

Abstract
The need for post-synthetic modifications and reactive prosthetic groups has long been a limiting factor in the synthesis and study of peptidic and peptidomimetic imaging agents. In this regard, the application of biologically and chemically orthogonal reactions to the design and development of novel radiotracers has the potential to have far-reaching implications in both the laboratory and the clinic. Herein, we report the synthesis and development of a series of modular and versatile building blocks for inverse electron-demand Diels–Alder copper-free click chemistry: tetrazine-functionalized artificial amino acids. Following the development of a novel peptide coupling protocol for peptide synthesis in the presence of tetrazines, we successfully demonstrated its effectiveness and applicability. This versatile methodology has the potential to have a transformational impact, opening the door for the rapid, facile, and modular synthesis of bioorthogonally reactive peptide probes.
Keywords: bioorthogonal chemistry, click chemistry, positron emission tomography (PET), solid-phase peptide synthesis, zirconium-89
Over the last two decades, low-molecular-weight peptides have received increasing interest as vectors for the delivery of diagnostic and therapeutic payloads to tumors. Indeed, the high specificity and selectivity, short blood half-life and rapid excretion profile have made short oligopeptides particularly attractive in the realm of nuclear medicine, specifically positron emission tomography (PET) imaging. Radiolabeled variants of several tissue-specific peptides have been successfully synthesized and validated in vitro and in vivo, including 18F-and 64Cu-labeled peptides that target the somatostatin receptor type 2 (sstr),[1] the gastrin-releasing peptide receptor (GRPr),[2] and the glucagon-like peptide 1 receptor (GLP1-R).[3]
However, even after a worthwhile biomarker and a specific and selective targeted peptide have been identified, one of the most common challenges in the development of novel PET imaging agents is the identification of a rapid, efficient, and high-yielding method for the conjugation of a radionuclide to the peptide in question. The most common synthetic strategy for the radiolabeling of peptides is the post-synthetic conjugation of a reactive prosthetic group or radiometal chelator to an intact peptide, followed by a subsequent radiolabeling reaction (Scheme 1). To this end, prosthetic groups and chelators have been appended to peptides by a wide variety of methods, yet three strategies for conjugation based on naturally occurring amino acids stand as the most common routes: (1) peptide bond formation between an activated carboxylic acid (e.g., a succinimidyl ester) and an amine, (2) thioether formation via reaction of a thiol and a maleimide, and (3) thiourea bond formation via reaction of an isothiocyanate and a primary amine. Each of these methods is dependent on the reactivity of either lysines (amines) or cysteines (thiols).[4], [5] While the post-synthetic modification of these naturally occurring amino acids has proven effective in a variety of settings, it is not without significant limitations. The late-stage incorporation of a prosthetic group or chelator requires additional synthetic steps that can add complexity and reduce yield. In addition, the use of naturally occurring amino acids as conjugation handles often renders the selective and site-specific attachment of radionuclides difficult, since multiple reactive amino acids can be present in a single biomolecule.[5], [6] Furthermore, the modification of natural amino acids in a peptide risks compromising the biological reactivity of the biomolecule. Finally, labeling naturally occurring amino acids necessarily reduces the number of biogenic residues that remain available for inclusion in a peptide, in turn putting strain on the design of targeted biomolecules.
Scheme 1.
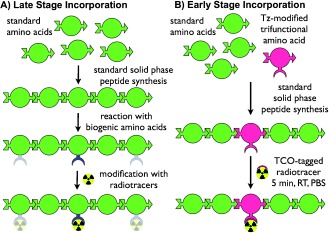
Conceptual synthesis of tetrazine-amino acid incorporation into peptides. A) The late stage incorporation of an orthogonal reactive group, a common synthetic route for the construction of targeted peptides: peptides are synthesized using SPPS and subsequently derivatized using bifunctional prosthetic groups that react selectively or semi-selectively with biogenic amino acids. B) The incorporation of an orthogonal reactive group into peptides via tetrazine-bearing amino acids: tetrazine-bearing amino acids are employed in a manner identical to biogenic amino acids and incorporated during SPPS. After cleavage of the peptide from the solid support, the tetrazine-bearing amino acids can serve as chemically orthogonal groups that react at radiochemically relevant concentrations, yielding targeted imaging agents quickly and selectively.
Recent years, however, have witnessed a move beyond naturally occurring amino acids, with an increasing amount of focus on the use of artificial amino acids bearing orthogonal functional groups. Along these lines, we have hypothesized that the inverse electron-demand Diels–Alder cycloaddition between tetrazine and trans-cyclooctene offers a near ideal strategy for the site-selective labeling of biomolecules.[7] Indeed, the rapidity, chemoselectivity, and bioorthogonality of this emergent click ligation make it particularly well suited to bioconjugation applications and especially radiochemistry.[8], [9]
Herein, we report the design, development, and validation of a synthetic strategy that inverts the traditional peptide modification paradigm: the incorporation of bioorthogonal tetrazine-bearing amino acids into peptides via solid-phase peptide synthesis (SPPS). We contend that the incorporation of a reactive tetrazine moiety into a peptide at the initial stage of peptide synthesis rather than in a post-synthetic modification offers maximal versatility, modularity, and facility. As an initial proof of concept, we have created a bioorthogonally reactive pentapeptide using a trifunctional tetrazine-containing amino acid derivative that is compatible with SPPS (Scheme 1). Indeed, this artificial amino acid can be incorporated into a peptide chain in a manner identical to naturally occurring amino acids via the coupling, deprotection, and cleavage reactions of SPPS.
This technology offers a number of critical advantages over nonspecific methodologies that rely on the late-stage incorporation of an orthogonal moiety or prosthetic group. Specifically, this strategy eliminates an additional step in the synthesis of the radiolabeled probe: the conjugation of the orthogonal group to the peptide. The incorporation of an orthogonal group at such an early stage also allows for precise control over the molecular location of the radiolabel in the primary sequence of the peptide. And finally, the use of a tetrazine-bearing amino acid precludes the need to dedicate specific amino acids for derivatization, thereby freeing all of the biologically available amino acids for use in the peptide in question.
The first step in the creation of this system was the synthesis of a series of tetrazine-bearing amino acid derivatives. To this end, all of the functionalized amino acids were derived from l-lysine and fully characterized using HPLC, LC–ESI-MS, HRMS and NMR spectroscopy. To synthesize the tert-butoxycarbonyl (Boc)-protected amino acid Boc-τ-OH (1 Figure 1), a previously published carboxylic acid-bearing tetrazine[10] (Tz-COOH) was reacted with N-hydroxysuccinimide (NHS) in the presence of the coupling agent N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDCI). The resulting Tz-NHS ester was isolated using silica gel chromatography and reacted with Boc-Lys-OH, affording 1 as a pink film in 50 % yield.
Figure 1.
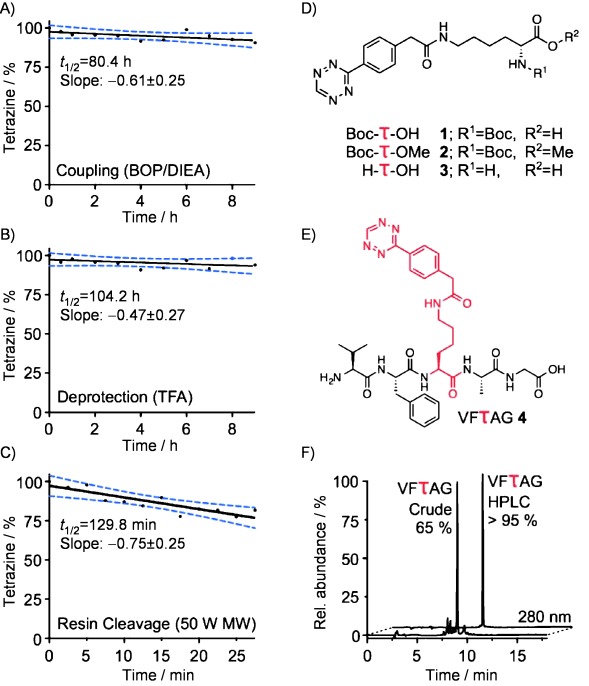
Stability of tetrazine-bearing amino acids under SPPS conditions: A) amino acid coupling conditions; B) N-Boc deprotection conditions; C) resin cleavage conditions. E) Structure of the novel tetrazine-pentapeptide VFτAG. F) HPLC trace of the pentapeptide VFτAG before and after HPLC purification.
The stability of the tetrazine-bearing amino acids under the various reaction conditions of SPPS was paramount to the success of the system. Not surprisingly, potential pitfalls were found at each of the SPPS steps: deprotection, coupling, and cleavage from the resin. The two most common protecting group methodologies used to generate peptides using SPPS are the Boc-and 9-fluorenylmethoxycarbonyl (Fmoc)-based strategies. For the synthesis of tetrazine-labeled peptides, Boc-protected amino acids are the most suitable option, for their removal is based on non-reducing acids (typically trifluoroacetic acid, TFA), and solvents in which reduction-prone tetrazines are stable. Fmoc-based strategies, in contrast, commonly require piperidine or other nucleophilic bases for the removal of the Fmoc protecting group, reagents that are generally incompatible with the tetrazine moiety. Further, we found that tetrazines decompose in the presence of the common coupling agents 1-hydroxybenzotriazole (HOBt) and N,N′-diisopropylcarbodiimide (DIC) but are stable in the presence of Hünigs base (N,N-diisopropylethylamine, DIEA) and (benzotriazol-1-yloxy)tris(dimethylamino)phosphonium hexafluorophosphate (BOP). Finally, SPPS via the Boc-based strategy often requires strong acids for the cleavage of the synthesized peptides from the resin, most commonly HF, HBr, or trimethylsilyl trifluoromethanesulfonate. Unfortunately, none of these options represents a suitable choice. HF is highly toxic, and the necessary associated specialized equipment might hamper the synthesis of tetrazine-labeled peptides in biomedical research laboratories. The two other cleavage reagents lead to complete decomposition of the tetrazine. In order to prevent this, we turned to microwave-assisted cleavage of the peptide from the resin. Microwave-assisted synthetic transformations have been used before to mediate resin-associated reactions,[11] and they have shown to be successful for peptide cleavage as well.[12]
To test the stability of the tetrazines under amino acid coupling conditions, Tz-NHS ester was reacted with methoxy-protected Boc-Lys-OMe, preventing the activation and thus the derivatization of the carboxylic acid. The resulting Boc-τ-OMe (2, Figure 1) was dissolved in a freshly prepared mixture of BOP (10 mm) and DIEA (100 mm) in N,N-dimethylformamide (DMF). The decomposition of 2 was measured using HPLC against a coumarin standard. The decomposition rate was found to be linear with a half-life (t1/2) of 80.4 h (Figure 1). Under these conditions, each coupling reaction (60 min) would cause a decomposition of only 0.61±0.25 %.
The stability of the tetrazine under deprotection conditions was measured using H-τ-OH (3), which was generated from Boc-τ-OH (1) in 72 % isolated yield via deprotection with TFA/CH2Cl2 (1:1). Similar to the coupling conditions test, the stability of 3 was measured using HPLC against a coumarin standard. In 95 % TFA, the half-life (t1/2) was 104.2 h (Figure 1). This, in turn, results in each deprotection reaction (25 min) causing 0.20±0.11 % of the tetrazine to decompose.
For the cleavage of tetrazine-containing peptides from the resin, microwave-assisted acidic cleavage was chosen, a technique which has been previously employed in SPPS as a mild cleavage technique.[12] The deprotected tetrazine-bearing amino acid H-τ-OH (3) was dissolved in a mixture of TFA and water (97.5 % TFA) and subjected to 50 MW microwave irradiation. Again, the results were analyzed by HPLC, and coumarin served as an internal standard. Under these conditions, the half-life (t1/2) of 3 was observed to be 129.8 min (Figure 1). Consequently, during a cleavage procedure employing these conditions (2×5 min), an estimated 7.5±2.5 % of the orthogonal group on the immobilized peptide would decompose. Coupling efficiencies using only naturally occurring amino acids and standard protocols are typically around 99 %, which illustrates that the measured decomposition rates are well within an acceptable range. Based on these decomposition rates, a 14 amino acid peptide (e.g., α-melanocyte-stimulating hormone, α-MSH) with a tetrazine in position 7 could be synthesized, deprotected, and removed from the resin with 85 % of the tetrazines intact. If, however, the tetrazine were placed in position 14 of the peptide, 81 % of the tetrazine in the peptide would remain impact. Alternatively, if the tetrazine-amino acid were the last residue incorporated, 92 % of the cleaved peptide would contain the reactive functional group. Importantly, this decomposition would represent a small reduction in the overall yield of the synthesized peptide but would not compromise the purity of the peptide after HPLC purification (Figure 1).
Using these reaction conditions, we subsequently synthesized a model tetrazine-containing pentapeptide, VFτAG (4, Figure 1). The peptide represents a non-targeted amino acid sequence, chosen to illustrate that the tetrazine-bearing amino acid can be incorporated centrally in the backbone of a peptide rather than simply at one of the two termini. It was synthesized using a standard Merrifield-resin, which was preloaded with glycine (0.01–0.02 mmol scale). The deprotection, coupling, and resin cleavage was conducted according to the conditions delineated above and in Figure 1. After the cleavage of the peptide, the crude product was purified using HPLC and obtained as a light pink solid.
In order to illustrate the potential applications of this technology in the synthesis of constructs for PET imaging, we next sought to radiolabel the peptide using the bioorthogonal chemistry made possible by the tetrazine moiety. To this end, we first synthesized a trans-cyclooctene (TCO)-bearing derivative of the 89Zr4+ chelator desferrioxamine (DFO) via the coupling of an NHS-activated ester of a TCO-COOH moiety with desferrioxamine mesylate. After the purification of DFO–TCO, we then radiolabeled the TCO-modified chelator with 89Zr under standard 89Zr4+ radiolabeling conditions: incubation in phosphate-buffered saline at pH 7.2–8.0 for 30 min at 37 °C. Following purification via reversed-phase HPLC (C18), the radiolabeled 89Zr-DFO–TCO construct (5) was isolated in relatively high yield (39 %), high radiochemical purity (>98 %), and high specific activity (5.59 mCi μmol−1). Finally, the rapid and clean bioorthogonal conjugation between 89Zr-DFO–TCO and VFτAG was performed in an acidified solution of acetonitrile and water (1:1, 0.1 % TFA, Figure 2), producing 89Zr-VFτAG (6) in >95 % radiochemical purity and 65 % uncorrected isolated radiochemical yield (RCY; Figure 2).
Figure 2.
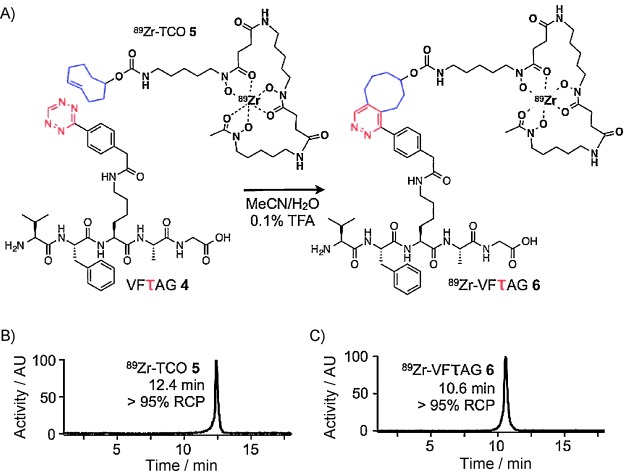
Chemically orthogonal synthesis of 89Zr-VFτAG (6). A) Synthesis and structure of 6, product of the chemically orthogonal reaction of VFτAG (4) with 89Zr-TCO (5); Reaction conditions: CH3CN/H2O (1:1, 0.1 % TFA), 30 min. B) Radio-HPLC trace of 5. C) Radio-HPLC trace of 6.
Ultimately, we believe that this chemical technology has the potential to have a transformational impact on the synthesis, development, and optimization of molecular imaging probes for PET and beyond. In all but the simplest of cases, the incorporation of the bioorthogonal tetrazine functionality during SPPS will reduce complexity, increase yields, and provide significant flexibility and modularity. In essence, the tetrazine-bearing amino acid—a tri-functional moiety bearing a carboxylic acid, an amine, and a bioorthogonal tetrazine—can be seen as an addition to the library of already available natural amino acids used during SPPS and thus, after cleavage, will yield the reactive target molecule without further chemical modification or derivatization.
Perhaps more excitingly, tetrazine-bearing amino acids could eventually be used as building blocks in the combinatorial synthesis of potential biologically active peptides, facilitating the development of libraries of probes on a fully automated basis without the need for multiple purification steps after synthesis. Further, the modularity of the system does not end with the tetrazine side of the reaction; just as the tetrazine can be incorporated into a myriad of different peptide sequences, its trans-cyclooctene reaction partner can be appended with a tremendous variety of cargoes ranging from radionuclides to fluorophores to chemotherapeutics. Currently, efforts are underway to optimize the system to increase yields, to utilize this technology to produce longer, biomarker-targeting, tetrazine-bearing peptides for PET imaging applications, and to apply this technology to develop peptides for other molecular imaging modalities, including single-photon emission computed tomography (SPECT) and fluorescence imaging.
Experimental Section
Unless otherwise noted, all reagents were purchased from Sigma–Aldrich (St. Louis, MO, USA) and used without further purification. Boc-Gly, Merrifield resin and Boc-Lys-OMe were purchased from Bachem (Torrance, CA, USA). Boc-Ala-OH, Boc-Lys-OH, Boc-Phe-OH and Boc-Val-OH were purchased from Novabiochem (Merck KGaA, Darmstadt, Germany). 3-(4-Phenylacetic acid)-1,2,4,5-tetrazine (Tz-COOH) and (E)-cyclooct-4-enyl 2,5-dioxopyrrolidin-1-yl carbonate (TCO-NHS) were synthesized as described before.[10], [13] HPLC analyses were performed using a Shimadzu HPLC equipped with 2 LC-10AT pumps, a SPD-M10AVP photodiode array detector, a Flow Count PIN diode radiodetector from BioScan and a Waters Atlantis T3 column (6×250 mm, 5 μm). A gradient of 95:5→0:100 H2O/CH3CN supplemented with 0.1 % TFA over 18 min at 1 mL min−1 was used. For preparative HPLCs, a Phenomenex Jupiter (250×100 mm, 5 μm), and flowrates of 5 mL min−1 were used. Radioactivity measurements were performed with a Capintec CRC1243 Dose Calibrator (Capintec, Ramsay, NJ, USA). For TLC measurements with radioactive materials, a Bioscan AR2000 (Bioscan, Inc., Washington, DC, USA) was used. Microwave irradiations were carried out using a Biotage Initiator microwave synthesizer (Biotage, LLC, Charlotte, NC, USA). LRMS were recorded with a Waters Acquity UPLC with electrospray ionization SQ detector (ESI). HRMS were recorded with a Waters LCT Premier system (ESI). 1H NMR spectra were recorded on a Bruker AVIII (500 MHz) spectrometer. Chemical shifts for protons are reported in parts per million (ppm) and are referenced against the residual proton resonance of deuterated solvents (CDCl3: 1H, 7.26 ppm; [D4]MeOH: 1H, 3.31 ppm; [D4]DMSO: 1H, 2.50 ppm). NMR data are reported as follows: chemical shift, multiplicity (s=singlet, d=doublet, t=triplet, m=multiplet), coupling constants (Hz) and integration.
Synthesis
3-(4-Phenylacetic acid)-1,2,4,5-tetrazine succinimidyl ester (Tz-NHS): N-Hydroxysuccinimide (163 mg, 1.42 mmol) and Et3N (495 μL, 3.55 mmol) were added to a mixture Tz-COOH (150 mg, 0.71 mmol) and N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDCI, 544 mg, 2.84 mmol) in CH2Cl2 (15 mL), and the reaction mixture was stirred for 6 h at RT. The mixture was extracted with acetic acid (1 m, 2×10 mL) and H2O (2×10 mL), dried (MgSO4), and volatiles were removed in vacuo. The crude product was purified by column chromatography (CH2Cl2/MeOH, 97.5:2.5), yielding Tz-NHS as a pink solid (67.3 mg, 0.22 mmol, 31 %): 1H NMR (500 MHz, [D4]MeOH): δ=10.60 (s, 1 H), 8.51 (d, J=8.3, 2 H), 7.55 (d, J=8.3, 2 H), 7.20 (d, J=7.7, 1 H), 4.32 (s, 2 H), 2.36 (s, 4 H); LC–MS (ESI−): m/z (%): 313.1 (100) [M−H]−, 312.1 (100) [2 M−H]−.
N2-(tert-Butoxycarbonyl)-N6-(tetrazine)-l-lysine (Boc-τ-OH, 1): Tz-NHS (55.0 mg, 0.18 mmol) was dissolved in a mixture of Boc-l-Lys-OH (53.2 mg, 0.36 mmol) and Et3N (63 μL, 0.45 mmol) in MeOH (6 mL) and stirred for 2 h. The reaction mixture was dried in vacuo, and the resulting crude product purified by column chromatography (5–25 % MeOH in CH2Cl2), yielding 1 as a pink film (40.3 mg, 0.09 mmol, 50 %): 1H NMR (500 MHz, [D4]MeOH): δ=10.31 (s, 1 H), 8.55 (d, J=8.4 Hz, 2 H), 7.56 (d, J=8.4 Hz, 2 H), 4.05 (dd, J=8.7, 4.7 Hz, 1 H), 3.22 (t, J=6.9 Hz, 2 H), 1.86–1.61 (m, 2 H), 1.58–1.38 (m, 13 H); LC–MS (ESI+): m/z (%): 467.3 (30) [M+Na]+, 911.6 (10) [2 M+Na]+; LC–MS(ESI−): m/z (%): 443.3 (10) [M−H]−, 887.6 (20) [2 M−H]−; HRMS (ESI) m/z [M+Na]+calcd for C21H28N6O5Na: 467.2005, found: 467.2019.
Methyl N2-(tert-butoxycarbonyl)-N6-(tetrazine)-l-lysinate (Boc-τ-OMe, 2): N-(3-Dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (51.8 mg, 0.27 mol) was added to a mixture of 3-(4-phenylacetic acid)-1,2,4,5-tetrazine succinimidyl ester (20 mg, 0.09 mmol), Boc-l-Lys-OH (28.6 mg, 0.11 mmol), Et3N (63 μL, 0.45 mmol) in CH2Cl2 (5 mL), and the reaction mixture was stirred over night at RT, before it was dried in vacuo. The resulting crude product was purified by column chromatography (5 % MeOH in DCM), yielding 2 as a pink film (20.8 mg, 0.05 mmol, 56 %): 1H NMR (500 MHz, [D6]DMSO): δ=10.57 (s, 1 H), 8.44 (d, J=8.2, 2 H), 8.13 (t, J=5.5 Hz, 1 H), 7.55 (d, J=8.2 Hz, 2 H), 7.20 (d, J=7.7 Hz, 1 H), 3.93–3.88 (m, 1 H), 3.61 (s, 3 H), 3.55 (s, 2 H), 3.05 (q, J=6.5 Hz, 2 H), 1.65–1.52 (m, 2 H), 1.43–1.27 (m, 13 H); LC–MS (ESI+) m/z (%): 359.3 (75) [M−Boc+H]+, 459.3 (40) [M+H]+, 917.7 (30) [2 M+H]+; HRMS (ESI): m/z [M+H]+ calcd for C22H30N6O5Na: 481.2171, found: 481.2175.
N6-(tetrazine)-l-lysine (H-τ-OH, 3): Boc-τ-OH 1 (10 mg, 22.5 μmol) was dissolved in a mixture of TFA/CH2Cl2 (1:1) and vigorously stirred for 1.5 h. Volatiles were removed in vacuo, and the resulting crude material purified using HPLC, yielding 3 as a pink solid (5.6 mg, 16.3 μmol, 72 %): 1H NMR (500 MHz, [D4]MeOH): δ=10.32 (s, 1 H), 8.55 (d, J=8.3, 2 H), 7.57 (d, J=8.3, 2 H), 3.92 (t, J=6.3, 2 H), 3.65 (s, 2 H), 3.24 (dt, J=6.3, 3.2 Hz, 2 H), 2.00–1.84 (m, 2 H), 1.62–1.42 (m, 2 H); LC–MS (ESI+): m/z (%): 345.2 (100) [M+H]+, 367.2 (15) [M+Na]+; HRMS (ESI): m/z [M+H]+ calcd for C16H21N6O3: 345.1668, found: 345.1675.
Pentapeptide GAτFV (4): The synthesis was carried out on a 0.01–0.02 mmol scale in a fritted syringe (3 mL volume), using a Merrifield resin, which was preloaded with Boc-Gly-OH (20 mg, 0.5–1.0 mmol g−1). The resin was swollen by washing with DMF (2×2 mL) and CH2Cl2 (2×2 mL). Nα-Boc groups were removed by addition of a solution of TFA/CH2Cl2 (1:1; 1×5 min, 2 mL and 1×20 min, 2 mL), followed by washing with CH2Cl2 (2×2 mL) and DMF (2×2 mL). Amino acids were coupled (1 h, RT) without prior neutralization of the resin using a freshly prepared mixture of BOP (0.04 mmol, 18 mg), Hünigs base (N,N′-diisopropylethylamine, DIEA; 0.12 mmol, 16 mg) and amino acid (0.08 mmol) in DMF (2.5 mL). After coupling, the resin was washed with DMF (3×2 mL) and CH2Cl2 (3×2 mL). After completion of the deprotection/coupling sequence, the peptide was transferred to a microwave vessel, suspended in a solution of TFA/H2O (97.5:2.5, 500 μL), and microwaved (2×5 min, 50 Hz). The resin was removed by filtration and Et2O (10 mL) added to the supernatant. The resulting crude precipitate was filtered and purified using HPLC to yield 4 as a pink solid (0.9 mg, 1.3 μmol, 7 % yield): LC–MS (ESI+): m/z (%): 719.5 (20) [M+H]+; LC–MS (ESI−): m/z (%): 717.5 (10) [M−H]−; HRMS (ESI): m/z calcd for [M+H]+ C35H47N10O7: 719.3633, found: 719.3629.
trans-Cyclooctene–desferrioxamine conjugate (TCO–DFO): TCO-NHS (12 mg, 45 µmol) was added to a solution deferrioxamine mesylate salt (20 mg, 30 μmol) and Et3N (21 μL, 0.15 mmol) in CH3CN/H2O (1:1, 1 mL) and stirred at RT for 90 min. Subsequently, volatiles were removed in vacuo, and the crude product was purified using HPLC, yielding TCO–DFO as a white solid (6.6 mg, 9 μmol, 30 %): 1H NMR (500 MHz, [D6]DMSO): δ=9.68–9.54 (m, 2 H), 7.78–7.74 (m, 1 H), 6.93–6.88 (m, 1 H), 5.60–5.40 (m, 2 H), 4.21–4.17 (m, 1 H), 3.48–3.41 (m, 6 H), 3.03–2.87 (m, 6 H), 2.61–2.53 (m, 4 H), 2.29–2.21 (m, 7 H), 1.96 (s, 3 H), 1.91–1.85 (m, 4 H), 1.56–1.15 (m, 22 H); LC–MS (ESI+): m/z (%): 357.4 (10) [M+2 H]2+, 713.6 (100) [M+H]+, 735.5 (70) [M+Na]+, 759.4 (10) [M+HCOO]+; HRMS (ESI): m/z calcd for [M+H]+ C34H61N6O10: 713.4474, found: 713.4449.
89Zr-trans-cyclooctene (89Zr-TCO, 5): [89Zr]Zr-oxalate (59.2–74 MBq, 1.600–2.000 μCi) in oxalic acid (1.0 m, 150 μL) was adjusted to pH 7.2–8.0 with Na2CO3 (1.0 m, ≈150 μL). After the evolution of CO2(g) stops, the 89Zr was added to a solution of TCO–DFO (1 mm, 100 μL) in PBS/DMSO (9:1). The reaction solution was stirred at 37 °C for 30 min, before the reaction progress was assayed using instant thin-layer chromatography (ITLC) with an eluent of 1 m citric acid. In the ITLC experiments, 89Zr-TCO remains at the baseline, while free 89Zr4+ ions and other [89Zr]-citrate complexes elute with high Rf values. The crude reaction mixture was purified using HPLC, and volatiles were removed in vacuo, yielding 5 in >98 % radiochemical purity, 39 % uncorrected isolated radiochemical yield (RCY), and a specific activity of >5.59 mCi μmol−1 (n=3).
89Zr-GAτFV (6): GAτFV (4, 20 nmol, 20 μL, 1 mm in DMSO) was added to 89Zr-TCO (56 μCi, 10 μL, 5.56 mCi mL−1 in 1:1 CH3CN/H2O with 0.05 % TFA), and the mixture was agitated at RT for 20 min, before purification using HPLC, yielding 6 in >95 % purity, 65 % uncorrected isolated RCY, and a specific activity of >3.00 mCi μmol−1 (n=3).
Tetrazine stability determination
Boc-deprotection conditions: TFA (760 μL) was added to a solution of 3 (5 mm, 20 μL, DMSO) and coumarin (5 mm, 20 μL, DMSO) and the resulting mixture stirred at room temperature. A control sample consisted of DMSO (760 μL), which was added to a solution of 3 (5 mm, 20 μL, DMSO) and coumarin (5 mm, 20 μL, DMSO). The amount of 3 relative to coumarin was measured at t=0, 0.5, 1, 2, 3, 4, 5, 6, 7, 8 and 9 h using HPLC analysis, and the results were compared to the control sample. All measurements were performed in triplicate.
Coupling conditions: Solutions of (benzotriazol-1-yloxy)tris(dimethylamino)phosphonium hexafluorophosphate (BOP, 10 mm, 25 μL, DMF) and DIEA (100 mm, 25 μL, DMF) were added to a solution of 2 (5 mm, 25 μL, DMF) and coumarin (5 mm, 25 μL, DMF). A control sample consisted of DMF (50 μL), which was added to a solution of 2 (5 mm, 25 μL, DMF) and coumarin (5 mm, 25 μL, DMF). The amount of 3 relative to coumarin was measured at t=0, 0.5, 1 h, 2 h, 3 h, 4 h, 5 h, 6 h, 7 h, 8 h and 9 h using HPLC analysis, and the results were compared to the control sample. All measurements were performed in triplicate.
Resin-cleavage conditions: TFA (950 μL) and deionized H2O (25 μL) were added to a solution of 3 (5 mm, 20 μL, DMSO) and coumarin (5 mm, 5 μL, DMSO), and the resulting mixture was exposed to sequential microwave irradiation intervals (0–11×2.5 min, 50 W). A control sample consisted of DMSO (975 μL), which was added to a solution of 3 (5 mm, 20 μL, DMSO) and coumarin (5 mm, 5 μL, DMSO). At the end of each microwave irradiation interval, a sample was drawn, the amount of 3 relative to coumarin was measured using HPLC analysis, and the results were compared to the control sample. All measurements were performed in triplicate.
Acknowledgments
The authors thank Dr. Wolfgang Weber (Memorial Sloan-Kettering Cancer Center) for helpful discussions. The authors also thank the US National Institute of Health (NIH) (JSL: R01A138468 and TR: K25EB016673) and the Brain Tumor Center of Memorial Sloan-Kettering Cancer Center (TR) for their generous funding.
References
- [1].Guhlke S, Wester HJ, Bruns C, Stocklin G. Nucl Med Biol. 1994;21:819. doi: 10.1016/0969-8051(94)90161-9. [DOI] [PubMed] [Google Scholar]; Fani M, Del Pozzo L, Abiraj K, Mansi R, Tamma ML, Cescato R, Waser B, Weber WA, Reubi JC, Maecke HR. J Nucl Med. 2011;52:1110. doi: 10.2967/jnumed.111.087999. [DOI] [PubMed] [Google Scholar]
- [2].Zhang X, Cai W, Cao F, Schreibmann E, Wu Y, Wu JC, Xing L, Chen X. J. Nucl. Med. 2006;47:492. [PubMed] [Google Scholar]; Honer M, Mu L, Stellfeld T, Graham K, Martic M, Fischer CR, Lehmann L, Schubiger PA, Ametamey SM, Dinkelborg L, Srinivasan A, Borkowski S. J. Nucl. Med. 2011;52:270. doi: 10.2967/jnumed.110.081620. [DOI] [PubMed] [Google Scholar]; Yang M, Gao H, Zhou Y, Ma Y, Quan Q, Lang L, Chen K, Niu G, Yan Y, Chen X. Theranostics. 2011;1:220. doi: 10.7150/thno/v01p0220. [DOI] [PMC free article] [PubMed] [Google Scholar]; Lears KA, Ferdani R, Liang K, Zheleznyak A, Andrews R, Sherman CD, Achilefu S, Anderson CJ, Rogers BE. J. Nucl. Med. 2011;52:470. doi: 10.2967/jnumed.110.082826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Kiesewetter DO, Gao H, Ma Y, Niu G, Quan Q, Guo N, Chen X. Eur J Nucl Med Mol Imaging. 2012;39:463. doi: 10.1007/s00259-011-1980-0. [DOI] [PMC free article] [PubMed] [Google Scholar]; Wang Y, Lim K, Normandin M, Zhao X, Cline GW, Ding YS. Nucl Med Biol. 2012;39:167. doi: 10.1016/j.nucmedbio.2011.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]; Wu Z, Todorov I, Li L, Bading JR, Li Z, Nair I, Ishiyama K, Colcher D, Conti PE, Fraser SE, Shively JE, Kandeel F. Bioconjugate Chem. 2011;22:1587. doi: 10.1021/bc200132t. [DOI] [PubMed] [Google Scholar]
- [4].Fani M, Maecke HR, Okarvi SM. Theranostics. 2012;2:481. doi: 10.7150/thno.4024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Zeglis BM, Mohindra P, Weissmann GI, Divilov V, Hilderbrand SA, Weissleder R, Lewis JS. Bioconjugate Chem. 2011;22:2048. doi: 10.1021/bc200288d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Sletten EM, Bertozzi CR. Angew. Chem. 2009;121:7108. doi: 10.1002/anie.200900942. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2009;48:6974. [Google Scholar]
- [7].Devaraj NK, Weissleder R. Acc. Chem. Res. 2011;44:816. doi: 10.1021/ar200037t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Keliher EJ, Reiner T, Turetsky A, Hilderbrand SA, Weissleder R. ChemMedChem. 2011;6:424. doi: 10.1002/cmdc.201000426. [DOI] [PMC free article] [PubMed] [Google Scholar]; Reiner T, Lacy J, Keliher EJ, Yang KS, Ullal A, Kohler RH, Vinegoni C, Weissleder R. Neoplasia. 2012;14:169. doi: 10.1593/neo.12414. [DOI] [PMC free article] [PubMed] [Google Scholar]; Zeglis BM, Sevak KK, Reiner T, Mohindra P, Carlin SD, Zanzonico P, Weissleder R, Lewis JS. J Nucl Med. 2013;54:1389. doi: 10.2967/jnumed.112.115840. [DOI] [PMC free article] [PubMed] [Google Scholar]; Wu Z, Liu S, Hassink M, Nair I, Park R, Li L, Todorov I, Fox JM, Li Z, Shively JE, Conti PS, Kandeel F. J. Nucl. Med. 2013;54:244. doi: 10.2967/jnumed.112.109694. [DOI] [PubMed] [Google Scholar]
- [9].Keliher E, Reiner T, Thurber GM, Upadhyay R, Weissleder R. ChemistryOpen. 2012;1:177. doi: 10.1002/open.201200014. [DOI] [PMC free article] [PubMed] [Google Scholar]; Selvaraj R, Liu S, Hassink M, Huang CW, Yap LP, Park R, Fox JM, Li Z, Conti PS. Bioorg. Med. Chem. Lett. 2011;21:5011. doi: 10.1016/j.bmcl.2011.04.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Yang J, Karver MR, Li W, Sahu S, Devaraj NK. Angew. Chem. 2012;124:5312. doi: 10.1002/anie.201201117. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2012;51:5222. [Google Scholar]
- [11].Kappe CO. Curr Opin Chem. Biol. 2002;6:314. doi: 10.1016/s1367-5931(02)00306-x. [DOI] [PubMed] [Google Scholar]
- [12].Kluczyk A, Rudowska M, Stefanowicz PSZ, Szewczuk Z. J. Pept. Sci. 2010;16:31. doi: 10.1002/psc.1191. [DOI] [PubMed] [Google Scholar]
- [13].Devaraj NK, Upadhyay R, Haun JB, Hilderbrand SA, Weissleder R. Angew. Chem. 2009;121:7147. doi: 10.1002/anie.200903233. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2009;48:7013. [Google Scholar]