

Abstract
AZD3783, a cationic amphiphilic drug and a potent inhibitor of the 5-hydroxytryptamine (5-HT1B) receptor, was explored as a potential treatment for depression. To support clinical trials, repeat dose toxicity studies in rats and dogs were conducted. Here we report toxicity findings in dogs after dosing from 1 to 3 months. In the 1-month study, there were minimal neuronal vacuolation in the brain, a marked increase in liver enzymes accompanied by hepatocellular degeneration/necrosis and phospholipidosis (PLD), and PLD/cholecystitis in the gallbladder of animals dosed at 47 mg/kg/day. In the 3-month study, neurotoxicity resulted in euthanasia of one animal dosed at 30 mg/kg/day after 86 days. Extensive pathologic changes were seen in all animals in retina epithelium (inclusion bodies), brain (neuronal vacuolation, degeneration, or necrosis and nerve fiber degeneration), spinal ganglia (vacuolation, degeneration, or necrosis), as well as sciatic and optic nerves (degeneration). Pigment-laden macrophages were observed in the lung, kidney, liver, gallbladder, bone marrow, gastrointestinal tract, and lymphoid tissues. Also seen were vitrel and retinal hemorrhage in the eyes. A brain concentration and pathology study showed that the concentration of AZD3783 in the brain was approximately 4 times higher than in the plasma after 4 weeks of dosing, however, they were similar in all regions examined, and did not correlate with areas with pathologic findings. Our findings with AZD3783 in dogs have not been reported previously with other CNS compounds that effect through serotonergic pharmacology.
Keywords: pathology; neurotoxicity, preclinical safety assessment; phospholipidosis; cationic amphiphilic drugs (CADs), dog
Introduction
AZD3783 is a potent and selective antagonist of the human 5-hydroxytryptamine 1B (serotonin, or 5-HT1B) receptor, with a Ki of 12.5 nM1. Serotonin is a central nervous system (CNS) neurotransmitter that is involved in physiological functions including thermoregulation, modulation of neurotransmitter release, as well as anxiety and mood regulation2. The pre-synaptic 5-HT1B receptor regulates synaptic release of serotonin, thus 5-HT1B antagonists have been explored as an alternative therapy to selective serotonin re-uptake inhibitors (SSRIs) for treatment of depression3,4. In guinea pig models, AZD3783 (0.6 µmol/kg. p.o.) was shown to increase extracellular serotonin in the brain, block hypothermia induced by 5-HT1B-agonists, and elicit effects indicative of anxiolytic and anti-depressant efficacy1. In positron emission tomography (PET) studies, AZD3783 was demonstrated to bind dose-dependently to brain 5-HT1B receptors in non-human primates and human subjects5.
AZD3783 binds to the 5-HT1B receptors in the rat cerebral cortex with a Ki of 51 nM, which is approximately 4-fold lower than its affinity for the guinea pig or human 5-HT1B receptors. No binding characterization has been conducted with the dog receptors, however, given the receptor’s structural conservation6, dogs would also be pharmacologically responsive. Besides the 5-HT1B receptor, AZD3783 binds to adrenergic α1D receptor and is an antagonist of the adrenergic α1A and α1B receptors in secondary pharmacology screens at 10 µM.
AZD3783 contains a hydrophilic ring and a hydrophobic region and is regarded as a cationic amphiphilic drug (CAD). Many CADs have been shown to cause phospholipidosis (PLD) in vitro and in animals and man7,8. Although PLD is considered an adaptive response rather than a manifestation of toxicity, questions remain as to the toxicological significance of PLD in affected tissues, since it is sometimes associated with concurrent toxicities clinically and preclinically9,10. Examples include amidarone and liver toxicity11, fluoxetine and pulmonary alveolitis that progresses to fibrosis12, peripheral neuropathies from perhexiline13, and corneal opacities from amidarone14. Thus PLD is regarded as a potential safety liability by regulatory agencies15,16. For drugs intended to treat CNS disorders, the safety concern maybe greater, because they are designed to cross the blood brain barrier, increasing the potential for PLD and associated toxicities in the brain, which are difficult to monitor.
In an attempt to minimize safety concern and the risk of attrition, an in vitro PLD screen was developed to screen compounds with CAD structures for their ability to cause PLD17. AZD3783 was selected from a list of potential drug candidates based on a number of criteria - in vitro potency to cause PLD, in vivo repeat dose toxicity, and pharmacokinetic properties. In the dog, AZD3783 is readily absorbed after oral dosing (F=58%), plasma clearance is moderate (18 mL/min/kg, after IV dosing), with a steady state volume of distribution of 4.3 L/kg. AZD3783 readily crosses the blood brain barrier and is not likely to be a p-glycoprotein substrate1.
AZD3783 was deemed a weak phospholipogenic compound in vitro (EC50=164 µM) compared to reference compounds such as amiodarone, chloroquine, and perhexiline (EC50s<20 µM), and not a direct inhibitor of mitochondrial oxidative phosphorylation (unpublished data). It caused dose-dependent PLD in a limited number of tissues in a 14-day rat study, but at an incidence and severity less than the other comparison compounds (unpublished data). In the dog, it did not cause treatment-related pathology in a 14-day study in which limited tissues including dorsal root ganglion were examined (data not shown). However, it caused neurotoxicity and progressive pathologic changes in the nervous tissues after repeat dosing for 3 months. Herein we describe toxicologic findings in dogs after repeat dose exposure to AZD3783 for 1 or 3 months. Some of our findings have not been reported with other CADs in dogs, nor have they been seen for other compounds that modulate the serotonin pharmacology.
Materials and Methods
Chemicals
AZD3783, [(2R)-6-methoxy-8-(4-methylpiperazin-1-yl)-N-(4-morpholin-4-ylphenyl) chromane-2-carboxamide;MW=466.6 g/mol; for structure, see Reference 1] was supplied by AstraZeneca’s Pharmaceutical Development Department in Macclesfield, UK. The test compound was fully characterized (for content, impurities, solvents and appearance), the identity was confirmed, and the storage condition was determined. The purity of the micronized test substance was ≥ 99% in the two batches used in the studies.
Animals and studies
Three studies are described here: 1- and 3-month toxicity studies to evaluate systemic effects, and a 1-month study to exam brain concentration and brain pathology. The 1-month toxicity and brain concentration/pathology studies were conducted at AstraZeneca sites in Alderley Park, UK, and Wilmington, DE, US, respectively. The 3-month toxicity study was conducted at Charles River Laboratories in Montreal, Canada. Laboratory-reared beagle dogs were obtained from the dog breeding unit, AstraZeneca, Alderley Park (1-month toxicity study) or Marshall Farms, New York (1-month brain concentration and 3-month toxicity studies), and maintained in compliance with applicable animal welfare and regulatory guidance. The dogs were housed individually during feeding and after dosing for observations, but were otherwise group housed (2–3 dogs per sex and group). The husbandry, enrichment, experimental use and methods of euthanasia met the internal and external ethical guidelines for the humane care and treatment of laboratory animals. The dogs were ≥10 months of age on the first day of dosing in the 1-month study, and were 9-month old at study start in the 3-month study. Upon allocation to the studies, the dogs were assigned randomly to experimental groups; littermates were assigned to different groups as much as possible.
A total of 18 males and 18 females were used in each of the toxicity studies, while 7 males and 5 females were used in the 1-month brain concentration study. The group sizes for these studies conform to the regulatory guidelines and AstraZeneca guideline, and are the minimum that allows for statistical evaluation.
Dosing procedure
AZD3783 dosing formulation was administered via oral gavage once daily in the morning during the dosing period. The dosing volume was 5 mL/kg. Dosing formulation concentrations ranged from 0.5 to 9.3 mg/mL. Control dogs received the vehicle, 0.1 M lactic acid in water (pH 3.0). Both the vehicle and AZD3783 dosing formulations were prepared weekly, and used within the stability period. To prepare the dosing formulation, an appropriate amount of AZD3783 was added to a pre-calibrated beaker. An appropriate volume of 0.1 M lactic acid (not adjusted for pH) was then added and the mixture was sonicated and stirred. The mixture was then brought to volume using 0.1 M lactic acid (pH adjusted to 3), and stirred until a solution was obtained. The concentrations of AZD3783 in the dosing formulations were acceptable (± 2% of nominal). Absence of AZD3783 was confirmed for the vehicle samples.
Experimental design
In the 1- and 3-month toxicity studies, groups of 3 males and 3 females per dose were included for evaluation and were necropsied at the end of the dosing period. Additional animals (3 males and 3 females per group) were allocated to the control and high dose groups for evaluation of recovery after a dosing-free recovery period. The recovery period was 4 weeks for the 1-month study and 5 weeks for the 3-month study. The doses used in the 1-month toxicity study were: 0 (vehicle control), 2.3, 14, and 47 mg/kg/day, whereas the doses used in the 3-month toxicity study were: 0 (vehicle control), 7, 15, and 30 mg/kg/day. In the 1-month brain concentration/pathology study, the doses and group size were 0 (vehicle, 1 male), 2.3 (2 males and 1 female), 14 (3 males and 1 female), and 47 mg/kg/day (3 males and 1 females). All animals were given a full clinical examination before study start and during the study. Multiple toxicology parameters were assessed, including mortality/morbidity, clinical observations, body weight, food consumption, ophthalmology, ECG and blood pressure, neurologic examination (3-month toxicity study only), hematology, clinical chemistry, urinalysis, toxicokinetics, necropsy, light microscopic pathology on a full list of tissues (including dorsal root ganglia in the 3-month study) and electron microscopy (on liver and gallbladder form the 1-month study).
In-life examinations: Neurological examination: Neurological examination included an assessment of general attitude and behavior, gait, postural reactions (assessments of proprioceptive positioning, hemihopping and hemistanding, wheelbarrowing, hopping, visual and tactile placing reactions, and righting reaction). The cranial nerve function assessment included head movement and symmetry, head muscle tone, eye reactions, eye symmetry, vestibular nystagmus, eye position, corneal reflex, papillary light reflex, nasal septum, mouth, tongue and pharynx. The spinal nerve function assessment included muscle tone, patellar reflex, flexor reflex, panniculus reflex and perineal reflex.
Ophthalmological examination: Ophthalmological examinations were performed during pretest, end of dosing, and end of recovery in the 1-month toxicity study. In the 3-month toxicity study, ophthalmological examination was conducted during pretest, Weeks 6, and 13 of dosing period, as well as Weeks 3 and 5 of the recovery period. The eyes were examined after application of a mydriatic agent (tropicamide solution, 1% MydriacylTM, Alcon), using an indirect ophthalmoscope and also a slit lamp (3 month study only).
Electrocardiogram (ECG) and blood pressure: ECG and direct blood pressure were recorded from all dogs during pretest, Weeks 1, 2 and 4 of dosing period and end of the recovery period in the 1-month toxicity study. In the 3-month toxicity study, ECGs were recorded during pretests, Weeks 6 and 13 of dosing period, and end of the recovery period. Heart rate, PR, QRS, RR, QT and QTc intervals were measured or calculated.
Clinical pathology: Hematology, clinical chemistry and urinalysis were conducted on samples collected at pretest, Days 15, 28 of dosing period, and end of recovery in the 1-month toxicity study. In the 3-month toxicity study, measurements were conducted on samples collected during pretest, Weeks 6 and 13 of dosing period, and end of recovery.
Toxicokinetics and brain exposure: To assess systemic exposure to AZD3783, blood samples were collected from all animals during Days 1 and end of dosing period in the 1-month toxicity study. In the 3-month toxicity study, blood samples were collected on Days 1, 23, 56, and 91 from all surviving animals; limited samples were also collected from one decedent on Day 86 prior to euthanasia. In both toxicity studies, serial blood samples were collected at pre-dose and from 0.5 to 24 h after dose administration, and analyzed for concentration of AZD3783.
In the 1-month brain concentration/pathology study, blood samples were collected on Days 1, 7 and 28 for determination of AZD3783 concentration. Different brain regions (brain stem, hippocampus, frontal lobe) were obtained for determination of AZD3783.
Pharmacokinetic parameters were derived for time of maximum plasma concentration (tmax), maximum plasma concentration, Cmax, area under the curve (AUC), half-life (t1/2).
Necropsy and histology: All necropsies were conducted under the supervision of a pathologist. Animals were anesthetized with sodium pentobarbitone or ketamine HCl for injection + Xylazine, and exsanguinated. A complete list of tissues including gross lesions was collected for the 1- and 3-month studies. In the brain concentration/pathology study, only brain sections were obtained and processed.
Following collection at necropsy, eyes and optic nerves were fixed in Davidson’s, testes and sciatic nerves were fixed in Bouin’s, and all other tissues were fixed in 10% buffered formalin. The tissues were then processed into wax block, sectioned at approximately 5 µm, stained with hematoxylin and eosin (H&E) and examined under light microscope. A peer review of pathology findings was conducted for both the 1- and 3-month toxicity studies (A-L B).
Sections of liver from 3 dogs in the 1-month toxicity study were prepared from the wax blocks and subsequently stained by standard techniques by the PBR, Schmorl’s and Fouchet’s methods for the detection of hemosiderin, lipofuscin and bile, respectively.
For electron microscopy, samples of formalin fixed livers (N=5) and gallbladder (N=4) in the 1-month toxicity study were processed by standard techniques to araldite blocks. Following the preparation of semi-thin (0.5 µm) toluidine blue stained sections for selection of tissue area, ultra-thin (0.1 µm) sections were prepared and stained with lead citrate and uranyl acetate for ultrastructural examination using an Hitachi H7100 transmission electron microscope.
Statistical analysis
No statistical analysis was performed in the 1-month toxicity study. In the 3-month toxicity study, numerical data were subjected to calculation of group means and standard deviations. For each parameter of interest, group variances were compared using Levene’s test at the 0.05 significance level. When differences between group variances were not found to be significant, a parametric two-sample t-test was used to compare the group mean between the control and treated groups.
Whenever Levene’s test indicated heterogeneous group variances (P≤0.05), then the control group was compared to the treated group using the non-parametric Wilcoxon rank-sum test. For each group comparison done with the t-test or the Wilcoxon test, significance was reported at the 0.05, 0.01 and 0.001 levels.
Results
Toxicokinetic data
A summary of the toxicokinetic data is shown in Table 1. AZD3893 was rapidly absorbed after oral administration and tmax occurred between 0.5 and 3 h post-dose. At doses ≥14 mg/kg/day, plasma t1/2 appeared to increase with dose, especially after 3 months of dosing.
Table 1. Pharmacokinetic Parameters in Dogs Administered AZD3783 for 1 or 3 Months.
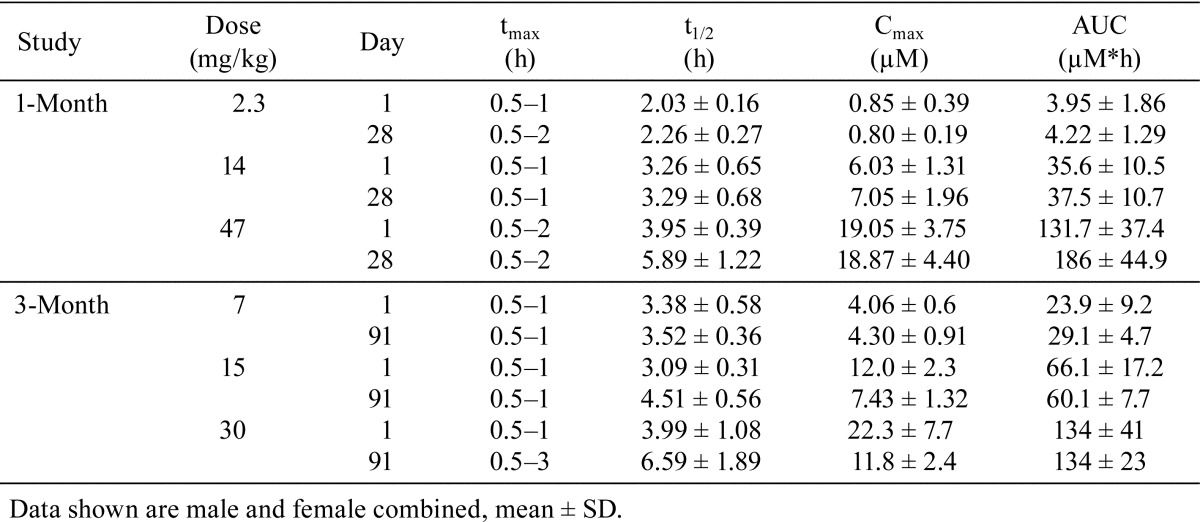
In the 1-month toxicity study, Cmax were comparable between Day 1 and Day 28 for all dose groups, whereas t1/2 for the 47 mg/kg/day group was slightly longer on Day 28 than on Day 1. In the 3-month toxicity study, Cmax was lower and t1/2 was longer on Day 91 than on Day 1 in the 15 and 30 mg/kg/day groups, while AUCs were similar between Day 1 and Day 91. These results indicate a decrease or saturation in absorption and a slowdown in elimination with repeat dosing. However, the mechanisms behind these pharmacokinetic changes are not clear.
In-life observations
1-month toxicity study: All animals survived to scheduled necropsy. The high dose, 47 mg/kg/day, induced CNS signs (ataxia, tremor, decreased motor activity, eyes half shut, and subdued behavior) after dosing, with recovery between dose administration. Subdued behavior was observed during Week 1 only, and the frequency of clinical observations decreased as the study progressed. No treatment-related observations were seen in the eyes of any dog.
There was a trend for treatment-related increase in heart rate (correspond to tmax) in animals dosed at 47 mg/kg/day, but heart rate and blood pressure tended to be lower 24 h post dose in all dose groups. When compared to pretest data, the only consistent reductions in blood pressure were observed for systolic blood pressure in males dosed at 47 mg/kg/day and in females dosed at 14 and 47 mg/kg/day. There was no treatment-related effect on ECG.
Platelet counts were increased 10–15% in males and 24–52% in females dosed at 47 mg/kg/day. Group mean values were elevated approximately 3-fold for alanine aminotransferase (ALT), and 7-fold for glutamate dehydrogenase (GLDH), in 47 mg/kg/day animals from Week 2 until end of dosing, attributable to 4 of 12 dogs, which had mild to marked increases in ALT (up to 17 fold vs. pretest), Aspartate aminotransferase (AST, up to 7 fold vs. pretest), and GLDH (up to 37-fold vs. pretest).
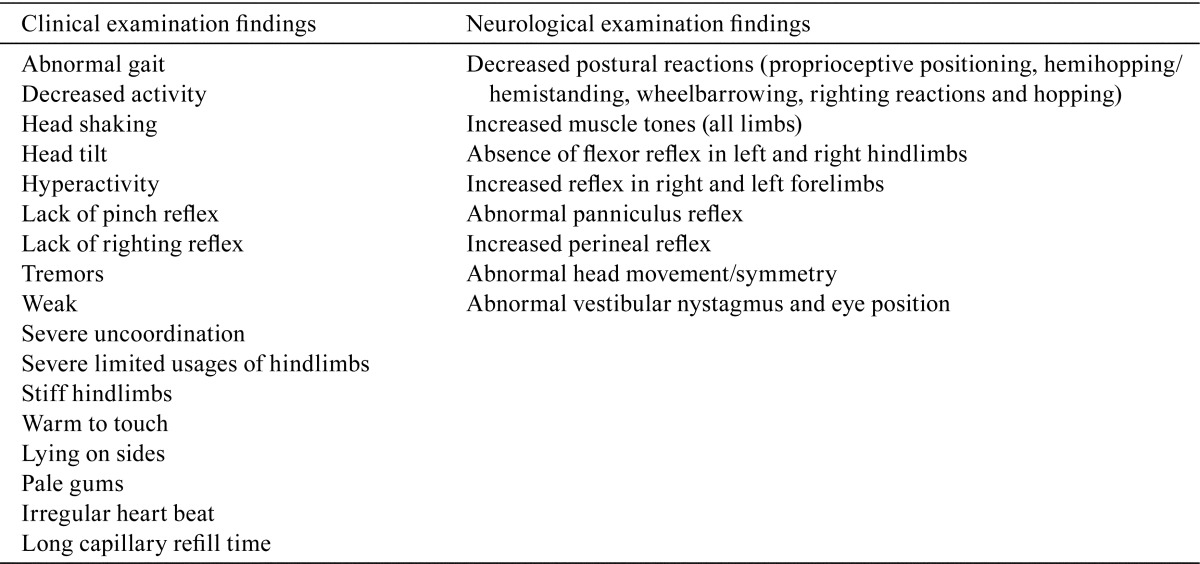
3-month toxicity study: Swelling and redness of the pinnas, periorbital region, cranium muzzle, lower jaw, abdominal area and/or limbs was noted in the 15 and 30 mg/kg/day groups during the first few weeks of dosing with the severity and incidences slightly higher in males than females. These clinical signs subsided as dosing continued. Partly closed eyes were observed in animals from the 15 and 30 mg/kg/day groups on most days during the study. One 30 mg/kg/day female was euthanized on Day 86 of the study, due to severe adverse clinical signs. The animal was noted to have reduced appetite, abnormal gait and was slightly uncoordinated starting on Day 83. Neurological examinations performed prior to termination revealed abnormal responses. A summary of the clinical and neurological findings from this dog is shown in Table 2. For all other animals, there were no treatment-related clinical or neurological findings.
Table 2. Clinical and Neurological Examination Findings in a Female Dog Dosed at 30 mg/kg/day AZD3783 for 86 Days.
Platelet counts were slightly increased (24–30%) and reticulocytes (% and total count) were decreased (42–50%) in the 30 mg/kg/day group after 3 months. Group mean values were elevated from Week 3 onward until end of dosing for ALT (ca. 2-fold) and GLDH (ca. 4-fold) vs. pretest for 30 mg/kg/day males. The decedent female in the 30 mg/kg/day group had slightly increased WBC (<2-fold), neutrophils (2-fold), platelet (1.6-fold), GLDH (2-fold), and mild to moderately to markedly increased bile (5-fold on Day 84 and 64-fold before necropsy) compared to pretest values.
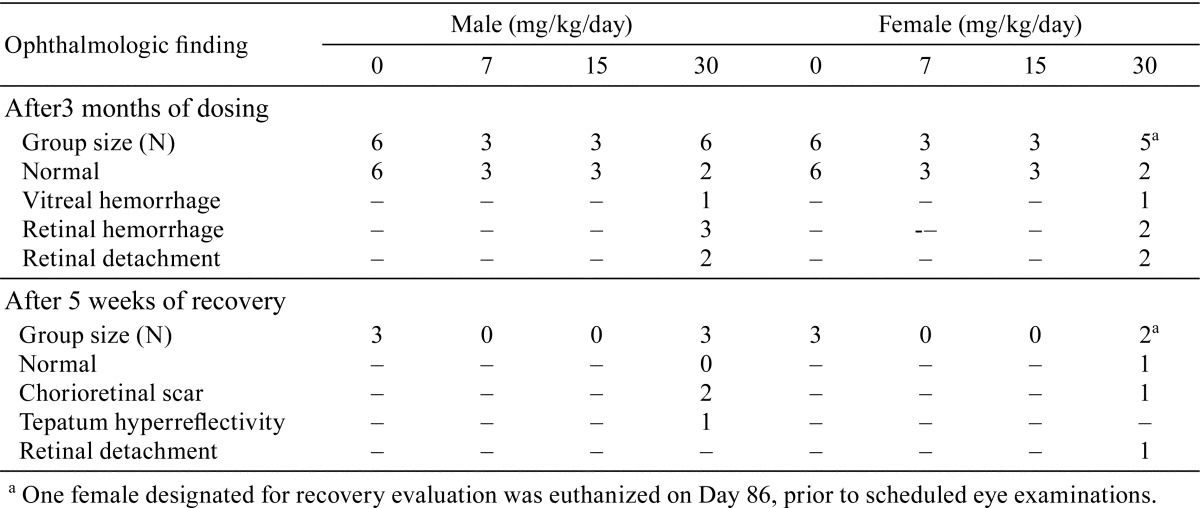
No treatment-related ophthalmologic observations were recorded during Week 6 examination. However, at Week 13 examination, 7/11 dogs (12 eyes total) dosed at 30 mg/kg/day had findings including retinal hemorrhage (slight in one or both eyes), vitreous hemorrhage (moderate, 1 eye), or retinal detachment (both eyes, multifocal, small circular areas of retinal separation scattered throughout tapetal area). During recovery, vitreal hemorrhage and retinal detachment improved and resolved while sequelae such as chorioretinal scar or tapetum hyperreflectivity or hyperpigmentation was observed. A summary of the incidence of ophthalmologic findings is shown in Table 3.
Table 3. Ophthalmologic Findings in Dogs Administered AZD3783 for 3 Months, followed by 5 Weeks of Recovery.
Macroscopic and microscopic pathology
1-month toxicity study: One female dosed at 47 mg/kg/day and necropsied after the recovery period had depressed areas on the surface of all lobes of the liver and an enlarged bile duct, although the organ weight was normal. Treatment-related microscopic changes, in order of severity, were observed in the liver, gallbladder, lymphoid tissue, and brain, in the 47 mg/kg/day group. In the brain, minimal fine cytoplasmic vacuolation of occasional neuronal cell bodies consistent with PLD was noted in 5/6 animals dosed at 47 mg/kg/day. There were no pathological findings in the spinal cord or any CNS tissues from animals dosed at 2.3 mg/kg/day or 14 mg/kg/day, or in recovery animals previously dosed at 47 mg/kg/day.
Pathological lesions in the liver and gallbladder were present prominently in those 4 dogs which had elevated liver biomarkers. In the liver, microscopic findings included inflammatory cell infiltration, degenerative and necrotic changes in the centrilobular region and moderate pigments in histiocytes (Fig. 1), which were confirmed as hemosiderin and lipofuscin. In the gallbladder, the mucosa showed vacuolation and hypertrophy of the epithelium and inflammation ranging from minimal polymorphonuclear cell infiltration to frank cholecystitis. Electron microscopy of the liver and gallbladder tissues showed lamellar bodies consistent with PLD in hepatocytes (Fig. 2), intrahepatic bile duct epithelial cells and gallbladder epithelial cells. The bile duct and gallbladder epithelial cells also had increased numbers and size of lipid droplets as compared to controls.
Fig. 1.
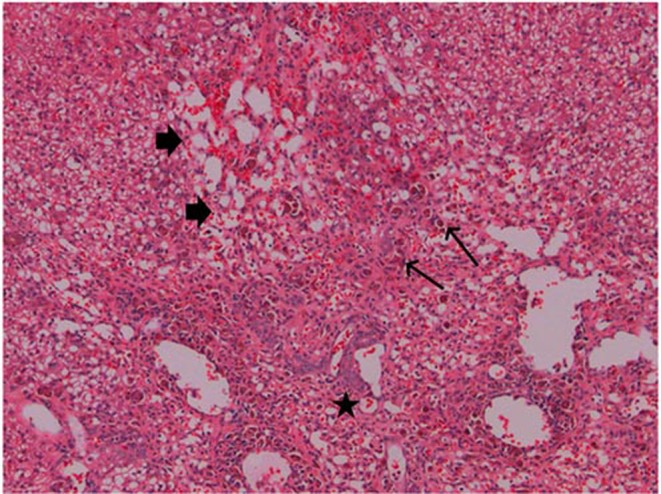
Treatment with AZD3783 for 1 month at 47 mg/kg/day caused marked loss of hepatocytes with fibrosis (thick arrow), bile duct proliferation (star), and pigments in Kupffer cells (thin arrow) in male dog (M20). H&E staining, 200×.
Fig. 2.
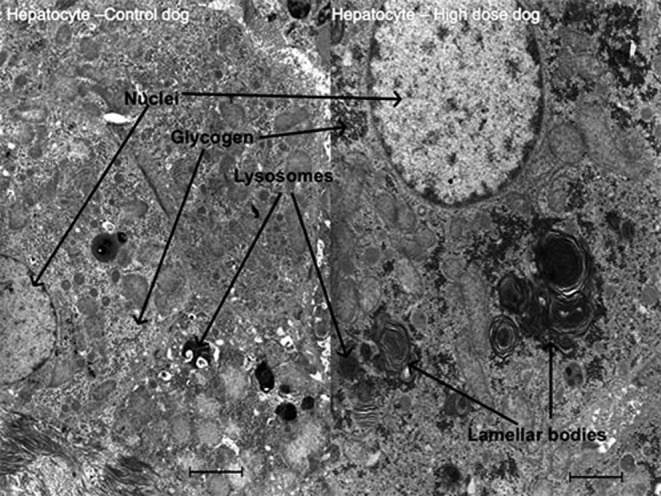
Treatment with AZD3783 for 1 month at 47 mg/kg/day caused phospholipidosis in the liver. Shown here are lamellar bodies in the hepatocyte as seen by electron microscopy. Bar = 2 µm.
Lymphoid tissues (lymph nodes, spleen, Peyer’s patches in the intestines, and lymphoid follicles in the mucosa of the gallbladder and stomach) showed an increased incidence and severity of lymphocytolysis, which was attributable to PLD.
3-month toxicity study: Among dogs from the 15 and 30 mg/kg/day groups which were necropsied after 3 months of dosing, the major treatment-related microscopic findings were seen in the nervous system, including the eye, optic nerve, brain, spinal ganglia and sciatic nervous. Other tissues with microscopic findings included liver, gallbladder, bone marrow, GI tract, kidney, lung, lymph nodes, spleen, and thymus. Pathological findings from the decedant animal were similar in character and severity to those observed in other dogs dosed at 30 mg/kg/day. A summary of the pathological findings in central and peripheral nervous tissues after 3 months of dosing is shown in Table 4. Findings from the decedant female which was initially designated for recovery evaluation but was killed preterminally on Day 86 are included in the summary with the 3 other females from the group that were necropsied on Day 91.
Table 4. Incidence of Nervous Tissue Pathology in Dogs Given AZD3783 for 3 Months.
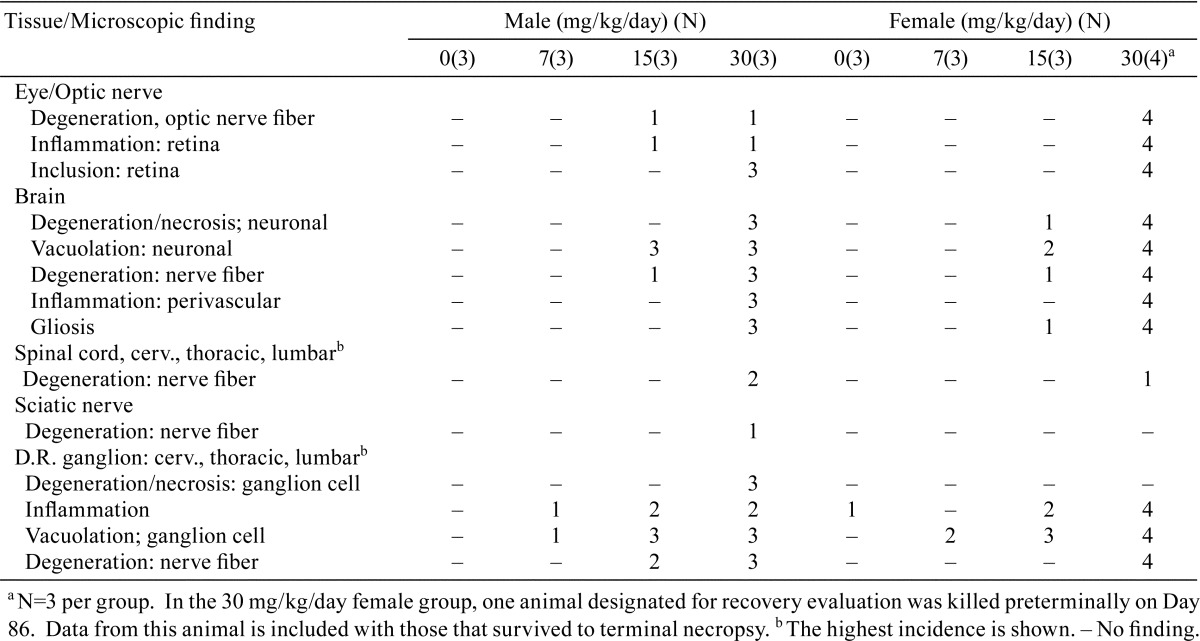
In the spinal cord, minimal to slight nerve fiber degeneration was present in the cervical segment in 2 males and 1 female at 30 mg/kg/day. Nerve fiber degeneration involving the thoracic and lumbar spinal cord segments was present in 2 males at 30 mg/kg/day. In the dorsal root ganglion, varying combinations of ganglion cell degeneration/necrosis, inflammation, ganglion cell vacuolation, and/or nerve fiber degeneration were present in at least one dog from all treatment groups (Fig. 3A). In the sciatic nerve, nerve fiber degeneration was present in one 30 mg/kg/day male dog, which was normal in the neurologic examination.
Fig. 3.

Treatment with AZD3783 for 3 months at 15 or 30 mg/kg/day caused degenerative and inflammatory lesions in the central and peripheral nervous tissues. (A) Ganglion cell vacuolation and mononuclear cell infiltration in the DRG; (B) Necrosis and inflammation in the cochlear nuclei; (C) Neuronal degeneration/necrosis and inflammation in the CA2 region of the hippocampus; (D) Perivascular mononuclear cell infiltration in the retina. H&E staining, 200×. In the insert, the arrow points to eosinophilic inclusion body.
In the brain, one or more of the microscopic findings of neuronal degeneration/necrosis, neuronal vacuolation (fine cytoplasmic vacuoles), nerve fiber degeneration, perivascular inflammation, and/or gliosis were present in all dogs dosed at 30 mg/kg/day, and in 2 females dosed at 15 mg/kg/day (Fig. 3B, C). One 15 mg/kg/day male had neuronal vacuolation and nerve fiber degeneration in the brain. The brain regions that were typically affected with one or more microscopic findings included the visual/optical tracts/pathways (retina, optic nerve, optic tracts, and lateral geniculate body), brain stem (cochlear nuclei and superior olivary complex), and the CA2, CA3, and CA4 region of the hippocampus. The severity of lesions in the brain was dose-related and ranged from slight to moderate.
In the eye, perivascular inflammation of the retina was present in 1 male and 4 females dosed at 30 mg/kg/day (Fig. 3D). Small, retinal eosinophilic inclusions were present in all animals dosed at 30 mg/kg/day (Fig. 3D, insert). In the optic nerve, nerve fiber degeneration was present in animals dosed at 15 and 30 mg/kg/day (Table 4).
In the liver, microscopic findings consisted of infiltration of mononuclear cells or neutrophils, pigment deposits (golden-brown, within macrophages) in all but one treated male (7 mg/kg/day), and hepatocellular single cell necrosis (30 mg/kg/day). In the gallbladder, pigment deposit (lamina propria) was noted in all animals in the treated groups.
Increased pigment deposits (golden-brown) in macrophages and/or epithelial cells was present in several organs including bone marrow, GI tract, kidney, lung, lymph nodes, spleen, and thymus. These findings were present at all dose levels of AZD3783.
The majority of the pathological lesioncs, except those in thymus and bone marrow, were still present after the 5-week recovery period. In the brain, there was extensive neuronal loss accompanied by marked astrogliosis in affected areas (Fig. 4). Based on the extensive pathological findings, a NOEL was not established for the study.
Fig. 4.
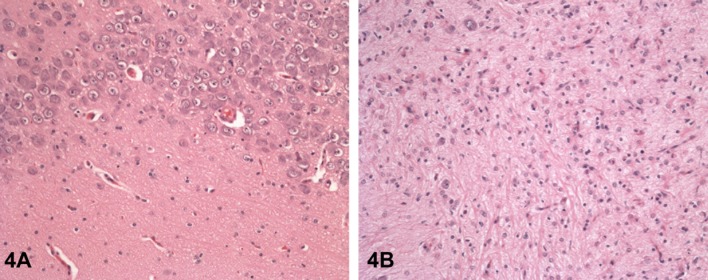
Compound-induced lesions in the brain of dogs treated with AZD3783 are irreversible. (A) Hippocampus CA4 region of vehicle-treated control dog; (B) Extensive neuronal loss and prominent astrogliosis in the hippocampus CA4 region of a dog treated with AZD3783 at 30 mg/kg/day for 3 months and then being dose-free for 5 weeks. H&E staining, 200×.
4-week brain exposure/pathology study
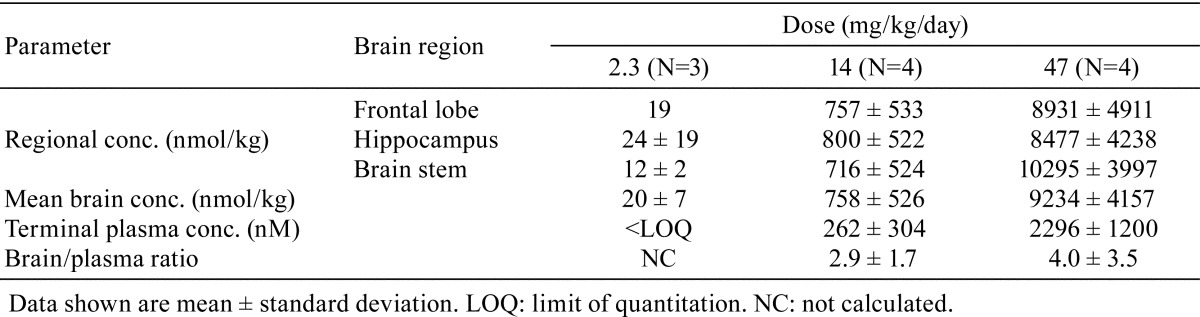
Plasma and brain concentration: AZD3783 was detected in plasma 24 h after dosing from dogs dosed at 14 or 47 mg/kg/day, but not from dogs dosed at 2.3 mg/kg/day. However, appreciable concentrations of AZD3783 were detected in the brains of dogs from all groups, with greater than dose-proportional increases between 14 and 47 mg/kg (Table 5). The mean brain concentrations at 14 and 47 mg/kg were 38- and 460-fold of the concentration detected at 2.3 mg/kg, a supra-proportional increase relative to dose. However, at each dose level, there was no difference in regional brain concentrations. The brain/plasma concentration ratio appears to be constant (approximately 4) across the dose range, indicating that there is no saturation of clearance from the brain.
Table 5. Terminal Plasma and Brain Concentrations of AZD3783 in Dogs after 4 Weeks of Dosing.
Mortality: A male in the 47 mg/kg/day group was euthanized on Day 20. This dog had exhibited severe clinical signs including dragging/knuckling of the left forelimb, a series of clonic/tonic postures with stiff limbs in lateral recumbancy and inability to right itself. The brain and plasma concentrations of AZD3783 in this dog were not remarkably different from the other dogs within the group.
Pathologic findings: The brain of the decedant showed neuronal degeneration and necrosis microscopically, accompanied by minimal gliosis in the medulla oblongata (dorsal nucleus of trapezoid body). There was also slight foamy cytoplasmic vacuolation of neurons in the cochlear nucleus. The other three dogs in the 47 mg/kg/day group had minimal to moderate foamy cytoplasmic vacuolation of neurons and two of them also showed slight to moderate neuronal degeneration and necrosis, accompanied by slight to moderate gliosis, and minimal to slight mononuclear cell infiltration, in the medulla oblongata.
Two dogs in the 14 mg/kg/day group showed minimal granular, yellowish pigmentation in the cytoplasm of neurons in the medulla oblongata. No treatment-related microscopic change in the brain was seen in the 2.3 mg/kg/day group.
Discussion
AZD3783, a CAD, was designed to cross the blood brain barrier and exert its pharmacologic effects on the serotonergic pathway. Many CADs cause PLD in animals and some induce PLD in humans. CAD-induced PLD is characterized by appearance of foamy macrophages or foamy cells (vacuolation) in tissues under light microscopy and by appearance of ultrastructural lamellar bodies under electron microscopy18,19,20. While some CADs only induce PLD in individual tissues, such as liver or lungs, others may induce generalized PLD in various tissues. The reason for these differences is unclear, but it may be related to difference in tissue distribution, and compound lipophilicity or basicity21.
AZD3783 caused PLD in dogs, which was identified by electron microscopy the presence of lamellar bodies in the liver and gallbladder of some treated dogs in the 1-month study. Although no electron microscopy was conducted on any tissues to confirm PLD in the 3-month toxicity study, various tissues showed PLD-like vacuolation under examination by light microscopy. Since AZD3783 is a CAD, it is highly likely that the PLD-like vacuolation seen in the various tissues, including the nervous tissues are due to PLD.
In the eye, inclusion bodies in the retina, perivascular inflammation, and degeneration of optic nerve were observed in dogs dosed at 30 mg/kg/day for 3 months. The retina inclusions are also consistent with PLD, which with associated myeloid bodies or inclusion bodies are a common CAD-induced retinal change in the rat22,23, and was seen in dogs administered fluoxetine24. However, degeneration of optic nerve due to PLD has not been reported previously. For example, amiodarone, which causes generalized PLD, did not cause any alteration in the retina or optic nerve in dogs after 11 weeks of dosing, although it did induce corneal microdeposits in 1 of 6 dogs after 6 weeks25. Thus, microscopic findings in the eye after AZD3783 treatment are more severe than those previously reported with other CADs. The reason for these differences is not clear.
In the 3-month study, one female dosed at 30 mg/kg/day exhibited severe clinical signs near the end of the dosing period. Since the intended pharmacologic action of AZD3783 is to increase brain serotonin concentration, and serotonin toxicity has been reported in dogs after incidental ingestions of SSRI or hydroxytriptophan, a precursor of serotonin26, we first considered whether the dog was experiencing serotonin toxicity. Serotonin toxicity in dogs is acute in onset and characterized by behavioral change (agitation, lethargy) accompanied by autonomic signs (e.g., vomiting, mydriasis, hypersalivation, hyperthermia, tachycardia), and neuromuscular signs (e.g., ataxia, tremors, myoclonus, hyperreflexia, nystagmus, and seizure). In severe cases, coma, hyperpyrexia, and generalized seizure can be rapidly fatal27. However, we discounted that possibility for the reasons described here. The high dose, 30 mg/kg/day, was selected based on results from the 1-month study, so as not to elicit severe liver toxicity and intolerable CNS effects. Indeed, the clinical signs observed in both 1- and 3-month studies were not remarkable and no seizure was ever noted. In addition, the decedent did not exhibit any of the autonomic signs, such as emesis, hypertension, or hyperthermia. Although its plasma concentration of AZD3783, 17 µM, obtained prior to euthanasia on Day 86, was higher than Cmax from other surviving females on Day 91 (which ranged from 11 to 15.7 µM), it was lower than Cmax on Day 1 (which ranged from 23.7 to 33.7 µM). As shown in Table 1, Cmax appeared to decrease as dosing progressed. Since CNS clinical signs are generally Cmax driven, and no adverse CNS signs were seen in any of the treated dogs during the early weeks of treatment, this suggests that the neurologic effects in the decedant developed over time with repeat dosing, and is most likely related to the pathological changes observed in the nervous tissues.
The pathologic changes in the nervous tissues induced by AZD3783 were time and dose dependent. Whereas minimal vacuolation (but no degeneration) was observed after 1 month of dosing at 47 mg/kg/day, vacuolation plus degeneration or necrosis were seen after 3 months at 15 or 30 mg/kg/day, while only vacuolation in the spinal ganglia was observed at 7 mg/kg/day. This pattern of morphologic changes suggests that vacuolation due to PLD is a leading event to the degeneration or necrosis. PLD in the brain, nerves, or peripheral ganglia has not been reported with very many chemicals28. Even among antidepressant drugs or drug candidates, which as a class has the highest potential to induce generalized PLD29, few were reported to cause widespread PLD in the nervous system. Furthermore, most observations were made in rats. In rats, imipramine (a tricyclic) caused PLD in retina and DRG30; citalopram (an SSRI) caused PLD in 1 sympathetic ganglion, none or very weak PLD in retinal and trigeminal ganglia, and hypothalamic neurosecretory perikarya31; fluoxetine (an SSRI) caused PLD in retinas24 as well as PLD-like vacuolation in nerve cells in the thalamus, cerebellum, spinal cord and ganglion or rats32. In dogs, citalopram did not cause PLD33, whereas fluoxetine caused generalized PLD, with EM confirmation in the retina and cerebellar cortex after 1 year of dosing; however, no neuronal degeneration or neurological findings were observed. Clinical signs observed in the 1-year study included convulsions, tremors, transient nystagmus and slow/incomplete papillary responses24. Among the non-CNS drugs, posaconazole (an antifungal) caused PLD in DRG and medulla oblongata neurons in the brain of dogs after chronic administration. However, there was no evidence of neuronal degeneration or necrosis, nor was there any alteration in the amplitude, or latency of the auditory, visual, or somatosensory evoked potentials28. Clearly, there are differences in the physical and chemical properties between AZD3783 and these aforementioned CADs that contributed to the differences in the toxicity and pathologic findings in the nervous tissues. One notable difference is that, in the study with posaconazole28 inflammatory changes were not observed in any tissues with PLD. In contrast, in our studies with AZD3783, inflammation and gliosis was present in the nervous tissue (eye, ganglion, and brain). Based on the work by Wada et al.34 and assuming similarity in the pathogenesis of toxicity induced by PLD and lysosomal storage disease, one would hypothesize that the inflammatory response secondary to PLD was one of the trigger events to the neurodegeneration. Wada et al.34, using a mouse model of Sandhoff’s disease, a lysosomal storage disorder characterized by storage of gangliosides in the CNS, showed that microglia activation and inflammation preceded the neurodegeneration. The activated microglia produced TNF-α, a cytokine which is both neuroprotective and neurotoxic. In the mouse model, although glycolipid storage occurred in all neurons, apoptotic cell death was concentrated in the caudal region of the CNS, in spinal cord, brainstem, and thalamus, where microglia activation was indicated by overexpression of gene activation.
In the 4-week brain exposure/pathoogy study, one dog dosed at 47 mg/kg was euthanized after exhibiting severe neuromuscular effects. This dog and other dogs dosed at 47 mg/kg/day had, in addition to neuronal vacuolation, also neuronal degeneration and necrosis accompanied by gliosis in the medulla oblongata. The manifestation of the neurological effect after 20 days of dosing, and the degenerative changes in the brain tissues in this investigative study contrast with the minimum brain vacuolation observed in the 1-month toxicity study. The differences between the two studies could be due to differences in dog sources and individual animal sensitivity. Nonetheless, the mean group plasma concentrations of AZD3783 after administration of various doses were comparable across studies.
The brain concentration study reveals that after 4 weeks of dosing, disproportionately high concentrations of AZD3783 relative to plasma were present in various brain regions. Whereas the plasma concentrations were generally dose-proportional (see Table 1), the mean brain concentrations of AZD3783 were 38- and 460-fold higher at 14 and 47 mg/kg/day dose, respectively, relative to that at 2.3 mg/kg/day. Since CADs, e.g., amidarone, have been shown to accumulate in tissues in association with PLD35, one may hypothesize that the high brain to plasma concentration ratio is due to PLD, following accumulation of AZD3783 in lysosomes. On the other hand, at each dose, the concentrations of AZD3783 in the different brain regions were similar, i.e., there was no measureable preferential distribution of AZD3783 in select regions of the brain where dose-dependent PLD-like vacuolation was observed (e.g. brainstem and hippocampus), or where degeneration was observed, relative to non-responding regions (e.g. frontal cortex; data not shown). These results, while suggesting that the pathological changes may not be caused by direct cytotoxicity of AZD3783, do not rule out if it is due to differential sensitivity in different brain regions. The mechanisms leading to neuronal injury and cell death in the visual/optical tracts/pathways, brain stem and the CA2, CA3, and CA4 regions of the hippocampus remain to be elucidated.
After 3 months of dosing, 7 of 11 dogs dosed at 30 mg/kg/day were noted to have retinal or vitreous hemorrhage, or retinal edema or detachment upon ophthalmologic examinations. These findings are possibly related to the exaggerated pharmacologic effects of AZD3783 on serotonin and platelet function. Vitreous hemorrhage, which can occur in beagle dogs as a spontaneous variant, is most frequently caused by trauma, or a systemic clotting disorder, and can occur secondary to retinal detachment, which is often associated with increased ocular pressure. Retinal hemorrhage is induced by compounds acting directly or indirectly on the clotting mechanisms, e.g., coumarin anticoagulants22 and is the most common ocular lesion in dogs with systemic hypertension36. The SSRIs and serotonin and norepinephrine re-uptake inhibitors (SNRIs) drugs have effects on cardiovascular and coagulation mechanisms. Increases in heart rate and blood pressure have been seen clinically with fluoxetine overdose. Altered or abnormal bleeding has been observed in the upper GI, and rare hemorrhage in retina or subjunctiva has been noted with fluoxetine, paroxetane, or venlafaxine (a SNRI) in man. However, to our knowledge, these ophthalmological findings have not been reported in preclinical studies with these aforementioned compounds. Besides a slight increase in platelet counts, which was also observed in fluoxetine dog studies, there was no change in coagulation parameters, nor was hemorrhage detected in any tissues after 13 weeks of dosing. In cardiovascular telemetry study in dogs, AZD3783 causes modest tachycarda with decrease in blood pressure at 47 mg/kg, but no effect at 14 mg/kg/day. Thus, mechanisms underlying the ophthalmological effect from AZD3783 in dogs remain to be confirmed.
In the rat, AZD3783 treatment also caused generalized PLD; whereas the microscopic changes in many visceral organs were similar to those seen in the dogs, the pathologic changes in the nervous tissues were limited and less severe. After 1 month of dosing at 126 mg/kg/day, minimal neuronal vacuolation in the DRG and brain stem were observed. After 3 months of dosing at 100 mg/kg/day, minimal to slight neuronal vacuolation with eosinophilic deposits were seen in the DRG, brain stem, and additionally, spinal cord (unpublished results). Inflammation was not seen in any of these aforementioned tissues and no microscopic changes were seen in the eyes, optic nerve, ventral root ganglion, sciatic nerve or in other brain regions. No ophthalmologic changes similar to those in the dogs were observed (unpublished results). The systemic exposures at the observed dose for neuropathology, 100 mg/kg/day, were 30 µM for Cmax and 402 µM.h for AUC; the exposures at the NOEL for neuropathology, 25 mg/kg/day, were 11.4 µM for Cmax and 84 µM.h for AUC (unpublished results). Whereas the dose normalized plasma concentrations were comparable between rats and dogs, the rat had approximately 2-fold higher dose-normalized AZD3783 concentration in the brain stem or hippocampus. Thus species differences in nervous tissue sensitivity and responses, e.g., inflammation or glia cell activation, to PLD may underlie some of the differences in neurotoxicity observed between dogs and rats after exposure to AZD3783.
In AZD3783 Phase 1 trials in healthy volunteers, single ascending oral doses from 1 to 40 mg was well tolerated and did not elicit any severe adverse effects5. In a PET study in man, 5 mg dose resulted in 50% receptor occupancy in brain region5, and is projected to be the efficacious dose, where the estimated Cmax is 33 nM and AUC is 383 nM*h1. Compared to the predicted therapeutic concentration based on 50% receptor occupancy in the brain, the AUC exposure achieved in the 3-month study was 76-fold higher at 7 mg/kg/day, where only low incidences of vacuolation and inflammation were seen in the ganglion cell. At 30 mg/kg/day, where pathological changes were seen in brain, spinal ganglia, as well as sciatic and optic nerves, the exposure was 350-fold of the predicted therapeutic exposure. Thus, there appeared to be large safety margins to the observed effects. Nevertheless, a comparison of findings from the 1- and 3-month studies with AZD3783 vs. those reported in the literature raised these questions: 1) Are the more severe brain lesions (degeneration, necrosis, or inflammation) a direct or indirect consequence of PLD? 2) Is there an additional AZD3783-unique neurotoxic mechanism at play apart from PLD that lead to the severe lesions? 3) Will PLD and the neurotoxicity get progressively worse with chronic dosing, such that a NOEL will not be identified and thus no safety margin is attainable? And 4) Why and how is AZD3783 different from other SSRIs or SNRIs? These questions, together with the lack of recovery in effects in the 3-month study raised significant safety concerns and a decision to discontinue the project before multiple dose phase in man was initiated.
In summary, we observed unexpected neurotoxicity and progressive pathologic changes in various tissues in dogs after repeat dosing with AZD3783 from 1 to 3 months. The observation of vacuolation in the various tissues suggests generalized PLD, which is consistent with the chemical structure of AZD3783. Questions remain if the increased brain concentration and the observed neuropathology are attributable to PLD or chemical toxicity unique to AZD3783.
Acknowledgments
The authors thank the scientific staff at AstraZeneca R&D in the US, UK and Sweden, as well as Carmela Parente and her teams at Charles River Laboratories, Montreal, Canada, whose dedication and excellent efforts made these investigations possible.
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution Non-Commercial No Derivatives (by-nc-nd) License <http://creativecommons.org/licenses/by-nc-nd/3.0/>.
References
- 1.Zhang M, Zhou D, Wang Y, Maier DL, Widzowski DW, Sobotka-Briner CD, Brockel BJ, Potts WM, Shenvi AB, Bernstein PR, and Pierson ME. Preclinical pharmacology and pharmacokinetics of AZD3783, a selective 5-hydroxytryptamine1B receptor antagonist. J Pharmacol Exp Ther. 339: 567–578 2011. [DOI] [PubMed] [Google Scholar]
- 2.Fink KB, and Göthert M. 5-HT receptor regulation of neurotransmitter release. Pharmacol Rev. 59: 360–417 2007. [DOI] [PubMed] [Google Scholar]
- 3.Moret C, and Briley M. The possible role of 5-HT1B/1D receptors in psychiatric disorders and their potential as a target for therapy. Eur J Pharmacol. 404: 1–12 2000. [DOI] [PubMed] [Google Scholar]
- 4.Slassi A. Recent advances in 5-HT1B/1D receptor antagonists and agonists and their potential therapeutic applications. Curr Top Med Chem. 2: 559–574 2002. [DOI] [PubMed] [Google Scholar]
- 5.Varnäs K, Nybert S, Karlsson P, Pierson ME, Kågedal M, Cselényi Z, McCarthy D, Xiao A, Zhang M, Halldin C, and Farde L. Dose-dependent binding of AZD3783 to brain 5- HT1B receptors in non-human primates and human subjects: a positron emission tomography study with [11C]AZ10419369. Psychopharmacology. 213: 533–545 2011. [DOI] [PubMed] [Google Scholar]
- 6.Peroutka SJ, and Howell T. The molecular evolution of G protein-coupled receptors. Neuropharmacology. 33: 319–324 1994. [DOI] [PubMed] [Google Scholar]
- 7.Lüllmann H, Lüllmann-Rauch R, Wassermann O, and de la Lglesia FA. Drug-induced phospholipidosis. CRC Crit Rev Toxicol. 2: 185–218 1975 [DOI] [PubMed] [Google Scholar]
- 8.Kodavanti UP, and Mehendale HM. Cationic amphiliphilic drugs and phospholipid storage disorder. Pharmacol Rev. 42: 327–354 1990. [PubMed] [Google Scholar]
- 9.Reasor MJ. A review of the biology and toxicologic implications of the induction of lysosomal lamellar bodies by drugs. Toxicol Appl Pharmacol. 97: 47–56 1989. [DOI] [PubMed] [Google Scholar]
- 10.Reasor MJ, and Kacew S. Drug-induced phospholipidosis: are there functional consequences? Exp Biol Med (Maywood). 226: 825–830 2001. [DOI] [PubMed] [Google Scholar]
- 11.Lewis JH, Ranard RC, Caruso A, Jackson LK, Mullick F, Ishak KG, Seeff LB, and Zimmer HJ. Amidarone hepatotoxicity: prevalence and clinicopathologic correlation among 104 patients. Hepatology. 9: 679–685 1989. [DOI] [PubMed] [Google Scholar]
- 12.Gonzalez-Rothi RJ, Zander DS, and Ros PR. Fluoxetine hydrochloride (Prozac)-induced pulmonary disease. Chest. 107: 1763–1765 1995. [DOI] [PubMed] [Google Scholar]
- 13.Laplane D, and Bousser MG. Polyneuropathy during perhexiline maleate therapy. Int J Neurol. 15: 293–300 1981. [PubMed] [Google Scholar]
- 14.Mäntyjärvi M, Tuppurainen K, and Ikäheimo K. Ocular side effects of amiodarone. Surv Ophthalmol. 42: 360–366 1998. [DOI] [PubMed] [Google Scholar]
- 15.Reasor MJ, Hastings KL, and Ulrich RG. Drug-induced phospholipidosis: issues and future directions. Expert Opin Drug Saf. 5: 567–583 2006. [DOI] [PubMed] [Google Scholar]
- 16.FDA. The pink sheet: Safety a concern. FDA phospholipidosis guidance being developed. Vol. 66, No. 133, pp. 24. 2004. [Google Scholar]
- 17.Morelli JK, Buehrle M, Pognan F, Barone LR, Fieles W, and Ciaccio PJ. Validation of an in vitro screen for phospholipidosis using a high-content biology platform. Cell Biol Toxicol. 22: 15–27 2006. [DOI] [PubMed] [Google Scholar]
- 18.Hruban Z. Pulmonary changes induced by amphiphilic drugs. Environ Health Perspect. 16: 111–118 1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lüllmann-Rauch R. Drug-induced lysosomal storage disorders. Front Biol. 48: 49–130 1979. [PubMed] [Google Scholar]
- 20.Robison RL, Visscher GE, Roberts SA, Engstrom RG, Hartman HA, and Ballard FH. Generalized phospholipidosis induced by an amphiphilic cationic psychotropic drug. Toxicol Pathol. 13: 335–348 1985. [DOI] [PubMed] [Google Scholar]
- 21.Hanumegowda UM, Wenke G, Regueiro-Ren A, Yordanova R, Corradi JP, and Adams SP. Phospholipidosis as a function of basicity, lipophilicity, and volume of distribution of compounds. Chem Res Toxicol. 23: 749–755 2010. [DOI] [PubMed] [Google Scholar]
- 22.Haywood R. The eye. In: Target Organ Toxicity, Vol. II. GM Cohen (ed). CRC Press, Boca Raton. 109–124. 1986 [Google Scholar]
- 23.Drenckhahn D, and Lüllmann-Rauch R. Drug-induced retinal lipidosis: differential susceptibilities of pigment epithelium and neuroretina toward several amphiphilic cationic drugs. Exp Mol Pathol. 28: 360–371 1978. [DOI] [PubMed] [Google Scholar]
- 24.Summary Basis of Approval, FDA, NDA18-936 (Fluoxetine) Dec 7, 1987
- 25.Bicer S, Fuller GA, Wilkie DA, Yamaguchi M, and Hamlin RL. Amidarone-induced keratopathy in healthy dogs. Vet Ophthalmol. 5: 35–38 2002. [DOI] [PubMed] [Google Scholar]
- 26.Gwaltney-Brant SM, Albretsen JC, and Khan SA. 5-Hydroxytryptophan toxicosis in dogs: 21 cases (1989-1999). J Am Vet Med Assoc. 216: 1937–1940 2000. [DOI] [PubMed] [Google Scholar]
- 27.Mohammad-Zaddeh LF, Moses L, and Gwaltney-Brant SM. Serotonin: a review. J Vet Pharmacol Therap. 31: 187–199 2008. [DOI] [PubMed] [Google Scholar]
- 28.Cartwright ME, Petruska J, Arezzo J, Frank D, Litwak M, Morrissey RE, MacDonald J, and Davis TE. Phospholipidosis in neurons caused by posaconazole, without evidence for functional neurologic effects. Toxicol Pathol. 37: 902–910 2009. [DOI] [PubMed] [Google Scholar]
- 29.Sadrich N. The regulatory challenge of drug-induced phospholipidosis. Presented in: FDA Pharmaceutical Science and Clinical Pharmacology Advisory Committee meeting, April 14, 2010
- 30.Spaet RH, Sullivan DJ, and Diener RM. Occurrence of myeloid bodies in rats following two-year administration of imipramine hydrochloride. Toxicol Pathol. 11: 3–11 1983. [DOI] [PubMed] [Google Scholar]
- 31.Lüllmann-Rauch R, and Nässberger L. Citalopram-induced generalized lipidosis in rats. Acta Pharmacol Toxicol (Copenh). 52: 161–167 1983. [DOI] [PubMed] [Google Scholar]
- 32.Nonoyama T, and Fukuda R. Drug-induced phospholipidosis – pathological aspects and its prediction. J Toxicol Pathol. 21: 9–24 2008. [Google Scholar]
- 33.Summary Basis of Approval, FDA, NDA 20-822 (Citalopram Hydrobromide) June 2, 1998.
- 34.Wada R, Tifft CJ, and Proia RL. Microglial activation precedes acute neurodegeneration in Sandhoff disease and is suppressed by bone marrow transplantation. Proc Natl Acad Sci USA. 97: 10954–10959 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kannan R, Sarma SM, Guha M, and Venkataraman K. Tissue drug accumulation and ultrastructural changes during amiodarone administration in rats. Fundam Appl Toxicol. 13: 793–803 1989. [DOI] [PubMed] [Google Scholar]
- 36.Leblanc NL, Steven RL, and Bentley E. Ocular lesions associated with systemic hypertension in dogs: 65 cases (2005-2007). J Am Vet Med Assoc. 238: 915–921 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]