

Abstract
Our research aims to evaluate the function of the STAMP2 gene, an important trigger in insulin resistance (IR), and explore its role in macrophage apoptosis in diabetic atherosclerotic vulnerable plaques. The characteristics of diabetic mice were measured by serial metabolite and pathology tests. The level of STAMP2 was measured by RT-PCR and Western blot. The plaque area, lipid and collagen content of brachiocephalic artery plaques were measured by histopathological analyses, and the macrophage apoptosis was measured by TUNEL. Correlation of STAMP2/Akt signaling pathway and macrophage apoptosis was validated by Ad-STAMP2 transfection and STAMP2 siRNA inhibition. The diabetic mice showed typical features of IR, hyperglycaemia. Overexpression of STAMP2 ameliorated IR and decreased serum glucose level. In brachiocephalic lesions, lipid content, macrophage quantity and the vulnerability index were significantly decreased by overexpression of STAMP2. Moreover, the numbers of apoptotic cells and macrophages in lesions were both significantly decreased. In vitro, both mRNA and protein expressions of STAMP2 were increased under high glucose treatment. P-Akt was highly expressed and caspase-3 was decreased after overexpression of STAMP2. However, expression of p-Akt protein was decreased and caspase-3 was increased when STAMP2 was inhibited by siRNA. STAMP2 overexpression could exert a protective effect on diabetic atherosclerosis by reducing IR and diminishing macrophage apoptosis.
Keywords: STAMP2/Akt signalling pathway, apoptosis of macrophage, Type 2 diabetes mellitus, insulin resistance, vulnerable plaque, vascular
Introduction
The type 2 diabetes mellitus forms a growing public health problem worldwide. In Western countries, about 60% patients with coronary heart disease also have diabetes mellitus, and the number has reached to 80% in China. Even with many medical therapies, the mortality is still as high as 70%. Therefore, atherosclerotic cardiovascular disease has become the most serious complication of diabetes 1,2. Patients with diabetes are at a high risk for developing acute coronary syndrome (ACS) 3. Diabetic patients with ACS suffer from increased mortality due to rupture of vulnerable atherosclerotic plaques compared with their non-diabetic peers 4,5. The patients with diabetes have more serious vulnerable plaques and higher incidence. However, the mechanism remains to be elucidated. Diabetes mellitus and atherosclerotic cardiovascular disease increase the medical cost directly, also increase the economic burden of society indirectly. With the increasing health consciousness and development of preventive medicine in recent years, the mortality of coronary heart disease has declined. However, the mortality of diabetic coronary heart disease increased year by year. Therefore, it is necessary to explore the mechanism for ACS treatment and take advantage of medical resources reasonably.
Insulin resistance (IR) plays a causal role in the pathogenesis and development of type 2 diabetes 6,7 and atherosclerotic cardiovascular disease 8. Studies have shown that IR is involved in impairment of glucolipid metabolism 9, thus increasing atherosclerotic plaque formation and decreasing plaque stabilization 10. In advanced lesions, macrophage apoptosis promotes the development of the necrotic core, a key factor in rendering plaques vulnerable to disruption 11–13. Analysis of culprit lesions from diabetic patients of sudden coronary death has shown that the majority of cells at the rupture site were macrophages and that the apoptosis in macrophages was more frequent at the rupture site as opposed to areas of intact fibrous cap 14. Akt signalling pathway is a key step in signal transductions of apoptosis in macrophages 15. Akt inhibition leads to macrophage apoptosis, abnormal glucose tolerance and IR 15–17. Therefore, insulin-resistance macrophages are more susceptible to apoptosis in atherosclerotic lesions, probably predisposing to plaque rupture 9,18,19. Carlos suggests that special treatment of activated Akt1 could increase stability and reduce formation of atherosclerotic plaques in vivo. On the other hand, Akt1 reduction could reduce the LDL intake of macrophages, suggesting Akt1 may have a role in atherogenesis 15. PI3K/Akt activation enhances macrophage survival in lesions, and the persistent activation of Akt promotes cellular hypertrophy and hyperplasia, thereby promoting atherogenesis 15. Therefore, exploring a new biomarker to regulate Akt signalling pathway exactly through adjusted insulin pathway is necessary.
Studies have shown that six-trans membrane protein STAMP2 20,21 may be the key molecule. STAMP2 is involved in regulation of glucose metabolism. Its deficiency induces impairment of Akt signalling pathway and insulin exocytosis that lead to glucose metabolism disorder and IR 22. Moreover, STAMP2 is expressed in human and mouse macrophages, and regulates foam cell formation. STAMP2 is expressed in both human and mouse atherosclerotic plaques, and its deficiency promotes atherosclerosis in mice 23. Therefore, we propose that STAMP2/Akt signalling pathway may be the common intermediary among macrophage apoptosis, IR and vulnerable plaques in the diabetic state.
Here, we investigated the role of STAMP2 in macrophage apoptosis of vulnerable atherosclerotic plaques and determined the molecular mechanism whereby STAMP2 modulates Akt signalling pathways in RAW264.7. Regulating STAMP2 appears a promising novel therapeutic approach in patients with ACS.
Materials and methods
Mice
All animal procedures were in accordance with the institutional guidelines of Qilu Hospital of Shandong University and approved by Shandong University Institutional Animal Care and Use Committee.
Induction of diabetes and atherosclerosis in mice
Three-week-old male ApoE−/−/LDLR−/− mice were fed a high-fat diet (HFD; 20% fat, 20% sugar, 1.25% cholesterol; Beijing HFK Bio-Technology, China). At the age of 9 weeks, any mouse who exhibited IR was injected once with low-dose streptozotocin (STZ, Sigma-Aldrich, St. Louis, MO, USA; 75–80 mg/kg i.p. in 0.1 mol/l citrate buffer, pH 4.5). After 2 weeks, most mice displayed hyperglycaemia, IR and glucose intolerance, as previously reported 24. At age 11 weeks, similar animals were randomly divided into two groups, one will be treated with the STAMP2-expressing adenoviruses, referred to as the diabetes model (DM) STAMP2 group (n = 10), and the other treated with the expressing vector control adenovirus, referred to as the DM Vehicle group (n = 10; see later for the details of the adenovirus used). The mice fed a normal diet were used as non-diabetic controls, divided into two groups. One will be treated with the STAMP2-expressing adenovirus, referred to as the Control STAMP2 group (n = 10), and the other treated with the expressing vector control adenovirus, referred to as the Control Vehicle group (n = 10).
Intraperitoneal glucose tolerance test (IPGTT)
Glucose tolerance was assessed by IPGTT. Mice fasted overnight were challenged intraperitoneally with glucose at 1.5 g/kg bodyweight. Blood samples were collected sequentially from the tail vein at 0, 15, 30, 60 and 120 min. and tested for glucose. Plasma glucose of every animal was measured with the OneTouch SureStep glucometer (LifeScan Inc., Milpitas, CA, USA). The mean area under the receiver operating characteristic curve (AUC) was calculated for glucose.
Construction of STAMP2-expressing adenovirus
The recombinant pAdxsi adenovirus constitutively expressing STAMP2 was constructed using the pAdxsi Adenoviral System (SinoGenoMax, Beijing, China). The STAMP2 cDNAs from mouse were inserted into pShuttle-CMV-EGFP vector. The pAdxsi vector adenovirus was used as the control vehicle virus. The STAMP2-expressing adenovirus of 5 × 109 plaque-forming units (PFU) were injected to the mice (the DM STAMP2 group and the Control STAMP2 group) by jugular vein injection at 20 weeks and another 5 × 109 PFU of virus by jugular vein injection at 22 weeks. The control vehicle virus of the same PFU was also injected to the mice (the DM Vehicle group and the Control Vehicle group) by jugular vein injection. All mice were killed for further biochemical and histological study at 24 weeks.
Blood analyses
At the end of all experiment, we collected the murine samples to measure the levels of fasting blood glucose, total cholesterol, triglyceride and free fatty acids. Fasting serum insulin level was measured by enzyme-linked immunosorbent assay. The homeostasis model assessment (HOMA) method was used to calculate IR 25.
Histology and morphometric analysis
After paraformaldehyde (4%)-fixed mice aortas overnight, the aortas were cut open longitudinally and stained with Oil Red O (Sigma-Aldrich) to calculate the lesions area. Brachiocephalic arteries (BCA) were embedded in optimal cutting temperature compound (OTC; Sakura Finetek, Beijing, China). Sections were cut every 5 μm along the BCA and stained with haematoxylin and eosin, Picrosirius red, Masson' trichrome staining and Oil Red O. A Nikon microscope (Nikon, Melville, NY, USA) was used to capture the images. The mean was obtained by quantitative morphometry with automated image analysis (Image-Pro Plus, Version 5.0; Media Cybernatics, Houston, TX, USA).
Immunohistochemical staining and TUNEL
The following primary antibodies were used: rabbit polyclonal anti-SMCs (α-actin, 1:150; Abcam, Cambridge, MA, USA), and anti-macrophage antibodies (MOMA-2, 1:100; Abcam). Paraffin sections underwent immunohistochemistry by a microwave-based antigen retrieval method. The results were viewed under a confocal FV 1000 SPD laser scanning microscope (Olympus, Tokyo, Japan).
For the detection of oligonucleosomal DNA cleavage, a stringent TUNEL (terminal deoxynucleotidyl transferase end labelling) technique was used (Roche, Mannheim, Germany). In brief, sections were immersed in PBS for 15 min. at room temperature, incubated in permeabilization solution for 2 min. on ice, then transferred into the TUNEL reaction mixture, and incubated for 60 min. at 37°C in a humidified atmosphere in the dark.
Adhesion assay
The Costar culture plates were precoated with poly-lysine (50 μg/ml, Sigma-Aldrich). Added 5 × 106 macrophages into plates, after 10 min. incubation at 37°C, the adherent cells were stained by DAPI and counted under a fluorescence microscope.
Migration assay
Cell migration assays were performed with 24-well Costar Transwell plates (5 μm). After overnight starvation in DMEM with 2% foetal bovine serum, 7.5 × 104 cells were loaded into the upper chambers. The lower chambers were filled with solution of monocyte chemoattractant protein-1 (100 ng/ml; PeproTech Inc, Rochy Hill, NJ, USA) in DMEM. After 6 hrs incubation at 37°C, macrophages were stained with crystal violet (Sigma-Aldrich). The macrophages on the upper side of the membrane were removed by a cotton swab. Then we counted the average number of macrophages on the lower side.
Phagocytosis assay
Cells (1 × 106 cells/well) were seeded to glass bottom microwell dishes (MatTek Corporation, Ashland, MA, USA). After macrophages were incubated 10 min. at 37°C in dark with Alexa Fluor 488 Ac-LDL (10 μg/ml; Molecular Probes, Invitrogen, Eugene, OR, USA), excessive Ac-LDL was washed away and cells stained by DAPI in 5 min. Then the cells were observed under a laser scanning confocal microscopy (Leica TCS SP2; Leica, Wetzlar, Germany) and analysed by a flow cytometry (FACSCaliber; BD Biosciences Franklin Lakes, NJ, USA).
Apoptosis assay
Cells (1 × 106 cells/well) were incubated 15 min. at 4°C in dark with Annexin-V (10 μg/ml). After added propidium iodide (PI; 5 μg/ml) to DMEM 5 min., the Annexin-V-positive and PI -positive cells were analysed by a flow cytometry.
Cell Culture
The murine monocyte/macrophage cell-line Raw264.7 were grown in DMEM medium (Gibco, Carlsbad, CA, USA) supplemented with 10% foetal bovine serum, 100 U/ml penicillin, 100 μg/ml streptomycin. The Raw264.7 macrophages were cultured in vitro and divided into three groups: low glucose group (LG; 5.5 mmol/l glucose), high glucose group (HG; 25 mmol/l glucose) and hypertonic group (5.5 mmol/l glucose+19.5 mmol/l mannitol, HO). The Raw264.7 macrophages were harvested for mRNAs and proteins at different time points of treatment by the PI3K inhibitor LY294002, STAMP2 siRNAs or recombinant STAMP2-expressing adenovirus.
SiRNA transfection
Transfection was performed with Lipofectamine 2000 reagent (0.5 μl, Invitrogen). For reporter assays, 1.5 × 105 cells were seeded in 12-well plates at least 12 hrs before transfection. Cells were transfected with 2 μg of siRNA-STAMP2 or 2 μg of negative control (NC) siRNA. Transfection was performed using the following primers: STAMP2, forward 5′-GCA GCA UCC AAG UCU GAC ATT-3′ and reverse 5′-UGU CAG ACU UGG AUG CUG CTT3-3′; NC, forward 5′-UUC UCC GAA CGU GUC ACG UTT-3′ and reverse 5′-ACG UGA CAC GUU CGG AGA ATT3-3′. After incubation for 6 hrs, culture medium should be changed. Then the cells were observed under a laser scanning confocal microscopy (Leica TCS SP2; Leica) and the transfection efficiency was calculated. Twenty-four hours after transfection, cells were re-treated for 16 hrs with HG and harvested for further study.
Transfection of STAMP2 overexpressing adenovirus
The cells (1 × 106 cells/well) were administered virus in 200 MOI. The culture medium was changed after 12 hrs. Then the cells were observed under a laser scanning confocal microscopy. Twenty-four hours after transfection, cells were re-treated for 16 hrs with HG and harvested for further study.
Quantitative real-time RT-PCR
Total RNA was isolated from each aorta or 1 × 106 cells with the TRIzol reagent (Invitrogen, Carlsbad, CA, USA). RT-PCR was performed using the following primers: STAMP2, forward 5′-TCA AAT GCG GAA TAC CTT GCT-3′ and reverse 5′-GCA TCT AGT GTT CCT GAC TGG A-3′; β-actin, forward 5′-CAA CTT GAT TGA AGG CTT TGG T-3′ and reverse 5′-ACT TTT ATT GGT CTC AAG TCA GTG TAC AG-3′. Reactions were carried out on a real-time PCR thermocycler (IQ5 Real-Time PCR cycles; Bio-Rad, Hercules, CA, USA), using SYBR green as fluorescence dye. Relative expression analysis involved the 2−ΔΔCT method.
Western blot analysis
Western blot analysis was performed as described previously 26. We used antibodies against STAMP2 (Abcam), p-PI3K/PI3K, p-Akt/Akt, caspase-3 and Bcl-2 (Cell Signaling Technology, Beverly, MA, USA), followed by anti-IgG horseradish peroxidase-conjugated secondary antibody. STAMP2, p-PI3K, PI3K, p-Akt, Akt, caspase-3 and Bcl-2 protein levels were normalized to that of β-actin as an internal control and phosphospecific proteins to that of total protein.
Statistical analysis
Data are presented as mean ± SD (n is noted in the fig legends), and the statistical significance of differences was evaluated with an anova. Significance was accepted at the level of P < 0.05.
Results
Diabetic atherosclerosis is induced in ApoE−/−/LDLR−/− mice. We generated a non-genetic rodent model closely resembling human diabetic atherosclerosis disease by feeding male ApoE−/−/LDLR−/− mice with high fat diet and by the STZ treatment (referred to as DM mice hereafter), and normal diet (chow diet) fed mice serve as controls, as previously reported [24] (Materials and methods). Multiple metabolic parameters of the mice were examined to confirm the model.
Insulin resistance in the DM mice was confirmed by IPGTT. The levels of blood glucose in the DM group were similar to that of the control group at the age of 3 weeks (P > 0.05; Fig. 1A). However, at the age of 9 and 11 weeks, the levels of blood glucose in the DM group were significantly higher than the control mice at all of the time points tested (P < 0.05; Fig. 1A). Similarly, at the age of 9 and 11 weeks, the AUC for glucose level of the DM mice was higher compared with the control mice compared to that at baseline of 3 weeks (P < 0.05; respectively; Fig. 1B). Intraperitoneal glucose tolerance test results in the control mice had no significant changes between week-3 and week-11.
Figure 1.
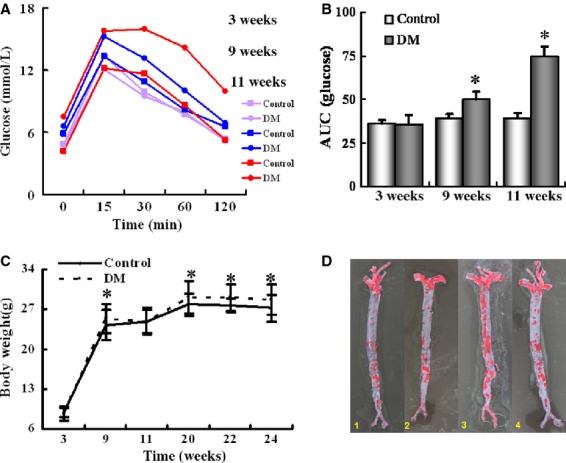
Generation of diabetic atherosclerosis mouse model. (A) ApoE−/−/LDLR−/− mice were fed on chow diet (Chow) or high-fat diet (DM) and analysed by IPGTT at 3, 9 and 11 weeks of feeding. (B) Area under curve in 3, 9 and 11-week-old ApoE−/−/LDLR−/− mice. (C) Bodyweight at the ages of 3, 9, 11, 20, 22 and 24 weeks. (D) Representative en face staining of brachiocephalic artery of 24-week-old mice (red indicates positive staining). Experimental groups are indicated as follows: 1–4: Control+ Vehicle mice (Column 1), Control+ STAMP2 mice (Column 2), DM+ Vehicle mice (Column 3), DM+ STAMP2 mice (Column 4). Data are mean ± SD; *P < 0.05 versus chow (3 weeks).
As expected, the bodyweight was significantly higher in the DM group than in the control group at and after week-9 except at week-11 (P < 0.05 for all pair-wise comparisons; Fig. 1C).
We also confirmed atherosclerosis as previously reported 27 in the DM mice at the age of 24 weeks after high fat diet and STZ treatment (Fig. 1D).
In summary, the diabetic model induced by a HF diet and STZ showed typical features of IR, hyperglycaemia, obesity and aorta lipid accumulation, resembling the state of human diabetic atherosclerosis disease.
Endogenous STAMP2 expression is suppressed in atherosclerotic aorta
To study the local functional role of STAMP2 in atherogenesis, we investigated the mRNA and protein expression levels of STAMP2 in the aorta of the diabetic mice and the non-diabetic mice. The mRNA level of STAMP2 in aorta was significantly decreased in the DM Vehicle group compared with that in the Control Vehicle group, as assessed by the RT-PCR (0.27 ± 0.08 versus 1.00 ± 0.00; P < 0.01; Fig. 2A). Consistent with the mRNA expression levels, the protein level of STAMP2 was also significantly decreased by 28%, in the DM Vehicle group compared with in the Control (non-diabetic) vehicle group, as assessed by Western blot analysis (0.55 ± 0.02 versus 0.76 ± 0.04; P < 0.01; Fig. 2B and C). These data demonstrate the correlation between the atherosclerotic aorta and low levels of STAMP2 expression.
Figure 2.
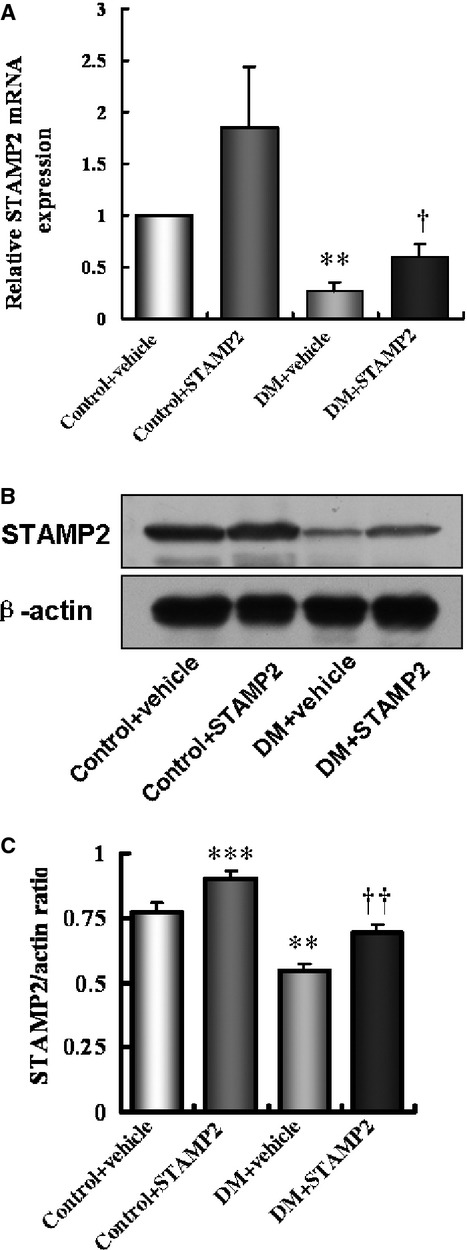
Overexpression of STAMP2 in aorta. Experimental groups are indicated as follows: (A–C) Control+ Vehicle mice (Column 1), Control+ STAMP2 mice (Column 2), DM+ Vehicle mice (Column 3), DM+ STAMP2 mice (Column 4). (A) Relative mRNA expression of STAMP2. (B and C) Western blot analysis of STAMP2. Data are mean ± SD;n = 4 per group; **P < 0.01, ***P < 0.001 versus Control + Vehicle mice, †P < 0.05, † †P < 0.01 versus DM + Vehicle mice.
Overexpression of STAMP2 in aorta
Although we showed a lower STAMP2 expression in atherosclerotic aorta as compared with normal aorta, it is not known if it contributes to atherogenesis or merely one of the consequences of atherogenesis. Thus, we asked if overexpression of STAMP2 in aorta would be sufficient to reduce atherogenesis.
As expected, the mRNA expression of STAMP2 in aorta was up-regulated in the DM STAMP2 mice group by 122% compared to the DM Vehicle mice group (0.60 ± 0.12 versus 0.27 ± 0.08; P < 0.05; Fig. 2A), and the STAMP2 mRNA level was increased in the Control STAMP2 mice group compared with the Control Vehicle mice group (1.85 ± 0.60 versus 1.00 ± 0.00; P = 0.22; Fig. 2A). Consistent with the STAMP2 mRNA level changes, the STAMP2 protein level was also increased in the DM STAMP2 mice group by 27% compared with the DM Vehicle mice group (0.70 ± 0.04 versus 0.55 ± 0.02; P < 0.001; Fig. 2B and C), and the STAMP2 protein level was increased in the Control STAMP2 mice group by 18% compared to the Control Vehicle mice group (0.90 ± 0.03 versus 0.76 ± 0.04; P < 0.001; Fig. 2B and C). The magnitude of protein level changes was smaller compared with that of the mRNA level changes.
Overexpression of STAMP2 improves metabolism in ApoE−/−/LDLR−/− mice
STAMP2 was shown previously to play an important role in regulating metabolism, IR and atherogenesis. We investigated the global effect of STAMP2 by systemic overexpression of STAMP2 in our mouse model of diabetes and atherosclerosis. STAMP2-expressing adenovirus was used to treat diabetic and non-diabetic ApoE−/−/LDLR−/− mice by jugular vein injection and the expression vehicle adenovirus was used as control.
As expected from our previous results, several metabolic indices were substantially altered in the diabetic mice. Comparing the DM Vehicle group to the Control Vehicle group, the bodyweight, the level of fasting serum glucose and cholesterol were significantly increased (Table 1). After overexpression of STAMP2, the indices decreased numerically in the DM STAMP2 group versus DM Vehicle group, although for most part not reaching statistically significance (Table 1). Calculation of the HOMA index as a measure of IR in the end of this experiment revealed an increase in the DM Vehicle group versus the Control Vehicle group (Table 1). In contrast, HOMA decreased in the DM STAMP2 group versus the DM Vehicle group (Table 1).
Table 1.
Metabolic parameters of ApoE−/−/LDLR−/− mice
| Control + Vehicle (n = 10) | Control + STAMP2 (n = 10) | DM + Vehicle (n = 10) | DM + STAMP2 (n = 10) | P | |
|---|---|---|---|---|---|
| 22w | |||||
| Bodyweight (g) | 27.82 ± 0.92 | 27.23 ± 1.58 | 31.06 ± 2.97** | 30.18 ± 3.37* | 0.003 |
| 24w | |||||
| Bodyweight (g) | 27.32 ± 1.69 | 27.34 ± 2.57 | 30.90 ± 2.61* | 29.36 ± 1.72* | 0.002 |
| FBG (mmol/l) | 6.67 ± 2.51 | 5.01 ± 2.01 | 20.42 ± 12.10*** | 7.06 ± 2.42††† | 0.000009 |
| Insulin (μIU/ml) | 28.59 ± 14.43 | 24.51 ± 10.31 | 41.09 ± 18.15 | 27.90 ± 10.35 | 0.053 |
| HOMA-IR | 9.56 ± 7.67 | 6.06 ± 3.62 | 35.07 ± 25.37*** | 8.27 ± 3.28††† | 0.00005 |
| TG (mmol/l) | 1.52 ± 0.45 | 1.53 ± 0.42 | 2.17 ± 1.33 | 1.56 ± 0.76 | 0.233 |
| TC (mmol/l) | 16.77 ± 2.98 | 17.78 ± 4.29 | 34.94 ± 17.82*** | 20.99 ± 4.60†† | 0.0004 |
| FFA (mmol/l) | 61.70 ± 32.24 | 69.86 ± 39.14 | 93.54 ± 53.40 | 49.20 ± 23.03 | 0.089 |
P < 0.05
P < 0.01
P < 0.001 versus Control+ Vehicle
P < 0.01
P < 0.001 versus DM + Vehicle.
Data are expressed as mean ± SD. Serum sampling was taken under fasting condition and measured.
FBG: fasting blood glucose; TG: total triglycerides; TC: total cholesterol; FFA: free fatty acids.
Taken together, these data suggest that systemic STAMP2 overexpression does not have obvious deleterious effect, and can modestly reverse the metabolic disease state of the diabetic ApoE−/−/LDLR−/− mice.
Overexpression of STAMP2 stabilizes lesions in the brachiocephalic artery
Spontaneous plaque rupture has been demonstrated in the BCA of mice in recent research 28. We asked whether the overexpression of STAMP2 is able to affect the stability of atherosclerotic plaque.
The area, lipid content and macrophage quantity in BCA were known previously to be associated with the plaque stability. We found that each of the three parameters in DM Vehicle group was significantly increased compared with those in the Control Vehicle group (Fig. 3A1–A3; Table 2). STAMP2 overexpression lead to significant less area, lipid content and macrophage quantity in the DM STAMP2 group compared with those in the DM Vehicle group (Fig. 3A1–A3; Table 2). Similarly, STAMP2 overexpression also reduced each of the three parameters in the non-diabetic mice, the Control STAMP2 group, compared with the Control Vehicle group (Fig. 3A1–A3; Table 2).
Figure 3.

STAMP2 increases the stability of brachiocephalic plaques in 24-week-old diabetic ApoE−/−/LDLR−/− mice. Experimental groups are indicated as follows: (A–C) Control + Vehicle mice (Column 1), Control + STAMP2 mice (Column 2), DM + Vehicle mice (Column 3), DM + STAMP2 mice (Column 4). (A) Pathology features of ApoE−/−/LDLR−/− mice with or without STAMP2 overexpression. Five-micrometer sequential sections from the brachiocephalic artery (BCA) are shown. Representative haematoxylin and eosin staining of brachiocephalic lesions sections (A1). Representative Oil-Red-O-positive staining of brachiocephalic lesions sections for lipid (A2). Representative immunohistochemistry for macrophage (MOMA-2) of brachiocephalic lesions sections (A3). Representative masson staining of brachiocephalic lesions sections (collagen is green; A4). Representative Picrosirius Red staining of brachiocephalic lesions sections for collagen (A5). Representative immunohistochemistry for SMC (α-actin) brachiocephalic lesions sections (A6); scale bar = 100 μm; n = 5 per group. (B) Fluorescent imaging of cell apoptosis. Five-micrometer sequential sections from the BCA were stained for apoptosis (×103 μm2). Red, TUNEL; blue, DAPI nuclear stain; scale bar = 100 μm; n = 5 per group. (C) Fluorescent imaging of macrophage apoptosis (×103 μm2). Five-micrometer sequential sections from the BCA were stained for apoptosis. Red, TUNEL; green, macrophage; blue, DAPI nuclear stain; scale bar = 100 μm; n = 5 per group.
Table 2.
Histology analyses of brachiocephalic atherosclerotic plaques
| Control + Vehicle (n = 5) | Control + STAMP2 (n = 5) | DM + Vehicle (n = 5) | DM + STAMP2 (n = 5) | P | |
|---|---|---|---|---|---|
| Plaque area (%) | 29.56 ± 0.42 | 18.24 ± 4.61*** | 45.68 ± 0.89*** | 30.98 ± 3.98††† | 3 × 10−9 |
| Lipid (%) | 33.98 ± 3.26 | 24.32 ± 2.72** | 51.82 ± 1.19*** | 40.72 ± 2.67**,††† | 1 × 10−10 |
| Macrophage (%) | 14.84 ± 0.68 | 13.72 ± 1.25 | 28.38 ± 2.61*** | 17.71 ± 1.78*,††† | 1 × 10−9 |
| Collagen content (%) | 16.29 ± 2.11 | 17.50 ± 0.89 | 7.98 ± 1.25*** | 10.32 ± 0.23***,† | 5 × 10−9 |
| I–III collagen ratio | 1.39 ± 0.17 | 2.08 ± 0.28*** | 1.20 ± 0.05 | 1.61 ± 0.22†† | 2 × 10−5 |
| SMC (%) | 10.21 ± 1.69 | 14.39 ± 1.35*** | 7.12 ± 1.34** | 8.24 ± 0.98* | 1 × 10−6 |
| Vulnerability index | 1.87 ± 0.31 | 1.20 ± 0.13* | 5.37 ± 0.63*** | 3.16 ± 0.31***,††† | 1 × 10−10 |
P < 0.05
P < 0.01
P < 0.001 versus Control+ Vehicle
P < 0.01
P < 0.001 versus DM + Vehicle.
Data are expressed as mean ± SD. SMC, smooth muscle cell. Vulnerability index = (macrophages + extracellular lipids)/(SMC + collagen fibres).
Brachiocephalic arteries collagen content was known to affect the stability of atherosclerotic plaques. We used Masson's trichrome staining and Picrosirius red staining to measure BCA collagen content. Comparing to the BCA plaques of the Control Vehicle group, the BCA plaques of the DM Vehicle group had lower collagen (51% reduction), collagen I–III ratio (14% reduction, P = 0.15) and smooth muscle cell (SMC) content (30% reduction; Fig. 3A4–A6; Table 2), indicating a highly instable state. STAMP2 overexpression appears to be able to correct the situation. STAMP2 overexpression increased the collagen content by 29%, increased the collagen I–III ratio by 34% and SMC content by 16% (P = 0.21) in the BCA plaques, when comparing the DM STAMP2 group with the DM Vehicle group (Fig. 3A4–A6; Table 2). STAMP2 overexpression also increased the collagen content by 7% (P = 0.16), the collagen I–III ratio by 50% and SMC content by 41% in the BCA plaques of the non-diabetic mice, the Control STAMP2 group, when compared with the Control Vehicle group (Fig. 3A4–A6; Table 2).
The vulnerability index (VI) was used to describe atherosclerotic plaque stability in previous studies 29. In our study, VI was significantly higher, by about threefold, in the DM Vehicle group than in the Control Vehicle group (Table 2). STAMP2 overexpression decreased VI in the DM STAMP2 group by 41% compared with the DM Vehicle group (Table 2).
Taken together, these results showed that the BCA plaques are destabilized in the diabetic mice and that STAMP2 overexpression is sufficient to increase the stability of the BCA plaques, thereby likely preventing plaque rupture.
Overexpression of STAMP2 reduces macrophages apoptosis in the BCA
Cell apoptosis, especially macrophage apoptosis, has significant roles in the atherosclerosis process. We investigated whether the STAMP2 overexpression exhibited an anti-apoptotic effect.
Thin sections of mice BCA were stained by TUNEL which labels apoptotic cells. We found that cell apoptosis was significantly higher, by 86%, in the DM Vehicle group than the Control Vehicle group (174.16 ± 13.55 versus 93.71 ± 0.78 × 103 μm2; P < 0.001; Fig. 3B). STAMP2 overexpression significantly reduced TUNEL staining in the BCA of the DM STAMP2 group compared with the DM Vehicle group (98.88 ± 12.52 versus 174.16 ± 13.55 × 103 μm2; P < 0.001; Fig. 3B). The Control STAMP2 group had less cell apoptosis compared with the Control Vehicle group (82.78 ± 1.01 versus 93.71 ± 0.78 × 103 μm2; P < 0.001; Fig. 3B).
We further analysed the macrophage apoptosis in the BCA plaques by double staining the BCA plaques with TUNEL and an antibody against MOMA-2, a macrophage marker. There was a significantly higher number of TUNEL+ and MOMA-2+ macrophages in the DM Vehicle group than the Control Vehicle group (136.50 ± 13.26 versus 52.63 ± 4.83 × 103 μm2; P < 0.001; Fig. 3C). Overexpression STAMP2 significantly reduced macrophage apoptosis in the BCA plaques of the DM STAMP2 group compared with the DM Vehicle group (65.42 ± 6.26 versus 136.50 ± 13.26 × 103 μm2; P < 0.001; Fig. 3C) and in the Control STAMP2 group compared with the Control vehicle (35.24 ± 7.45 versus 52.63 ± 4.83 × 103 μm2; P < 0.01; Fig. 3C).
These data showed that macrophage apoptosis is substantially increased in the atherosclerotic plaques of diabetic mice. The result suggested that one of the cellular mechanisms by which STAMP2 overexpression increases the stability of atherosclerotic plaques is to decrease the macrophage apoptosis.
Overexpression of STAMP2 activates the Akt signalling pathway in aorta plaques of ApoE−/−/LDLR−/− mice
It was known previously that STAMP2 regulates Akt signalling pathway 22 and Akt pathway controls cell survival by regulating the apoptotic machinery such BCL family of anti-apoptotic factors. Thus, we investigated whether one of the molecular mechanisms by which STAMP2 affects atherogenesis and cell apoptosis in plaques is through Akt pathway regulation.
We have shown that the STAMP2 mRNA and protein levels in aorta were significantly decreased in the DM Vehicle group compared with the Control Vehicle group (Fig. 2A–C). This appears to cause the down-regulation of Akt activity in aorta. Phosphorylation of Akt, the measure of Akt activation, was decreased in the DM Vehicle group by 28% compared with the Control Vehicle group (0.26 ± 0.01 versus 0.36 ± 0.00; P < 0.001; Fig. 4A and B). Since Akt phosphorylation activates the BCL family of anti-apoptotic factors, as expected, Bcl-2 was down-regulated by 32% in the DM Vehicle group compared with the Control Vehicle group (0.38 ± 0.01 versus 0.56 ± 0.03; P < 0.01; Fig. 4A and C). Consistent with the Bcl-2 level changes, caspase-3 protein level was up-regulated by 59% in the DM Vehicle group compared with the Control Vehicle group (0.89 ± 0.00 versus 0.56 ± 0.02; P < 0.001; Fig. 4A and D), which may explain the 86% increase in BCA cell apoptosis observed (Fig. 3B; see above section). STAMP2 overexpression significantly increased the aorta p-Akt level by 27% in the DM STAMP2 group compared with the DM Vehicle group (0.33 ± 0.02 versus 0.26 ± 0.01, P < 0.001; Fig. 4A and B). As expected, comparing the DM STAMP2 group to the DM Vehicle group, the Bcl-2 protein level was increased by 26% (0.48 ± 0.01 versus 0.38 ± 0.01, P < 0.001; Fig. 4A and C), but caspase-3 protein level was decreased by 13% (0.77 ± 0.02 versus 0.89 ± 0.00; P < 0.001; Fig. 4A and D). Similarly, p-Akt level was up-regulated by 8% in the Control STAMP2 group compared with the Control Vehicle group (0.39 ± 0.02 versus 0.36 ± 0.00; P < 0.05; Fig. 4A and B). Bcl-2 protein level was increased (0.75 ± 0.03 versus 0.56 ± 0.03; P < 0.001; Fig. 4A and C) and caspase-3 protein level was decreased (0.42 ± 0.01 versus 0.56 ± 0.02; P < 0.001; Fig. 4A and D) in the Control STAMP group compared with the Control Vehicle group.
Figure 4.
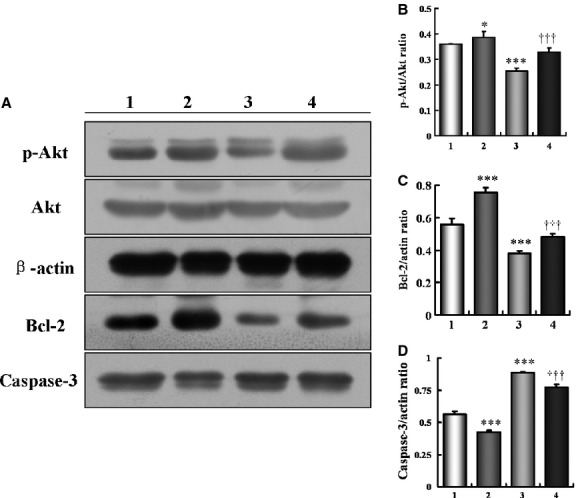
Overexpression of STAMP2 activates STAMP2/Akt signalling pathway in diabetic ApoE−/−/LDLR−/− mice. Experimental groups are indicated as follows: (A–D) Control + Vehicle mice (Column 1), Control + STAMP2 mice (Column 2), DM + Vehicle mice (Column 3), DM + STAMP2 mice (Column 4). (A–D) Western blot analysis of p-Akt/Akt, Bcl-2 and caspase-3. Data are mean ± SD;n = 4 per group; *P < 0.05, ***P < 0.001 versus Control + Vehicle mice, †††P < 0.001 versus DM + Vehicle mice.
These results suggested that the STAMP2 reduces the pathogenesis of atherosclerosis by activating the Akt signalling pathway and preventing cell apoptosis, including macrophage apoptosis, in the plaques of diabetic mice.
Macrophage cell line model in hyperglycaemic condition
To further investigate the molecular mechanisms underlining STAMP2 function in diabetic mice, we established a macrophage cell line model in hyperglycaemic condition in vitro which recapitulates some of the characteristics of macrophages in the diabetic mouse model.
RAW 264.7 is a mouse monocyte/macrophage cell line. The cells were cultured under a LG (5.5 mmol/l), HG (25 mmol/l glucose) and the control hypotonic (5.5 mmol/l glucose+19.5 mmol/l mannitol, HO) conditions.
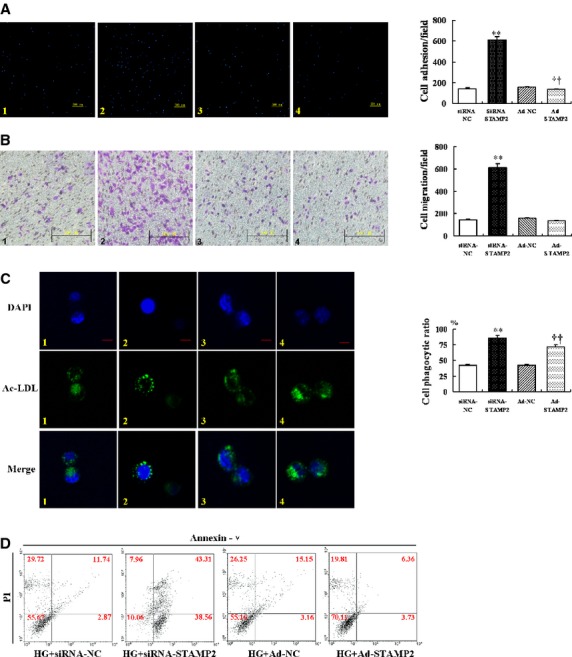
We found that cells from the HG treated group had significantly higher abilities of adhesion, migration and phagocytosis than the LG and the HO treated group (P < 0.05; Fig. 5A–C).
Figure 5.
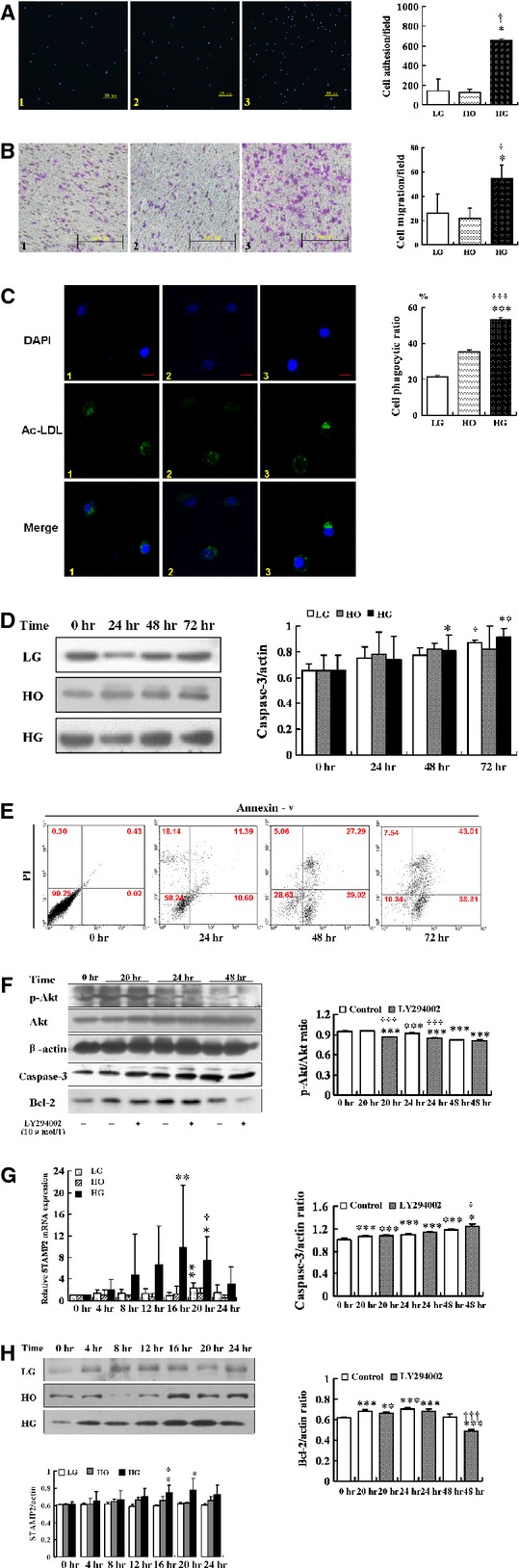
The effects of hyperglycaemic treatment on macrophage apoptosis and functions. Experimental groups are indicated as follows: (A–C) Low glucose (LG, 5.5 mmol/l) group (Column 1); Hypertonic (HO, 19.5 mmol/l mannitol + 5.5 mmol/l glucose) group (Column 2); High glucose (HG, 25 mmol/l) group (Column 3). Images and quantification of macrophages adhesion (A), migration (B) and phagocytosis (C). (A) High glucose can increase adhesion ability in RAW264.7. Blue, DAPI nuclear stain; scale bar = 200 μm. Data are mean ± SD; *P < 0.05 versus LG; †P < 0.05 versus HO. (B) High glucose can increase migration ability in RAW264.7; scale bar = 100 μm; data are mean ± SD; *P < 0.05 versus LG; †P < 0.05 versus HO. (C) High glucose can increase phagocytosis in RAW264.7. Green, Ac-LDL-488; blue, DAPI nuclear stain; scale bar = 5 μm. Data are mean ± SD; ***P < 0.001 versus LG; †††P < 0.001 versus HO. (D) Western blot analysis of caspase-3 by high glucose treatment. Data are mean ± SD; *P < 0.05, **P < 0.01 versus HG (0 hr); †P < 0.05 versus LG (0 hr). (E) RAW264.7 stained for Annexin V-PI to test apoptosis assay according to the instructions of the manufacturer. Data were analysed using WinMDI software. Representative dot plots are shown for each group. (F) After inhibited PI3K activation by LY294002 (10 μmmol/l), western blot analysis of p-Akt/Akt, caspase-3 and Bcl-2 in RAW264.7. Data are mean ± SD; *P < 0.05, **P < 0.01, ***P < 0.001 versus Control (0 hr); †P < 0.05, †††P < 0.001 versus Control (20, 24, 48 hrs). (G) Relative mRNA expression of STAMP2 in RAW264.7. Data are mean ± SD; *P < 0.05, **P < 0.01 versus LG/HG (0 hr); †P < 0.01 versus LG (20 hrs). (H) Western blot analysis of STAMP2 in RAW264.7. Data are mean ± SD; *P < 0.05 versus HG (0 hr); †P < 0.05 versus LG (16 hrs).
High glucose treatment also increased RAW 264.7 cell apoptosis. Caspase-3 protein level showed a time-dependent increase with a maximum at 72 hrs in the HG treated cells compared with 0 hr (P < 0.01; Fig. 5D). Flow cytometry analysis based on Annexin-V and PI staining showed that the HG treatment induced higher level of apoptosis at different time points compared with the control cells (time 0 hr; Fig. 5E).
High glucose treatment effect on cell apoptosis is mediated by the Akt signalling pathway. As western blot analysis showed, LY294002 inhibited Akt phosphorylation under HG treatment in 20 hrs (0.86 ± 0.01 versus 0.95 ± 0.00, P < 0.001), 24 hrs (0.84 ± 0.00 versus 0.92 ± 0.01, P < 0.001), 48 hrs (0.81 ± 0.01 versus 0.81 ± 0.01, P = 0.44), respectively, compared with no LY294002 treatment (Fig. 5F). Western blot analysis showed that the protein level of caspase-3 increased in 20 hrs (1.32 ± 0.02 versus 1.30 ± 0.03, P = 0.29), 24 hrs (1.36 ± 0.01 versus 1.34 ± 0.03, P = 0.25), 48 hrs (1.25 ± 0.07 versus 1.38 ± 0.03, P < 0.05), respectively, compared with no LY294002 treatment (Fig. 5F). However, the level of protective protein Bcl-2 decreased compared with no LY294002 treatment in 20 hrs (0.66 ± 0.02 versus 0.68 ± 0.02, P = 0.15), 24 hrs (0.68 ± 0.02 versus 0.71 ± 0.02, P = 0.11), 48 hrs (0.49 ± 0.01 versus 0.63 ± 0.03, P < 0.001; Fig. 5F), respectively.
These data would suggest that HG could suppress Akt signalling pathway to induce macrophages apoptosis. We showed that in our diabetic mouse model, endogenous STAMP2 expression was reduced in aorta. However, it was not known if STAMP2 expression was directly affected in macrophage cells by hyperglycaemia and if down-regulation of STAMP2 is sufficient to induce the phenotypic changes of macrophages seen in diabetic mice. To answer these questions, we studied STAMP2 function in the RAW264.7 cell line model under the hyperglycaemic condition.
The relative mRNA expressions of STAMP2 were significantly increased by 16 hrs HG treatment compared with 0 hr HG treatment (9.90 ± 11.45 versus 1.00 ± 0.00, P < 0.01; Fig. 5G). Although LG treatment slightly up-regulated STAMP2 mRNA level with a maximum at 20 hrs (2.32 ± 0.98 versus 1.00 ± 0.00, P < 0.01; Fig. 5G), in comparison, the effect of HG treatment is much higher. As expected, there was no significant change STAMP2 expression by hypertonic (HO) treatment (P > 0.05). Consistently, the STAMP2 protein levels under HG treatment also increased at different time points compared with at 0 hr time point with maximum reached at 20 hrs treatment (P < 0.05; Fig. 5H). The LG treatment had minimal effect and HO treatment had no effect (P > 0.05; Fig. 5H).
We investigated the role of STAMP2 in regulating the Akt signalling pathway to control cell apoptosis under HG treatment. Western blot analysis showed that the protein level of STAMP2 was 20% lower in the RNAi-STAMP2 group than the RNAi-Normal Control (RNAi-NC) group (0.78 ± 0.10 versus 0.97 ± 0.03; P < 0.05; Fig. 6A and B). After silence of STAMP2, the phosphorylated PI3K and phosphorylated Akt were reduced by 1% (0.81 ± 0.06 versus 0.82 ± 0.01; P = 0.84) and 11% (0.76 ± 0.06 versus 0.85 ± 0.03; P < 0.05; Fig. 6A, C and D), respectively, in RNAi-STAMP2 group compared with RNAi-NC group. However, the caspase-3 protein level resulted in a significant increase by 4-fold in RNAi-STAMP2 group compared with RNAi-NC group (0.90 ± 0.02 versus 0.65 ± 0.05; P < 0.001; Fig. 6A and E). The Bcl-2 protein level was reduced by 57% (0.30 ± 0.23 versus 0.70 ± 0.16; P < 0.05; Fig. 6A and F). Taken together, these data showed that STAMP2 silencing suppresses the PI3K/Akt signalling pathway and increases cell apoptosis.
Figure 6.
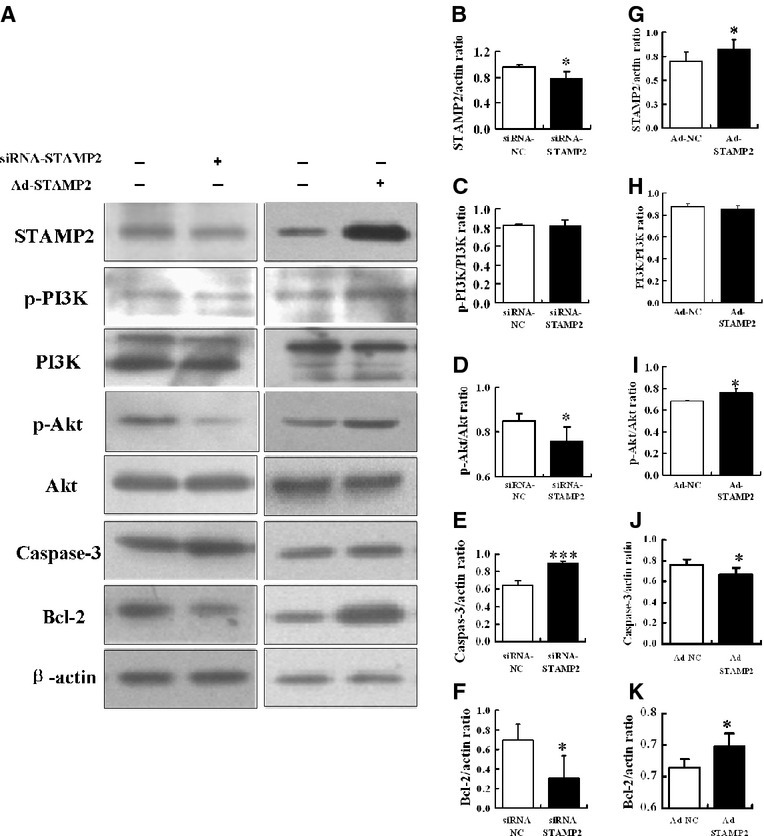
Silence or overexpression of STAMP2 under HG treatment. (A–F) After silence of STAMP2 in RAW264.7, western blot analysis of STAMP2, p-PI3K/PI3K, p-Akt/Akt, caspase-3 and Bcl-2. Data are mean ± SD; *P < 0.05, ***P < 0.001 versus siRNA-NC. (A, G–K) After overexpression of STAMP2 in RAW264.7, western blot analysis of STAMP2, p-PI3K/PI3K, p-Akt/Akt, caspase-3 and Bcl-2. Data are mean ± SD; *P < 0.05 versus Ad-NC.
STAMP2 silencing in the RNAi-STAMP2 group significantly increased cell adherence, migrate, phagocytosis and apoptosis abilities compared with the RNAi-NC group (P < 0.01 for all; Fig. 7 A1, A2; B1, B2; C1, C2; D).
Figure 7.
The macrophage functions after silence or overexpression of STAMP2 under HG treatment. Experimental groups are indicated as follows: (A–C) siRNA-Normal Control group (siRNA-NC, Column 1); siRNA-STAMP2 group (Column 2); Ad-Normal Control group (Ad-NC, Column 3); Ad-STAMP2 group (Column 4). Images and quantification of RAW264.7 adhesion (A), migration (B), phagocytosis (C) and apoptosis (D). (A) Silence or overexpression of STAMP2 is associated with adhesion ability in RAW264.7. Blue, DAPI nuclear stain; scale bar = 200 μm. Data are mean ± SD; **P < 0.01 versus siRNA-NC, ††P < 0.01 versus Ad-NC. (B) Silence or overexpression of STAMP2 is associated with migration in RAW264.7; scale bar = 100 μm. Data are mean ± SD; **P < 0.01 versus siRNA-NC. (C) Silence or overexpression of STAMP2 is associated with phagocytosis ability in RAW264.7. Green, Ac-LDL-488; blue, DAPI nuclear stain; scale bar = 10 μm. Data are mean ± SD; **P < 0.01 versus siRNA-NC; ††P < 0.01 versus Ad-NC. (D) Silence or overexpression of STAMP2 is associated with apoptosis in RAW264.7. RAW264.7 stained for Annexin V–PI to test apoptosis assay according to the instructions of the manufacturer. Data were analysed using WinMDI software.
Overexpression of STAMP2 on cell apoptosis under HG treatment
As seen in Figure 6A and G, western blot analysis showed that the protein level of STAMP2 was increased by 17% in the STAMP2-expressing adenovirus (Ad-STAMP2) group compared to Normal Control-expressing adenovirus (Ad-NC) group (0.82 ± 0.06 versus 0.70 ± 0.04; P < 0.05). The phosphorylated PI3K was not changed in Ad-STAMP2 group compared with Ad-NC group (0.87 ± 0.03 versus 0.86 ± 0.03; P = 0.48; Fig. 6A and H). However, the phosphorylated Akt was significantly enhanced in Ad-STAMP2 group (0.76 ± 0.04 versus 0.69 ± 0.00; P < 0.05; Fig. 6A and I). Meanwhile, caspase-3 protein level resulted in a significantly decrease in Ad-STAMP2 group compared with Ad-NC group (0.67 ± 0.06 versus 0.76 ± 0.06; P < 0.05; Fig. 6A and J). The protein level of Bcl-2 was increased in the Ad-STAMP2 group (0.72 ± 0.02 versus 0.68 ± 0.02; P < 0.05; Fig. 6A and K). The results indicate that the activation of STAMP2/PI3K/Akt signalling pathway is responsible to reduce cell apoptosis.
Overexpression of STAMP2 and macrophage functions
Results have shown that cells from Ad-STAMP2 group showed significantly lower adhesion ability and higher phagocytosis ability compared with Ad-NC group (P < 0.01; Fig. 7A3, 4 and C3, 4). However, migration ability had no change in Ad-STAMP2 group compared with Ad-NC group (P = 0.09; Fig. 7B3, 4). The cells from Ad-STAMP2 group showed less apoptosis compared with Ad-NC group (P < 0.001; Fig. 7D). These results suggested that silence or overexpression of STAMP2 affect cells abilities of adhesion, migration, phagocytosis and apoptosis.
Discussion
The major finding of this study is that overexpression of STAMP2 could effectively improve metabolic indices, reduce IR and stabilize the atherosclerotic plaques in the diabetic ApoE−/−/LDLR−/− mice. Mechanistically, this phenotype is due to STAMP2 regulation of the Akt signalling pathway, thus diminishing macrophage apoptosis and stabilizing the vulnerable plaques.
STAMP2 may be causally associated with IR, playing an important role in the regulation of glucolipid metabolism. For example, after STAMP2 is suppressed, glucose transport stimulated by insulin and Glut4 inversion to membrane mediated by insulin is significantly reduced 22. In addition, STAMP2−/− mice showed IR, glucose intolerance and lipid metabolism disorder 22. We found a significant increase in bodyweight, blood glucose levels, total serum cholesterol and triglyceride levels in diabetic ApoE−/−/LDLR−/− mice. Overexpression of STAMP2 effectively improves metabolic indices and reduces IR in diabetic ApoE−/−/LDLR−/− mice, although there was no significant effect on serum insulin, FFA and TG level. Our data provide evidence that STAMP2 effectively reduces risk factors of atherosclerosis in the diabetic mouse model.
The relationship between macrophage apoptosis and atherosclerosis is complex. Advanced macrophage apoptosis is an important contributing factor in vulnerable atherosclerotic lesions 13. STAMP2 was expressed in both human and mouse atherosclerotic plaques 15. In line with our observations in the brachiocephalic plaques of ApoE−/−/LDLR−/− mice, as assessed by staining for TUNEL, macrophage apoptosis was significantly higher in diabetes group than in chow diet group. With the overexpression of STAMP2, the macrophage apoptosis in lesions of diabetic ApoE−/−/LDLR−/− mice was diminished. The lesions in brachiocephalic artery of diabetic ApoE−/−/LDLR−/− mice were demonstrated to be vulnerable. Some phenotypic characteristics of atherosclerotic plaques such as area of plaque, lipid content and macrophage quantity significantly increased. However, collagen content, SMC content in lesions were reduced. Usually increased content of lipid and macrophage or decreased content of collagen and SMC in patches have been widely used as indicators of plaque instability. We confirmed that overexpression of STAMP2 could lead to more contents of collagen, more SMC, less contents of lipid and less macrophage, thus increasing the stability of the plaques. Consequently, overexpression of STAMP2 could reduce the vulnerability of atherosclerotic plaques through diminishing macrophage apoptosis effectively.
Akt signalling pathway involved in regulation of many cell functions is a key component in the regulation of macrophage apoptosis 30,31. We showed in a cellular model that the expressions of STAMP2 mRNA and protein are increased initially by HG treatment but eventually decreased after extended treatment. We also showed in a diabetic mouse model where serum glucose level is persistently high, STAMP2 expression is also suppressed. In vitro, macrophage cell line RAW264.7 exhibited significantly increased adhesion, migration and phagocytosis abilities and apoptosis with HG treatment. Cell apoptosis induced by HG treatment is also induced by the PI3K inhibitor LY294002, indicating that the STAMP2/PI3K/Akt pathway play an important role in macrophage apoptosis under HG.
We showed that in a macrophage cellular model, STAMP2 expression is necessary and sufficient to regulate the capabilities of migration, adhesion, phagocytosis and apoptosis by the loss-of-function experiment via STAMP2 siRNA and by the gain-of-function experiment via STAMP2 overexpression.
Conclusions
This study revealed that STAMP2 activity is important in increasing plaque stability in diabetic atherosclerotic mice, which is likely to function by activating the Akt signal pathway and suppressing apoptosis machinery to decrease the macrophage apoptosis in the atherosclerotic plaques. In addition to providing important information about the pathophysiology of atherosclerosis, the data from these studies might form the basis of novel therapeutic approaches to combat vulnerable plaque.
Acknowledgments
This work was supported by the research grants from the Independent Innovation Foundation of Shandong University (2012JC034), the Natural Science Foundation of Shandong Province (ZR2009CM022 and ZR2009CM025), the National Natural Science Foundation of China (30971215, 81070141, 81070192, 81100605, 81270352 and 81270287), the National Basic Research Program of China (973 Program, grant no. 2013CB530700), the Key Technologies R & D Program of Shandong Province (2010G0020262).
Conflicts of interest
The authors declare that there is no duality of interest associated with this manuscript.
Author contribution
Jia Wang and Lu Han researched data and wrote the manuscript. Zhi-hao Wang researched data and contributed to discussion. Wen-yuan Ding, Yuan-yuan Shang, Meng-xiong Tang and Wen-bo Li researched data. Wei Zhang and Ming Zhong wrote, reviewed and edited the manuscript. Yun Zhang reviewed and edited the manuscript.
References
- 1.Howard BV, Rodriguez BL, Bennett PH, et al. Prevention conference VI: diabetes and cardiovascular disease. Writing group I: epidemiology. Circulation. 2002;105:132–7. doi: 10.1161/01.cir.0000013953.41667.09. [DOI] [PubMed] [Google Scholar]
- 2.Hurst RT, Lee RW. Increased incidence of coronary atherosclerosis in type 2 diabetes mellitus: mechanisms and management. Ann Intern Med. 2003;139:824–34. doi: 10.7326/0003-4819-139-10-200311180-00010. [DOI] [PubMed] [Google Scholar]
- 3.Sethi SS, Akl EG, Farkouh ME. Diabetes mellitus and acute coronary syndrome: lessons from randomized clinical trials. Curr Diab Rep. 2012;12:294–304. doi: 10.1007/s11892-012-0272-9. [DOI] [PubMed] [Google Scholar]
- 4.Moreno PR, Murcia AM, Palacios IF, et al. Coronary composition and macrophage infiltration in atherectomy specimens from patients with diabetes mellitus. Circulation. 2000;102:2180–4. doi: 10.1161/01.cir.102.18.2180. [DOI] [PubMed] [Google Scholar]
- 5.Naghavi M, Libby P, Falk E, et al. From vulnerable plaque to vulnerable patient: a call for new definitions and risk assessment strategies: part I. Circulation. 2003;108:1664–72. doi: 10.1161/01.CIR.0000087480.94275.97. [DOI] [PubMed] [Google Scholar]
- 6.Reaven GM. Banting Lecture 1988. Role of insulin resistance in human disease. Diabetes. 1988;37:1595–607. doi: 10.2337/diab.37.12.1595. [DOI] [PubMed] [Google Scholar]
- 7.Kahn CR. Banting Lecture. Insulin action, diabetogenes, and the cause of type II diabetes. Diabetes. 1994;43:1066–84. doi: 10.2337/diab.43.8.1066. [DOI] [PubMed] [Google Scholar]
- 8.Verschuren L, Kooistra T, Bernhagen J, et al. MIF deficiency reduces chronic inflammation in white adipose tissue and impairs the development of insulin resistance, glucose intolerance, and associated atherosclerotic disease. Circ Res. 2009;105:99–107. doi: 10.1161/CIRCRESAHA.109.199166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Han S, Liang CP, DeVries-Seimon T, et al. Macrophage insulin receptor deficiency increases ER stress-induced apoptosis and necrotic core formation in advanced atherosclerotic lesions. Cell Metab. 2006;3:257–66. doi: 10.1016/j.cmet.2006.02.008. [DOI] [PubMed] [Google Scholar]
- 10.Van Gaal LF, Mertens IL, De Block CE. Mechanisms linking obesity with cardiovascular disease. Nature. 2006;444:875–80. doi: 10.1038/nature05487. [DOI] [PubMed] [Google Scholar]
- 11.Lim WS, Timmins JM, Seimon TA, et al. Signal transducer and activator of transcription-1 is critical for apoptosis in macrophages subjected to endoplasmic reticulum stress in vitro and in advanced atherosclerotic lesions in vivo. Circulation. 2008;117:940–51. doi: 10.1161/CIRCULATIONAHA.107.711275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Seimon T, Tabas I. Mechanisms and consequences of macrophage apoptosis in atherosclerosis. J Lipid Res. 2009;50:382–7. doi: 10.1194/jlr.R800032-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tabas I. Consequences and therapeutic implications of macrophage apoptosis in atherosclerosis: the importance of lesion stage and phagocytic efficiency. Arterioscler Thromb Vasc Biol. 2005;25:2255–64. doi: 10.1161/01.ATV.0000184783.04864.9f. [DOI] [PubMed] [Google Scholar]
- 14.Kolodgie FD, Narula J, Burke AP, et al. Localization of apoptotic macrophages at the site of plaque rupture in sudden coronary death. Am J Pathol. 2000;157:1259–68. doi: 10.1016/S0002-9440(10)64641-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fernández-Hernando C, Ackah E, Yu J, et al. Loss of Akt1 leads to severe atherosclerosis and occlusive coronary artery disease. Cell Metab. 2007;6:446–57. doi: 10.1016/j.cmet.2007.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ono H, Sakoda H, Fujishiro M, et al. Carboxy-terminal modulator protein induces Akt phosphorylation and activation, thereby enhancing antiapoptotic, glycogen synthetic, and glucose uptake pathways. Am J Physiol Cell Physiol. 2007;293:1576–85. doi: 10.1152/ajpcell.00570.2006. [DOI] [PubMed] [Google Scholar]
- 17.Cho H, Mu J, Kim JK, et al. Insulin resistance and a diabetes mellitus-like syndrome in mice lacking the protein kinase Akt2 (PKB beta) Science. 2001;292:1728–31. doi: 10.1126/science.292.5522.1728. [DOI] [PubMed] [Google Scholar]
- 18.Senokuchi T, Liang CP, Seimon TA, et al. Forkhead transcription factors (FoxOs) promote apoptosis of insulin-resistant macrophages during cholesterol-induced endoplasmic reticulum stress. Diabetes. 2008;57:2967–76. doi: 10.2337/db08-0520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liang CP, Han S, Okamoto H, et al. Increased CD36 protein as a response to defective insulin signaling in macrophages. J Clin Invest. 2004;113:764–73. doi: 10.1172/JCI19528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Korkmaz CG, Korkmaz KS, Kurys P, et al. Molecular cloning and characterization of STAMP2, an androgen-regulated six transmembrane protein that is overexpressed in prostate cancer. Oncogene. 2005;24:4934–45. doi: 10.1038/sj.onc.1208677. [DOI] [PubMed] [Google Scholar]
- 21.Ohgami RS, Campagna DR, McDonald A, et al. The Steap proteins are metalloreductases. Blood. 2006;108:1388–94. doi: 10.1182/blood-2006-02-003681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wellen KE, Fucho R, Gregor MF, et al. Coordinated regulation of nutrient and inflammatory responses by STAMP2 is essential for metabolic homeostasis. Cell. 2007;129:537–48. doi: 10.1016/j.cell.2007.02.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.ten Freyhaus H, Calay ES, Yalcin A, et al. Stamp2 controls macrophage inflammation through nicotinamide adenine dinucleotide phosphate homeostasis and protects against atherosclerosis. Cell Metab. 2012;16:81–9. doi: 10.1016/j.cmet.2012.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mu J, Woods J, Zhou YP, et al. Chronic inhibition of dipeptidyl peptidase-4 with a sitagliptin analog preserves pancreatic beta-cell mass and function in a rodent model of type 2 diabetes. Diabetes. 2006;55:1695–704. doi: 10.2337/db05-1602. [DOI] [PubMed] [Google Scholar]
- 25.Matthews DR, Hosker JP, Rudenski AS, et al. Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia. 1985;28:412–9. doi: 10.1007/BF00280883. [DOI] [PubMed] [Google Scholar]
- 26.Sun M, Chen M, Dawood F, et al. Tumor necros is factor-alpha mediates cardiac remodeling and ventricular dysfunction after pressure overload state. Circulation. 2007;115:1398–407. doi: 10.1161/CIRCULATIONAHA.106.643585. [DOI] [PubMed] [Google Scholar]
- 27.Wang ZH, Shang YY, Zhang S, et al. Silence of TRIB3 suppresses atherosclerosis and stabilizes plaques in diabetic ApoE-/-/LDL receptor-/- mice. Diabetes. 2012;61:463–73. doi: 10.2337/db11-0518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Johnson JL, Jackson CL. Atherosclerotic plaque rupture in the apolipoprotein E knockout mouse. Atherosclerosis. 2001;154:399–406. doi: 10.1016/s0021-9150(00)00515-3. [DOI] [PubMed] [Google Scholar]
- 29.Shiomi M, Ito T, Hirouchi Y, et al. Fibromuscular cap composition is important for the stability of established atherosclerotic plaques in mature WHHL rabbits treated with statins. Atherosclerosis. 2001;157:75–84. doi: 10.1016/s0021-9150(00)00708-5. [DOI] [PubMed] [Google Scholar]
- 30.Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002;296:1655–7. doi: 10.1126/science.296.5573.1655. [DOI] [PubMed] [Google Scholar]
- 31.Whiteman EL, Cho H, Birnbaum MJ. Role of Akt/protein kinase B in metabolism. Trends Endocrinol Metab. 2002;13:444–51. doi: 10.1016/s1043-2760(02)00662-8. [DOI] [PubMed] [Google Scholar]