She was healthy until age 6, when she began to toe-walk. She was diagnosed with “cerebral palsy” and later HSP, undergoing multiple orthopedic foot surgeries. Her toes (except for the big toes) were flexed and made activities such as walking or bicycling painful. She was dependent on crutches by age 18 and later developed mild functional impairment of her hands, dysarthria, diurnal variation of symptoms, shortness of breath and anxiety. On examination, she walked with a spastic gait, deep tendon reflexes were increased without clonus, big toes were extended at rest and remaining toes flexed tonically (Fig 1). She had undergone extensive but unsuccessful medical evaluations, including genetic testing for HSP and brain/spine MRI studies (Fig 1). She was enrolled in a research study of WES[1] and an informed consent was signed.
Figure 1.
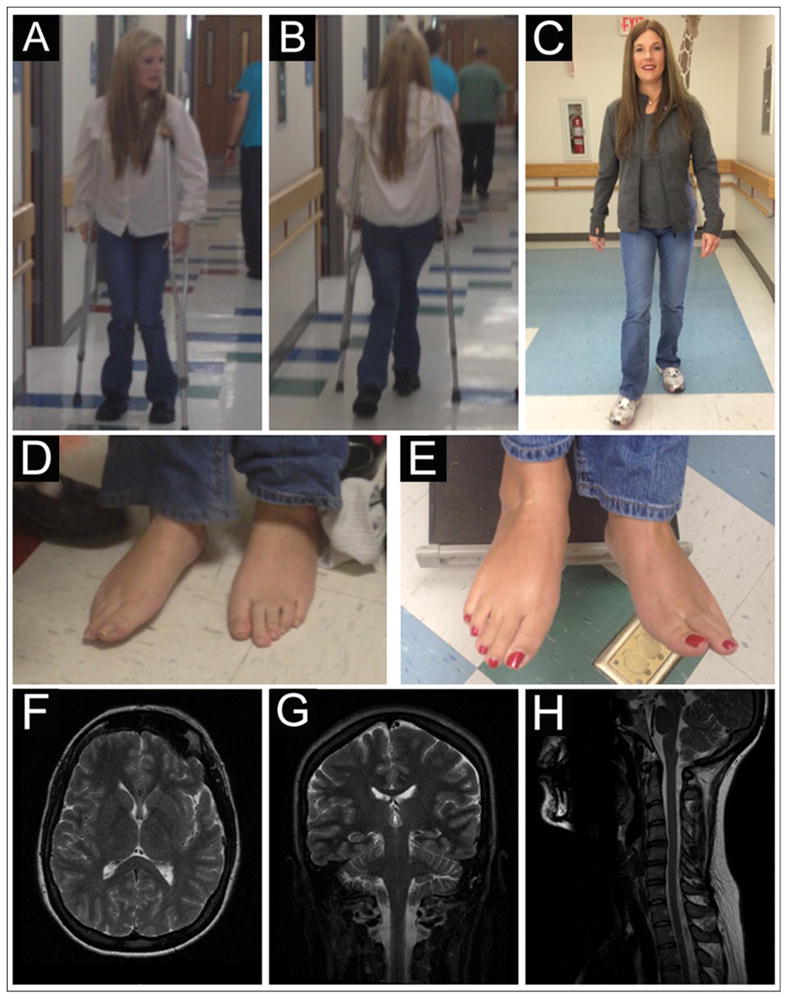
The patient uses crutches prior to the L-dopa trial, note the hip adduction and flexion and knee flexion while she was walking (A and B). Nine weeks after the initiation of L-dopa trial, the patient is able to walk without crutches (C). The patient’s toes (except the big toes) are flexed and painful prior to the L-dopa trial (arrow in D). Her toes are relaxed and in neutral position, a few weeks after starting L-dopa (arrow in E). T2-weighted brain/spine images in axial (F), coronal (G) and sagittal (H) view.
Initial WES analysis, focused on variants in genes known to be associated with HSP, was negative. However, all participants in the study undergo analysis for rare “medically actionable incidental findings” that are presumably unrelated to the participant’s diagnosis but would provide well-defined medical benefit if discovered in a pre-symptomatic individual[2, 3]. This analysis revealed a heterozygous nonsense variant (NM_000161.2; c.646C>T, p.R216X) in the GCH1 (GTP cyclohydrolase 1) gene, confirmed by clinical Sanger sequencing. The R216X variant had been previously reported as causal for dopa-responsive dystonia DRD (MIM #128230)[4–6], a phenotype that can resemble HSP[7–9]. Despite the reported phenotypic variation and incomplete penetrance associated with GCH1 [4, 7–11], this variant was considered likely causal of the patient’s condition, given her presentation and was reported as a positive diagnostic finding.
The patient was started on carbidopa/levodopa (25/100) at a dose of 0.5 tablet daily. On day 3 she noted improvement in her pain and movement of her toes. She noticed clear improvement in leg stiffness within the first week. She continued to improve during upward titration over 8 weeks to an optimal dose of 1, 1 and 0.75 tablet, at which time she walked independently without crutches for the first time in 17 years (Fig 1). Her dysarthric speech improved and periodic muscle cramping disappeared. She reports feeling like a “whole new person with new legs.”
As with many genetic conditions, the phenotypic spectrum of DRD has expanded and includes presentations without apparent dystonia. It is generally agreed that individuals with childhood onset dystonic/spastic symptoms of unknown etiology should be treated with a trial of levodopa[12]. Moreover, other conditions such as spastic paraplegia 8 (MIM#610657) have been reported to respond to levodopa treatment[13]. Identifying the genetic etiology of a condition can guide management and provide valuable information about the risks of the condition for family members. The emerging availability of efficient DNA sequencing, coupled with difficulties inherent to establishing a diagnosis for many complex neurological presentations, compels consideration of broad strategies for genetic testing. Restricting the focus to a single family of genes (i.e. HSP) will miss treatable conditions such as DRD. Thus, it may be increasingly worthwhile to consider broad genetic testing through a clinical panel or WES, in order to provide an accurate diagnosis and correct treatment for patients with complex neurological conditions.
Acknowledgments
We want to thank the patient and her family for their dedication to research and their permission to use the pictures for publication.
This work was supported by a grant from the NIH/NHGRI, 1UO1HG006487-01 (Evans).
Footnotes
Conflict of Interest Statement: The authors declare that they have no conflict of interest.
References
- 1.Evans JP. In: NCGENES: North Carolina Clinical Genomic Evaluation by Next-generation Exome Sequencing. James Evans PI MD, PhD, editor. NHGRI, NIH; 2011–2015. IRB approval #11-1865. [Google Scholar]
- 2.Berg JS, Adams M, Nassar N, Bizon C, Lee K, Schmitt CP, Wilhelmsen KC, Evans JP. An informatics approach to analyzing the incidentalome. Genet Med. 2013;15(1):36–44. doi: 10.1038/gim.2012.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Berg JS, Khoury MJ, Evans JP. Deploying whole genome sequencing in clinical practice and public health: meeting the challenge one bin at a time. Genet Med. 2011;13(6):499–504. doi: 10.1097/GIM.0b013e318220aaba. [DOI] [PubMed] [Google Scholar]
- 4.Bandmann O, Nygaard TG, Surtees R, Marsden CD, Wood NW, Harding AE. Dopa-responsive dystonia in British patients: new mutations of the GTP-cyclohydrolase I gene and evidence for genetic heterogeneity. Hum Mol Genet. 1996;5(3):403–6. doi: 10.1093/hmg/5.3.403. [DOI] [PubMed] [Google Scholar]
- 5.Hagenah J, Saunders-Pullman R, Hedrich K, Kabakci K, Habermann K, Wiegers K, Mohrmann K, Lohnau T, Raymond D, Vieregge P, Nygaard T, Ozelius LJ, Bressman SB, Klein C. High mutation rate in dopa-responsive dystonia: detection with comprehensive GCHI screening. Neurology. 2005;64(5):908–11. doi: 10.1212/01.WNL.0000152839.50258.A2. [DOI] [PubMed] [Google Scholar]
- 6.Mayahi L, Mason L, Bleasdale-Barr K, Donald A, Trender-Gerhard I, Sweeney MG, Davis MB, Wood N, Mathias CJ, Watson L, Pellerin D, Heales S, Deanfield JE, Bhatia K, Murray-Rust J, Hingorani AD. Endothelial, sympathetic, and cardiac function in inherited (6R)-L-erythro-5,6,7,8-tetrahydro-L-biopterin deficiency. Circ Cardiovasc Genet. 2010;3(6):513–22. doi: 10.1161/CIRCGENETICS.110.957605. [DOI] [PubMed] [Google Scholar]
- 7.Furukawa Y, Shimadzu M, Rajput AH, Shimizu Y, Tagawa T, Mori H, Yokochi M, Narabayashi H, Hornykiewicz O, Mizuno Y, Kish SJ. GTP-cyclohydrolase I gene mutations in hereditary progressive amd dopa-responsive dystonia. Ann Neurol. 1996;39(5):609–17. doi: 10.1002/ana.410390510. [DOI] [PubMed] [Google Scholar]
- 8.Grimes DA, Barclay CL, Duff J, Furukawa Y, Lang AE. Phenocopies in a large GCH1 mutation positive family with dopa responsive dystonia: confusing the picture? J Neurol Neurosurg Psychiatry. 2002;72(6):801–4. doi: 10.1136/jnnp.72.6.801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tassin J, Durr A, Bonnet AM, Gil R, Vidailhet M, Lucking CB, Goas JY, Durif F, Abada M, Echenne B, Motte J, Lagueny A, Lacomblez L, Jedynak P, Bartholome B, Agid Y, Brice A. Levodopa-responsive dystonia. GTP cyclohydrolase I or parkin mutations? Brain. 2000;123( Pt 6):1112–21. doi: 10.1093/brain/123.6.1112. [DOI] [PubMed] [Google Scholar]
- 10.Nygaard TG, Wilhelmsen KC, Risch NJ, Brown DL, Trugman JM, Gilliam TC, Fahn S, Weeks DE. Linkage mapping of dopa-responsive dystonia (DRD) to chromosome 14q. Nat Genet. 1993;5(4):386–91. doi: 10.1038/ng1293-386. [DOI] [PubMed] [Google Scholar]
- 11.Steinberger D, Korinthenberg R, Topka H, Berghauser M, Wedde R, Muller U. Dopa-responsive dystonia: mutation analysis of GCH1 and analysis of therapeutic doses of L-dopa. German Dystonia Study Group. Neurology. 2000;55(11):1735–7. doi: 10.1212/wnl.55.11.1735. [DOI] [PubMed] [Google Scholar]
- 12.Furukawa Y. GTP Cyclohydrolase 1-Deficient Dopa-Responsive Dystonia. GeneReviews™. 2002 Feb 21; Updated 2012 May 3; Available from: http://www.ncbi.nlm.nih.gov/books/NBK1508/ [PubMed]
- 13.Bettencourt C, Morris HR, Singleton AB, Hardy J, Houlden H. Exome sequencing expands the mutational spectrum of SPG8 in a family with spasticity responsive to L-DOPA treatment. J Neurol. 2013;260(9):2414–6. doi: 10.1007/s00415-013-7044-6. [DOI] [PMC free article] [PubMed] [Google Scholar]