

Abstract
A major limitation to the use of microelectrode technologies in both research and clinical applications is our inability to consistently record high quality neural signals. There is increasing evidence that recording instability is linked, in part, to neuroinflammation. A number of factors including extravasated blood products and macrophage released soluble factors are believed to mediate neuroinflammation and the resulting recording instability. However, the roles of other inflammatory stimuli, such as residual endotoxin contamination, are poorly understood. Therefore, to determine the effect of endotoxin contamination we examined the brain tissue response of C57/BL6 mice to non-functional microelectrodes with a range of endotoxin levels. Endotoxin contamination on the sterilized microelectrodes was measured using a limulus amebocyte lysate test following FDA guidelines. Microelectrodes sterilized by autoclave, dry heat, or ethylene oxide gas, resulted in variable levels of residual endotoxins of 0.55 EU/mL, 0.22 EU/mL, and 0.11 EU/mL, respectively. Histological evaluation at two weeks showed a direct correlation between microglia/macrophage activation and endotoxin levels. Interestingly, astrogliosis, neuronal loss, and blood brain barrier dysfunction demonstrated a threshold-dependent response to bacterial endotoxins. However, at sixteen weeks, no histological differences were detected, regardless of initial endotoxin levels. Therefore, our results demonstrate that endotoxin contamination, within the range examined, contributes to initial but not chronic microelectrode associated neuroinflammation. Our results suggest that minimizing residual endotoxins may impact early recording quality. To this end, endotoxins should be considered as a potent stimulant to the neuroinflammatory response to implanted intracortical microelectrodes.
1. Introduction
Microelectrodes have proven to be a critical basic science tool for improving our understanding of how the nervous system works1–3. Additionally, recorded action potentials from individual or small populations of neurons using intracortical microelectrodes have been shown to be a promising source of control signals for various rehabilitation applications4, 5. A major hurdle to the use of microelectrode technologies in both research and clinical applications is our inability to consistently record high quality neural signals over time6–11. While a variety of factors are believed to impact recording quality, it is widely accepted that a major contributing factor is the brain tissue response to intracortical microelectrodes8,9,11.
More than 100 studies have been performed to determine how the brain tissue response might impact recording quality. This large body of work has identified several aspects of the brain tissue response that could adversely impact recording performance. Early hypotheses focused on the encapsulating astroglial scar, which may limit ion transport and electrically isolate the microelectrode from nearby neuronal targets12, 13. Further, the viability and functionality of neuronal targets themselves may also impact recording performance. For example, after microelectrode implantation, the density of neuronal targets is reduced within the critical recording range and many of the remaining neurons within the recording range show evidence of degeneration14, 15. Additionally, recent work has indicated that normal neuronal function may be further impaired by alterations in the local ionic environment due to blood-brain barrier dysfunction16–18.
Underlying the observed astrogliosis, loss of neuronal viability and blood-brain barrier dysfunction is a well-documented chronic neuroinflammatory state involving both resident microglia and blood-born macrophages19–21. Increasing evidence indicates that activated macrophages and microglia may serve as the key cellular mediator of the brain tissue response that limits recording quality. Once activated, both microglia and macrophages release a range of pro-inflammatory/cytotoxic molecules, which can damage healthy neurons or activate astrocytes22–26. For example, we have recently shown that reactive oxygen species play a dominant role in neuronal viability and blood-brain barrier permeability at the microelectrode-tissue interface15. Beyond acting as a potent source of cytotoxic soluble factors, persistent inflammatory cell trafficking between the brain tissue and the local vasculature may explain observations of chronic blood-brain barrier dysfunction27.
The direct connection between neuroinflammation and recording function was first established by Rennekar et al., who showed that administration of the anti-inflammatory drug minocycline improved recording performance9. Unfortunately, prolonged administration of minocycline has been shown to result in decreased renal function, vertigo, bone discoloration/loss, fatal colitis, or intracranial hypertension28–30. Building off the primary hypothesis that neuroinflammation impacts recording quality, several groups have investigated strategies to minimize the neuroinflammatory response and improve recording function with varying degrees of success9, 27, 31–36. Further developing our understanding of the factors that initiate and perpetuate macrophage/microglial activation surrounding implanted microelectrodes will lead to advanced strategies for improving recording performance.
Fortunately, a large body of literature exists describing endogenous danger and distress signals that serve as pro-inflammatory stimuli for macrophage and microglia activation (Figure 1A). Examples of endogenous signals include cellular debris, infiltrating blood-derived cells, and denatured serum proteins37, 38. Additionally, extravasated fibrinogen, plasma soluble fibronectin, complement factors, and other blood products have been shown to be potent mediators of macrophage and microglial activation39. Following extravasation, blood-products perpetuate inflammatory cell activation through Toll Like Receptor (TLR), Cluster of Differentiation 14 (CD14), and other receptor-mediated pathways40,41. Resulting from either the initial damage of microelectrode implantation into the cortex or possibly motion-induced damage at later time points, several endogenous signals are likely present at the electrode-tissue interface and may perpetuate the inflammatory response.
Figure 1.
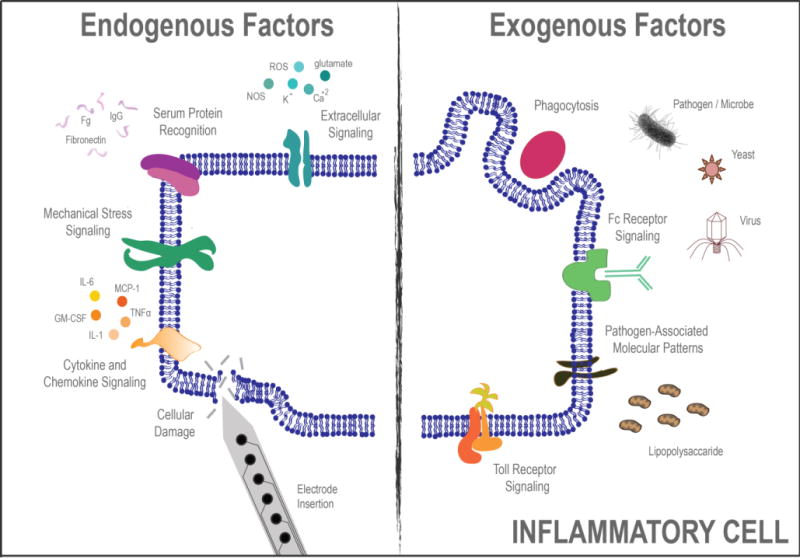
Cartoon depiction of several endogenous and exogenous factors known to stimulate the inflammatory response to microglia and macrophages.
Beyond endogenous signals for microglia and macrophage activation there are also a range of exogenous signals that may play a role in microelectrode-associated neuroinflammation (Figure 1B). For example, most microbes have pathogen-associated molecular patterns (PAMPs) that are recognized by macrophages/microglia. Following PAMP recognition, intracellular signaling cascades direct microglia and macrophages down classic inflammatory pathways42. Importantly, the impact of PAMPs, in particular the endotoxin lipopolysaccharide (LPS), has been recently demonstrated on inflammation and recording function of intracortical microelectrodes43. Specifically, LPS administration significantly increased local inflammation while lowering signal to noise ratios and the number of recorded units compared to saline-only control animals43. While the work by Harris et al., has demonstrated that high levels of purposefully administered endotoxins impacts neuroinflammation and recording function43, the impact of low-level residual endotoxin contamination that may remain even after traditional sterilization techniques is unclear.
Interestingly, recent work by Gorbet and Sefton indicates that even low-levels of endotoxin contamination can significantly affect the biological tissue response to other types of medical devices hindering device performance44. Gorbet and Sefton’s findings suggest that endotoxin contamination should be investigated as a potential stimulus of the inflammatory response to implanted medical devices and biomaterials44. Therefore, to examine the role of endotoxin contamination on the neuroinflammatory response to implanted microelectrodes, we studied the brain tissue response of C57/BL6 mice to microelectrodes with a range of low-level, residual endotoxins left on the microelectrodes after a variety of standard sterilization procedures were performed.
2. Experimental Methods
2.1 Method of Microelectrode Sterilization
Non-functional single shank ‘Michigan-style’ microelectrodes were fabricated in the Advanced Platform Technology Center’s microfabrication facility. Electrode shanks were 2 mm long × 123 μm wide × 15 μm thick with a tapered tip and a 1 mm × 1mm bond tab. Prior to sterilization, all microelectrodes were cleaned and disinfected by soaking for 5 minutes in 70% ethanol, followed by three rinses in deionized water (ddH2O). To create a tiered array of residual endotoxin levels, microelectrodes were then sterilized by one of three common methods (autoclave, dry heat, or ethylene oxide gas). Briefly, microelectrodes were autoclaved in a Market Forge Sterilmatic Autoclave (Model: STM-EL) with 15 psi pressure at 121°C for 35 minutes. Microelectrodes sterilized by dry heat were placed in a Thermo Scientific Precision Oven (Model: 6912) at 180°C for 4 hours. Finally, ethylene oxide gas sterilization was performed at University Hospitals – Case Western Reserve Medical Center. All ethylene oxide sterilized samples underwent a 12 hour aeration and degassing cycle prior to testing or implantation.
2.2 Measurement of Residual Endotoxins
A Kinetic-QCL® chromogenic limulus amebocyte lysate (LAL) endotoxin assay (Lonza, Inc Catalog Number: 50–650U) was used to determine the level of residual endotoxins on the microelectrode surface after the various sterilization protocols. The Kinetic-QCL® assay is a quantitative assay for the detection of Gram-negative endotoxins, where endotoxin level is indirectly measured as a function of the activation of the proenzyme45. Endotoxin activity on a given microelectrode sample was calculated from a standard curve generated from the Kinetic-QCL kit following methods described by the vendor (Lonza). Six microelectrodes were analyzed for each sterilization method (n = 6).
2.3 Surgical Implantation of Intracortical Microelectrodes
To assess the impact of endotoxin contamination on the initial and chronic phases of the neuroinflammatory response, male C57/BL6 mice received a single implant for either 2 or 16 weeks, respectively. All mice were obtained from Jackson Laboratory (strain #000664) and age-matched to 6 weeks of age (~20g) at the time of surgery. All animal practices were performed in accordance to protocols established by Case Western Reserve University Institution of Animal Care and Use Committee.
Surgical procedures were performed with slight modifications made to previously described methods. To limit subsequent bacterial contamination, all surgical procedures were performed under sterile conditions using micro-isolator techniques in a class II sterile hood. All surgical procedures were performed by the same surgeon to minimize intra-surgeon variability. Mice were anesthetized with isoflurane (5% induction, 1–2% maintenance) and mounted onto a stereotaxic frame. A local anesthetic (Marcaine 100μl, 0.25%), was given under the incision site by subcutaneous (SQ) injection. A sterile field was created and maintained throughout the surgery by shaving the animals’ head and scrubbing the shaved area with betadine followed by 70% isopropanol for antiseptic removal. Ophthalmic ointment was used throughout the surgery to avoid retinal drying. Additionally, Meloxicam (0.7mL, 1.15 mg/ml) was administered SQ at the start of surgery and once the following day for pain management. The antibiotic Cefazolin (16 mg/kg) was administered SQ at the start of surgery and twice the following day to prevent post-surgical infection.
Next, a midline incision was made and the tissue was retracted to expose the skull. To expose the brain tissue, a 3mm hole was made 1mm lateral to the midline and 2mm caudal to bregma using a biopsy punch (PSS Select). A single microelectrode was then manually implanted to a depth of 2 mm, taking precaution to avoid major surface vasculature. Following microelectrode implantation, the microelectrode’s bond tab was tethered to the skull with a cap of Kwik-sil (World Precision Instruments) and uv-cured liquid dentin (Fusio/Flow-it ALC, Pentron Dental). The surgical site was then closed using 5-0 monofilament polypropylene suture (Butler Schein). Triple antibiotic ointment was applied following suturing to prevent drying and localized infection around the incision.
Post-surgery animals were monitored daily for signs of pain and distress throughout the implant duration. No signs of infection were observed in any animal.
2.4 Tissue Processing
Inflammatory events were assessed at controlled time points of 2 or 16 weeks post implantation correlating with initial and chronic phases of neuroinflammation17,46–48. Mice were anesthetized with a 200μl interperitonial (IP) injection of rodent cocktail. Rodent cocktail (1.75ml total volume) was prepared by mixing 150μl Ketamine HCl (100 mg/ml), 150μl Xylazine HCl (20 mg/ml), 50μl Acepromazine (10 mg/ml), and 1.4ml Sterile Saline. Following induction, mice were transcardially perfused with phosphate buffered saline (PBS) until exudate was clear (~30mL). Mice then underwent perfusion fixation with 4% paraformaldehyde (PFA). Following perfusion fixation, brains were carefully extracted, placed in fresh 4% PFA, and post-fixed at 4 °C for an additional 2 days. For cryopreservation, the brains were then equilibrated in 30% Sucrose in PBS. The brain tissue was then frozen at −80 °C in optimal cutting temperature compound (OCT, Tissue-Tek). Horizontal sections (16 μm thick) were collected using a cryostat (Microm HM525) and directly mounted onto glass slides (Superfrost Plus, Fisher Scientific). Mounted sections were then stored at −80 °C until immunohistochemistry was performed.
2.5 Assessment of Neuroinflammation
Immunohistological analysis of common cellular markers (microglia/macrophages and astrocytes) as well as blood protein Immunoglobulin G (IgG), was performed to quantify extent of the neuroinflammatory response at the tissue-electrode interface as previously described49. Briefly, tissue sections were equilibrated to room temperature (RT) for 30 minutes and remaining OCT was removed with 3 rinses of PBS. Tissue sections were then permeated with a 15 minute incubation in PBS containing 0.1% Triton-X (PBS-T). Next, the sections were blocked in 4% chicken serum containing 0.3% Triton-X and 0.1% sodium azide for 1 hour at RT. Primary antibodies targeting specific antigens (Table 1) were then added to respective tissue samples and incubated overnight at 4 °C. The following day, tissue samples were washed six times with PBS-T to remove unbound primary antibody. Tissue samples were then incubated in AlexaFluor™ conjugated secondary antibodies (Table 1) specific to the corresponding primary antibodies (1:1000 dilution, 2 hrs at RT). To remove unbound secondary antibody, samples were washed six times in PBS-T and remaining detergent was removed with three additional rinses of PBS. Tissue autofluorescence was minimized with a 10 min incubation in 0.5mM copper sulfate buffer following previous methods (50mM Ammonium Acetate, pH 5.0)49. Following CuSO4 treatment, all slides were washed thoroughly with ddH2O to prevent prolonged quenching by copper sulfate. Finally, all slides were coverslipped using Fluoromount-G, and stored in the dark at 4 °C.
TABLE 1. Primary and Secondary Antibodies used for Immunohistological Analysis.
List of primary and secondary antibodies used for immunohistological analysis of common cellular markers including activated microglia/macrophages, astrocytes, neuronal nuclei as well as blood protein Immunoglobulin G.
| Cell Type | Antigen | Blocking, Buffer |
Primary Antibody | Secondary Antibody | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Name | Vendor | Catalog Number |
Primary Antibody Dilution |
Name | Vendor | Catalog Number |
Secondary Antibody Dilution |
|||
| Activated Microglia and Macrophages |
CD68 | Chicken | Rat mAb to CD68 [FA-11] |
Abcam | ab53444 | 1:500 | Chicken anti-Rat 488 |
Life Technologies |
A-21470 | 1:1000 |
| Astrocytes | GFAP | Chicken | GFAP Polyclonal Rabbit Antiserum |
Neuromics | RA22101 | 1:500 | Chicken anti-Rabbit 594 |
Life Technologies |
A-21442 | 1:1000 |
| Neurons | NeuN | Goat | Mouse anti Neuronal Nuclei Clone A60 |
Millipore | MAB377 | 1:250 | DAB Staining Kit |
Life Technologies |
87-9163 | Protocol included by vendor |
| Blood Protein Immunoglobulin G |
IgG | Chicken | Rb anti-Ms IgG | AbD Serotec |
STAR26B | 1:1000 | Chicken anti-Rabbit 488 |
Life Technologies |
A-21441 | 1:1000 |
2.6 Assessment of Neuronal Nuclei at Interface
Neuron viability was assessed at the interface using diaminobenzidine (DAB) histochemistry following the methods described by the vendor (Life Technologies). Tissue sections were prepared and permeated for staining as described in Section 2.5. Following tissue permeation, sections were blocked in 4% goat serum containing 0.3% Triton-X and 0.1% sodium azide for 1 hour at RT. Next, to specifically label neuronal nuclei, mouse anti-NeuN Clone A60 primary antibody (Table 1) was added at a 1:250 dilution for 1 hour at RT. Unbound primary antibody was removed with three washes of PBS and 100μl horse-radish peroxidae (HRP) polymer conjugate was added to each tissue section for 10 minutes. Next, unbound HRP polymer conjugate was removed with three washes of PBS. Following washing, 100μl of DAB chromagen (provided in DAB staining kit) was added to each tissue section for 5 minutes. The tissue sections were then rinsed thoroughly with ddH2O, counterstained with hematoxylin for enhanced tissue contrast, and mounted using Histomount mounting solution (Life Technologies).
2.7 Imaging and Quantitative Analysis
All images were acquired using a Carl Zeiss AxioObserver.Z1 (Zeiss Inc) inverted epifluorescence microscope and a 10X objective. Fluorescent markers (GFAP, CD68, and IgG) were imaged using an AxioCam MRm monochrome camera. Exposure times were optimized and held constant per stain for all analyzed time points. DAB stained neuronal nuclei sections were imaged under brightfield using an AxioCam ERc5 color camera. For a more complete analysis of the inflammatory response at the interface without compromising image resolution, 16 separate 10X images were acquired using an automated stage and stitched together using MosaiX software (Zeiss Inc) for all stained sections. For clarity of presentation, images reported here were cropped, pseudo-colored, and slightly enhanced (uniformly across all conditions) to improve visual display. However, all quantification, analysis, and statistical comparisons were performed on raw images.
Following acquisition, unaltered images of fluorescent markers were converted to 16-bit tagged imaging files using Axiovision LE software and analyzed using a custom MATLAB program (MINUTE V1.5, Microelectrode Interface Universal Tool for Evaluation). Briefly, the implant region of each image was manually designated. The program was then designed to extract fluorescent intensity measurements in expanding 2μm concentric intervals out to 1000μm from the user-defined interface. Raw fluorescent intensities were then normalized to background signal for each labeled antigen. Plots and images were truncated where the antigen detection reached background levels for presentation.
The area under the curve was calculated from the intensity profile for each image to allow for statistical comparisons between conditions. For clarity, the data is reported as normalized fluorescent intensity as a function of distance from the implant. To quantify cellular/protein distribution, exponential curve fitting was performed on normalized fluorescent intensity curves for each tissue section using Ezyfit, a publically available curve fitting toolbox for Matlab (http://www.fast.u-psud.fr/ezyfit/). Using the Nelder-Mead algorithm, curves were fit such that the sum of squared residuals was minimized with respect to two variables λ and a (Equation 1).
| (1) |
Here, a represents the peak fluorescent intensity, x (μm) is the distance from the implant site and y is the normalized fluorescent intensity. Lambda values, denoting the distance (μm) at which 63.2% of the total observed fluorescent intensity had returned to background expression levels, were recorded for each tissue section and are presented as an average ± standard error of the mean.
To quantify the neuronal populations surrounding the implant, a custom MATLAB code (N.E.R.D., Neurons Encompassed aRound the Device) was designed. Our custom code was designed to draw expanding concentric rings at fixed radial distances from the implant. The number of neurons in each ring was calculated through user-defined location of neuronal cell bodies. Neuronal density within each ring was then calculated within the code. Finally, neuronal nuclei cell counts were normalized to background counts from non-surgical aged-matched control animals. For clarity, data is reported as a percentage of neuronal density normalized to non-surgical age-matched control animals as a function of distance from the implant.
2.8 Statistical Analysis
All statistical analyses were performed using Minitab software (Minitab, Inc). Six microelectrodes were evaluated for each condition to preform statistical comparisons of endotoxin levels between sterilization methods. To account for dependencies in cellular/protein expression between slices from the same animal, measurements from all slices for a given animal were first averaged together. Statistical comparisons were then performed using the independent animal averages (n = 4–7 animals/condition). All statistical comparisons were performed using a general linear one-way analysis of variance (ANOVA) model. Pair-wise comparisons were conducted using a post-hoc Tukey testing, where significance was considered as p<0.05.
3. Results
3.1 Endotoxin Levels Resulting from Each Sterilization Method
A Kinetic-QCL Chromogenic LAL Endotoxin Assay was used in order to determine the level of residual endotoxins on non-functional intracortical microelectrodes before and after sterilization. The LAL test is an accepted method by the Food and Drug Administration to evaluate endotoxin contamination50. As there is a large disparity in the per weight potency of specific endotoxins, we quantified residual endotoxin contamination levels using the standardized metric of Endotoxin Units, where 1 EU is equivalent to the pyrogenic impact of 100pg of purified E coli LPS or roughly the same amount of LPS found on 100,000 E coli cells51.
Sterilization of microelectrodes by autoclave, dry heat, and ethylene oxide gas resulted in controlled low-level residual endotoxin contamination. All sterilized microelectrodes showed significantly lower endotoxin levels compared to unsterilized microelectrodes that were only disinfected using ethyl alcohol (Figure 2). Of the sterilized microelectrodes, autoclaved microelectrodes had the highest level of endotoxins (0.55 ± 0.09 EU/mL) followed by microelectrodes sterilized by dry heat (0.22 ± 0.04 EU/mL) and lastly ethylene oxide gas sterilized microelectrodes (0.11 ± 0.03 EU/mL). Further comparisons of endotoxin levels (Figure 2-Inset) showed that autoclaved electrodes had significantly higher endotoxin levels compared to microelectrodes sterilized by dry heat or ethylene oxide gas. Additionally, microelectrodes sterilized by dry heat had significantly higher residual endotoxin levels compared to microelectrodes sterilized by ethylene oxide gas.
Figure 2.
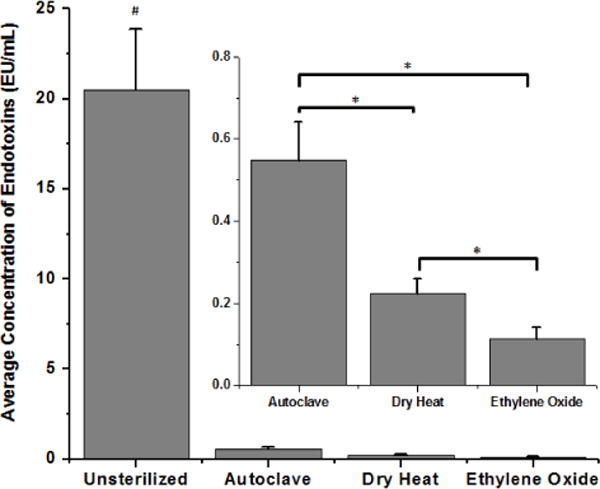
Residual endotoxin contamination present after cleaning and sterilization of microelectrodes by autoclave, dry heat or ethylene oxide gas. Microelectrodes that did not undergo sterilization had significantly higher residual endotoxin levels compared to all sterilized microelectrodes (#p<0.0002). Inset: autoclaved microelectrodes had significantly higher residual endotoxin levels when compared to microelectrodes sterilized by dry heat and ethylene oxide gas (*p<0.001). Microelectrodes sterilized by dry heat had significantly higher residual endotoxins compared to microelectrodes sterilized by ethylene oxide gas (*p<0.03).
3.2. Microglia/Macrophage Activation at the Tissue-Electrode Interface
To identify activated microglia and macrophages near the implant site, we used an anti-mouse CD68 antibody (FA/11 clone), specific to macrosialin, a sialoglycoprotein confined to activated, murine mononuclear phagocytes52 Increased CD68+ immunoreactivity of macrosialin is typically attributed to the increased presence of lysosomal vacuoles53, 54. Sections from non-surgical age-matched control animals showed minimal CD68+ immunoreactivity (data not shown), indicating negligible activation of microglia and macrophages without microelectrode implantation. In contrast, punctate CD68+ immunoreactivity concentrated at the interface between 0–50μm was seen surrounding all three microelectrode cohorts at both 2 and 16 weeks post implantation (Figure 3A–F).
Figure 3.
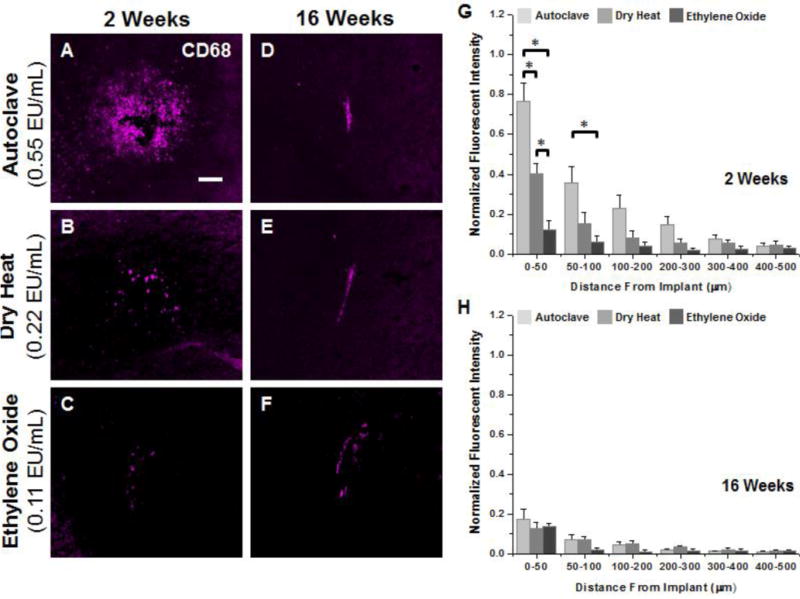
Representative immunohistochemical images of CD68+ immunoreacivity surrounding microelectrodes sterilized by autoclave, dry heat or ethylene oxide gas at 2 weeks (A–C) and 16 weeks (D–F) post implantation. Quantification of CD68+ immunoreactivity at 2 weeks post implantation (G) shows significantly higher levels between 0–100μm for microelectrodes sterilized by autoclave compared to microelectrodes sterilized by dry heat or ethylene oxide gas. Further, significantly higher CD68+ immunoreactivity was observed for microelectrodes sterilized by dry heat compared to ethylene oxide gas within the first 50μm. At 16 weeks post implantation (H), similar levels of CD68+ immunoreactivity was seen around all implant, regardless of initial endotoxin levels. Scale Bar = 100μm; * p<0.02
Two weeks post implantation, microglia and macrophage activation (CD68+ immunoreactivity) at the tissue-electrode interface directly correlated with residual endotoxin levels present on microelectrodes after sterilization (Figure 3G). Autoclaved microelectrodes, having the greatest level of residual endotoxin contamination, showed significantly more CD68+ immunoreactivity within the first 100μm compared to microelectrodes sterilized by either dry heat or ethylene oxide. Furthermore, microelectrodes sterilized by dry heat showed significantly more microglia and macrophage activation between 0–50μm compared to microelectrodes sterilized by ethylene oxide. However, quantification of activated microglia and macrophages at 16 weeks post implantation (Figure 3H) showed no significant difference between residual endotoxin levels.
Curve fitting analysis to assess activated microglia and macrophage distribution is shown in Table 2. Changes in lambda over time are indicative of either a change in the distribution of the antigen, or an overall change in the intensity profile of the detectable antigen. At 2 weeks post implantation, autoclaved microelectrodes had significantly larger lambda values for CD68+ immunoreactivity compared to dry heat or ethylene oxide gas sterilized microelectrodes, indicating a broader distribution of CD68+ immunoreactivity to autoclaved implants. At 16 weeks post implantation, ethylene oxide gas sterilized microelectrodes, which had the lowest level of residual endotoxins, continued to have significantly lower lambda values compared to autoclaved microelectrodes.
TABLE 2. Average Lambda Values ± S.E.M for IHC Analysis Curves.
Average lambda values ± S.E.M for CD68, GFAP and IgG analysis curves for each sterilization method at 2 and 16 weeks post implantation. a p<0.02; significance between autoclave and dry heat b p<0.02; significance between autoclave and ethylene oxide c p<0.05; significance between dry heat and ethylene oxide d p<0.02; significance between 2 and 16 weeks within cohort. n = 4–7 animals for each cohort.
| 2 Weeks | 16 Weeks | |
|---|---|---|
| CD68 | ||
| Autoclave | 100.18 ± 19.11a,b | 85.63 ± 17.22b |
| Dry Heat | 38.12 ± 5.79 | 88.48 ± 14.72c,d |
| Ethylene Oxide | 38.94 + 15.38 | 24.64 ± 1.25 |
| GFAP | ||
| Autoclave | 166.10 + 23.09a,b | 130.55 ± 20.50 |
| Dry Heat | 58.26 ± 10.21 | 154.73 ± 10.76d |
| Ethylene Oxide | 51.18 ± 1.19 | 157.89 ± 37.21d |
| IgG | ||
| Autoclave | 107.63 ± 20.84 | 69.96 ± 12.99c |
| Dry Heat | 61.45 ± 15.25 | 59.04 ± 24.04 |
| Ethylene Oxide | 76.75 + 15.37 | 48.06 ± 11.25 |
Comparisons between initial (2 weeks) and chronic (16 weeks) neuroinflammation showed a significant decrease in CD68+ immunoreactivity for autoclaved electrodes up to 400μm from the interface at 16 weeks post implantation. On the other hand, dry heat sterilized microelectrodes showed significantly lower CD68+ immunoreactivity at 16 weeks post implantation only over the first 50μm from the interface. Ethylene oxide sterilized microelectrodes had comparable levels of CD68+ immunoreactivity between 2 and 16 weeks. Our results suggest that lower residual endotoxin levels correlate with lower levels of CD68+ immunoreactivity, at two weeks post implantation.
3.3 Astrocyte Expression at the Tissue-Electrode Interface
The astrocyte-specific intermediate filament marker (GFAP) was used to measure the impact of residual endotoxin contamination on astrogliosis surrounding intracortical microelectrodes. Sections from non-surgical age-matched control animals showed ubiquitous low-level GFAP+ mmunoreactivity (data not shown), indicating a uniform presence of astrocytes throughout the cortical tissue prior to implantation. Here, increased GFAP+ immunoreactivity (indicating astrogliosis) was observed at the interface after implantation for all microelectrode cohorts at both 2 and 16 weeks post implantation (Figure 4A–F).
Figure 4.
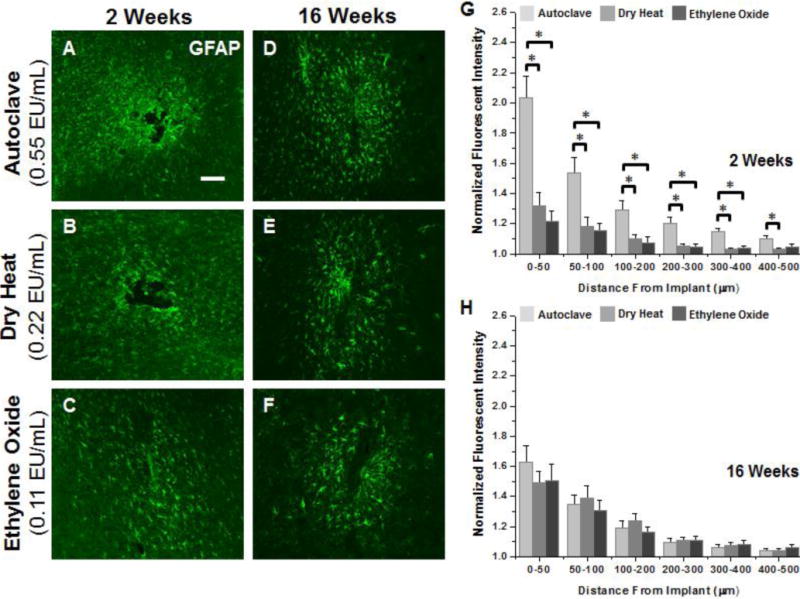
Representative immunohistochemical images of GFAP+ immunoreactivity surrounding microelectrodes sterilized by autoclave, dry heat or ethylene oxide gas at 2 weeks (A–C) and 16 weeks (D–F) post implantation. Quantification of GFAP at 2 weeks (G) shows significantly heightened astrogliosis up to 400μm from the interface for microelectrodes sterilized by autoclave compared to microelectrodes sterilized by dry heat or ethylene oxide gas. At 16 weeks post implantation (H) no difference was seen regardless of initial endotoxin levels. Scale Bar = 100μm; * p<0.02
Quantification of GFAP+ immunoreactivity at two weeks post implantation (Figure 4G) showed significantly more astrogliosis surrounding the tissue-electrode interface in microelectrodes sterilized by autoclave compared to dry heat (0–400μm) and ethylene oxide gas (0–500μm). In contrast to correlations found between endotoxin levels and microglia/macrophage activation, similar levels of astrogliosis were observed around microelectrodes sterilized by dry heat and ethylene oxide (no statistical significance between conditions). At 2 weeks post implantation, lambda values for distribution of astrocytes surrounding the implant were significantly higher for microelectrodes sterilized by autoclave compared to microelectrodes with lower residual endotoxin levels, indicating a more widespread/diffuse astroglial scar. Additionally, no significant difference was observed in lambda values between microelectrodes with lower residual endotoxin levels.
However, by 16 weeks post-implantation, astrogliosis surrounding the implant was comparable in both intensity (Figure 4H) and lambda (Table 2) between all microelectrode cohorts, with no significant differences. All microelectrode cohorts demonstrated heightened levels of GFAP+immunoreactivity at the interface compared to any other binned interval at the interface at 16 weeks post implantation. Comparisons between 2 and 16 weeks showed a significant decrease (0–50μm and 300–500μm) in astrogliosis at the interface for autoclaved sterilized implants at 16 weeks post implantation. In contrast, a significant increase in GFAP+ immunoreactivity was seen at 16 weeks for electrodes sterilized by dry heat (100–200μm) and ethylene oxide (0–50μm). Further, when comparing lambda values between 2 and 16 weeks, a significant increase in lambda was observed for microelectrodes sterilized by dry heat or ethylene oxide at 16 weeks.
3.4 Disruption of Blood-brain Barrier at the Tissue-Electrode Interface
To assess the impact of residual endotoxin contamination on blood-brain barrier dysfunction associated with microelectrode implantation, sections were reacted with antisera against mouse-IgG, a blood protein typically not found in native cortical tissue55. Sections from non-surgical age-matched control animals showed minimal IgG+ immunoreactivity in cortical tissue (data not shown), confirming negligible vascular damage prior to implantation. In contrast, we observed elevated IgG+ immunoreactivity surrounding implanted microelectrodes at both time points, regardless of sterilization method (Figure 5A–F).
Figure 5.
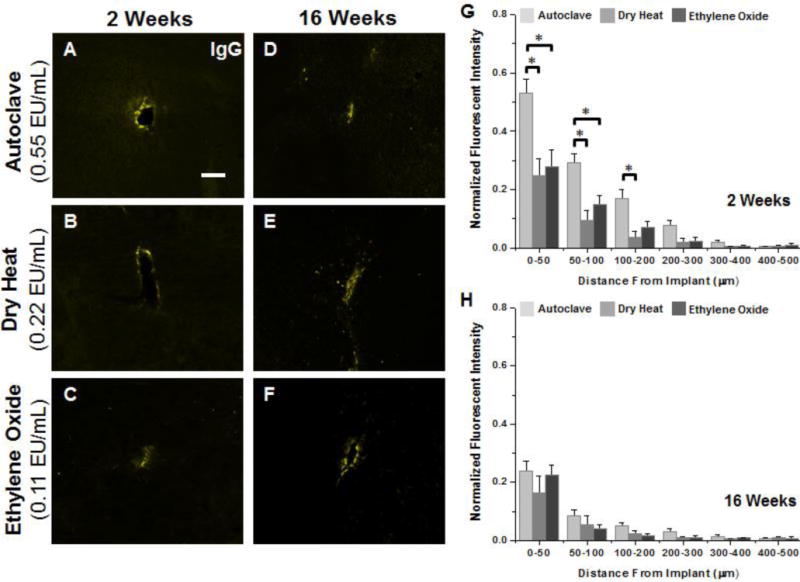
Representative immunohistochemical images of IgG+ immunoreactivity at the interface of microelectrodes sterilized by autoclave, dry heat or ethylene oxide gas at 2 weeks (A–C) and 16 weeks (D–F) post implantation. Quantification of IgG at 2 weeks (G) shows significantly higher accumulation of IgG was observed between 0–100μm for microelectrodes sterilized by autoclave compared to microelectrodes sterilized by dry heat or ethylene oxide gas. (H) No statistical difference was observed at 16 weeks post implantation regardless of initial endotoxin levels. Scale Bar = 100μm; * p<0.02
Quantification of IgG+ immunoreactivity at two weeks post implantation (Figure 5G) showed a significantly increased presence of IgG for autoclaved microelectrodes compared to dry heat (0–200μm) or ethylene oxide gas sterilized microelectrodes (0–100μm). Similar to GFAP+ immunoreactivity at the interface, no significant difference in IgG+ immunoreactivity was found at the interface between microelectrodes with lower residual endotoxin levels. Further, analysis of lambda values at 2 weeks showed no significant difference between conditions. Here, higher IgG+ immunoreactivity for autoclaved microelectrodes suggests an increase serum protein accumulation at the interface. However, similar lambda values for all cohorts suggest a similar rate of decay in IgG+ immunoreactivity around the interface.
At 16 weeks post implantation, no statistical differences were found in either IgG+ immunoreactivity (Figure 5H) or lambda values (Table 2) across all three microelectrode cohorts, which suggests a similar IgG profile for all cohorts at 16 weeks. Comparisons of the quantification of IgG+ immunoreactivity between 2 and 16 weeks post implantation demonstrated a significant reduction in IgG+ immunoreactivity for autoclaved implants (0–300μm) and ethylene oxide sterilized implants (50–100μm). Dry heat sterilized implants on the other hand, demonstrated consistent IgG+ immunoreactivity between 2 and 16 weeks. At both time points, elevated IgG+ immunoreactivity between 0–50μm and relatively small lambda values suggests that vascular damage was localized at the interface.
3.5 Neuronal Nuclei Survival at the Tissue-Electrode Interface
Antibodies against NeuN A60 clone were used to examine the density of neuronal nuclei near implanted microelectrodes with varying levels of residual endotoxin contamination after sterilization. Representative images of neuronal nuclei at the tissue-electrode interface for each method of sterilization is shown in Figure 6A–F at both 2 and 16 weeks post implantation. Density of neuronal nuclei in non-surgical aged-matched control animals was 1944 ± 28 cells/mm2 and 1841 ± 52 cells/mm2 at 2 and 16 weeks, respectively.
Figure 6.
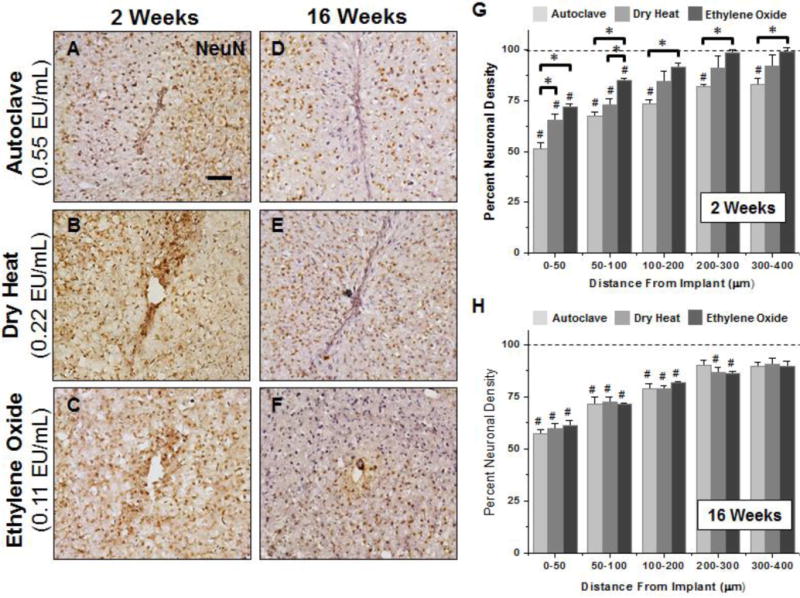
Representative immunohistochemical images of neuronal nuclei density surrounding microelectrodes sterilized by autoclave, dry heat or ethylene oxide gas at 2 weeks (A–C) and 16 weeks (D–F) post implantation. For all binned intervals at 2 weeks (G), microelectrodes sterilized by autoclave had significant reduced neuronal density compared to microelectrodes sterilized by ethylene oxide gas as well as background neuronal density of non-surgical age-matched controls. Further, significant decreases in neuronal densities were seen between microelectrodes sterilized by autoclave compared to microelectrodes sterilized by dry heat and ethylene oxide gas between 0–50μm. At 16 weeks, neuronal density remained significantly lower than background neuronal density up to 200μm, regardless of sterilization method. However, at 16 weeks post implantation we observed no significant difference based on initial endotoxin levels. Scale Bar = 100μm; * p<0.02; # p<0.05
A significant reduction in neuronal cell density compared to background was observed at the interface across all microelectrode cohorts at two weeks post implantation (Figure 6G). On the other hand, both dry heat and ethylene oxide gas sterilized microelectrodes recovered to background neuronal densities at distances greater than 100μm from the interface. At 2 weeks post implantation, statistical comparisons revealed that microelectrodes sterilized by autoclave had significantly decreased neuronal densities compared to background at all binned intervals up to 400μm from the interface. Further, neuronal density inversely correlated with the levels of endotoxins on the microelectrode surface. Microelectrodes sterilized by ethylene oxide gas showed significantly higher neuronal cell counts compared to autoclaved electrodes at all binned intervals up to 400μm from the interface. Additionally, dry heat sterilized electrodes showed significantly higher neuronal densities between 0–50μm when compared to autoclaved microelectrodes.
Quantification at 16 weeks post implantation (Figure 6H) showed no significant difference in neuronal density between cohorts, regardless of initial endotoxin levels. However, within the first 50μm neuronal densities remained less than 60% across all microelectrode cohorts at 16 weeks when compared to nonsurgical age-matched control animals. Further, at 16 weeks, autoclaved microelectrodes had significantly lower neuronal densities compared to background up to 200μm from the interface, while dry heat and ethylene oxide gas sterilized microelectrodes had significantly lower neuronal densities compared to background up to 300μm from the interface. Comparisons of neuronal nuclei density between 2 and 16 weeks post implantation demonstrated a consistent level of neurons over time for autoclaved implants across all binned intervals up to 400µm from the interface. However, a significant reduction in neurons was seen from 2 to 16 weeks for the dry heat sterilized implants (0–50μm, 100–300μm). Additionally, ethylene oxide sterilized implants showed a significant reduction in neuronal nuclei density from 2 to 16 weeks post implantation across all binned intervals up to 400 μm from the implant surface.
4. Discussion
In this study, we examined the role of residual endotoxin contamination on the neuroinflammatory response to implanted intracortical microelectrodes. We found that endotoxin contamination, within the range examined, contributed to initial but not chronic neuroinflammation. At two weeks post implantation, histological evaluation showed that the level of microglia/macrophage activation directly correlated with residual endotoxin levels present on non-functional microelectrodes after sterilization. Additionally, at two weeks post implantation, autoclaved implants had the highest levels of astrogliosis, neuronal loss, and blood-brain barrier dysfunction, with no differences between dry heat or ethylene oxide sterilized implants. However, at sixteen weeks postimplantation, no histological differences were detected, regardless of the initial endotoxin levels.
In evaluating the complex cascade of neuroinflammatrory events that occur after intracortical microelectrode implantation, several groups have suggested a strong correlation between the presence of activated microglia and macrophages and neuronal dieback at the interface14, 15, 17, 18, 20, 27, 36, 56–58. Further, recent evidence also suggests the role of inflammation in facilitating stability of chronic neural recordings18. Several pro-inflammatory factors including endogenous and exogenous signals (Figure 1) can directly cause microglia and macrophage activation38, 59. Therefore, understanding the factors that initiate and perpetuate microglia/macrophage activation surrounding implanted microelectrodes could lead to advanced strategies to promote high-quality recorded signals.
Consistent with previously reported literature, our results showed elevated microglia/macrophage activation (CD68+ immunoreactivity) at the tissue-electrode interface across all three microelectrode cohorts at both 2 and 16 weeks post implantation. Further, our results directly correlated increased residual endotoxin levels present on microelectrodes to elevated CD68+ immunoreactivity at two but not sixteen weeks post implantation (Figure 3). We hypothesize that CD68+ immunoreactivity was comparable across sterilization conditions at 16 weeks post implantation because endotoxins are likely removed from the surface from the initial response to the injury. Therefore, by chronic time points, neuroinflammation is likely to be mediated by material properties of the device, self-perpetuating and chronic blood brain barrier dysfunction, or possibly by chronic motion-induced damage. Analysis of lambda values between microelectrode cohorts indicated smaller lambda values for ethylene oxide gas sterilized microelectrodes with the lowest levels of residual endotoxins. Our results suggest that with increased levels of residual endotoxins, there is an increase in cellular infiltration, cellular recruitment, and/or cell trafficking of activated microglia/macrophages surrounding the implant, which may lead to a wider distribution of cellular activation.
Expression and diffusion of microglia-derived pro-inflammatory molecules have been implicated in many aspects of the reactive tissue response to implanted intracortical microelectrodes14, 60. It has been suggested that lowering the concentration of cytokines around intracortical microelectrodes may correlate with both decreased blood brain barrier leakiness and reduced neuronal cell loss27. Specifically, reduction of reactive oxygen species accumulation using resveratrol, a natural antioxidant, has been recently shown to promote blood brain barrier stability and promote neuronal survival at the microelectrode interface15. We have also previously demonstrated that microglia stimulated with increasing concentrations of nanoparticles elicit a neurotoxic response above a given stimulus-dependent threshold; a response which was prevented with antioxidant treatment61. Together, these studies suggest that microglia respond to stimuli in a dose-dependent fashion. However, down-stream gliosis and neuronal viability are likely threshold dependent, and can be regulated by either chemical or mechanical inhibition of pro-inflammatory molecule accumulation. In this study, threshold can be defined as 0.22 EU/mL. However, identifying the exact threshold for endotoxin-mediated neuroinflammation was beyond the scope of this study. Further, identification of an exact threshold level for a given antigen would require the unlikely assumption that only one antigen was stimulating the inflammatory response.
Microglia and macrophages have been shown to play an important role in promoting astrocyte hypertrophy in areas of injury and distress through extracellular signaling22–26. In the present study, we found that endotoxin levels not only correlated with elevated CD68+ immunoreactivity, but also resulted in a more pronounced glial scar formation at the interface (Figure 4). Lambda values suggest a strong correlation between increased levels of residual endotoxins and a more widespread distribution of astrocytes around the interface, at 2 weeks post implantation. Consistent with previously reported findings, our results also showed that microelectrode cohorts with a more diffuse astroglial scar had increased neuronal dieback at the interface17. Our results suggest that cellular compaction of astrogliosis at the interface may play a critical role in protecting native tissue from injury. It is likely that more compact astrocytic scars limit the diffusion of microglia and macrophage-derived pro-inflammatory molecules that have been suggested to be critical mediators of the brain’s response to implanted devices14, 60, 62.
In this study, we observed a threshold dependent response between residual endotoxins on intracortical microelectrodes and neuronal loss at the interface at two weeks post implantation. Further, loss in neuronal populations surrounding the implant correlated with threshold-dependent astrocytic scarring (Figure 4). Highest levels of endotoxin contamination from autoclaved implants correlated with the highest levels of neuronal loss and scar formation. Dry heat and ethylene oxide sterilized implants showed similar loss of neuronal populations surrounding the implant, yet both displayed higher neuronal densities compared to autoclaved implants (Figure 6). These results suggest that endotoxins are an effective stimulus to initiate density-dependent effects on neuronal populations around the intracortical microelectrode-tissue interface at two weeks post implantation. Since loss of viable neurons within the critical recording range (50–140μm) can directly cause loss in high-quality recorded signals63, it is likely that increased endotoxin contamination may directly impact the quality and stability of acute intracortical neural recordings.
Recent work has demonstrated a correlation between chronic intracortical neural recordings and blood-brain barrier stability18. In our study, we observed elevated levels of IgG around the interface at both time points indicating continued blood brain barrier permeability at 2 and 16 weeks (Figure 5). The highest levels of blood-brain barrier permeability (Figure 5) were consistent with the highest levels of reactive microglia (Figure 3) and neuronal loss (Figure 6), and support previous studies15, 16, 18, 27 Pro-inflammatory molecules secreted from activated microglia/macrophages have been shown to facilitate the breakdown of the blood-brain barrier, in an attempt to mediate inflammation64. Alterations in the local ionic milieu caused by blood brain barrier disruption have been suggested to lead to neuronal silencing, decreases in neuron conduction velocity, and/or compromised synaptic stability16, 27. Our observations support current literature that suggests a self-perpetuating neuroinflammatory response to pro-inflammatory molecules released by stimulated microglia and macrophages through continued influx of blood-derived components14, 15.
The common link between each of the neuroinflammatory markers examined in this study was the density dependent innate immune response to increasing levels of endotoxin contaminants. The activation of subsequent inflammatory-mediated events appears to be threshold dependent. Microglia and macrophages are capable of responding to many stimuli, including endotoxins. Therefore, in order to prevent threshold level activation of subsequent self-perpetuating neuroinflammatory events, each microglia stimuli, including endotoxins, should be minimized. Thus, our findings support previous studies by from Gorbet and Stefon, who advocate that endotoxin contamination should not be ignored in biomedical studies. Specifically, Gorbet and Stefon show that exotoxin contamination can significantly affect the biological tissue response, which can confound or mask the effect of the material and/or device44.
Furthermore, we agree with Gorbet and Sefton that routine testing of endotoxin levels on biomaterials is the responsibility of every scientist to ensure the validity of biomaterial studies44. To begin to examine the impact that residual endotoxins may have had on the brain-tissue response to intracortical microelectrodes, we surveyed the vast body of literature that exists for descriptions of the sterilization methods used across the field (Figure 7A). Our findings indicate a wide range of sterilization methods used across the field including autoclave, ultraviolet light, ethylene oxide gas treatment, gamma radiation, dry heat sterilization, and ethyl alcohol treatment, among others. Interestingly, of the 108 papers we surveyed, approximately 20% of studies do not mention any type of sterilization. Further, no single study specifically quantifies endotoxin levels prior to implantation. In a similar manner, we also surveyed a representative population of published manuscripts, which have performed chronic intracortical recording studies, and evaluated the sterilization method used in these studies (Figure 7B). Within the 58 chronic recording studies we surveyed, there existed a wide range of sterilization methods. Interestingly, over 80% of groups did not mention any kind of sterilization, and no single study specifically reported quantitation of endotoxin levels prior to implantation.
Figure 7.
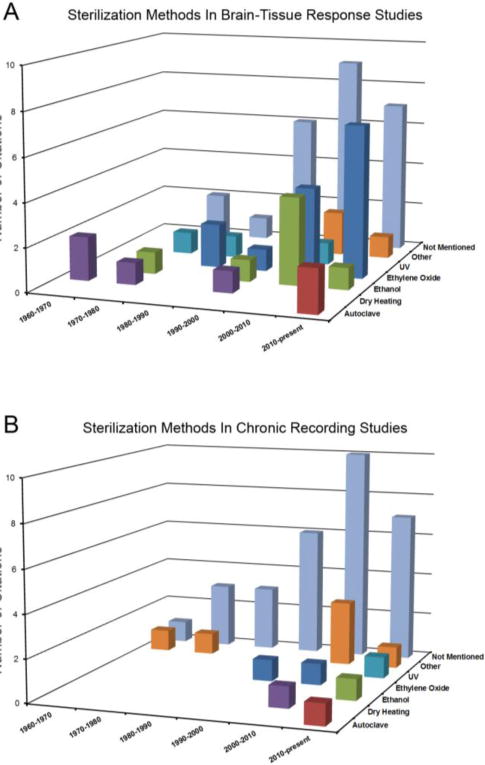
Quantification of the number of citations as a function of both time interval and sterilization method (if reported) for both histological studies of the brain-tissue response to non-functional intracortical microelectrodes (A) and electrophysiological studies of chronically implanted intracortical microelectrodes (B).
The choice of sterilization method is often governed by the surface properties as well as material and chemical composition of the device. Therefore, the high variability in sterilization methods used across the tissue-response and recording literature is not unexpected. However, as no study has ever reported quantification of residual endotoxin levels prior to implantation, we must do more to ensure that endotoxins are sufficiently removed from the device in order to limit activation of inflammatory cascades. Specifically, current FDA guidelines state that endotoxin levels must be at or below 0.5 EU/ml for blood contacting medical devices and be at or below 0.06 EU/mL for devices that contact cerebrospinal fluid (CNS devices)65, 66.
It is important to note that residual endotoxin levels on sterilized devices can greatly vary by changing properties including method of sterilization, time, temperature, pressure, and device composition65, 66. Therefore, importance should be given to determine the level of residual endotoxins resulting from each method of sterilization rather than the just the choice in the method of sterilization. For example, ethylene oxide gas sterilization as performed at our facilities exhibited lower endotoxin levels than autoclave or dry heat sterilization. However, this may not hold true at other institutions with altered procedural and environmental conditions. Furthermore, as even standard sterilization procedures were not sufficient to achieve FDA–recommended limits for residual endotoxins for CNS implants, cyclic sterilization or combination of several sterilization methods may be necessary to ensure the validity of histological examination of mechanisms contributing to the neuroinflammatory response to intracortical microelectrodes.
5. Conclusions
In the present study, we explored the neuroinflammatory response to residual endotoxin contamination on non-functional microelectrodes sterilized by autoclave, dry heat or ethylene oxide gas. Microelectrodes sterilized by autoclave, dry heat, or ethylene oxide gas, resulted in variable levels of residual endotoxins of 0.55 EU/mL, 0.22 EU/mL, and 0.11 EU/mL, respectively. Histological evaluation at two weeks post implantation showed a direct correlation between microglia/macrophage activation and residual endotoxin levels. Interestingly, astrogliosis, neuronal loss, and blood brain barrier dysfunction demonstrated a threshold-dependent response to bacterial endotoxins at the interface. At sixteen weeks post implantation, no histological differences were detected, regardless of the sterilization method employed. The present results demonstrate that endotoxin contamination contributes to variability in the initial but not chronic neuroinflammatory response to implanted intracortical microelectrodes. Therefore, endotoxins should be considered a potent stimulant to the neuroinflammatory response to implanted intracortical microelectrodes.
Acknowledgments
This work was supported by the Department of Biomedical Engineering at Case Western Reserve University through laboratory start-up funds, and NIH Neuroengineering Training Grant (5T32EB004314-14). Additional funding for this research program was provided in part by Rehabilitation Research and Development, Department of Veterans Affairs Merit Review (B7122R), and the Presidential Early Career Award for Scientists and Engineers (PECASE). None of the funding sources aided in the collection, analysis and interpretation of data, in the writing of the report, or in the decision to submit the paper for publication. The authors have no conflicts of interest related to this work to disclose.
References
- 1.F A, Renshaw B, Morison BR. Journal of neurophysiology. 1940;3:74–105. [Google Scholar]
- 2.C B, Grundfest H. Journal of neurophysiology. 1942;5:275–294. [Google Scholar]
- 3.S R, Grundfest H, Oettinger WH, Gurry RW. Review of Scientific Instruments. 1950;21:360–362. [Google Scholar]
- 4.Nicolelis MAL. Nature Reviews Neuroscience. 2003;4:417–422. doi: 10.1038/nrn1105. [DOI] [PubMed] [Google Scholar]
- 5.Schwartz AB. Annual review of neuroscience. 2004;27:487–507. doi: 10.1146/annurev.neuro.27.070203.144233. [DOI] [PubMed] [Google Scholar]
- 6.Burns BD, Stean JP, Webb AC. Electroencephalography and clinical neurophysiology. 1974;36:314–318. doi: 10.1016/0013-4694(74)90175-8. [DOI] [PubMed] [Google Scholar]
- 7.Liu X, McCreery DB, Carter RR, Bullara LA, Yuen TG, Agnew WF. IEEE transactions on rehabilitation engineering: a publication of the IEEE Engineering in Medicine and Biology Society. 1999;7:315–326. doi: 10.1109/86.788468. [DOI] [PubMed] [Google Scholar]
- 8.Liu X, McCreery DB, Bullara LA, Agnew WF. IEEE transactions on neural systems and rehabilitation engineering: a publication of the IEEE Engineering in Medicine and Biology Society. 2006;14:91–100. doi: 10.1109/TNSRE.2006.870495. [DOI] [PubMed] [Google Scholar]
- 9.Rennaker RL, Miller J, Tang H, Wilson DA. Journal of neural engineering. 2007;4:L1–5. doi: 10.1088/1741-2560/4/2/L01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ludwig KA, Miriani RM, Langhals NB, Joseph MD, Anderson DJ, Kipke DR. Journal of neurophysiology. 2009;101:1679–1689. doi: 10.1152/jn.90989.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ward MP, Rajdev P, Ellison C, Irazoqui PP. Brain research. 2009;1282:183–200. doi: 10.1016/j.brainres.2009.05.052. [DOI] [PubMed] [Google Scholar]
- 12.Collias JC, Manuelidis EE. Journal of neurosurgery. 1957;14:302–328. doi: 10.3171/jns.1957.14.3.0302. [DOI] [PubMed] [Google Scholar]
- 13.Schultz RL, Willey TJ. Journal of neurocytology. 1976;5:621–642. doi: 10.1007/BF01181577. [DOI] [PubMed] [Google Scholar]
- 14.Biran R, Martin DC, Tresco PA. Experimental neurology. 2005;195:115–126. doi: 10.1016/j.expneurol.2005.04.020. [DOI] [PubMed] [Google Scholar]
- 15.Potter KA, Buck AC, Self WK, Callanan ME, Sunil S, Capadona JR. Biomaterials. 2013;34:7001–7015. doi: 10.1016/j.biomaterials.2013.05.035. [DOI] [PubMed] [Google Scholar]
- 16.Winslow BD, Christensen MB, Yang WK, Solzbacher F, Tresco PA. Biomaterials. 2010;31:9163–9172. doi: 10.1016/j.biomaterials.2010.05.050. [DOI] [PubMed] [Google Scholar]
- 17.Potter KA, Buck AC, Self WK, Capadona JR. Journal of neural engineering. 2012;9:046020. doi: 10.1088/1741-2560/9/4/046020. [DOI] [PubMed] [Google Scholar]
- 18.Saxena T, Karumbaiah L, Gaupp EA, Patkar R, Patil K, Betancur M, Stanley GB, Bellamkonda RV. Biomaterials. 2013;34:4703–4713. doi: 10.1016/j.biomaterials.2013.03.007. [DOI] [PubMed] [Google Scholar]
- 19.Gehrmann J, Matsumoto Y, Kreutzberg GW. Brain research Brain research reviews. 1995;20:269–287. doi: 10.1016/0165-0173(94)00015-h. [DOI] [PubMed] [Google Scholar]
- 20.Polikov VS, Tresco PA, Reichert WM. Journal of neuroscience methods. 2005;148:1–18. doi: 10.1016/j.jneumeth.2005.08.015. [DOI] [PubMed] [Google Scholar]
- 21.Tresco PA, Winslow BD. Critical Reviews in Biomedical Engineering. 2011;39:29–44. doi: 10.1615/critrevbiomedeng.v39.i1.30. [DOI] [PubMed] [Google Scholar]
- 22.Chao CC, Hu S, Peterson PK. Crit Rev Neurobiol. 1995;9:189–205. [PubMed] [Google Scholar]
- 23.Hanisch UK, Kettenmann H. Nat Neurosci. 2007;10:1387–1394. doi: 10.1038/nn1997. [DOI] [PubMed] [Google Scholar]
- 24.Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Trends Immunol. 2002;23:549–555. doi: 10.1016/s1471-4906(02)02302-5. [DOI] [PubMed] [Google Scholar]
- 25.Quagliarello VJ, Wispelwey B, Long WJ, Jr, Scheld WM. J Clin Invest. 1991;87:1360–1366. doi: 10.1172/JCI115140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Clark IA, Alleva LM, Vissel B. Pharmacol Ther. 2010;128:519–548. doi: 10.1016/j.pharmthera.2010.08.007. [DOI] [PubMed] [Google Scholar]
- 27.Skousen J, Merriam S, Srivannavit O, Perlin G, Wise K, Tresco P. Progress in brain research. 2011;194C:167–180. doi: 10.1016/B978-0-444-53815-4.00009-1. [DOI] [PubMed] [Google Scholar]
- 28.Friedman DI. Am J Clin Dermatol. 2005;6:29–37. doi: 10.2165/00128071-200506010-00004. [DOI] [PubMed] [Google Scholar]
- 29.Ochsendorf F. Am J Clin Dermatol. 2010;11:327–341. doi: 10.2165/11319280-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 30.Reed DN, Gregg FO, Corpe RS. Orthopedics. 2012;35:e737–739. doi: 10.3928/01477447-20120426-30. [DOI] [PubMed] [Google Scholar]
- 31.Harris JP, Capadona JR, Miller RH, Healy BC, Shanmuganathan K, Rowan SJ, Weder C, Tyler DJ. Journal of neural engineering. 2011;8:066011. doi: 10.1088/1741-2560/8/6/066011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Spataro L, Dilgen J, Retterer S, Spence AJ, Isaacson M, Turner JN, Shain W. Experimental neurology. 2005;194:289–300. doi: 10.1016/j.expneurol.2004.08.037. [DOI] [PubMed] [Google Scholar]
- 33.Zhong Y, Bellamkonda RV. Journal of Controlled Release. 2005;106:309–318. doi: 10.1016/j.jconrel.2005.05.012. [DOI] [PubMed] [Google Scholar]
- 34.Azemi E, Lagenaur CF, Cui XT. Biomaterials. 2011;32:681–692. doi: 10.1016/j.biomaterials.2010.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ware T, Simon D, Arreaga-Salas DE, Reeder J, Rennaker R, Keefer EW, Voit W. Advanced Functional Materials. 2012;22:3470–3479. [Google Scholar]
- 36.Misra A, Kondaveeti P, Nissanov J, Barbee K, Shewokis P, Rioux L, Moxon KA. Journal of neural engineering. 2013;10:016011. doi: 10.1088/1741-2560/10/1/016011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pineau I, Lacroix S. Glia. 2009;57:351–361. doi: 10.1002/glia.20763. [DOI] [PubMed] [Google Scholar]
- 38.Anderson JM, Rodriguez A, Chang DT. Semin Immunol. 2008;20:86–100. doi: 10.1016/j.smim.2007.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Davalos D, Ryu JK, Merlini M, Baeten KM, Le Moan N, Petersen MA, Deerinck TJ, Smirnoff DS, Bedard C, Hakozaki H, Gonias Murray S, Ling JB, Lassmann H, Degen JL, Ellisman MH, Akassoglou K. Nat Commun. 2012;3:1227. doi: 10.1038/ncomms2230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Block ML, Zecca L, Hong JS. Nat Rev Neurosci. 2007;8:57–69. doi: 10.1038/nrn2038. [DOI] [PubMed] [Google Scholar]
- 41.Dheen ST, Kaur C, Ling EA. Curr Med Chem. 2007;14:1189–1197. doi: 10.2174/092986707780597961. [DOI] [PubMed] [Google Scholar]
- 42.Saijo K, Glass CK. Nat Rev Immunol. 2011;11:775–787. doi: 10.1038/nri3086. [DOI] [PubMed] [Google Scholar]
- 43.Harris JP. Doctor of Philosophy. Case Western Reserve Universtiy; 2011. [Google Scholar]
- 44.Gorbet MB, Sefton MV. Biomaterials. 2005;26:6811–6817. doi: 10.1016/j.biomaterials.2005.04.063. [DOI] [PubMed] [Google Scholar]
- 45.Berzofsky RN. Altex. 1995;12:93–97. [PubMed] [Google Scholar]
- 46.Szarowski DH, Andersen MD, Retterer S, Spence AJ, Isaacson M, Craighead HG, Turner JN, Shain W. Brain research. 2003;983:23–35. doi: 10.1016/s0006-8993(03)03023-3. [DOI] [PubMed] [Google Scholar]
- 47.McConnell GC, Rees HD, Levey AI, Gutekunst CA, Gross RE, Bellamkonda RV. Journal of neural engineering. 2009;6:056003. doi: 10.1088/1741-2560/6/5/056003. [DOI] [PubMed] [Google Scholar]
- 48.Winslow BD, Tresco PA. Biomaterials. 2010;31:1558–1567. doi: 10.1016/j.biomaterials.2009.11.049. [DOI] [PubMed] [Google Scholar]
- 49.Potter KA, Simon JS, Velagapudi B, Capadona JR. Journal of neuroscience methods. 2012;203:96–105. doi: 10.1016/j.jneumeth.2011.09.024. [DOI] [PubMed] [Google Scholar]
- 50.Hochstein HD. Prog Clin Biol Res. 1985;189:221–239. [PubMed] [Google Scholar]
- 51.Williams KL. Endotoxins: pyrogens, LAL testing and depyrogenation. 3. Informa Healthcare; New York: 2007. [Google Scholar]
- 52.Rabinowitz SS, Gordon S. J Exp Med. 1991;174:827–836. doi: 10.1084/jem.174.4.827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Smith MJ, Koch GL. J Cell Sci. 1987;87(Pt 1):113–119. doi: 10.1242/jcs.87.1.113. [DOI] [PubMed] [Google Scholar]
- 54.Verbeek MM, Otte-Holler I, Wesseling P, Van Nostrand WE, Sorg C, Ruiter DJ, de Waal RM. Acta neuropathologica. 1995;90:493–503. doi: 10.1007/BF00294811. [DOI] [PubMed] [Google Scholar]
- 55.Seitz RJ, Heininger K, Schwendemann G, Toyka KV, Wechsler W. Acta neuropathologica. 1985;68:15–21. doi: 10.1007/BF00688950. [DOI] [PubMed] [Google Scholar]
- 56.Woolley AJ, Desai HA, Otto KJ. Journal of neural engineering. 2013;10:026007. doi: 10.1088/1741-2560/10/2/026007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kozai TDY, Langhals NB, Patel PR, Deng X, Zhang H, Smith KL, Lahann J, Kotov NA, Kipke DR. Nature Materials. 2012;11:1065–1073. doi: 10.1038/nmat3468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Clark GA, Ledbetter NM, Warren DJ, Harrison RR. Conference proceedings: … Annual International Conference of the IEEE Engineering in Medicine and Biology Society IEEE Engineering in Medicine and Biology Society Conference. 2011;2011:4641–4644. doi: 10.1109/IEMBS.2011.6091149. [DOI] [PubMed] [Google Scholar]
- 59.White GE, Greaves DR. Blood. 2009;113:767–768. doi: 10.1182/blood-2008-11-189860. [DOI] [PubMed] [Google Scholar]
- 60.He W, Bellamkonda RV. In: Indwelling Neural Implants: Strategies for Contending with the In Vivo Environment. Reichert WM, editor. 2008. ch. 6. [PubMed] [Google Scholar]
- 61.Ravikumar M, Jain S, Miller RH, Capadona JR, Selkirk SM. Journal of neuroscience methods. 2012;211:280–288. doi: 10.1016/j.jneumeth.2012.09.004. [DOI] [PubMed] [Google Scholar]
- 62.Skousen J, Bolick K, Bridge M, Tresco P. IFESS. 2012 [Google Scholar]
- 63.Buzsaki G. Nat Neurosci. 2004;7:446–451. doi: 10.1038/nn1233. [DOI] [PubMed] [Google Scholar]
- 64.Abbott NJ. Cell Mol Neurobiol. 2000;20:131–147. doi: 10.1023/A:1007074420772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.United States Department of Health and Human Services – Office of Regulatory Affairs (ORA) Food and Drug Administration. 2012. pp. 1–13. June 2012 Compliance edn. [Google Scholar]
- 66.Rutala WA, Weber DJ, Weinstein RA, Siegel JD, Pearson ML, Chinn RYW, DeMaria A, Lee JT, Rutala WA, Scheckler WE, Stover BH, Underwood MA, editors. C f D Control. 2008. pp. 1–158. [Google Scholar]