

Abstract
Children exposed in utero to cannabis present permanent neurobehavioral and cognitive impairments. Psychoactive constituents from Cannabis spp., particularly Δ9-tetrahydrocannabinol (THC), bind to cannabinoid receptors in the fetal brain. However, it is unknown whether THC can trigger a cannabinoid receptor-driven molecular cascade to disrupt neuronal specification. Here, we show that repeated THC exposure disrupts endocannabinoid signaling, particularly the temporal dynamics of CB1 cannabinoid receptor, to rewire the fetal cortical circuitry. By interrogating the THC-sensitive neuronal proteome we identify Superior Cervical Ganglion 10 (SCG10)/stathmin-2, a microtubule-binding protein in axons, as a substrate of altered neuronal connectivity. We find SCG10 mRNA and protein reduced in the hippocampus of midgestational human cannabis-exposed fetuses, defining SCG10 as the first cannabis-driven molecular effector in the developing cerebrum. CB1 cannabinoid receptor activation recruits c-Jun N-terminal kinases to phosphorylate SCG10, promoting its rapid degradation in situ in motile axons and microtubule stabilization. Thus, THC enables ectopic formation of filopodia and alters axon morphology. These data highlight the maintenance of cytoskeletal dynamics as a molecular target for cannabis, whose imbalance can limit the computational power of neuronal circuitries in affected offspring.
Keywords: axon, growth cone, marijuana, neurite outgrowth, synaptogenesis
See also: L Cristino & V Di Marzo (April 2014)
Introduction
The prevalence of recreational cannabis use continues to increase with adolescents and young adults being primary affected (Substance Abuse & Mental Health Service Administration, 2010). Cannabis exposure of infants in utero or cannabis use in teenagers [peaking at 15–17 years of age (Substance Abuse & Mental Health Service Administration, 2010)] can coincide with critical periods of brain development when neuronal connectivity is prenatally established (Kostovic & Jovanov-Milosevic, 2006) or postnatally refined to increase modularity and integrative capacity (Dennis et al, 2013). Accordingly, prospective longitudinal assessments (Goldschmidt et al, 2004; Willford et al, 2010; Day et al, 2011) suggests that cannabis use during pregnancy can increase the risk for ill-behaviors (Goldschmidt et al, 2004; Day et al, 2011), cognitive deficit (Huizink & Mulder, 2006), drug seeking (Day et al, 2006), attention deficit (Leech et al, 1999), and anxiety and depression (Leech et al, 2006) among affected neonatal or adolescent offspring. Large-scale population analysis associates growth retardation with maternal cannabis use (El Marroun et al, 2009), particularly since there is an efficient cross-placental transfer of Δ9-tetrahydrocannabinol (THC) (Grotenhermen, 2003), the major psychoactive constituent of Cannabis spp. Nevertheless, a gap of knowledge exists regarding the neuronal basis of cannabis-induced developmental deficits in the nervous system.
THC promiscuously binds to members of the cannabinoid receptor family (Howlett, 2002), G protein-coupled receptors (GPCRs), whose cell-type-specific segregation furnishes intercellular interactions using endocannabinoids as physiological ligands (Kano et al, 2009; Pertwee et al, 2010; Di Marzo, 2011). Yet THC's mode of action on neuronal fate decisions, including the identity of the cannabinoid receptor mediating THC's effect in the developing nervous system, remains unknown (Keimpema et al, 2011). Considering that neurogenesis precedes the production of astrocytes and oligodendrocytes (Schmechel & Rakic, 1979; Kessaris et al, 2008) [the latter becoming molecularly specified in the forebrain only during late gestation (Hardy & Friedrich, 1996)], the type 1 cannabinoid receptor (CB1R) expressed by developing cortical neurons (Berghuis et al, 2007; Keimpema et al, 2011), particularly during the axonal growth process (Berghuis et al, 2007), emerges as a likely candidate. sn-1-Diacylglycerol lipases α and β (Bisogno et al, 2003) in growth cones generate 2-arachidonoylglycerol (2-AG) to focally activate CB1Rs (Keimpema et al, 2010), allowing protrusive endocannabinoid signalling upon activating Rho-family GTPases (Berghuis et al, 2007), a prerequisite of axonal motility (Yuan et al, 2003). Therefore, THC, a partial CB1R agonist as predicted by postnatal pharmacology (Pertwee et al, 2010), could disable the spatial specificity of endocannabinoid cues in the developing brain by indiscriminately activating CB1Rs, even at unfavorable subcellular positions (Keimpema et al, 2011). In doing so, THC might provoke spatially and temporally segregated signalling events to disrupt neuronal fate decisions and specification, particularly synaptic wiring, thus recapitulating corticofugal axon fasciculation and targeting defects in CB1R−/− mice (Berghuis et al, 2007; Diaz-Alonso et al, 2012). However, the molecular configuration of signalling cascades hijacked by THC to imprint permanent wiring errors in the fetal cerebrum remains elusive.
We set ourselves the goal to establish mouse models amenable to study the molecular underpinnings of life-long circuit modifications in offspring upon maternal THC exposure. We took note of the fact that cannabis can introduce epigenetic modifications (Dinieri et al, 2011), attenuating synaptic neurotransmission. If THC impedes the structural organization of neuronal networks then synapse re-positioning (i.e. the erroneous recruitment of synaptic afferents to somatodendritic domains on postsynaptic neurons) is likely an early developmental event (Keimpema et al, 2011), which might stabilize over time. Precluding the attainment of cell-type-specific neuronal structure would inevitably rely on cannabinoid receptor-mediated signalling events (Berghuis et al, 2007) with profound modifications to the cytoskeleton, particularly microtubule integrity. We have mapped Superior Cervical Ganglion 10/stathmin-2 (SCG10) and identified its loss as THC's molecular target in the mouse and human fetal nervous systems, and used SCG10 to acutely reset cytoskeletal dynamics upon THC-induced CB1R activation.
Results
Altered assembly of cortical networks in Δ9-tetrahydrocannabinol-exposed offspring
We hypothesized that if THC impairs endocannabinoid-mediated mechanisms of cortical development then this would manifest as the altered distribution of CB1R+ afferents and synapses (Berghuis et al, 2007; Keimpema et al, 2010, 2011; Diaz-Alonso et al, 2012), which adopt strict layer (L) specificity physiologically (Bodor et al, 2005). Therefore, we began to administer THC at a dose of 3 mg/kg (i.p., daily), which did not change maternal behavior or physical measures (Mato et al, 2004) as would be predicted for high-dose THC intoxication, from embryonic day (E)5.5–17.5 (Berghuis et al, 2005), and allowed offspring to mature under conventional husbandry conditions (without postnatal re-introduction of the drug) until postnatal day (P) 10 or 120. THC did not affect maternal bodyweight, the male:female sex ratio of the offspring [50.9% males (THC) versus 50.7% males (vehicle)] or the size of litters [5.9 ± 0.4 (THC) versus 6.3 ± 0.5 (vehicle)]. Male fetuses and offspring were analyzed because of their preferential sensitivity to cannabis (Hurd et al, 2005). By P10, THC-exposed offspring showed a reduced area of superficial LI/II otherwise receiving peak density of CB1R+ inputs (Bodor et al, 2005) (84.8 ± 2.6% of control, P < 0.01, Supplementary Fig S1A and A1). CB1Rs are expressed by both perisomatically-targeting cholecystokinin-containing interneurons (Bodor et al, 2005) and pyramidal cells (Kano et al, 2009) in the postnatal cerebral cortex. At P120, we first showed the disruption of perisomatic baskets (Fig S1A and A1), confinements of inhibitory synapses around pyramidal cell somata (Fig 1B and B1), recapitulating earlier findings in interneuron-specific CB1R knock-outs (Berghuis et al, 2007). Second, and reminiscent to the cortical reorganization of CB1R+ inputs, we found a significant increase in the density of CB1R+ boutons in the stratum radiatum of the hippocampal CA1 subfield in mice prenatally exposed to THC [7782 ± 409 (THC) versus 6860 ± 150 (vehicle) boutons/mm2, P < 0.05, Fig 1C–C2], suggesting synapse mistargeting in THC-exposed brains (Keimpema et al, 2011). Since many of these boutons are terminal specializations of Schaffer collaterals, a major glutamatergic pathway originating from CA3 pyramidal cells and whose activity is modulated by endocannabinoids (Takahashi & Castillo, 2006), we tested whether the sign of synaptic plasticity upon Schaffer collateral stimulation is altered in THC-exposed offspring. Upon using 900 pulses at 1 Hz to induce long-term depression (LTD) (Dudek & Bear, 1993), orthodromic stimulation-evoked LTD significantly diminished in THC-exposed offspring relative to controls [n = 7 (vehicle) versus n = 6 (THC); Fig 1D–D2], as measured in the CA1 stratum radiatum (Supplementary Fig S1E). Likewise, the long-term depression of neuronal population activity, measured in the CA1 stratum pyramidale and expressed as the population spike amplitude, was occluded upon low-frequency Schaffer collateral stimulation [n = 6 (vehicle) versus n = 6 (THC); Supplementary Fig S1E1–E3]. Moreover, we found increased paired-pulse facilitation at Schaffer collateral inputs (Fig 1E, Supplementary Fig S1F), suggesting increased and deregulated presynaptic activity, compatible with the hypothesis of long-lasting modifications to CB1R signaling in THC-exposed brains. These data suggest that administration of THC during pregnancy can induce long-term structural and functional modifications of the cortical circuitry.
Figure 1. In utero Δ9-tetrahydrocannabinol (THC) exposure modifies synaptic connectivity and responsiveness in the cortical circuitry of adult offspring.
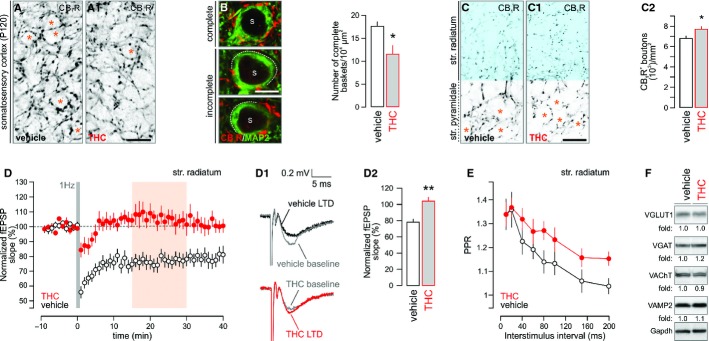
A, A1 Prenatal THC exposure significantly decreased the density of perisomatic CB1R+ boutons, presumed pre-synapses, within superficial cortical laminae receiving prominent CB1R+ innervation by P120 (* denotes the positioning of postsynaptic neurons in layer II/III).
B, B1 This deficit has precipitated the formation of fragmented perisomatic CB1R+ “baskets” (s denotes the location of neuronal somata).
C–C2 Similarly, THC exposure misrouted hippocampal CB1R+ afferents, significantly increasing their density in the CA1 stratum radiatum [blue in (C) and (C1)(* indicates the position of neuronal somata)]. Quantitative densitometry of CB1R+ boutons in the stratum radiatum is shown in (C2).
D–D2 Summary graphs of the time-course of field excitatory postsynaptic potentials (fEPSPs) from CA1 pyramidal cells showed impaired LTD induction upon low-frequency stimulation of Schaffer-collaterals (Dudek & Bear, 1993) in THC-exposed mice (n = 6–7 males/group; P120). Orange shading denotes data binned in (D2). Representative sample traces of experiments in vehicle or THC-exposed material recorded 15–30 min after LTD induction (D1). The degree of synaptic depression as revealed by binning data 15–30 min after LTD induction (D2).
E Paired-pulse facilitation revealed altered presynaptic neurotransmitter release upon prenatal THC exposure (n = 6 animals/group; P 120).
F Rewiring and reduced synaptic plasticity in the cortical circuitry were not associated with long-lasting modifications of synaptic protein expression in the hippocampus of offspring prenatally exposed to THC (P120). Fold changes were normalized to gapdh and expressed as percentages (n = 4/group; see Supplementary Fig S1B and C).
Data information: Data were expressed as means ± s.e.m.; ***P < 0.001, **P < 0.01, *P < 0.05. Scale bars, 20 μm (A1,C1) or 8 μm (B).
Source data are available online for this figure.
Maternal THC exposure can induce epigenetic modifications, such as repressive histone methylation (Dinieri et al, 2011). Here, we demonstrate by quantitative PCR and Western blotting that maternal THC administration did not alter the expression of vesicular glutamate, GABA and acetylcholine transporters, the SNARE component vesicle-associated membrane protein-2 (Fig 1F, Supplementary Fig S1B and C) or CB1R mRNA (Supplementary Fig S1D) in the hippocampus and neocortex of adult THC-exposed offspring. In sum, retained presynaptic protein expression levels and CB1R mRNA expression favor the conceptual framework of developmentally regulated circuit reorganization upon maternal THC exposure.
Δ9-tetrahydrocannabinol impairs axonal development in the corticofugal system
In developing neurons, THC might act as a “functional antagonist” since it can displace the binding of high-efficacy endocannabinoids, dampening their signaling efficacy (Paronis et al, 2012). If so, THC could modify the intrinsic program of neuronal fate specification via a receptor-mediated mechanism. To determine the molecular identity of THC-activated cannabinoid receptor(s) and the downstream signal transduction machinery impairing the cortical wiring map, we administered THC (3 mg/kg; i.p.), WIN55,212-2 (5 mg/kg; CB1R agonist) and AM 251 (5 mg/kg; CB1R antagonist) in gravid mice from E5.5-17.5. Since genetic disruption or pharmacological manipulation of CB1Rs introduces corticofugal axon fasciculation errors [i.e. enlargement with reduced myelination (Mulder et al, 2008)], we sampled the diameter of corticofugal axons in drug-exposed fetuses at E18.5. THC significantly increased the diameter of first-order fascicles relative to vehicle controls (P < 0.001, n > 300 fascicles from n = 4–7 fetuses/group; Fig 2A and A1). AM 251, but not WIN55,212-2, promoted the formation of enlarged fascicles in vivo (Supplementary Fig S2A and A1), which was recapitulated by in vitro AM 251 exposure of cortical neurons producing endocannabinoids (Keimpema et al, 2010) (Supplementary Fig S2B and B1). The AM 251-induced axonal redistribution suggests CB1R involvement.
Figure 2. Δ9-tetrahydrocannabinol (THC) affects directional growth of corticofugal axons in the fetal cerebrum.
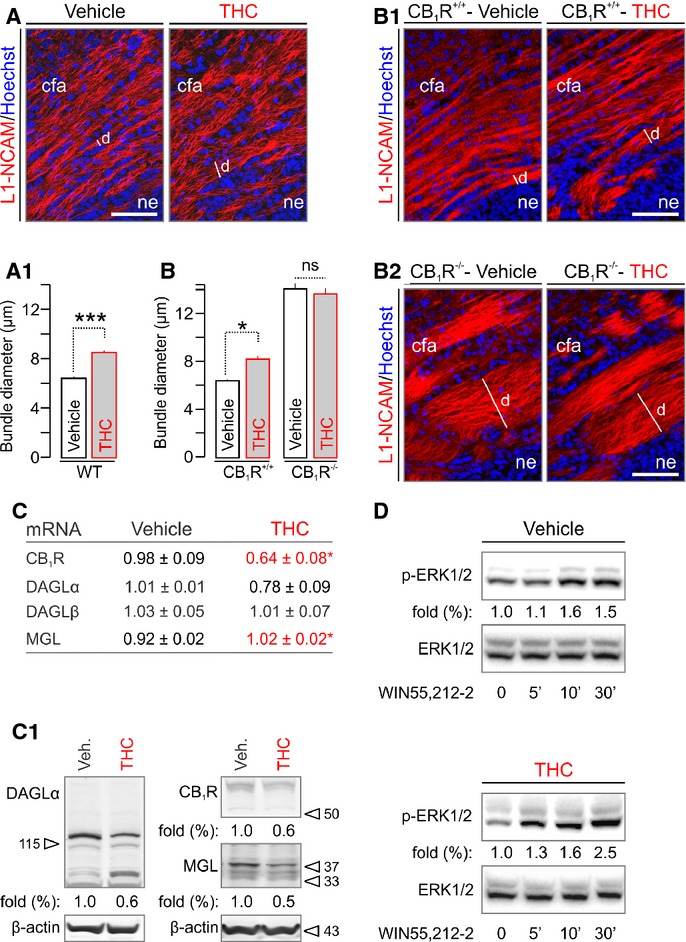
A, A1 Prenatal exposure to a non-aversive dose of THC (Mato et al, 2004) (3 mg/kg, embryonic day E5.5–17.5) altered the coalescing of axons committed to the corticofugal (cfa) pathway (Keimpema et al, 2010), as revealed by quantifying the transverse diameter (d) of individual fascicles labeled for L1-NCAM on E18.5 (A1) in the cortical intermediate zone of n = 4–7 male fetuses/group; n > 300 observations/group.
B–B2*Tightly constrained cfa axons formed enlarged bundles in CB1R−/− mice, whose diameter remained unchanged upon THC administration.
C, C1 mRNA profiling of the CB1R and enzymes involved in 2-AG metabolism showed significantly reduced CB1R but increased MGL expression in THC-exposed offspring. Note that DAGLα levels were also markedly reduced (n = 3 male fetuses/group). Western analysis confirmed reduced protein expression of the 2-AG signaling cassette (C1). Fold data represent the means from n = 3–4 fetal cortices/group, normalized to the vehicle-treated group.
D WIN55,212-2 induced Erk1/2 phosphorylation to a similar degree in vehicle- and THC-treated offspring, suggesting that in vivo THC administration in the present experimental paradigm did not desensitize CB1Rs. Representative Western blots from duplicate experiments are shown.
Data information: Data were expressed as means ± s.e.m.; ***P < 0.001, *P < 0.05. Scale bars, 100 μm (A,B). For a lits of abbreviations, see the Supplementary information.
Source data are available online for this figure.
CB1R−/− fetuses present enlarged corticofugal axon fascicles indistinguishable from AM 251-treated fetuses (Mulder et al, 2008) (Fig 2B–B2). Therefore, we hypothesized that CB1R−/− fetuses must develop a compound phenotype if THC acts via cannabinoid receptors other than CB1R. We tested this by quantitative morphometry of CB1R−/− and wild-type littermate brains (from heterozygous crosses). THC failed to modify the corticofugal axon phenotype of CB1R−/− fetuses [fascicle diameter: 13.73 ± 0.11 (THC) versus 14.17 ± 0.62 μm (vehicle), P = 0.60; Fig 2B–B2]. Our data imply that THC can impair the establishment of the corticofugal tract by disrupting endocannabinoid signaling at CB1Rs. This concept is supported by results of mRNA and protein analysis for CB1Rs, DAGLα (Bisogno et al, 2003) and monoacylglycerol lipase (MGL) (Dinh et al, 2002), metabolizing 2-AG, in THC-exposed fetuses, demonstrating coincidently reduced receptor and ligand availability upon THC administration (Fig 2C and C1). Nevertheless, CB1Rs in THC-exposed brains did not desensitize, as suggested by unchanged Erk1/2 phosphorylation levels upon acute WIN55,212-2 (500 nM) challenge (Fig 2D) relative to control.
Unbiased proteomics identifies disrupted SCG10 as a candidate effector of Δ9-tetrahydrocannabinol-induced circuit wiring defects
We sought to identify the molecular effector(s) whose altered expression in THC-exposed fetuses can impair axonal growth and guidance. We used isobaric tagging for relative and absolute quantification (iTRAQ) to carry out quantitative proteomics (Shirran & Botting, 2010) on cortices from male fetuses at E18.5 (Fig 3A), analysing the resultant peptides by both LC-MALDI/MS/MS and nLC-ESI/MS/MS mass spectrometry to profile THC-sensitive proteins. We identified 35 functionally heterogeneous proteins (out of 837 identified hits), as suggested by their classification according to protein function ontology (Fig 3A1; Supplementary Fig S3A), whose levels changed significantly upon THC treatment.
Figure 3. Δ9-tetrahydrocannabinol (THC) regulates SCG10 expression in the fetal cerebrum.
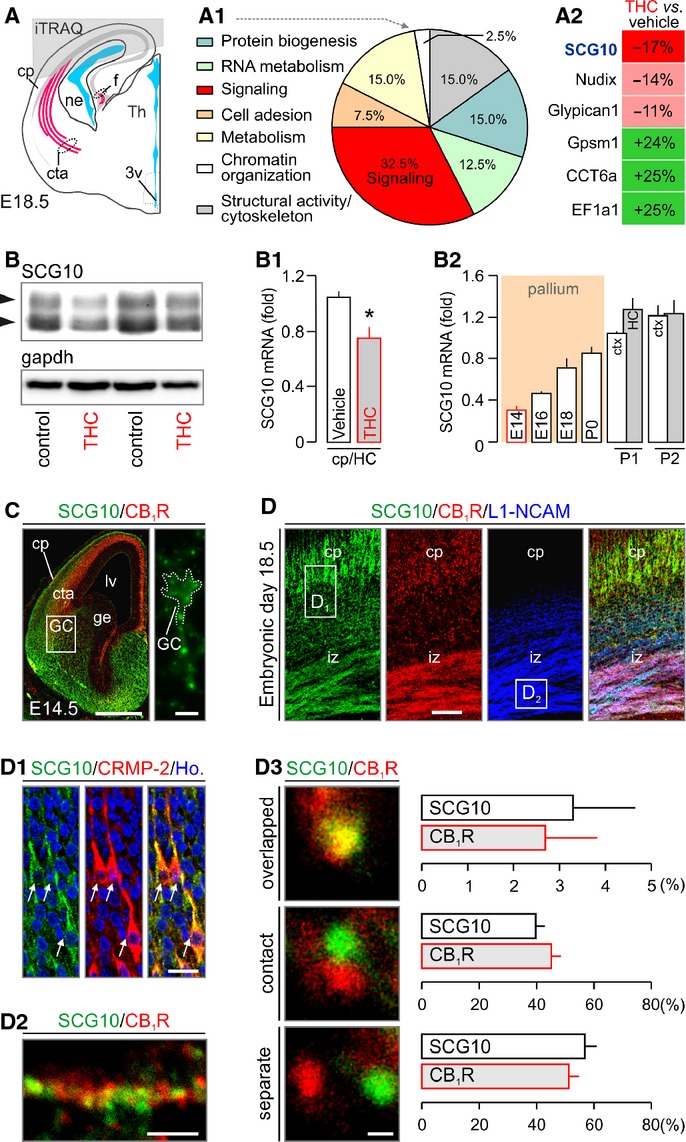
A–A2 Grey overlay indicates the origin of cortical tissues used for iTRAQ/mass spectrometry target discovery. Ontology classification of the 35 protein hits based on primary function assignments is shown in (A1). Topmost modified protein targets are listed in (A2). SCG10 was significantly down-regulated in THC-exposed fetuses (see also Supplementary Fig S3A).
B–B2 Reduced SCG10 protein (B) and mRNA levels (B1) in THC-exposed fetuses (E18.5) were verified by Western blotting and qPCR, respectively. Temporal profile of SCG10 mRNA expression during cortical development is shown in (B2). Grey bars identify the hippocampus (HC). SCG10 protein and mRNA levels were normalized to Gapdh (B, B1) or TATA-binding protein (B2), respectively.
C SCG10 protein is enriched in axonal pathways and accumulates in growth cone-like structures traversing the pallio-subpallial boundary by E14.5 (inset).
D–D3 SCG10 was found co-distributed with CB1Rs in pyramidal-like cells populating the cortical plate (cp) and corticofugal axons. Open rectangles denote the general localization of insets in (D1) and (D2). SCG10 co-existed with collapsing response mediator protein 2 (CRMP-2), a key mediator of appropriately targeted axonal extension (Goshima et al, 1995) in functional antagonism (Fukata et al, 2002) with SCG10 (D1). High-resolution confocal imaging revealed CB1Rs and SCG10 in close proximity in corticofugal axons (D2). Quantitative analysis of the physical proximity relationship of CB1Rs and SCG10 showed approximately 50% co-localization including overlapping and contacting signals (D3) (> 100 proximity relationships per axon in n > 25 axons/group from ≥ 3 animal/group were determined).
Data information: Data were expressed as means ± s.e.m.; *P < 0.05. Scale bars, 250 μm (C), 50 μm (D), 20 μm (D1), 5 μm (D2) or 1 μm (D3). For a list of abbreviations, see the Supplementary information.
Brain-specific SCG10 was found particularly reduced in THC-exposed brains (Fig 3A2), with a simultaneous reduction of its protein (Fig 3B) and mRNA expression (Fig 3B1). SCG10 is an appealing target since its microtubule destabilizing activity (Morii et al, 2006; Manna et al, 2007) chiefly contributes to the maintenance of cytoskeletal instability required for axonal growth (Stein et al, 1988; Grenningloh et al, 2004; Tararuk et al, 2006), and synaptic plasticity at Schaffer collaterals (Peng et al, 2004). SCG10 is regulated by Rho-family GTPases (e.g. Rho6/Rnd1) (Li et al, 2009), which were previously implicated in CB1R-induced growth cone collapse (Berghuis et al, 2007).
SCG10 is expected to be broadly expressed during corticogenesis, coincident with the onset of THC administration, if its loss is to underpin THC-induced modifications of axonal extension. Indeed, we detected SCG10 mRNA by E14 with its expression level gradually increasing until birth (Fig 3B2). SCG10 mapped to long-range forebrain projections (Supplementary Fig S3B–D), was enriched in growth cone-like structures (Fig 3C), and co-distributed with CB1Rs in both the intermediate zone of the cerebral cortex (Fig 3D–D2) and the primordial hippocampus (Supplementary Fig S2C and C1). CB1Rs are expressed during the radial migration and morphogenesis of pyramidal cells in the cerebral cortex (Mulder et al, 2008). Here, we validated SCG10 localization by showing its enrichment in cells with morphologies reminiscent of pyramidal cells in the cortical plate (Westerlund et al, 2011) (Fig 3D and D1). SCG10 is localized in the cytosol (Grenningloh et al, 2004). By using high-resolution laser scanning microscopy we show that approximately 40% of SCG10+ and CB1R+ puncta are closely associated, with approximately 3% directly overlapping in corticofugal axons (Fig 3D2 and D3). This finding establishes that SCG10 is proximal to CB1Rs in corticofugal axons, and could be a downstream target of this GPCR in particular neurite domains. These data, together with retained cerebral SCG10 mRNA and protein expression postnatally in the brain of THC-exposed fetuses (Supplementary Fig S1G and G1), highlight SCG10 as a developmentally regulated candidate protein whose loss can confer THC effects on neuronal morphology.
Maternal cannabis smoking reduces SCG10 mRNA expression in the fetal human hippocampus
Experimental models in mice might carry evolutionary bias, curtailing translational significance by being of limited relevance to human nervous system development. Therefore, we used in situ hybridization to first show the growth-associated increase of SCG10 mRNA expression, irrespective of maternal cannabis use, in the fetal human hippocampus (F = 8.579, P = 0.008; Supplementary Fig S4A and B). Next, we assessed the distribution and density of SCG10 mRNA in the primordial hippocampus and parahippocampal gyrus of electively aborted second trimester human fetuses exposed prenatally to cannabis [n = 12; confirmed by routine meconium toxicology (Hurd et al, 2005)] versus age-matched controls (n = 12). We find highest SCG10 mRNA expression in the CA1-CA3 hippocampal subfields with moderate mRNA hybridization signal detected in the dentate gyrus in normal fetuses (Fig 4A). In contrast, SCG10 mRNA expression was significantly reduced in cannabis-exposed subjects (4.32 ± 0.31 [cannabis] versus 5.33 ± 0.28 ln(dpm/mg tissue) [control], P < 0.001; Fig 4A1). Fetal growth retardation is a critical consequence of maternal cannabis smoking (Hurd et al, 2005). Therefore, we used multivariate analysis to control for covariates (fetal foot length and body weight). Both fetal foot length (F = 17.725, P < 0.001; Fig 4B) and fetal body weight (F = 11.566, P = 0.002; Supplementary Fig S4B) exhibited significant correlation with hippocampal SCG10 mRNA expression. Nevertheless, even with the consideration of these covariates in the statistical model there remained a significant difference in SCG10 mRNA expression levels between cannabis-exposed and control subjects (F = 14.669, P = 0.012). Subsequently, we asked whether SCG10 protein content is also reduced in cannabis-exposed fetuses. After successfully extracting proteins from tissue samples of the above subject cohort, we found significantly reduced SCG10 protein levels in the fetal hippocampus upon maternal cannabis smoking [0.27 ± 0.06 (cannabis, n = 11) versus 0.50 ± 0.05 (control, n = 14), integrated and normalized density (arbitrary units), P < 0.01; Fig 4C). Inclusion of fetal foot length in our multivariate model did not change group significance (F1,23 = 8.33, P < 0.01; Fig 4C1). Next, non-parametric correlation analysis was carried out to reveal a close positive relationship between SCG10 mRNA and protein levels (Spearman's ρ = 0.52, P < 0.02), lending further support to the cannabis-induced developmental deregulation of SCG10 expression in the fetal human cerebrum. Cumulatively, our findings in experimental models and human fetal brains identify SCG10 as a bona fide target of THC. Moreover, our data are compatible with the hypothesis that SCG10 expression in the developing human cerebrum coincides with the formation of intra- and extracortical axonal trajectories during weeks 20–29 (Kostovic & Judas, 2010), and is mandatory to maintain dynamic microtubule instability required for axonal growth (Grenningloh et al, 2004).
Figure 4. Maternal cannabis use during pregnancy reduces SCG10 in human fetal cerebrum.
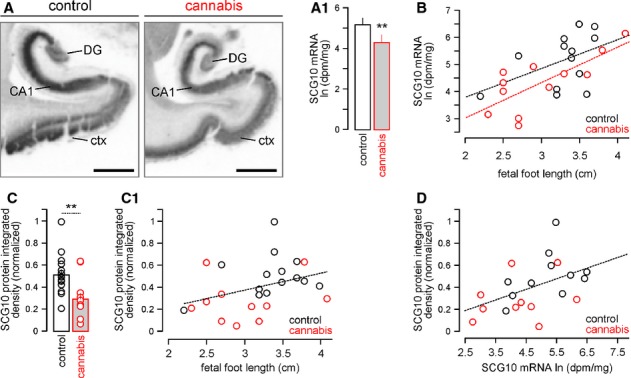
A, A1 In situ hybridization showing reduced mRNA levels in the cerebral cortex (ctx), CA1 subfield of the hippocampus and dentate gyrus (DG) of second trimester fetuses exposed in utero to cannabis. Quantitative data on SCG10 mRNA expression from the CA1 subfield is shown in (A1) (n = 12 fetuses/group).
B Scatter plot of SCG10 mRNA correlated with fetal foot length as developmental covariate.
C, C1 SCG10 protein levels were significantly reduced in human fetal cortices exposed to cannabis in utero. Smaller fetal foot length, a measure of general growth retardation, associated with reduced SCG10 levels in cannabis-exposed fetuses (C1).
D SCG10 mRNA and protein levels positively correlated, reinforcing data from mouse models (Fig 3B and B1).
Data information: Data in (A1) and (C) were expressed as means ± s.d.; dashed lines are subgroup-related regression plots. **P < 0.01. Scale bars, 2 mm.
Δ9-Tetrahydrocannabinol induces rapid axonal SCG10 degradation
The dynamic instability of microtubules (i.e. the innate ability of microtubules to abruptly switch between states of growth and shortening) (Grenningloh et al, 2004; Manna et al, 2007) and the integrity of the microtubule network (Shin et al, 2012) in developing neurons are paramount for the growing axon to maintain forward growth and execute steering decisions. SCG10 is a neuron-specific member of the stathmin family, and triggers microtubule disassembly by binding tubulin dimers in a ternary complex, and by promoting minus end disassembly (Manna et al, 2007). SCG10 is increasingly recognized as an axonal substrate of c-Jun N-terminal kinase (JNK) (Westerlund et al, 2011; Shin et al, 2012). Phosphorylation by JNK1, the active brain-specific JNK isoform (Bjorkblom et al, 2005; Westerlund et al, 2011), at Ser62 and Ser73 negatively regulates SCG10 activity (Grenningloh et al, 2004; Tararuk et al, 2006), and promotes its proteasomal degradation in mechanically-injured axons (Shin et al, 2012).
The maintenance of microtubule instability in elongating axons is a prerequisite of growth advance (Grenningloh et al, 2004). The THC-induced loss of SCG10 in vivo is mechanistically appealing since it could limit the rate of microtubule reorganization, introduce errors to axonal morphology and slow neurite outgrowth. Therefore, we hypothesized that THC-induced CB1R activation can recruit JNK (Rueda et al, 2000) [or extracellular signal-regulated kinase 1/2 (Erk1/2), (Derkinderen et al, 2003; Berghuis et al, 2007)] to phosphorylate and direct SCG10 to proteasomal degradation (Fig 5A). Consistent with these expectations, THC induced rapid (10 min) JNK and Erk1/2 phosphorylation [JNK: 1.4 ± 0.1 fold (THC), Erk1/2: 1.3 ± 0.1 (THC) fold relative to vehicle] in cultured cortical neurons (Fig 5B). JNK/Erk1/2 phosphorylation coincided with reduced SCG10 levels upon THC (10 μM) exposure (Fig 5C and C1). The THC-induced decrease of SCG10 (10 min: 73.8 ± 4.4%, 30 min: 77.2 ± 3.8% of control) was transient (Fig 5C1), likely reflecting the time course of THC-induced CB1R desensitization (Hsieh et al, 1999). In acute in vitro experiments, WIN55,212-2 (CB1R agonist) reproduced (84.6 ± 7.6%), while AM 251 (CB1R antagonist) blocked (103.5 ± 11.2%) the THC-induced loss of SCG10, confirming CB1R involvement. SP600125 (5 μM, JNK inhibitor), but not U0126 (10 μM, Erk1/2 inhibitor (Tararuk et al, 2006); Supplementary Fig S5A), prevented the rapid loss of SCG10 in cultured neurons exposed to THC [SP600125: 105.1 ± 13.1% of control, P < 0.05 versus THC (10 min); U0126: 89.5 ± 2.5% of control, P > 0.1 versus THC (10 min)]. Notably, lactacystin, which irreversibly inactivates the 26S proteasome (Keimpema et al, 2010), rescued SCG10, and stabilized both its non-phosphorylated and phosphorylated isoforms (Fig 5C and C1). These data are consistent with the molecular framework involving JNK recruitment to agonist-activated CB1Rs to promote SCG10 degradation. The accumulation of acetylated tubulin, a marker of long-lived, stable microtubules (Maruta et al, 1986), in THC-induced neurons (Fig 5C2; Supplementary Fig S5B and B1) supports that SCG10 degradation is a candidate mechanism for THC to impair axonal growth.
Figure 5. Δ9-tetrahydrocannabinol (THC)-induced phosphorylation drives SCG10 degradation.

A Schema of presumed signaling events and interactions. Grey color indicates the site of action for the various antagonists used in pharmacological experiments.
B THC stimulation (10 min) of cultured cortical neurons induced coordinated JNK and Erk1/2 phosphorylation. Fold changes were normalized to total (non-phosphorylated) JNK or ERK. Gapdh served as a loading control.
C–C2 THC and WIN55,212-2-induced SCG10 degradation is CB1R- and JNK-dependent, as shown in rescue experiments with co-applied AM 251 and SP600125. Moreover, lactacystin, an irreversible inhibitor of the 26S proteasome (Keimpema et al, 2010), prevented SCG10 loss, confirming its rapid proteasomal degradation downstream from CB1Rs. Increased tubulin acetylation in the same experiment suggests reduced SCG10 destabilizing activity, since acetylated tubulin accumulates in long-lived stable microtubules (C2) (Maruta et al, 1986).
D Time-resolved phosphoprotein profiling shows coincident Erk1/2, JNK and SCG10 phosphorylation after 10 min of THC exposure. Increased phospho-SCG10 levels persist up to 1 h after THC stimulation. Lactacystin (Keimpema et al, 2010) prevented phospho-SCG10 degradation. Note that CB1R phosphorylation is delayed until 3 h post-stimulation. Data were normalized to control.
E, E1 THC exposure (10 min) reduced SCG10 in neurite shafts. Quantitative fluorescence mapping of SCG10 in neurites (n = 35/condition). Representative images are shown in (E1).
F THC exposure for 24 h diminished neurite outgrowth (n ≥ 25 neurons/condition). Dynamic time-course analysis of neurite outgrowth using an IncuCyte automated imaging system (F1). Blue box indicates the period of THC application. Note the rapid growth arrest after introduction of the drug.
Data information: Data were expressed as means ± s.e.m. except for (E), which depicts the population mean. **P < 0.01 (including orange shading in E), *P < 0.05. Scale bar, 10 μm (E1).
Source data are available online for this figure.
Our data suggest that SCG10 undergoes JNK-dependent degradation. JNK can directly phosphorylate SCG10 (Tararuk et al, 2006) and target it for degradation (Shin et al, 2012). Alternatively, JNK effects may be more indirect. We addressed this question by exploiting the decreased gel mobility (increased molecular weight) of phosphorylated SCG10 (Shin et al, 2012). We argued that JNK inhibition should lead to the accumulation of non-phosphorylated SCG10. Conversely, proteasome inhibition should preserve phosphorylated SCG10. Indeed, SP600125 promoted the preferential accumulation of lower molecular weight, predominantly non-phosphorylated SCG10 (Shin et al, 2012) (Fig 5C). Moreover, by selective isolation of phosphoproteins we show that higher-molecular-weight (polyphosphorylated) SCG10 outweighs the relative abundance of mono- or non-phosphorylated SCG10 species in lactacystin-exposed neurons in a time-dependent fashion (Fig 5D). CB1R phosphorylation, marking GPCR internalization (Daigle et al, 2008), was only detected > 1 h, suggesting that SCG10 degradation is an early event downstream from CB1R activation (Fig 5D). The different time-course and extent of CB1R phosphorylation relative to elements of its signal transduction machinery served as negative control in phosphoprotein isolation. Cumulatively, phosphoprotein profiling demonstrates that phosphorylated SCG10 accumulates in THC-treated neurons upon proteasomal inhibition.
SCG10 undergoes fast anterograde axonal transport (Shin et al, 2012) and accumulates in the central domain of advancing growth cones (Grenningloh et al, 2004), where microtubules are highly dynamic. Therefore, we quantitatively sampled SCG10 distribution in THC-exposed neurites and growth cones. Consistent with previous findings (Grenningloh et al, 2004), SCG10 exhibits quasi-random distribution along the entire neurite under control conditions (Fig 5E). Both acute (10 μM, 10 min, Fig 5E and E1) and prolonged (2 μM, 24 h, Supplementary Fig S6A and A1) THC stimulation induced SCG10 loss from neurite shafts, with residual SCG10 localized to neurite branch forks particularly after 24 h THC treatment (Supplementary Fig S6A and A1). The above data on the molecular regulation of SCG10 availability and function is compatible with reduced neurite outgrowth of THC-treated cortical neurons [113.7 ± 9.5 μm (THC) versus 141.0 ± 10.2 μm (control), P < 0.05; Fig 5F and F1], and demonstrates the THC-induced reconfiguration of a key maintenance pathway for microtubule instability (Shin et al, 2012).
The above in vitro data suggest a central role for JNK1 in regulating CB1R activity-dependent SCG10 degradation. However, these data are limited in addressing THC-induced SCG10 degradation in the corticofugal projection system. Therefore, we prepared organotypic slices from E14.5 mouse forebrains to preserve cellular and axonal arrangements along the corticothalamic projection (Fig 6A) to test acute THC (10 μM, 30 min) on axonal SCG10 availability. We show that both SCG10 fluorescence intensity [56.17 ± 4.97% of vehicle-treated controls; n = 25 (THC) versus n = 29 (vehicle); P < 0.001] and spatial extent [“area coverage”: 75.62 ± 6.09% of vehicle-treated controls; n = 25 (THC) versus n = 29 (vehicle); P < 0.01] were significantly reduced upon THC application (Fig 6B,B1 and B3). SP600125 significantly attenuated the acute loss of SCG10 in corticofugal axons [fluorescence intensity: 81.75 ± 5.23% of control (THC+SP600125); area coverage: 97.76 ± 3.95% of control (THC+SP600125)]. L1-NCAM, which remained unaffected by either THC or SP600125 treatment (Fig 6B2 and B4), was used to reliably discern corticofugal axons and to normalize SCG10 fluorescence intensity.
Figure 6. THC-induced SCG10 degradation is mediated by JNK.

A Overview of organotypic slices prepared from E14.5 mouse embryos. Note the retained anatomical organization, as well as CB1R and SCG10 expression patterns of the cerebral cortex and thalamus. L1-NCAM was used as an axonal marker throughout.
B THC acutely (30 min) reduced SCG10 content in L1-NCAM+ corticofugal axons. JNK inhibition by SP600125 prevented the THC-induced loss of SCG10. Quantitative morphometry of SCG10 immunofluorescence signal intensity as a factor of L1-NCAM expression (B1), with the latter marker unchanged throughout (B2). We verified the above SCG10 loss by determining the extent of cortical surface occupied by SCG10+ puncta (B3), which reinforced the SP600125-induced rescue of SCG10 in THC-exposed cortices. SP600125 effects affected SCG10 but not L1-NCAM distribution (B4), suggesting specificity.
C, C1 JNK1−/− mice presented increased corticofugal axon fasciculation relative to wild-type littermates.
D, D1 The enlarged corticofugal fascicles in JNK1−/− mice harbored increased SCG10 immunoreactivity, supporting an upstream regulatory role for JNK1 in maintaining axonal SCG10 content.
Data information: Open and solid arrowheads denote the lack or elevated levels of SCG10 in L1-NCAM axons, respectively. Arrows in (B) point to enlarged axonal endings at cut surfaces, accumulating SCG10 proximally. Data were expressed as means ± s.e.m.; **P < 0.01, n.s.; non-significant. Scale bars, 500 μm (A), 50 μm (B,C,D,D1). For a list of abbreviations, see the Supplementary information.
Next, we determined SCG10 content in corticofugal axons of newborn JNK1−/− mice (Dong et al, 1998) to provide genetic evidence for the JNK1-mediated control of SCG10 availability. JNK1−/− mice presented altered corticofugal axon fasciculation (Fig 6C and C1). Notably, axonal SCG10 content was robustly increased in JNK1−/− mice relative to wild-type controls (Fig 6D and D1), reinforcing an upstream role for JNK1 in regulating SCG10 stability.
If the CB1R-JNK-SCG10 pathway alone is sufficient to account for the THC-induced cytoarchitectural modifications of cortical neurons then overexpression of a functionally inactive, pseudophosphorylated SCG10 mutant (Tararuk et al, 2006; Westerlund et al, 2011) (i.e. aspartate substitution of Ser62 and Ser73 (SCG10-DD)) would phenocopy THC effects. Consistent with this hypothesis, SCG10-DD overexpression appeared to outcompete endogenous SCG10 in neurites (Fig 7A), reduced neurite outgrowth and occluded THC-induced modifications to axonal morphology (84.0 ± 8.7 μm [THC+EGFP-C1] versus 92.6 ± 9.6 μm [THC+SCG10-DD], P > 0.5; Fig 7A and A1). PC12 cells transfected with SCG10-DD for 24-72 h were used to show that SCG10-DD is overexpressed at the expense of endogenous (wild-type) SCG10. By Western blotting PC12 cell lysates, we separated GFP-tagged SCG10-DD from unlabeled endogenous SCG10 and found the progressive reduction of the latter SCG10 form as a factor of time (Fig 7A2). In addition, siRNA-mediated knockdown of SCG10 in cultured cortical neurons reduced neurite outgrowth (Fig 7B–B2) to an extent similar to that seen upon SCG10-DD overexpression. Overall, these data highlight the central role of SCG10 degradation in axonal growth defects imposed by prenatal THC exposure.
Figure 7. Δ9-tetrahydrocannabinol (THC) promotes ectopic filopodiagenesis.

A,A2 Transient transfection of cortical neurons with a phospho-mimetic EGFP-SCG10S62D/S73D mutant (SCG10-DD) (Westerlund et al, 2011) led to a decrease in the primary neurite length, an effect equivalent to that of THC. SCG10-DD overexpression, which largely outcompeted endogenous SCG10 (insets), rendered THC ineffective to modify neurite elongation (n > 15 neurons/condition). Western analysis demonstrated a progressive increase in SCG10-DD protein expression in PC12 cells upon transient transfection(A2). Probing by an anti-GFP antibody differentiated endogenous SCG10 from overexpressed SCG10-DD. Note that SCG10-DD overexpression significantly reduced the endogenous SCG10 content (wild-type (WT) targets around ˜20 kDa). Membranes were stained with amido-back to control for equal protein loading.
B–B2 siRNA knockdown of SCG10 expression phenocopied and blunted the effects of THC on neurite outgrowth in cultured cortical neurons (n > 20 neurons/condition).
C SCG10 levels decreased in cortical growth cone particles exposed to THC for 10 min. SCG10 isoforms were numbered (Shin et al, 2012). Gapdh served as a loading control.
D, D1 In cultured cortical neurons, THC (10 min) allowed acetylated tubulin accumulation (arrowheads) and the formation of superfluous lamellipodium-like F-actin-rich cytoskeletal projections. Bar histograms of quantitative subcellular morphometry are shown in (D2). THC facilitated the morphological differentiation of growth cones (D3) including filopodiogenesis (n > 10 observations/condition; D4) without affecting the size of individual growth cones (D5). Growth cone morphology was classified as described by (Morii et al, 2006).
E, E1 Besides affecting the growth cone, THC acutely increased the formation and density of F-actin+ axial protrusions, likely filopodia, from distal neurite shafts.
Data information: Data were expressed as means ± s.e.m. **P < 0.01, *P < 0.05; n.s. = non-significant. Scale bars, 20 μm (A), 10 μm (B, D,D1,E)
SCG10 degradation in growth cones advances morphological growth cone differentiation
THC treatment for 24 h reduced SCG10 and to a lesser extent CB1R immunoreactivity (Berghuis et al, 2007) in growth cones (Supplementary Fig S6B–B2). If the loss of SCG10 in growth cones relates to diminished axonal growth then SCG10 degradation can be expected to progress in situ in the growth cone. We sought to address this hypothesis by isolating growth cone particles (GCPs) from late-gestational mouse cortices (Berghuis et al, 2007) and exposing intact GCPs to THC (10 min) in vitro. The selective loss of higher-molecular-weight SCG10 isoforms demonstrated that local SCG10 degradation persists in growth cones (Fig 7C). Similarly, acute THC treatment of cortical neurons diminished SCG10 in the central growth cone domain (Fig 7D and D1), which coincided with acetylated tubulin, a marker of stabilized and aged microtubules (Maruta et al, 1986), and F-actin accumulation (Fig 7D2). Overt cytoskeletal stability increased the appearance of growth cone with splayed out microtubules (Fig 7D3), and increased density of exploratory filopodia [11.9 ± 1.2 μm (THC) versus 7.3 ± 1.1 μm (control), P < 0.01; Fig 7D4], leaving the growth cone surface area unaffected (Fig 7D5). Notably, THC also increased the formation of F-actin-rich filopodia in the distal motile axon segment (Fig 7E and E1) suggesting that THC can enhance the formation of ectopic axon collaterals. These data demonstrate that THC modulation of CB1R activity in motile growth cones can inhibit forward motility and steering decisions.
CB1Rs are predominantly presynaptic in the adult brain (Kano et al, 2009). Likewise, cell-surface CB1Rs are only found on the axons of developing neurons (McDonald et al, 2007). The domain-specific cell-surface targeting of CB1Rs allowed us to assay whether the molecular pathway we uncovered is specific to elongating axons. To this end, we first determined THC-induced changes to SCG10 and acetylated tubulin contents in secondary, short neurites lacking CB1Rs, putative primordial dendrites (Supplementary Fig S6C). THC induced tubulin acetylation in both the growth cones and neurite stems (P < 0.05 versus vehicle-treated control; Supplementary Fig S6C1 and C2). In contrast, SCG10 levels remained unchanged (Supplementary Fig S6C3 and C4). Next, we co-localized acetylated tubulin and post-synaptic density protein 95 (PSD95), a marker of excitatory post-synapses (Tomasoni et al, 2013), to dissect THC effects at established synapses. THC did not induce tubulin acetylation in either PSD95+ post-synapses or apposing pre-synaptic terminals (Supplementary Fig S6D–D4). In sum, these experiments establish the axonal specificity of CB1R-dependent SCG10 degradation, and specifically implicate this mechanism in the modulation of axonal growth and guidance.
Discussion
Our results, for the first time, define a specific molecular target for THC in the developing central nervous system, whose modifications can directly and permanently impair the wiring diagram of neuronal networks during corticogenesis. These finding have direct and increasing human relevance since selective cultivation of Cannabis subspecies recently significantly altered their phytocannabinoid contents, with particularly pronounced increases in THC content (Pitts et al, 1992; Pijlman et al, 2005). We first mount compelling experimental support to the hypothesis that, when available for prolonged periods in vivo, THC disrupts endocannabinoid signaling at their cognate CB1R. Our mRNA and protein profiling of molecular components controlling 2-AG metabolism suggest that THC not only can act as a “functional antagonist” (i.e. displacement of endocannabinoid binding to the CB1R), but can disrupt 2-AG signaling by reducing both CB1R and DAGLα expression during cortical development. We attribute the discrepancy between MGL mRNA and protein expression levels to MGL being prone to posttranslational modifications facilitating its proteasomal degradation (Keimpema et al, 2013). Next, we identify SCG10 as a novel signaling node of morphogenic CB1R signaling since (i) SCG10 is ideally poised to link cell-surface CB1R activation and pleiotropic downstream mitogen-activated protein kinase (particularly JNK) activation (Rueda et al, 2000; Berghuis et al, 2007; Keimpema et al, 2011) to cytoskeletal instability (Keimpema et al, 2011), and (ii) the rapid degradation of SCG10 can allow ectopic growth with ensuing modifications to axodendritic morphology in vitro recapitulating corticofugal growth defects in vivo. This mechanism appears to be restricted to the THC-induced reorganization of axonal morphology, which is compatible with the sole localization of CB1Rs to growth cones and presynapses in the developing (Keimpema et al, 2010) and adult nervous systems (Kano et al, 2009), respectively. Nevertheless, we recognize that CB1R activation might engage multiple co-existent signaling pathways (e.g. RhoGTPases (Berghuis et al, 2007; Nithipatikom et al, 2012)) to couple cytoskeletal reorganization to the orchestration of axonal morphology.
THC exposure altered the levels of 35 proteins in the fetal cerebrum. Previously, a total of 49 differentially-regulated genes were identified by cDNA microarrays in nervous tissues from adult rats treated with THC for 24 h, 7 or 21 days (Kittler et al, 2000), including molecular constituents of endocannabinoid and lipid biosynthesis, and signal transduction machineries. Conspicuously, neural cell adhesion molecule and myelin basic protein, implicated in axonal growth (Yuan et al, 2003; Keimpema et al, 2011) and myelination by oligodendrocytes were also altered. Here, our protein profiling by mass spectrometry combined with cell biology provides a new perspective on developmentally-regulated candidates, and suggests that the reorganization of synaptic structure and plasticity is an inherent feature of THC action (Keimpema et al, 2011). This is further supported by the observation that SCG10 expression prevails during adulthood and aging in neuronal clusters with long-lasting synaptic plasticity (Peng et al, 2004). In using iTRAQ for quantitative candidate discovery we reasoned that mRNA may not be translated into biologically active protein. Moreover, we conceptually approached the 17% decrease in cortical SCG10 amount as an indication that a subset of cortical neurons selectively, rather than all of them indiscriminately, down-regulated SCG10 protein expression. Indeed, SCG10 is primarily expressed in a contingent of neurons in the fetal cortical plate that adopted pyramidal cell-like positioning and axodendritic morphology. Linking SCG10 deregulation to pyramidal cell development is not unexpected since genetic deletion of CB1Rs in pyramidal cells destined to superficial cortical layers leads to axon fasciculation and targeting errors (Mulder et al, 2008). Moreover, since CB1Rs are not expressed in cortical proliferative zones (Goncalves et al, 2008; Mulder et al, 2008), CB1R-mediated adverse THC effects will likely impact cortical wiring rather than neuronal diversification in the fetal cerebrum. Nevertheless, we find similarities in the molecular identity and functions of THC's differentially regulated targets in developmental and adult settings (Kittler et al, 2000), such as proteins implicated in secondary metabolism, protein folding and signal transduction (Supplementary Fig S3A). Our data in conjunction with large-scale analysis from adult brain (Kittler et al, 2000) reinforce that THC's mode of action involves the disrupted development and/or postnatal maintenance of synapses critical for highly ordered executive and cognitive functions.
Microtubules in developing neurons mediate growth cone steering and forward movement (Grenningloh et al, 2004; Morii et al, 2006; Tararuk et al, 2006; Manna et al, 2007). The dynamic instability of microtubules, and the integrity of the microtubule network determines the rate of axonal transport (Grenningloh et al, 2004; Shin et al, 2012). Within growth cones, microtubule plus end switching between growth and shortening states by the process of dynamic instability underpins receptor-mediated reorganization (Manna et al, 2007). The stathmin family of proteins share a C-terminal stathmin domain to bind α/β-tubulin dimers to form ternary T2S complexes (Manna et al, 2007). While stathmin is ubiquitously expressed, SCG10, SCLIP, RB3, RB3′ and RB3″ are neuron-specific proteins (Grenningloh et al, 2004; Tararuk et al, 2006). These predominantly cytosolic proteins can be differentially modulated to meet the cells' developmental potential and morphogenic demands. Notably, their complementary organ system distribution could constitute a first-order effector system diversifying endocannabinoid and cannabis effects downstream from CB1Rs. In fact, the signaling cascade we describe is, to the best of our knowledge, the first signaling axis directly linking a GPCR to SCG10 as molecular effector.
The considerably different subcellular distribution of stathmin and SCG10 (Grenningloh et al, 2004) could be permissive for the domain-specific modulation of CB1R-mediated signals on the neuronal cytoskeleton. Such a molecular platform is particularly relevant to brain development since advancing growth cones are a primary site for CB1R localization (Berghuis et al, 2007; Keimpema et al, 2010, 2011) and signaling (Berghuis et al, 2007) to promote forward axonal movement. SCG10, unlike stathmin, contains an N-terminal membrane-associating domain (Manna et al, 2007), which mediates its fast anterograde transport [at a velocity of ∼1 μm/s (Shin et al, 2012)] to growing tips of axons and dendrites. SCG10 accumulates in the central domain of growth cones where microtubules are highly dynamic (Grenningloh et al, 2004). Here, we define an integrated signaling axis: (i) triggered by THC, (ii) transduced by CB1R, (iii) via JNKs, and (iv) executed by SCG10. Biochemical indices of SCG10 degradation are supported by the loss of SCG10 immunosignal from THC-stimulated neurites and growth cones in vivo and in vitro, and stabilization of acetylated tubulin in the central growth cone domain. Since the frequency of posttranslational modification of tubulin directly correlates with its “age” (i.e. stability) (Maruta et al, 1986), and increased plus end activity in microtubules underpins ectopic filopodiagenesis (Grenningloh et al, 2004), our data identify CB1R activity as a driving force of SCG10 availability. Accordingly, depletion of SCG10 from the active growth cone domain increased the proportion of looped/spread growth cone morphologies, suggestive of their altered rate of motility, and reduced neurite outgrowth upon lessening of microtubule dynamics. Retention of residual SCG10 at branch forks can confer minimal essential cytoskeletal dynamics required to maintain cellular integrity and axonal arborization.
SCG10 exists in multiple phosphoisoforms in the neonatal rat brain (Tararuk et al, 2006; Westerlund et al, 2011). SCG10 activity is strongly regulated by phosphorylation of its four serine residues involving protein kinase A (Ser50/97), mitogen-activated protein kinases (Ser62/73) or CDK5/p25 (Ser73) (Grenningloh et al, 2004). JNKs phosphorylate SCG10 at residues Ser62/73 (Westerlund et al, 2011), likely generating mono- and polyphosphorylated SCG10 (Shin et al, 2012) to faciliate its degradation. In general, phosphorylation inhibits the microtubule destabilizing activity of SCG10 suggesting that this protein may link extracellular signals to the rearrangement of the neuronal cytoskeleton. Site-directed mutagenesis in which serines are replaced by phosphorylation-mimicking aspartate (D) residues (Tararuk et al, 2006; Westerlund et al, 2011) showed that the relative importance of each site to SCG10 function might vary, but that its overall activity decreases with increased phosphorylation (Grenningloh et al, 2004). SCG10-S62D/S73D is a functionally inactive, pseudophosphorylated mutant, mimicking JNK-phosphorylated SCG10 (Westerlund et al, 2011), whose dominant-negative action can consequently stabilize microtubules. Here, we find that SCG10-DD overexpression limits neurite outgrowth in competition with endogenous (wild-type) SCG10, and occludes THC effects. Our findings suggest that SCG10 is a key substrate whose phosphorylation downstream from agonist-activated CB1Rs reduces an essential level of cytoskeletal instability required to maintain the rate of neurite outgrowth. Overall, SCG10 loss is not the trigger for developmental axonal growth errors, but rather is a permissive factor that allows growth-associated protein stability pathways to initiate ectopic growth.
JNKs are essential regulators of morphogenesis during early development and, besides regulating transcription, are functional in the cytoplasm where SCG10 localizes (Tararuk et al, 2006). JNK is constitutively phosphorylated in the axon, with the kinetics of its spatiotemporal dephosphorylation considered rate limiting. Recently, MAP kinase phosphatase 1 was shown to mediate JNK dephosphorylation (Jeanneteau et al, 2010), allowing prolonged stathmin dephosphorylation, stabilization, and excess microtubule stability. Since cannabinoid receptors, particularly CB2Rs (Romero-Sandoval et al, 2009), can activate MAP kinase phosphatase 1 in a cell type-specific fashion, the balance of JNK phosphorylation/dephosphorylation will ultimately determine SCG10 availability and actions. JNK1 is the predominant physiologically active form of JNK in the brain (Tararuk et al, 2006) with JNK1−/− mice displaying severely disrupted commissure formation due to the loss of axonal microtubule integrity (Fig 6D and D1) (Chang et al, 2003; Westerlund et al, 2011). SCG10 is the preferred neuron-specific JNK1 substrate at low concentrations (Tararuk et al, 2006) because, unlike stathmin, its N-terminal extension allows for efficient JNK1 binding. Notably, SCG10 phosphorylation at Ser62/Ser73 is significantly depleted in JNK1−/− cortex (Tararuk et al, 2006), and disrupts cell cycle exit and radial migration of cortical neurons (Westerlund et al, 2011). Considering that CB1Rs affect long-distance neuronal migration, the present and previous (Tararuk et al, 2006; Berghuis et al, 2007; Keimpema et al, 2011; Westerlund et al, 2011) data cumulatively infer a signaling framework, which is sufficient to account for migration and connectivity deficits in CB1R−/− mice or upon exposure to cannabinomimetics or phytocannabinoids.
Recreational cannabis use is particularly prevalent in the young adult age group, including women of child-bearing age (Substance Abuse & Mental Health Service Administration, 2010). Although recent population statistics revealed significant fluctuations due to geoeconomic variables, > 10% of pregnancies in US and Europe are associated with maternal cannabis exposure (Substance Abuse & Mental Health Service Administration, 2010). Cannabis spp. reportedly contain > 400 bioactive components, with THC being its primary psychoactive constituent. During the past decades, selective agriculture of Cannabis spp. resulted in increased THC content at the expense of cannabidiol (Pitts et al, 1992; Pijlman et al, 2005). In the context of the present study, this is of particular concern since we predict that higher THC concentrations will be, upon efficient cross-placental transfer (Grotenhermen, 2003), increasingly detrimental for fetal development and postnatal health. Therefore, irrespective of the legal status of cannabis, caution must be exercised to hinder fetal cannabis exposure due to its unequivical impact on the establishment of synaptic connectivity in neuronal networks underpinning memory encoding, cognition and executive skills. Moreover, abnormal synaptic organization, even if remaining latent for long periods, might be prone to “circuit failure” if provoked. A “double hit” scenario of cortical failure when a labile network advances into a runaway cascade upon a secondary insult therefore might account for the increased incidence of schizophrenia, depression and addiction in offspring prenatally exposed to cannabis (Substance Abuse & Mental Health Service Administration, 2010; Keimpema et al, 2011).
Materials and Methods
Animals
Tissues from fetal (E14.5–E18.5), neonatal (P10) and adult (P120) wild-type and CB1R−/− mice were processed as described (Berghuis et al, 2007; Keimpema et al, 2010). Wild-type and CB1R−/− pregnant mice on C57Bl/6 background (Monory et al, 2006) were injected intraperitoneally with THC (3 mg/kg, THC Pharm) (Mato et al, 2004; Campolongo et al, 2007), WIN55,212-2 (5 mg/kg, Tocris) or AM 251 (5 mg/kg, Tocris) from E5.5 to E17.5 daily. Embryos were harvested on E18.5, their brains immersion fixed in 4% paraformaldehyde in 0.1 M phosphate buffer (PFA-PB), cryosectioned (16 μm) and glass-mounted coronal sections prepared for histochemistry (Keimpema et al, 2010). Postnatal offspring were perfusion-fixed (PFA-PB), their brains cryosectioned (40 μm) in the coronal plane, and processed as free-floating sections (Mulder et al, 2011). Given their preferential sensitivity to cannabis (Spano et al, 2007), male offspring were analyzed. JNK1−/− mice were produced as described (Dong et al, 1998) and bred conventionally (University Hospital of Schleswig-Holstein). We used JNK1−/− mice back-crossed for 10 generations onto a C57Bl/6N background, which served as controls. Where relevant, mouse genotyping was performed by amplifying genomic DNA using primer sets allowing the discrimination of the endogenous locus from targeted alleles. Experimental protocols on live animal conformed to the European Communities Council Directive (86/609/EEC) and were approved by the Home Office of the United Kingdom, as well as the Ministerium für Landwirtschaft und Naturschutz, Kiel, Germany. Efforts were made to minimize the number of animals and their suffering throughout the experiments.
Cortical cultures, transient transfection and siRNA knockdown
Cultured neurons were isolated from E16.5 mouse cortices (Keimpema et al, 2010). Neurons for morphological analysis were plated (25,000/well) onto poly-D-lysine-coated coverslips. On day 2 in vitro (DIV), neurons were stimulated by either 500 nM or 2 μM THC for 24 h. THC at 10 μM concentration was used in acute experiments (10 min). Cells were immersion fixed in PFA-PB for 20 min.
For transient transfection, neurons were seeded at a density of 50,000 cells/well, and transfected with EGFP-tagged SCG10S62D/S73D (SCG10-DD) or pEGFP-C1 (control) using Lipofectamine 2000 transfection agent (Invitrogen, for 6 h) 24 h later. THC (2 μM) stimulation for 24 h was carried out 1 day later. For the biochemical analysis of wild-type versus SCG10-DD contents, PC12 cells (500,000/well) were transfected with the constructs as above, lysed at the time points indicated in Fig 7A2, and probed for SCG10 and GFP. For siRNA knockdown of SCG10, cortical neurons (50,000 cells/well, 24-well format) were grown in DMEM/F12 (1:1) containing B-27 supplement (2%) and L-glutamine (2 mM) for 24 h. Subsequently, cultures were co-transfected with a GFP construct (0.5 μg; pEGFP-C1) used to identify positive transfectants, and either scrambled siRNAs (50 pmol; D-001810-10-05; Thermo Scientific) or a pool of SCG10 siRNAs (50 pmol; L-045412-01-0005; Thermo Scientific) with Lipofectamine 2000 (Invitrogen) for 45 min. Cells were washed once in full growth medium and returned to their original medium in presence of THC (2 μM) or vehicle for 24 h. Cultures were immersion fixed in 4% PFA-PB and processed as described above.
Organotypic slice culture
Organotypic cultures were prepared from C57Bl/6 mice on E14.5. Brains were rapidly dissected and subsequently embedded in 5% low-melt agarose (Promega). Coronal sections (300 μm) were cut on a vibratome (VT1200S, Leica), and transferred onto polytetrafluoroethylene culture membranes (30 mm in diameter; 0.4 μm pore size; Millipore) in 6-well plates containing DMEM supplemented with glutamine (2 mM), fetal bovine serum (10%) and penicillin-streptomycin (1%) in a total volume of 1 ml. After 1 h incubation, the medium was replaced with Neurobasal medium also containing glutamine (2 mM), B27 supplement (2%) and penicillin-streptomycin (1%) and slices allowed to equilibrate overnight. The next day, slices were pre-treated with SP600125 (5 μM) or vehicle for 2 h. Subsequently, THC (10 μM) was applied alone or in combination with SP600125 (5 μM) for an additional 30 min. Afterwards, slices were fixed with 4% PFA-PB and processed routinely for multiple-labeling immunofluorescence histochemistry. High-resolution images of corticofugal axons in E14.5 slices were captured at 20× primary magnification (Fig 6B). Mean intensities of SCG10 and L1-NCAM immunoreactive structures were measured with the ZEN2009 software (Zeiss; Fig 6B1). The area coverage of immunoreactive pixels was calculated by expressing the ratio of the immunofluorescent signal and the total surface area (Fig 6B3 and B4). Gray scale values were accepted between 10 and 245 to reduce unspecific noise.
Quantitative morphometry
Multiple immunofluorescence labeling of fetal and postnatal mouse brains and cultured neurons was performed by applying select cocktails of affinity-purified antibodies (Keimpema et al, 2010; Mulder et al, 2011) (Supplementary Table S1). Particular care was taken to assess the reliability of anti-SCG10 antisera, including simultaneous detection using two antibodies (from different hosts) raised against non-overlapping protein epitopes (Supplementary Fig S3D). Quantitative analysis at P10 was aided by acquiring tiled 12-bit greyscale images at 10× primary magnification on a VSlide slide-scanning microscope (MetaSystems) equipped with a CoolCube 1 camera. Hoechst 33342 nuclear stain was used to delineate cortical laminae. Alternatively, images were acquired on a Zeiss 710LSM confocal laser-scanning microscope using spectral detection tuned for maximal signal separation and digital zoom ranging from 1.5 to 3.0× (Keimpema et al, 2010; Mulder et al, 2011). The co-existence of immunosignals was verified by capturing serial orthogonal z image stacks at 40× primary magnification (up to 3× optical zoom) and accepted if these were present without physical signal separation in ≤1.0-μm optical slices, and overlapped in all three (x, y and z) dimensions within individual cellular domains. Co-localization between CB1Rs and SCG10 in vivo was performed at 63× primary magnification and 5× optical zoom (Zeiss 700LSM). The physical distance between individual CB1R+ and SCG10+ puncta was divided in three groups [overlap, contact (i.e. adjacent signals without overlap) and separate; Fig 3D2 and D3], and expressed as the percentage of the total number of immunoreactive puncta counted per axon.
Morphometric analysis of cultured neurons was aided by the ZEN2009 software and included: (i) filopodia number and density (expressed per 100 μm neurite segment), (ii) neurite branching (n), (iii) length of the primary neurite (μm), (iv) the diameter of neurite bundles (μm), (v) the morphology, surface area and filopodia number of growth cones, and (vi) fluorescence intensity distribution of SCG10 in growth cones and along neurite shafts (Keimpema et al, 2010). The longest process emanating from neuronal somata was analyzed and considered as the prospective axon. Similarly, the transverse diameter of L1-NCAM+ corticofugal axons in fetal wild-type and CB1R−/− brains after THC or vehicle administration was measured with the ZEN2009 software package on calibrated images. Only first-order fascicles of > 3 μm in diameter were analyzed. In P10 or P120 animals, synapse density and distribution were measured on high-resolution graphic images exported into the UTHSCSA ImageTool (version 3.0). CB1R+ “perisomatic baskets”, defined as CB1R+ terminal-like boutons engulfing MAP2+ somata in layers (L)II/III of the neocortex on at least three quadrants (Fig 1B), were counted in ImageJ 1.45s with their density expressed per 104 μm2. The density of terminal profiles (> 5 continuous pixels at 1670 × 945 pixel input resolution) was expressed per 103 μm2.
The intensity of acetylated tubulin in pre- and postsynaptic terminals of cortical cultures (4DIV) was measured by ImageJ 1.45s and expressed as arbitrary units (Supplementary Fig S6D–D3). Postsynaptic terminals were visualized by postsynaptic density protein 95 (PSD95) immunoreactivity and measured if a PSD95− process of another individual neuron (putative pre-synapse) made contact. The brightness or contrast of confocal laser-scanning micrographs was occasionally linearly enhanced. Multi-panel images were assembled in CorelDraw X5.
High-throughput neurite tracking
Primary cortical neurons were isolated and plated as above. After 10 h, cultures were treated with THC (2 μM) or vehicle for an additional 62 h. Drug treatments were performed in quadruplicates, and imaged live in parallel using an IncuCyte Zoom live-cell imaging devise (Essen Bioscience). Time-lapse images were acquired every 2 h. The growth rate of neurites in each well was obtained by measuring the surface area covered by neurites, and expressed as mm/mm2.
Electrophysiology
Transverse coronal slices (300 μm; Leica VT1200S vibratome) of the dorsal hippocampus were prepared in ice-cold solution (90 NaCl, 2.5 KCl, 1.25 Na2HPO4, 0.5 CaCl2, 8 MgSO4, 26 NaHCO3, 20 D-Glucose, 10 HEPES, 3 Na-pyruvate, 5 Na-ascorbate; in mM) on P120 after rapidly dissecting the brains of decapitated animals (Mulder et al, 2011). Experiments on equilibrated slices (> 60 min; pH7.4 under continuous oxygenation), which were continuously superfused (∽10 ml/min) with artificial cerebrospinal fluid (ACSF) and oxygenated at 33–34°C, were performed in a round 1.5-ml recording chamber. Field excitatory postsynaptic potentials (fEPSPs) from the CA1 subfield were recorded upon orthodromic stimulation of Schaffer collaterals using a voltage stimulus isolator (A360LA, World Precision Instruments) connected to a concentric bipolar platinum-iridium stimulating electrode (MX211ES, FHC & Co), positioned in the stratum radiatum at the CA2/CA1 border. The impulse intensity was set to 65% of the current that evoked minimum saturated fEPSPs, and delivered at 0.66 Hz. Recording borosilicate glass (Hilgenberg) capillary electrodes (3–4 MΩ, P-1000, Sutter) filled with ACSF were positioned 500-700 μm away from the stimulating electrode. One was placed in the str. radiatum of CA1 to sample fEPSPs, while another was inserted in the CA1 str. pyramidale to record population spikes (Supplementary Fig S1E). Both recording electrodes were connected to high input-impedance headstages (HEKA EPC-10). After a baseline period of > 10 min, long-term depression of Schaffer collateral-CA1 synaptic responses and extracellular potentials was induced by 900 pulses at 1 Hz (at 100% intensity of the test stimulus) with fEPSPs and population spikes recorded simultaneously for ≥ 40 min. Stimulation artifacts were continuously monitored, and recordings with > 20% change in the fiber volley amplitude after LTD induction were excluded. We recorded the paired-pulse ratio (PPR) with interstimulus intervals of 10, 20, 40, 60, 80, 100, 150 and 200 ms (at 100% intensity of the test stimulus) with recordings made simultaneously in str. radiatum (Fig 1E) and str. pyramidale (Supplementary Fig S1F). The slope of fEPSPs, as well as the amplitude of population spikes was analyzed. N = 5–6 mice/group, n = 2–3 slices per brain were analyzed. Figure 1D2 was generated by binning data on fEPSP suppression during a 15-min period of recordings (indicated by orange background, Fig 1D).
Western blotting
Primary cortical neurons cultured at a density of 106 cells/well (6-well plate format) for 48 h before being exposed to select combinations of: THC (0.5, 2 or 10 μM, THC Pharm), AM 251 (200 nM), lactacystin, (20 μM) (Keimpema et al, 2010), SP600125 (5 μM) (Westerlund et al, 2011), U0126 (10 μM) (Mikaels-Edman et al, 2003) and WIN55-212,2 (500 nM, all from Tocris) for 10, 30, 180 min or 24 h. Cortical neurons and fetal cortices were lysed in a modified radioimmunoprecipitation assay buffer (Mulder et al, 2011) and analyzed by SDS-PAGE. CB1R-induced Erk1/2 phosphorylation was probed in cortical lysates of vehicle and THC-exposed fetuses by applying WIN55,212-2 (500 nM) at 37°C for the periods indicated in Fig 2D. Membranes were exposed to primary antibodies (Supplementary Table S1) overnight at 4°C, washed, incubated with HRP-conjugated secondary antibodies (1:10,000; Sigma) for 1 h and processed for enhanced chemiluminescence detection (Bio-Rad XRS+).
Polymerase chain reaction
To discriminate the sex of mouse fetuses, we used primers to amplify Y and X chromosome-specific genomic DNA sequences. The Sry gene (forward: GTTCAGCCCTACAGCCACAT, reverse: CAGCTGCTTGCTGATCTCTG, amplicon size: 197 bp [Y chromosome]) and DXNds3 microsatellite (forward: GAGTGCCTCATCTATACTTACAG, reverse: TCTAGTTCATTGTTGATTAGTTGC, amplicon size: 244 bp [X chromosome]) were amplified. For quantitative PCR, RNA was extracted using RNeasy Plus Mini kit (Qiagen) with optional DNase I (Qiagen) treatment. The High-capacity cDNA reverse transcription kit (Applied Biosystems) was used to generate cDNA. Gene expression profiling (triplicate samples) was run on a MyiQ Apparatus using iQ SYBR Green Supermix (Bio-Rad). Custom-made primer pairs (Supplementary Table S2A) were designed to amplify short fragments for each gene, including glyceraldehyde-3-phosphate dehydrogenase (gapdh) and TATA-binding protein as reference genes (Keimpema et al, 2010), in mouse (Supplementary Table S2B,C).
iTRAQ proteomics, nLC-MALDI/MS/MS and nLC-ESI/MS/MS target discovery
Proteins were extracted from fetal cortices (n = 5/3 THC/vehicle from independent pregnancies) by homogenization in triethylammonium bicarbonate (25 mM), Na2CO3 (20 mM) and protease inhibitor cocktail (all from Sigma, pH10), with their concentration determined using the bichinconinic acid (BCA) protein assay (Novagen). Proteins were acetone precipitated, and 100 μg used for isobaric tag for relative and absolute quantitation in 8-plex layout (iTRAQ, ABSciex). Proteins were denatured, reduced, alkylated, trypsin digested, individually labeled with appropriate iTRAQ tags, pooled, concentrated (SpeedVac, Thermo Scientific), re-suspended in 1.4 ml load buffer (10 mM KH2PO4 pH 3.0 in 25% acetonitrile) and sonicated. The peptides were then separated by cation exchange chromatography on a PolySulfoethyl A column (PolyLC) over 30 min with a KCl gradient increasing up to 0.5 M, and 0.5 ml fractions collected. Twenty fractions across the elution profile of similar peptide concentration were generated and concentrated (SpeedVac). Fractions were re-suspended in 0.1% trifluoroacetic acid (TFA) and desalted on C18 spin columns (PepClean C18, Thermo Scientific). For each fraction half of the sample was separated using a Dionex UltiMate 3000 nanoLC (Dionex) equipped with a PepMap100 C18 300 μm × 5 mm trap and 75 μm × 15 cm column (Dionex), using a 3.5 h gradient of increasing acetonitrile concentration, containing 0.05% TFA (5–35% acetonitrile in 3 h, 35–50% in a further 30 min, followed by 95% acetonitrile). The eluent was spotted onto a MALDI target plate, along with α-cyano-4-hydroxycinnamic acid (2 mg/ml in 70% acetonitrile: 0.1% TFA matrix solution) using a Dionex Probot spotter. The nLC-MALDI/MS/MS runs were analyzed using an 4800 MALDI TOF/TOF Analyser (ABSciex) equipped with a Nd:YAG 355 nm laser in a plate wide data-dependent manner. All spots were initially analyzed in positive MS mode in the range of 800–4000 m/z by averaging 1,000 laser spots (Shirran & Botting, 2010). The MS ions that satisfied the precursor criteria (200 ppm fraction to fraction precursor exclusion, S/N ratio > 20) were selected for subsequent MS/MS from the spot in where the MS ion gave the highest counts, with up to 5 MS/MS being acquired from each spot, selecting the strongest precursor ion first. MS/MS spectra were acquired with a maximum of 3,000 laser shots or until the accumulated spectrum reached a S/N ratio of 35 for 10 peaks. All MS/MS data were acquired using 1 keV collision energy.
The remaining half of the desalted fractions were separated using an UltiMate nanoLC (Dionex) equipped with a PepMap C18 trap and column, using a gradient of increasing acetonitrile concentration, containing 0.1% formic acid (5–35% acetonitrile in 180 min, 35–50% in a further 30 min, followed by 95% acetonitrile). The eluent was sprayed into a Q-Star XL tandem mass spectrometer (ABSciex) and analyzed in Information Dependent Acquisition (IDA) mode, performing 1 s of MS followed by 3 s MS/MS analyses of the 2 most intense peaks seen by MS. These masses were then excluded from analysis for the next 60 s. Rolling collision energy was employed for fragmentation, set 10V higher than that normally used for peptides, to provide sufficient peptide fragmentation and generation of the iTRAQ reporter groups.
Mass spectrometric data analysis
For the nLC-ESI/MS/MS, MS/MS data for doubly and triply charged precursor ions was converted to centroid data, without smoothing, using the Analyst QS1.1 mascot.dll data import filter with default settings. The data files were processed by Mascot v2.2 (Matrix Science). All searches were performed against the NCBI database using a mouse taxonomy filter (137 038 sequences). Automatic isotope correction was carried out using the values supplied with the ABSciex reagents. The “MS/MS averaging of IDA dependents” had a precursor mass tolerance for grouping of 0.1 and the maximum number of cycles between groups and minimum number of cycles per group were both set to 1. The MS/MS settings included: spectra de-isotoped (except for the iTRAQ reporter region), peak areas reported, spectra rejected if they contained < 10 peaks, and peaks not removed if they were close to the precursor m/z. The nLC-ESI/MS/MS data were searched with a tolerance of 0.08 Da for the precursor ions and 0.2 Da for the fragment ions. The nLC-MALDI/MS/MS data were extracted using TS2Mascot 1.0.0 (Matrix Science) and the data saved to a peak list. The nLC-MALDI/MS/MS data were searched with tolerances of 100 ppm for the precursor ion and 0.5 Da for the fragment ions. For both ionization routes the following settings were used: trypsin was the cleavage enzyme, one missed cleavage, methylthio modification of cysteines and iTRAQ 8-plex modification of lysines and N-terminal amines were fixed modifications; methionine oxidation was selected as a variable modification. The following settings were used to manipulate the quantification results: the protein ratio type was the “weighted” geometric mean, there was no normalization, outlier removal was “automatic” (Dixon's method up to 25 data points, Rosner's method above 25 data points), the peptide threshold was “at least homology” (peptide score did not exceed the absolute threshold but was an outlier from the quasi-normal distribution of random scores), the minimum number of unique peptides was two, and peptides were required to be the top ranking peptide matches. An automatic decoy database search was also performed.
Human subjects, in situ hybridization and Western blotting
Midgestational fetal brain subjects (18–22 weeks of gestation) were collected after saline-induced elective abortions under Institutional Review Board approval at SUNY Downstate Medical Center, Brooklyn, New York (Hurd et al, 2005). Fetal brains were fixed in 1% PFA-PB for 24 h, frozen in dry ice-cooled isopentane, cryosectioned in the coronal plane (20 μm) and stored at −30°C. In situ hybridization to study SCG10 mRNA expression was performed as described (Hurd, 2003). Briefly, PCR-derived RNA probes spanning exons 2–4 of human SCG10 (NM_001199214; T7-SCG10 sense primer: CTGTAATACGACTCACTATAGGG-AGGAGCTGTCCATGCTGTCACTG, SP6-SCG10 anti-sense primer: GGGATTTAGGTGACACTATAGAA-AGCAGCTAGATTAGCCTCACGGT) were transcribed in the presence of UTPαS (1000–1500 Ci/mmol [35S] specific activity, Perkin Elmer). [35S]-labeled probe was applied to the brain sections at a concentration of 2 × 104 cpm/mm2. Two adjacent sections per subject were studied at the level of the caudal hippocampal fold. Slides were hybridized at 55°C overnight and apposed to Imaging Plates (Fujifilm) along with [14C] standards (American Radiolabeled Chemicals). Films were developed with a FLA-7000 phosphoimaging analyzer (Fujifilm). Images were analyzed using MultiGauge software (Fujifilm). Relative mRNA expression levels were measured along the CA1-CA3 subregions of the hippocampus. Values from duplicate brain sections for each subject were averaged and expressed as dpm/mg of tissue by reference to co-exposed standards. Background normalization was performed on each section, and set to subcortical white matter area lacking SCG10 mRNA expression.
To determine SCG10 protein levels, human fetal hippocampal tissues, which were partially immersion fixed (Hurd et al, 2005), were lysed using the procedure established by Nirmalan et al (Nirmalan et al, 2009) and analyzed by SDS-PAGE. Membranes were exposed to SCG10 primary antibody (Supplementary Table S1) overnight at 4°C, washed, incubated with IRDye-conjugated secondary antibodies (1:5,000, Li-Cor) for 1 h and imaged on an Odyssey CLx infrared imager (Li-Cor). Western blots were analyzed by using ImageJ 1.45s to measure integrated optical density values and then normalized to total protein (Aldridge et al, 2008).
Preparation of growth cone particles
Isolated growth cone particles (GCPs) were prepared from the cortices of E17.5 C57Bl/6 mouse embryos (Lockerbie et al, 1991). GCPs pooled from n = 17 embryos were diluted in 4 volumes of modified Krebs-Ringer solution containing (in mM): 180 sucrose, 50 NaCl2, 5 KCl, 22 HEPES, 10 glucose, 1.2 NaH2PO4, and 1.2 MgCl2 (pH 7.4). SCG10 degradation upon THC exposure for 10 min was determined by Western blotting (relative to 0.1% DMSO as vehicle). Gapdh was used as loading control. Data were expressed as fold decrease of the unstimulated control value.
Phosphoprotein isolation
Cultured cortical neurons (107 cells/sample) were lysed in a buffer containing 0.25% CHAPS, protease/phosphatase inhibitors and benzonase for 30 min at 4°C, then cleared by centrifugation (10,000 g at 4°C, 30 min). Total extracted proteins were diluted to a concentration of 0.1 mg/ml (25 ml total volume) and loaded onto PhosphoProtein purification columns (Qiagen). Phosphoproteins were eluted with 2 ml of PhosphoProtein Elution Buffer (Qiagen), and concentrated by ultrafiltration (10 kDa cut-off). The yield of phosphorylated proteins was determined by the Bradford assay (Bio-Rad), and 15 μg aliquots analyzed by SDS-PAGE using antibodies specifically recognizing phosphoproteins (Supplementary Table S1).
Statistics
iTRAQ data were statistically evaluated by the Wilcoxon rank-sum test after log transformation of unscaled input data exceeding a cut-off of ± 2× geometric s.d. relative to vehicle-treated controls. The fEPSP slope was expressed as a percentage of the average of 10-min baseline preceding LTD induction. In both data from electrophysiology studies and the cohort of human fetal samples for mRNA analysis, the Shapiro–Wilk and Levene tests failed to demonstrate normal distribution and equality of variances, respectively. Thus, non-parametric Mann–Whitney U-test (independent samples) was chosen for statistical analysis. The mRNA expression levels (dpm/mg) from human fetal brains were normalized by natural log transformation, and plotted (ln(dpm/mg); Fig 4A1 and B). To analyze both SCG10 mRNA and protein expression in human tissues, general linear model analysis was used to control for the effects of other confounding factors alongside cannabis exposure. Variables were clustered as (i) fetal age, (ii) fetal body weight and foot length (“developmental measures”) and (iii) cannabis exposure. A univariate model was first performed with each cluster of variables and any variable that showed P < 0.10 was included in the stepwise regression in addition to cannabis exposure (Hurd et al, 2005). Variables with P < 0.05 were included in the final regression model as covariates of cannabis exposure. In all other experiments, group comparisons were performed by Student's t-test unless otherwise stated. Results of acute pharmacological experiments on organotypic slices were analyzed using one-way ANOVA followed by Tukey's post-hoc comparisons. A P level of < 0.05 was considered statistically significant (SPSS 21.0). Data were expressed as means ± s.e.m.
Acknowledgments
This work was supported by the Scottish Universities Life Science Alliance (T.Har., A.A.), Vetenskapsrådet (T.Har.), Hjärnfonden (T.Har.), Novo Nordisk Foundation (T.Har.), the Wellcome Trust (equipment grants, C.H.B.), the Petrus and Augusta Hedlunds Stiftelse (T.Har.) and the National Institutes of Health (RO1-DA023214, T.Har. & Y.L.H.; F31-DA031559, C.V.M.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the US National Institutes of Health. We thank M. Watanabe (Hokkaido University) for antibodies, E.T. Coffey (University of Turku) for a vector containing point mutated EGFP-SCG10S62D/S73D, M. Zilberter (Karolinska Institute) for his contribution to the initial phase of electrophysiology experiments, O.K. Penz (Karolinska Institutet) for technical assistance, I. Parisi and J. Mulder (Science for Life Laboratory) for assistance with automated slide-scanning microscopy, and H.-C. Lu (Baylor College), I. Galve-Roperh (Complutense University), T. Hökfelt (Karolinska Institutet) and members of the Harkany laboratory for constructive discussions and feedback. Laser-scanning microscopy was performed at the Click Imaging Core Facility of the Karolinska Institutet.
Author contributions
THar and YLH conceived the general framework of this study; GT, THer, MC, YLH and THar designed experiments; GT, CVM, SLS, DC, EK, KR, THer, CHB, JF, AA performed experiments; KR, THer, MC provided unique reagents; GT, AA, JF, DC, EK analyzed data; GT and THar wrote the manuscript.
Conflict of Interest
The authors declare that they have no conflict of interest.
Supplementary information for this article is available online: http://emboj.embopress.org
References
- Aldridge GM, Podrebarac DM, Greenough WT, Weiler IJ. The use of total protein stains as loading controls: an alternative to high-abundance single-protein controls in semi-quantitative immunoblotting. J Neurosci Methods. 2008;172:250–254. doi: 10.1016/j.jneumeth.2008.05.00. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berghuis P, Dobszay MB, Wang X, Spano S, Ledda F, Sousa KM, Schulte G, Ernfors P, Mackie K, Paratcha G, Hurd YL, Harkany T. Endocannabinoids regulate interneuron migration and morphogenesis by transactivating the TrkB receptor. Proc Natl Acad Sci USA. 2005;102:19115–19120. doi: 10.1073/pnas.0509494102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berghuis P, Rajnicek AM, Morozov YM, Ross RA, Mulder J, Urban GM, Monory K, Marsicano G, Matteoli M, Canty A, Irving AJ, Katona I, Yanagawa Y, Rakic P, Lutz B, Mackie K, Harkany T. Hardwiring the brain: endocannabinoids shape neuronal connectivity. Science. 2007;316:1212–1216. doi: 10.1126/science.1137406. [DOI] [PubMed] [Google Scholar]
- Bisogno T, Howell F, Williams G, Minassi A, Cascio MG, Ligresti A, Matias I, Schiano-Moriello A, Paul P, Williams EJ, Gangadharan U, Hobbs C, Di Marzo V, Doherty P. Cloning of the first sn1-DAG lipases points to the spatial and temporal regulation of endocannabinoid signaling in the brain. J Cell Biol. 2003;163:463–468. doi: 10.1083/jcb.200305129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjorkblom B, Ostman N, Hongisto V, Komarovski V, Filen JJ, Nyman TA, Kallunki T, Courtney MJ, Coffey ET. Constitutively active cytoplasmic c-Jun N-terminal kinase 1 is a dominant regulator of dendritic architecture: role of microtubule-associated protein 2 as an effector. J Neurosci. 2005;25:6350–6361. doi: 10.1523/JNEUROSCI.1517-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodor AL, Katona I, Nyiri G, Mackie K, Ledent C, Hajos N, Freund TF. Endocannabinoid signaling in rat somatosensory cortex: laminar differences and involvement of specific interneuron types. J Neurosci. 2005;25:6845–6856. doi: 10.1523/JNEUROSCI.0442-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campolongo P, Trezza V, Cassano T, Gaetani S, Morgese MG, Ubaldi M, Soverchia L, Antonelli T, Ferraro L, Massi M, Ciccocioppo R, Cuomo V. Perinatal exposure to delta-9-tetrahydrocannabinol causes enduring cognitive deficits associated with alteration of cortical gene expression and neurotransmission in rats. Addict Biol. 2007;12:485–495. doi: 10.1111/j.1369-1600.2007.00074.x. [DOI] [PubMed] [Google Scholar]
- Chang L, Jones Y, Ellisman MH, Goldstein LS, Karin M. JNK1 is required for maintenance of neuronal microtubules and controls phosphorylation of microtubule-associated proteins. Dev Cell. 2003;4:521–533. doi: 10.1016/s1534-5807(03)00094-7. [DOI] [PubMed] [Google Scholar]
- Daigle TL, Kwok ML, Mackie K. Regulation of CB1 cannabinoid receptor internalization by a promiscuous phosphorylation-dependent mechanism. J Neurochem. 2008;106:70–82. doi: 10.1111/j.1471-4159.2008.05336.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day NL, Goldschmidt L, Thomas CA. Prenatal marijuana exposure contributes to the prediction of marijuana use at age 14. Addiction. 2006;101:1313–1322. doi: 10.1111/j.1360-0443.2006.01523.x. [DOI] [PubMed] [Google Scholar]
- Day NL, Leech SL, Goldschmidt L. The effects of prenatal marijuana exposure on delinquent behaviors are mediated by measures of neurocognitive functioning. Neurotoxicol Teratol. 2011;33:129–136. doi: 10.1016/j.ntt.2010.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis EL, Jahanshad N, McMahon KL, de Zubicaray GI, Martin NG, Hickie IB, Toga AW, Wright MJ, Thompson PM. Development of brain structural connectivity between ages 12 and 30: a 4-Tesla diffusion imaging study in 439 adolescents and adults. Neuroimage. 2013;64:671–684. doi: 10.1016/j.neuroimage.2012.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derkinderen P, Valjent E, Toutant M, Corvol JC, Enslen H, Ledent C, Trzaskos J, Caboche J, Girault JA. Regulation of extracellular signal-regulated kinase by cannabinoids in hippocampus. J Neurosci. 2003;23:2371–2382. doi: 10.1523/JNEUROSCI.23-06-02371.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Marzo V. Endocannabinoid signaling in the brain: biosynthetic mechanisms in the limelight. Nat Neurosci. 2011;14:9–15. doi: 10.1038/nn.2720. [DOI] [PubMed] [Google Scholar]
- Diaz-Alonso J, Aguado T, Wu CS, Palazuelos J, Hofmann C, Garcez P, Guillemot F, Lu HC, Lutz B, Guzman M, Galve-Roperh I. The CB1 cannabinoid receptor drives corticospinal motor neuron differentiation through the Ctip2/Satb2 transcriptional regulation axis. J Neurosci. 2012;32:16651–16665. doi: 10.1523/JNEUROSCI.0681-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinh TP, Carpenter D, Leslie FM, Freund TF, Katona I, Sensi SL, Kathuria S, Piomelli D. Brain monoglyceride lipase participating in endocannabinoid inactivation. Proc Natl Acad Sci USA. 2002;99:10819–10824. doi: 10.1073/pnas.152334899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinieri JA, Wang X, Szutorisz H, Spano SM, Kaur J, Casaccia P, Dow-Edwards D, Hurd YL. Maternal cannabis use alters ventral striatal dopamine D2 gene regulation in the offspring. Biol Psychiatry. 2011;70:763–769. doi: 10.1016/j.biopsych.2011.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong C, Yang DD, Wysk M, Whitmarsh AJ, Davis RJ, Flavell RA. Defective T cell differentiation in the absence of Jnk1. Science. 1998;282:2092–2095. doi: 10.1126/science.282.5396.2092. [DOI] [PubMed] [Google Scholar]
- Dudek SM, Bear MF. Bidirectional long-term modification of synaptic effectiveness in the adult and immature hippocampus. J Neurosci. 1993;13:2910–2918. doi: 10.1523/JNEUROSCI.13-07-02910.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Marroun H, Tiemeier H, Steegers EA, Jaddoe VW, Hofman A, Verhulst FC, van den BW, Huizink AC. Intrauterine cannabis exposure affects fetal growth trajectories: the generation R study. J Am Acad Child Adolesc Psychiatry. 2009;48:1173–1181. doi: 10.1097/CHI.0b013e3181bfa8ee. [DOI] [PubMed] [Google Scholar]
- Fukata Y, Itoh TJ, Kimura T, Menager C, Nishimura T, Shiromizu T, Watanabe H, Inagaki N, Iwamatsu A, Hotani H, Kaibuchi K. CRMP-2 binds to tubulin heterodimers to promote microtubule assembly. Nat Cell Biol. 2002;4:583–591. doi: 10.1038/ncb825. [DOI] [PubMed] [Google Scholar]
- Goldschmidt L, Richardson GA, Cornelius MD, Day NL. Prenatal marijuana and alcohol exposure and academic achievement at age 10. Neurotoxicol Teratol. 2004;26:521–532. doi: 10.1016/j.ntt.2004.04.003. [DOI] [PubMed] [Google Scholar]
- Goncalves MB, Suetterlin P, Yip P, Molina-Holgado F, Walker DJ, Oudin MJ, Zentar MP, Pollard S, Yanez-Munoz RJ, Williams G, Walsh FS, Pangalos MN, Doherty P. A diacylglycerol lipase-CB2 cannabinoid pathway regulates adult subventricular zone neurogenesis in an age-dependent manner. Mol Cell Neurosci. 2008;38:526–536. doi: 10.1016/j.mcn.2008.05.001. [DOI] [PubMed] [Google Scholar]
- Goshima Y, Nakamura F, Strittmatter P, Strittmatter SM. Collapsin-induced growth cone collapse mediated by an intracellular protein related to UNC-33. Nature. 1995;376:509–514. doi: 10.1038/376509a0. [DOI] [PubMed] [Google Scholar]
- Grenningloh G, Soehrman S, Bondallaz P, Ruchti E, Cadas H. Role of the microtubule destabilizing proteins SCG10 and stathmin in neuronal growth. J Neurobiol. 2004;58:60–69. doi: 10.1002/neu.10279. [DOI] [PubMed] [Google Scholar]
- Grotenhermen F. Pharmacokinetics and pharmacodynamics of cannabinoids. Clin Pharmacokinet. 2003;42:327–360. doi: 10.2165/00003088-200342040-00003. [DOI] [PubMed] [Google Scholar]
- Hardy RJ, Friedrich VL., Jr Oligodendrocyte progenitors are generated throughout the embryonic mouse brain, but differentiate in restricted foci. Development. 1996;122:2059–2069. doi: 10.1242/dev.122.7.2059. [DOI] [PubMed] [Google Scholar]
- Howlett AC. The cannabinoid receptors. Prostaglandins Other Lipid Mediat. 2002;68–69:619–631. doi: 10.1016/s0090-6980(02)00060-6. [DOI] [PubMed] [Google Scholar]
- Hsieh C, Brown S, Derleth C, Mackie K. Internalization and recycling of the CB1 cannabinoid receptor. J Neurochem. 1999;73:493–501. doi: 10.1046/j.1471-4159.1999.0730493.x. [DOI] [PubMed] [Google Scholar]
- Huizink AC, Mulder EJ. Maternal smoking, drinking or cannabis use during pregnancy and neurobehavioral and cognitive functioning in human offspring. Neurosci Biobehav Rev. 2006;30:24–41. doi: 10.1016/j.neubiorev.2005.04.005. [DOI] [PubMed] [Google Scholar]
- Hurd YL. In situ hybridization with isotopic riboprobes for detection of striatal neuropeptide mRNA expression after dopamine stimulant administration. Methods Mol Med. 2003;79:119–135. doi: 10.1385/1-59259-358-5:119. [DOI] [PubMed] [Google Scholar]
- Hurd YL, Wang X, Anderson V, Beck O, Minkoff H, Dow-Edwards D. Marijuana impairs growth in mid-gestation fetuses. Neurotoxicol Teratol. 2005;27:221–229. doi: 10.1016/j.ntt.2004.11.002. [DOI] [PubMed] [Google Scholar]
- Jeanneteau F, Deinhardt K, Miyoshi G, Bennett AM, Chao MV. The MAP kinase phosphatase MKP-1 regulates BDNF-induced axon branching. Nat Neurosci. 2010;13:1373–1379. doi: 10.1038/nn.2655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kano M, Ohno-Shosaku T, Hashimotodani Y, Uchigashima M, Watanabe M. Endocannabinoid-mediated control of synaptic transmission. Physiol Rev. 2009;89:309–380. doi: 10.1152/physrev.00019.2008. [DOI] [PubMed] [Google Scholar]
- Keimpema E, Barabas K, Morozov YM, Tortoriello G, Torii M, Cameron G, Yanagawa Y, Watanabe M, Mackie K, Harkany T. Differential subcellular recruitment of monoacylglycerol lipase generates spatial specificity of 2-arachidonoyl glycerol signaling during axonal pathfinding. J Neurosci. 2010;30:13992–14007. doi: 10.1523/JNEUROSCI.2126-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keimpema E, Mackie K, Harkany T. Molecular model of cannabis sensitivity in developing neuronal circuits. Trends Pharmacol Sci. 2011;32:551–561. doi: 10.1016/j.tips.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keimpema E, Tortoriello G, Alpar A, Capsoni S, Arisi I, Calvigioni D, Hu SS, Cattaneo A, Doherty P, Mackie K, Harkany T. Nerve growth factor scales endocannabinoid signaling by regulating monoacylglycerol lipase turnover in developing cholinergic neurons. Proc Natl Acad Sci USA. 2013;110:1935–1940. doi: 10.1073/pnas.1212563110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessaris N, Pringle N, Richardson WD. Specification of CNS glia from neural stem cells in the embryonic neuroepithelium. Philos Trans R Soc Lond B Biol Sci. 2008;363:71–85. doi: 10.1098/rstb.2006.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kittler JT, Grigorenko EV, Clayton C, Zhuang SY, Bundey SC, Trower MM, Wallace D, Hampson R, Deadwyler S. Large-scale analysis of gene expression changes during acute and chronic exposure to [Delta]9-THC in rats. Physiol Genomics. 2000;3:175–185. doi: 10.1152/physiolgenomics.2000.3.3.175. [DOI] [PubMed] [Google Scholar]
- Kostovic I, Jovanov-Milosevic N. The development of cerebral connections during the first 20–45 weeks' gestation. Semin Fetal Neonatal Med. 2006;11:415–422. doi: 10.1016/j.siny.2006.07.001. [DOI] [PubMed] [Google Scholar]
- Kostovic I, Judas M. The development of the subplate and thalamocortical connections in the human foetal brain. Acta Paediatr. 2010;99:1119–1127. doi: 10.1111/j.1651-2227.2010.01811.x. [DOI] [PubMed] [Google Scholar]
- Leech SL, Larkby CA, Day R, Day NL. Predictors and correlates of high levels of depression and anxiety symptoms among children at age 10. J Am Acad Child Adolesc Psychiatry. 2006;45:223–230. doi: 10.1097/01.chi.0000184930.18552.4d. [DOI] [PubMed] [Google Scholar]
- Leech SL, Richardson GA, Goldschmidt L, Day NL. Prenatal substance exposure: effects on attention and impulsivity of 6-year-olds. Neurotoxicol Teratol. 1999;21:109–118. doi: 10.1016/s0892-0362(98)00042-7. [DOI] [PubMed] [Google Scholar]
- Li YH, Ghavampur S, Bondallaz P, Will L, Grenningloh G, Puschel AW. Rnd1 regulates axon extension by enhancing the microtubule destabilizing activity of SCG10. J Biol Chem. 2009;284:363–371. doi: 10.1074/jbc.A808126200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lockerbie RO, Miller VE, Pfenninger KH. Regulated plasmalemmal expansion in nerve growth cones. J Cell Biol. 1991;112:1215–1227. doi: 10.1083/jcb.112.6.1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manna T, Grenningloh G, Miller HP, Wilson L. Stathmin family protein SCG10 differentially regulates the plus and minus end dynamics of microtubules at steady state in vitro: implications for its role in neurite outgrowth. Biochemistry. 2007;46:3543–3552. doi: 10.1021/bi061819d. [DOI] [PubMed] [Google Scholar]
- Maruta H, Greer K, Rosenbaum JL. The acetylation of alpha-tubulin and its relationship to the assembly and disassembly of microtubules. J Cell Biol. 1986;103:571–579. doi: 10.1083/jcb.103.2.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mato S, Chevaleyre V, Robbe D, Pazos A, Castillo PE, Manzoni OJ. A single in-vivo exposure to delta 9THC blocks endocannabinoid-mediated synaptic plasticity. Nat Neurosci. 2004;7:585–586. doi: 10.1038/nn1251. [DOI] [PubMed] [Google Scholar]
- McDonald NA, Henstridge CM, Connolly CN, Irving AJ. An essential role for constitutive endocytosis, but not activity, in the axonal targeting of the CB1 cannabinoid receptor. Mol Pharmacol. 2007;71:976–984. doi: 10.1124/mol.106.029348. [DOI] [PubMed] [Google Scholar]
- Mikaels-Edman A, Baudet C, Ernfors P. Soluble and bound forms of GFRalpha1 elicit different GDNF-independent neurite growth responses in primary sensory neurons. Dev Dyn. 2003;227:27–34. doi: 10.1002/dvdy.10280. [DOI] [PubMed] [Google Scholar]
- Monory K, Massa F, Egertova M, Eder M, Blaudzun H, Westenbroek R, Kelsch W, Jacob W, Marsch R, Ekker M, Long J, Rubenstein JL, Goebbels S, Nave KA, During M, Klugmann M, Wolfel B, Dodt HU, Zieglgansberger W, Wotjak CT, et al. The endocannabinoid system controls key epileptogenic circuits in the hippocampus. Neuron. 2006;51:455–466. doi: 10.1016/j.neuron.2006.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morii H, Shiraishi-Yamaguchi Y, Mori N. SCG10, a microtubule destabilizing factor, stimulates the neurite outgrowth by modulating microtubule dynamics in rat hippocampal primary cultured neurons. J Neurobiol. 2006;66:1101–1114. doi: 10.1002/neu.20295. [DOI] [PubMed] [Google Scholar]
- Mulder J, Aguado T, Keimpema E, Barabas K, Ballester Rosado CJ, Nguyen L, Monory K, Marsicano G, Di Marzo V, Hurd YL, Guillemot F, Mackie K, Lutz B, Guzman M, Lu HC, Galve-Roperh I, Harkany T. Endocannabinoid signaling controls pyramidal cell specification and long-range axon patterning. Proc Natl Acad Sci USA. 2008;105:8760–8765. doi: 10.1073/pnas.0803545105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulder J, Zilberter M, Pasquare S, Alpar A, Schulte G, Ferreira SG, Köfalvi A, Martin-Moreno AAM, Keimpema E, Tanila H, Watanabe M, Mackie K, Hortobagyi T, de Ceballos MAL, Harkany T. Molecular reorganization of endocannabinoid signalling in Alzheimer's disease. Brain. 2011;134:1041–1060. doi: 10.1093/brain/awr046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nirmalan NJ, Harnden P, Selby PJ, Banks RE. Development and validation of a novel protein extraction methodology for quantitation of protein expression in formalin-fixed paraffin-embedded tissues using western blotting. J Pathol. 2009;217:497–506. doi: 10.1002/path.2504. [DOI] [PubMed] [Google Scholar]
- Nithipatikom K, Gomez-Granados AD, Tang AT, Pfeiffer AW, Williams CL, Campbell WB. Cannabinoid receptor type 1 (CB1) activation inhibits small GTPase RhoA activity and regulates motility of prostate carcinoma cells. Endocrinology. 2012;153:29–41. doi: 10.1210/en.2011-1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paronis CA, Nikas SP, Shukla VG, Makriyannis A. Delta(9)-Tetrahydrocannabinol acts as a partial agonist/antagonist in mice. Behav Pharmacol. 2012;23:802–805. doi: 10.1097/FBP.0b013e32835a7c4d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng H, Derrick BE, Martinez JL., Jr Time-course study of SCG10 mRNA levels associated with LTP induction and maintenance in the rat Schaffer-CA1 pathway in vivo. Brain Res Mol Brain Res. 2004;120:182–187. doi: 10.1016/j.molbrainres.2003.10.009. [DOI] [PubMed] [Google Scholar]
- Pertwee RG, Howlett AC, Abood ME, Alexander SP, Di MV, Elphick MR, Greasley PJ, Hansen HS, Kunos G, Mackie K, Mechoulam R, Ross RA. International Union of Basic and Clinical Pharmacology. LXXIX. Cannabinoid receptors and their ligands: beyond CB and CB. Pharmacol Rev. 2010;62:588–631. doi: 10.1124/pr.110.003004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pijlman FT, Rigter SM, Hoek J, Goldschmidt HM, Niesink RJ. Strong increase in total delta-THC in cannabis preparations sold in Dutch coffee shops. Addict Biol. 2005;10:171–180. doi: 10.1080/13556210500123217. [DOI] [PubMed] [Google Scholar]
- Pitts JE, Neal JD, Gough TA. Some features of Cannabis plants grown in the United Kingdom from seeds of known origin. J Pharm Pharmacol. 1992;44:947–951. doi: 10.1111/j.2042-7158.1992.tb07072.x. [DOI] [PubMed] [Google Scholar]
- Romero-Sandoval EA, Horvath R, Landry RP, DeLeo JA. Cannabinoid receptor type 2 activation induces a microglial anti-inflammatory phenotype and reduces migration via MKP induction and ERK dephosphorylation. Mol Pain. 2009;5:25. doi: 10.1186/1744-8069-5-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rueda D, Galve-Roperh I, Haro A, Guzman M. The CB(1) cannabinoid receptor is coupled to the activation of c-Jun N-terminal kinase. Mol Pharmacol. 2000;58:814–820. doi: 10.1124/mol.58.4.814. [DOI] [PubMed] [Google Scholar]
- Schmechel DE, Rakic P. A Golgi study of radial glial cells in developing monkey telencephalon: morphogenesis and transformation into astrocytes. Anat Embryol. 1979;156:115–152. doi: 10.1007/BF00300010. [DOI] [PubMed] [Google Scholar]
- Shin JE, Miller BR, Babetto E, Cho Y, Sasaki Y, Qayum S, Russler EV, Cavalli V, Milbrandt J, DiAntonio A. SCG10 is a JNK target in the axonal degeneration pathway. Proc Natl Acad Sci USA. 2012;109:E3696–E3705. doi: 10.1073/pnas.1216204109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirran SL, Botting CH. A comparison of the accuracy of iTRAQ quantification by nLC-ESI MSMS and nLC-MALDI MSMS methods. J Proteomics. 2010;73:1391–1403. doi: 10.1016/j.jprot.2010.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spano MS, Ellgren M, Wang X, Hurd YL. Prenatal cannabis exposure increases heroin seeking with allostatic changes in limbic enkephalin systems in adulthood. Biol Psychiatry. 2007;61:554–563. doi: 10.1016/j.biopsych.2006.03.073. [DOI] [PubMed] [Google Scholar]
- Stein R, Mori N, Matthews K, Lo LC, Anderson DJ. The NGF-inducible SCG10 mRNA encodes a novel membrane-bound protein present in growth cones and abundant in developing neurons. Neuron. 1988;1:463–476. doi: 10.1016/0896-6273(88)90177-8. [DOI] [PubMed] [Google Scholar]
- Substance Abuse and Mental Health Service Administration. 2010. Treatment Episode Data Set 2000–2010.
- Takahashi KA, Castillo PE. The CB1 cannabinoid receptor mediates glutamatergic synaptic suppression in the hippocampus. Neuroscience. 2006;139:795–802. doi: 10.1016/j.neuroscience.2006.01.024. [DOI] [PubMed] [Google Scholar]
- Tararuk T, Ostman N, Li W, Bjorkblom B, Padzik A, Zdrojewska J, Hongisto V, Herdegen T, Konopka W, Courtney MJ, Coffey ET. JNK1 phosphorylation of SCG10 determines microtubule dynamics and axodendritic length. J Cell Biol. 2006;173:265–277. doi: 10.1083/jcb.200511055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomasoni R, Repetto D, Morini R, Elia C, Gardoni F, Di LM, Turco E, Defilippi P, Matteoli M. SNAP-25 regulates spine formation through postsynaptic binding to p140Cap. Nat Commun. 2013;4:2136. doi: 10.1038/ncomms3136. [DOI] [PubMed] [Google Scholar]
- Westerlund N, Zdrojewska J, Padzik A, Komulainen E, Bjorkblom B, Rannikko E, Tararuk T, Garcia-Frigola C, Sandholm J, Nguyen L, Kallunki T, Courtney MJ, Coffey ET. Phosphorylation of SCG10/stathmin-2 determines multipolar stage exit and neuronal migration rate. Nat Neurosci. 2011;14:305–313. doi: 10.1038/nn.2755. [DOI] [PubMed] [Google Scholar]
- Willford JA, Chandler LS, Goldschmidt L, Day NL. Effects of prenatal tobacco, alcohol and marijuana exposure on processing speed, visual-motor coordination, and interhemispheric transfer. Neurotoxicol Teratol. 2010;32:580–588. doi: 10.1016/j.ntt.2010.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan XB, Jin M, Xu X, Song YQ, Wu CP, Poo MM, Duan S. Signalling and crosstalk of Rho GTPases in mediating axon guidance. Nat Cell Biol. 2003;5:38–45. doi: 10.1038/ncb895. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.