

Abstract
A novel series of ligands for the recombinant human AT2 receptor has been synthesized utilizing a fast and efficient palladium-catalyzed procedure for aminocarbonylation as the key reaction. Molybdenum hexacarbonyl [Mo(CO)6] was employed as the carbon monoxide source, and controlled microwave heating was applied. The prepared N-aryl isoleucine derivatives, encompassing a variety of amide groups attached to the aromatic system, exhibit binding affinities at best with Ki values in the low micromolar range versus the recombinant human AT2 receptor. Some of the new nonpeptidic isoleucine derivatives may serve as starting points for further structural optimization. The presented data emphasize the importance of using human receptors in drug discovery programs.
Keywords: aminocarbonylation, AT2 receptor, medicinal chemistry, palladium catalysis, peptide mimics
Introduction
The octapeptide angiotensin II (Ang II) is the major effector peptide of the renin-angiotensin system (RAS). It acts via two receptors, the AT1 and the AT2 receptor (AT1R and AT2R). The effects mediated by AT1R are well known and include regulation of blood pressure and fluid/electrolyte balance.[1] When AT2R is expressed together with AT1R, its activation results in several effects that oppose those mediated by the latter. Thus, stimulation induces vasodilatation, antiproliferation and apoptosis. Conversely, when expressed alone in undifferentiated cells, AT2R stimulation is involved in cell differentiation.[1–2] In fact, AT2R is abundant in fetal tissues but its expression drops rapidly after birth, an observation in agreement with its role in cell differentiation. In the healthy adult, aside from a few specific tissues, the expression is at barely detectable levels.[2b, 3] However, a re-expression of the receptor occurs in some pathological states, such as heart and renal failure, myocardial infarction, hypertension, brain disorders or obesity disorders.[4] There is piling evidence that AT2R is involved in tissue repair. Therefore, AT2R has attracted special interest in connection with cardiac remodeling, and has now been addressed as a new target for drug intervention.[5]
We have conducted two projects in parallel with the common objective to identify selective drug-like agonists to AT2R. The first project commenced with the endogenous peptide ligand Ang II (A) and subsequent stepwise modifications, including minimizations/truncations, rigidifications and incorporation of turn mimetics resulted in series of AT2R-selective analogues, for example C (Figure 1).[6] This approach has led us to a new unique lead structure (E) that we anticipated could serve as a starting point for a new class of selective AT2R agonists (Figure 1).[6f] A parallel project focused on transforming the nonpeptidic but nonselective AT1R agonist L-162,313, disclosed by Merck, into a nonpeptidic AT2R-selective agonists.[7] We demonstrated that L-162,313 acts as an agonist also at the AT2R, and stepwise structural modifications of L-162,313 led to the identification of the first selective drug-like nonpeptide AT2R agonist C21/M024 (D) that has been extensively studied in various in vitro and in vivo models (Figure 1).[8] When comparing the two lead structures D and E, the structural similarities seem obvious, despite the different origins of the molecules. Hence, we hypothesize that both of the leads, D and E, mimic the C terminus of Ang II (A) and the truncated analogues B (Ang IV) and C (Figure 1).[6f] As indicated in Figure 1, the imidazole group would thus correspond to the histidine side chain, the sulfonyl carbamate would provide an acidic proton corresponding to the C-terminal carboxylic acid, and either the isobutyl or the n-butyl chain in C21/M024 would be able to mimic the hydrophobic Phe/Ile side chain of the C terminus of the peptide analogues.
Figure 1.
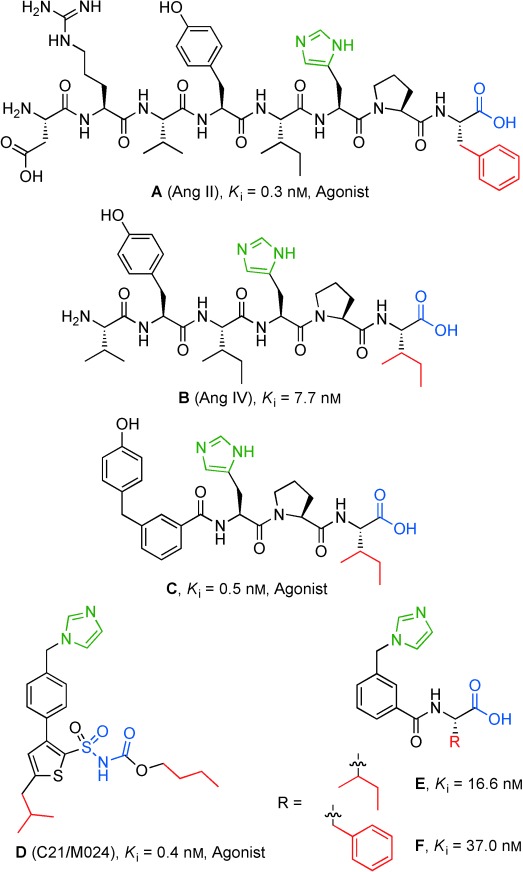
Selective AT2R ligands with common and potentially important structural motifs indicated, Ki values are derived from radioligand binding assay by displacement of radiolabeled Ang II from AT2R in pig uterus membranes. E originates from the endogenous peptide Ang II (A) while the nonpeptide C21/M024 (D) originates from the nonpeptide AT1/AT2 receptor agonist L-162,313.
Both lead structures comprise an imidazole group, which is frequently associated with undesired interactions with cytochrome P450 (CYP) enzymes. This issue was addressed, and CYP inhibition could successfully be minimized be replacement of the imidazole in C21/M024 (D) with various amide groups, providing ligands with retained activity and function.[9]
With the ambition to assess the potential of lead E, as an entry to a new class of selective AT2R agonists, we aimed at replacing the imidazole with a substituent less prone to bind to CYP enzymes, for example various amide groups. We decided to evaluate this new class of ligands towards the human AT2R using transfected HEK-293 cells (HEK293-hAT2R)[8e] rather than AT2R in pig myometrial membranes, which had been used previously.
Herein we report a convenient synthesis and pharmacological evaluation of a series of benzamides derived from E, comprising an isoleucine residue at the C terminus and with the generic structure depicted in Figure 2. We further conclude that the amides synthesized as well as E exhibit only a weak affinity towards human AT2R, while C21/M024 (D) binds with high affinity.
Figure 2.
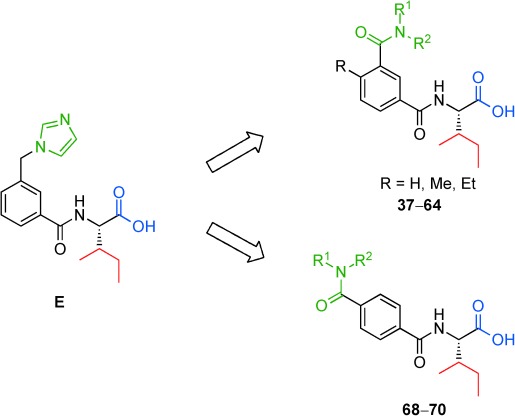
Lead structure E and the generic structures of the synthesized benzamides.
Results and Discussion
Chemistry
The synthesis of the new potential AT2R ligands was performed by palladium-catalyzed aminocarbonylation reactions starting from the corresponding iodo compounds under microwave heating. To allow this reaction to be conducted in sealed vials under CO gas-free conditions, molybdenum hexacarbonyl (Mo(CO)6) was chosen as the carbon monoxide source.[10] This rather recent method allows an efficient, fast and straightforward benzamide synthesis in air, and it has previously been used for the synthesis of biologically active compounds.[11] Although gaseous CO is advantageous for aminocarbonylations in large scale,[12] solid CO sources[13] such as Mo(CO)6[14] are safer and more convenient for lab-scale chemistry since no gas tubes and high pressure equipment are required.
The aryl iodides 1–4 were converted by a standard coupling with isoleucine-tert-butyl ester to afford 5–8 (Scheme 1). After purification, moderate to excellent yields of 38–98 % were achieved. The aryl iodides coupled with the isoleucine-tert-butyl ester residue were subsequently MW irradiated for 15 min at 100 °C in the presence of palladium catalyst with Mo(CO)6 and a selection of primary and secondary amines bearing aliphatic, aromatic, cyclic as well as heterocyclic groups with diverse steric and lipophilic properties (9–36 and 65–67, Table 1). The yields of the aminocarbonylation step varied much depending on the steric hindrance of the nucleophilic amine and its electronic properties and ranged from 14 % (19; Table 1, entry 11) to 85 % (17 and 22; entries 9 and 14). After hydrolysis with trifluoroacetic acid (TFA), target compounds 37–64 and 68–70 were afforded in mostly good to very good isolated yields, between 52 % (63, Table 1, entry 27) and 96 % (47, entry 11). In a few cases the hydrolysis resulted in yields below 50 % (entries 1, 14, 26, 28).
Scheme 1.
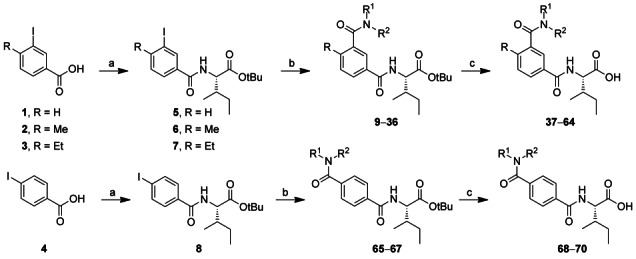
Synthesis of novel benzamides as AT2R ligands. Reagents and conditions: a) IleOtBu, HATU, DIEA, DMF, RT, 16 h; b) HNR1R2, Mo(CO)6, DBU, Pd(OAc)2, THF, MW 100 °C, 15 min; c) TFA/DCM (1:1), RT, 2 h.
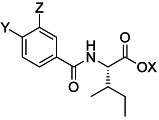
Table 1.
Yields of synthesized benzamides and affinity towards human AT2R in HEK-293 cells by displacement of either [125I]CGP-42112A or [125I]Sarile, and reference Ki values from displacement of [125I]Ang II from the AT2R in pig myometrial membrane.
 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | Y | Z | Yield[a] | Yield[b] | Inhibition [%] of | Ki AT2R [μm] | |||
| X=tBu | X=H | [125I]CGP binding | Human | Human | Pig | ||||
| 1 μm | 10 μm | [125I]CGP | [125I]Sarile | [125I]Ang II | |||||
| 1 | H | 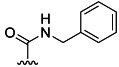 |
78 % 9 | 21 % 37[e] | 6 | 10 | |||
| 2 | H | 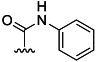 |
66 % 10 | 56 % 38[e] | 3 | 11 | |||
| 3 | H | 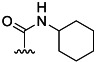 |
70 % 11 | 75 % 39[e] | 4 | 13 | |||
| 4 | H | 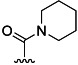 |
76 % 12 | 82 % 40[e] | –[c] | 12 | |||
| 5 | H | 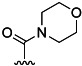 |
57 % 13 | 68 % 41[e] | 5 | 6 | |||
| 6 | H | 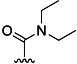 |
24 % 14 | 73 % 42[e] | 6 | 23 | 22.0 | 19.4 | |
| 7 | H | 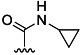 |
42 % 15 | 69 % 43[e] | 2 | 4 | |||
| 8 | H | 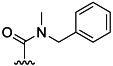 |
75 % 16 | 65 % 44[e] | 6 | 32 | 9.0 | 11.0 | |
| 9 | H | 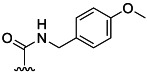 |
85 % 17 | 62 % 45[e] | 6 | 7 | |||
| 10 | H | 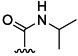 |
55 % 18 | 95 % 46[e] | 6 | 7 | |||
| 11 | H | 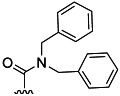 |
14 % 19 | 96 % 47[e] | 13 | 43 | 7.5 | 9.6 | |
| 12 | H | 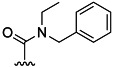 |
43 % 20 | 64 % 48 | 14[d] | 31[d] | 11.0 | 3.1 | |
| 13 | H | 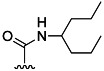 |
70 % 21 | 89 % 49 | –[c] | 12 | |||
| 14 | Me | 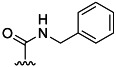 |
85 % 22 | 47 % 50[e] | –[c] | 7 | |||
| 15 | Me | 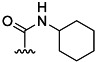 |
75 % 23 | 77 % 51[e] | 7 | 13 | |||
| 16 | Me | 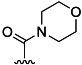 |
53 % 24 | 75 % 52[e] | 4 | 7 | |||
| 17 | Me | 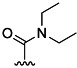 |
22 % 25 | 72 % 53[e] | 28 | 62 | 2.4 | 2.6 | |
| 18 | Me | 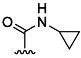 |
30 % 26 | 84 % 54[e] | 1 | 18 | |||
| 19 | Me | 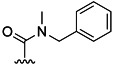 |
45 % 27 | 68 % 55[e] | 16 | 34 | 7.6 | 11.2 | |
| 20 | Me | 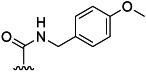 |
83 % 28 | 65 % 56[e] | 1 | 7 | |||
| 21 | Me | 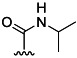 |
74 % 29 | 75 % 57[e] | 1 | 4 | |||
| 22 | Me | 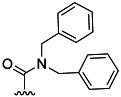 |
22 % 30 | 82 % 58[e] | 18 | 62 | 2.5 | 2.3 | |
| 23 | Me | 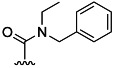 |
48 % 31 | 71 % 59 | 4[d] | 54[d] | 3.4 | 1.5 | |
| 24 | Me | 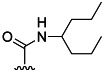 |
75 % 32 | 76 % 60 | –[c] | 6 | |||
| 25 | Et | 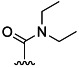 |
48 % 33 | 58 % 61 | 6[d] | 29[d] | 10.0 | ||
| 26 | Et | 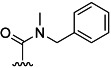 |
66 % 34 | 49 % 62 | –[c] | 26[d] | 16.0 | ||
| 27 | Et | 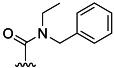 |
50 % 35 | 52 % 63 | 11[d] | 40[d] | 8.3 | ||
| 28 | Et | 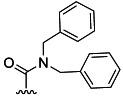 |
62 % 36 | 46 % 64 | 11[d] | 50[d] | 4.4 | ||
| 29 | 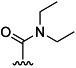 |
H | 68 % 65 | 74 % 68 | –[c] | –[c] | n.c.[f] | ||
| 30 | 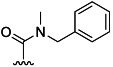 |
H | 73 % 66 | 88 % 69 | –[c] | 7[d] | >100 | ||
| 31 | 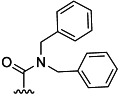 |
H | 16 % 67 | 57 % 70 | –[c] | 5[d] | 35.0 | ||
| Compound | |||||||||
| 32 | E | 110.0 | 47.0 | 0.0166 | |||||
| 33 | F | 63.0 | 0.0370 | ||||||
| 34 | C | 0.025 | 0.0005 | ||||||
| 35 | B (Ang IV) | 0.035 | 0.0077 | ||||||
| 36 | A (Ang II) | 0.044×10−3 | 0.0028 | 0.0003 | |||||
| 37 | D (C21/M024) | 0.0098 | 0.0076 | 0.0004 | |||||
Isolated yield after the aminocarbonylation reaction, >95 % purity.
Isolated yield after the deprotection, >95 % purity.
Not active.
Compound not included in the affinity screen, value taken from Ki determination.
Evaluated for AT1R affinity, none of the compounds exhibited any binding.
Not calculable—less than 25 % displacement at highest concentration.
In vitro pharmacology: Binding assays
All free acids shown in Table 1 (37–64 and 68–70) were evaluated in a first radioligand binding assay relying on the displacement of [125I]CGP-42112A (CGP-42112A; Nα-nicotinoyl-Tyr-(Nα-Cbz-Arg)-Lys-His-Pro-Ile), a selective but peptidic AT2 receptor agonist[15] from human AT2R expressed in HEK-293 cells (HEK293-hAT2R). Ang II was used as the reference substance.[16] The majority of the new benzamides shown in Table 1 (20 compounds) were also evaluated for binding toward human AT1R.[17] None of the evaluated compounds showed any affinity toward AT1R, and therefore the remaining compounds were evaluated only towards AT2R. In this first assay, the compounds were initially screened for binding activity (% inhibition of [125I]CGP-42112 A binding) at a concentration of 1 μm and 10 μm. The results from the initial compound screen indicated that noncyclic disubstituted benzamides (CONR1R2) showed better interaction with AT2R. More lipophilic substituents on the benzamide function led to higher affinity towards AT2R, and all compounds with activity in the initial screen had at least one benzyl group as substituent, except the diethyl benzamide, 42 and 53. Based on the activities found in the screen, compounds were selected for Ki value determinations. The ligands bearing a methyl substituent in para position showing a displacement of more than 30 % in the affinity screen [Table 1, entries 17 (53), 19 (55) and 22 (58)] were selected for Ki determinations. Additionally, the para-unsubstituted derivatives [Table 1, entries 6 (42), 8 (44) and 11 (47)] were also included to evaluate the influence of substitution in this position of the aromatic ring, even though they did not fully reach the same displacement. For the same reason, the para-ethyl derivatives [Table 1, entries 25 (61), 26 (62), 28 (64)] and the derivatives with the benzamides in the para position [Table 1, entries 29 (68), 30 (69), 31 (70)] were submitted directly for Ki determination, despite not being included in the initial screen. Furthermore, it was decided to include the benzylethyl benzamides (48, 59, 63) based on the preliminary results of the diethyl (42, 53) and dibenzyl compounds (47, 58).
The Ki values were determined from at least six data points with test concentrations ranging from 30 pm to 1 mm. The concentration range was adjusted to be appropriate for the expected Ki values. The results are summarized in Table 1.
The para-H and para-methyl benzamide analogues, compounds 42/53, 44/55, 47/58 and 48/59, were also evaluated in a second radioligand binding assay, relying on the displacement of the AT1R/AT2R balanced peptide [125I]Sarile (Sarile; [Sar1, Ile8]Ang II) instead of the AT2R selective [125I]CGP-42112 A from human AT2R (HEK293-hAT2R) as well as human AT1R (HEK293-hAT1R).[8e, 18] No binding to the AT1R was observed, and the Ki values towards AT2R are summarized and compared with Ki values from the first HEK-293 binding assay in Table 1. As can be seen, the Ki values from these two assays correspond very well.
When comparing the affinity results given in Table 1, two trends become apparent. First, within each of the three sets of compounds differing only in the substituent in para position (i.e., H, Me, Et), the methyl substituted ligands show the highest affinities. The second trend is that the larger the substituents on the benzamide, the smaller the difference in Ki value within each group. In the case of the diethyl substituted compounds, the values are 22.0 μm ([125I]CGP-42112A) for 42 as compared to 2.4 μm ([125I]CGP-42112A) for 53, and in the case of the dibenzyl substituted compounds, the difference is as low as 5 μm between the para-methylated ligand 58 and the unsubstituted compound 47. Moving the benzamide group to the para position leads to a significant loss in affinity. Only compound 70 still shows some affinity towards the AT2R with a Ki value of 35.0 μm, but the compound is inferior to its meta analogues (47, 58 and 64). The dibenzyl substituted benzamides were the most consistent in affinity in all sets of analogues. The benzamides showing the best Ki values correlate partly to the most potent benzamide analogues of C21/M024. The diethyl substituted analogues are among the compounds with highest affinities in both series, which could suggest that the amide functions of the two classes of compounds interact with the same environment in the receptor.[9b]
To be able to correlate the results from these new ligands targeting the human AT2R to our previous studies performed with AT2R in membranes from pig uterus, a few reference compounds were selected and included in the Ki determination. To our surprise, lead compound E (Table 1, entry 32) only exhibited a Ki value of 110.0 μm. In our previous studies using the pig AT2R, the Ki value was found to be 16.6 nm.[6f] Furthermore, reduced binding affinities were encountered also for the other peptide analogues, that is, F with 63.0 μm (pig AT2R 37.0 nm),[6f] C with 25 nm (pig AT2R 0.5 nm),[6e] and Ang IV (B) with a Ki value of 35 nm (pig AT2R 7.7 nm; see Table 1, entries 33–35). The nonpeptide agonist C21/M024 (D) displayed a reduced binding but not to the same extent (Ki human AT2R 9.8 nm versus Ki pig AT2R 0.4 nm). The values were verified by the second binding assay, also targeting the human AT2R, but with displacement of [125I]Sarile instead of [125I]CGP-42112A. In this assay, lead compound E exhibited a Ki value of 47.0 μm, C21 M024 (D) showed a Ki value of 7.6 nm, while Ang II (A) showed a 60 times higher Ki value (2.8 nm; see Table 1). C21/M024 (D) has also been evaluated for binding towards the human AT2R by Bosnyak et al. with a reported IC50=2.29 nm (Ang II (A); IC50=0.522 nm).[19] Thus, while the drug-like C21/M024 (D) binds with high affinity to AT2R both from human and pig, the lead E exhibits a remarkable difference in affinity in the two species.
The data suggest a species difference in the interaction of the peptide analogues and the nonpeptidic substances, respectively, with regard to their binding to AT2R, and emphasizes the importance of performing affinity studies on human AT2R. The differences could of course be due to experimental conditions, in addition to species variations, for example, cell and receptor origin (endogenous membranes from uterus versus transfected kidney cells) and/or differences in the experimental design to measure ligand binding. However, the assays seem comparable, as the reference compounds exhibit the same relative order of affinity in both assays (Table 1, entries 32–37).
A comparison of the sequence of AT2R from human and pig reveals that the receptors are very similar (95 % homology in the amino acid sequence),[20] and very fine-tuned receptor models on a molecular level or mutation studies are required to investigate whether the observed discrepancy in binding data originates from species differences at the receptor level. It is conceivable that binding could be modulated by various levels of interferences in the two assays with partner proteins as AT2R-interacting proteins (ATIP), the promyelocytic zinc finger protein (PLZF), the phosphatase SHP-1 or alpha subunit of G proteins. Such interactions might account for the incongruity reported herein.[21] Functional diversity of highly homologous proteins is rare (>90 % amino acid identity), although it has been shown for AT2R orthologues when comparing rabbit and human AT2R.[22] To verify if our results originates from functional diversity (i.e., species differences) much more studies must be performed that are outside of the scope of this report. The AT2R exists as a single copy, localized on the X chromosome and contains no intron in its coding region, and we hypothesize that it is more likely that the different binding data obtained are related to variations in tissues rather than species.
Georgsson et al. described the possibility to reduce the ligand size from C to the structures E and F without a major loss of affinity (Figure 1).[6f] Interestingly, these smaller ligands showed much higher Ki values when tested towards the human AT2 receptor. The herein presented new ligands are of comparable size as E and F but show improved affinity towards the human receptor. Thus, these new compounds will serve as a new starting point for further improvement of this new class of AT2R-selective ligands.
Conclusions
In summary, an efficient palladium-catalyzed procedure for aminocarbonylation of aryl iodides utilizing molybdenum hexacarbonyl (Mo(CO)6) as carbon monoxide source has been employed to make a series of AT2R ligands. Even though the Ki values presented in this work are comparably high (micromolar range), 13 of the 15 evaluated compounds demonstrate higher affinities towards the human AT2R (HEK293-hAT2R) than the original lead structure E. The large drop in affinity going from pig to human AT2 receptor assay was unexpected, and the discrepancy is more pronounced for the peptides and pseudopeptides than for the nonpeptidic drug-like structure D. The presented data emphasize the importance of using human receptors in drug discovery programs. However, with the synthesized benzamides, we have obtained new starting points for targeting the human AT2R, but significantly more efforts are required until equally high potency at the human AT2R as our previously reported selective nonpeptide AT2R agonist C21/M024 (D), is achieved.
Experimental Section
Chemistry
General information and materials: The microwave heating was performed in a Biotage Initiator single mode reactor, which produces controlled irradiation at 2450 MHz. The reaction temperature was determined using the built-in online IR sensor. Microwave mediated reactions were performed in sealed Smith process vials designed for 2–5 mL reaction volumes. Analytical TLC was performed using Merck aluminium-backed 0.2 mm silica gel 60 F-254 plates, and visualization was performed with UV light (λ=254 nm). Silica gel 60 was purchased from Merck. NMR spectra were recorded on a Varian Mercury plus at 25 °C and 400 MHz for 1H NMR and 100 MHz for 13C NMR. Chemical shifts (δ) are reported in ppm and referenced indirectly to TMS via the solvent (or residual solvent) signals. Analytical RP-HPLC–MS was performed on a Gilson-Finnigan ThermoQuest AQA system (Onyx monolithic C18 column, 50×4.6 mm) and a Dionex Ultimate 3000 (C18 column, 50×3 mm) using a MeCN/H2O gradient with 0.05 % HCOOH. Detection was performed using UV (λ=214 nm and 254 nm) and MS detection in ESI mode. Preparative RP-HPLC was performed on a Dionex Ultimate 3000 system (SB-C8 column, 21.2×150 mm; MeCN/H2O gradient with 0.05 % HCOOH) using UV detection (λ=214 nm and 254 nm). Molecular masses were determined on a mass spectrometer equipped with an electrospray ion source (ESI-HRMS; 7-T hybrid linear ion trap (LTQ) FT mass spectrometer modified with a nanoelectrospray ion source). The optical rotation was determined using a PerkinElmer 241 polarimeter. Specific rotations ([α] ) are reported in 10−1×deg×cm2 g−1, and the samples were prepared at a concentration of 1.0 g/100 mL in CHCl3. All starting materials, reagents and solvents are commercially available and were used as received.
) are reported in 10−1×deg×cm2 g−1, and the samples were prepared at a concentration of 1.0 g/100 mL in CHCl3. All starting materials, reagents and solvents are commercially available and were used as received.
General procedure A: Synthesis of iodoaryl OtBu-Ile derivatives 5–8: The iodo benzoic acid (1–4; 1 equiv), l-isoleucine tert-butyl ester hydrochloride (IleOtBu⋅HCl; 1.1 equiv) and 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate (HATU; 1.1 equiv) were dissolved in N,N-dimethylformamide (DMF; 4 mL mmol−1). N,N-Diisopropylethylamine (DIEA; 3.3 equiv) was added, and the reaction mixture was stirred at RT overnight. The mixture was poured into H2O and extracted with EtOAc (4×40 mL mmol−1). The combined organic layers were washed with saturated NH4Cl (1×40 mL mmol−1), H2O (4×40 mL mmol−1) and brine (2×40 mL mmol−1). After drying over Na2SO4, the solvent was evaporated in vacuo. Purification by column chromatography provided the pure products in moderate to excellent yields (5: 94 %, 6: 98 %, 7: 38 %, 8: 88 %).
(2S,3 R)-tert-Butyl 2-(3-iodobenzamido)-3-methylpentanoate (5): According to general procedure A, 3-iodo benzoic acid (1, 1.24 g, 5.0 mmol) was reacted with IleOtBu (1.23 g, 5.5 mmol), HATU (2.09 g, 5.5 mmol) and DIEA (2.9 mL, 16.5 mmol) in DMF (20 mL). Purification by column chromatography (i-hexane/EtOAc, 0–100 %) afforded 5 as a white semi-solid (1.96 g, 94 %): 1H NMR (CDCl3): δ=0.96 (d, J=6.9 Hz, 3 H), 0.95–1.00 (m, 3 H), 1.22–1.31 (m, 1 H), 1.45–1.58 (m, 1 H), 1.49 (s, 9 H), 1.94–2.02 (m, 1 H), 4.68 (dd, J=8.2 Hz, 4.4 Hz, 1 H), 6.68 (d, J=8.1 Hz, 1 H), 7.16 (t, J=7.8 Hz, 1 H), 7.74 (ddd, J=7.8 Hz, 1.6 Hz, 1.1 Hz, 1 H), 7.82 (ddd, J=7.9 Hz, 1.7 Hz, 1.1 Hz, 1 H), 8.12 ppm (t, J=1.6 Hz, 1 H); 13C NMR (CDCl3): δ=11.8, 15.4, 25.5, 28.1, 38.5, 57.1, 82.4, 94.2, 126.1, 130.2, 136.1, 136.3, 140.5, 165.3, 171.1 ppm.
(2S,3 R)-tert-Butyl 2-(4-methyl-3-iodobenzamido)-3-methylpentanoate (6): According to general procedure A, 3-iodo-4-methyl-benzoic acid (2, 1.31 g, 5.00 mmol) was reacted with IleOtBu (1.23 g, 5.50 mmol), HATU (2.09 g, 5.50 mmol) and DIEA (2.90 mL, 16.50 mmol) in DMF (20 mL). Purification by column chromatography (i-hexane/EtOAc, 0–100 %) afforded 6 as a white semi-solid (2.12 g, 98 %): 1H NMR (CDCl3): δ=0.93 (d, J=6.9 Hz, 3 H), 0.93 (t, J=7.3 Hz, 3 H), 1.18–1.32 (m, 1 H), 1.46 (s, 9 H), 1.47–1.55 (m, 1 H), 1.95 (ddd, J=9.0 Hz, 4.5 Hz, 2.1 Hz, 1 H), 2.41 (s, 3 H), 4.66 (dd, J=8.3 Hz, 4.6 Hz, 1 H), 6.79 (d, J=7.8 Hz, 1 H), 7.21 (d, J=7.9 Hz, 1 H), 7.61 (dd, J=7.9 Hz, 1.8 Hz, 1 H), 8.18 ppm (d, J=1.8 Hz, 1 H); 13C NMR (CDCl3): δ=11.6, 15.3, 25.5, 27.98, 28.02, 38.3, 57.0, 82.2, 100.8, 126.6, 129.4, 133.3, 137.5, 145.1, 165.0, 171.2 ppm.
(2S,3 R)-tert-Butyl 2-(4-ethyl-3-iodobenzamido)-3-methylpentanoate (7): According to general procedure A, 3-iodo-4-ethyl-benzoic acid (3, 2.00 g, 7.25 mmol) was reacted with IleOtBu (1.78 g, 7.97 mmol), HATU (3.03 g, 7.97 mmol) and DIEA (4.20 mL, 23.91 mmol) in DMF (25 mL). Purification by column chromatography (i-hexane/EtOAc, 0–100 %) afforded 7 as a white semi-solid (1.20 g; 38 %): 1H NMR (CDCl3): δ=0.98 (t, J=7.3 Hz, 6 H), 1.22–1.34 (m, 1 H), 1.29 (t, J=7.5 Hz, 3 H), 1.49 (s, 9 H), 1.54 (ddd, J=13.1 Hz, 7.6 Hz, 3.7 Hz, 1 H), 1.99 (dddd, J=11.4 Hz, 6.8 Hz, 4.5 Hz, 2.3 Hz, 1 H), 2.95 (q, J=7.5 Hz, 2 H), 4.70 (dd, J=8.2 Hz, 4.4 Hz, 1 H), 6.79 (d, J=7.9 Hz, 1 H), 7.43 (dd, J=8.1 Hz, 2.1 Hz, 1 H), 7.94 (dt, J=8.0 Hz, 1.7 Hz, 1 H), 8.27 ppm (d, J=1.8 Hz, 1 H); 13C NMR (CDCl3): δ=11.8, 14.7, 15.4, 25.6, 26.1, 28.1, 38.4, 57.2, 82.6, 110.0, 123.2, 131.1, 131.6, 133.3, 142.3, 164.5, 171.0 ppm.
(2S,3 R)-tert-Butyl 2-(4-iodobenzamido)-3-methylpentanoate (8): According to general procedure A, 4-iodo-benzoic acid (4, 2.48 g, 10.0 mmol) was reacted with IleOtBu (2.46 g, 10.0 mmol), HATU (4.18 g, 10.0 mmol) and DIEA (5.75 mL, 33.0 mmol) in DMF (30 mL). Purification by column chromatography (i-hexane/EtOAc, 0–100 %) afforded 8 as a white semi-solid (3.69 g, 88 %): 1H NMR (CDCl3): δ=0.94 (d, J=7.4 Hz, 3 H), 0.95 (t, J=7.5 Hz, 3 H), 1.20–1.32 (m, 1 H), 1.47 (s, 9 H), 1.49–1.57 (m, 1 H), 1.92–2.01 (m, 1 H), 4.66 (dd, J=8.2 Hz, 4.5 Hz, 1 H), 6.74 (d, J=8.1 Hz, 1 H), 7.48–7.52 (m, 2 H), 7.73–7.77 ppm (m, 2 H); 13C NMR (CDCl3): δ=11.8, 15.3, 25.6, 28.1, 38.4, 57.1, 82.3, 98.5, 128.6, 133.7, 137.7, 166.1, 171.1 ppm.
General procedure B: Aminocarbonylation reactions: Iodoaryl OtBu-Ile derivative (5–8; 1 equiv, 0.5–1.0 mmol), amine (3 equiv), Pd(OAc)2 (0.1 equiv) and Mo(CO)6 (1 equiv) were dissolved in tetrahydrofuran (THF; 2.5 mL mmol−1) in a 2–5 mL Smith process vial. The mixture was stirred for 2 min at RT. Diazabicycloundecene (DBU; 3 equiv) was added, and the vial was immediately sealed and irradiated in a microwave reactor at 100 °C for 15 min. After cooling to RT, MeOH was added, and the suspension was filtered through a short plug of Celite®. The solvent was evaporated, and the crude mixture was purified by column chromatography (i-hexane/EtOAc, 0–100 %), giving the desired products in yields between 14 % and 85 %.
(2S,3 R)-tert-Butyl 2-(3-(diethylcarbamoyl)benzamido)-3-methylpentanoate (14): According to general procedure B, reaction of derivative 5 afforded 14 as a colorless oil (45 mg, 24 %): 1H NMR (CDCl3): δ=0.93 (d, J=6.9 Hz, 3 H), 0.94 (t, J=7.4 Hz, 3 H), 1.02–1.15 (m, 3 H), 1.16–1.30 (m, 4 H), 1.45 (s, 9 H), 1.43–1.56 (m, 1 H), 1.95 (ddt, J=11.4 Hz, 6.8 Hz, 2.2 Hz, 1 H), 3.20 (s, 2 H), 3.51 (s, 2 H), 4.65 (dd, J=8.2 Hz, 4.5 Hz, 1 H), 6.73 (d, J=8.2 Hz, 1 H), 7.43 (t, J=7.5 Hz, 1 H, CHAr), 7.47 (ddd, J=7.6 Hz, 1.6 Hz, 1.1 Hz, 1 H, CHAr), 7.76 (t, J=1.6 Hz, 1 H, CHAr), 7.80 ppm (dt, J=7.3 Hz, 1.6 Hz, 1 H); 13C NMR (CDCl3): δ=11.7, 12.7, 14.1, 15.3, 25.5, 28.0, 38.3, 39.3, 43.3, 57.1, 82.2, 124.9, 127.7, 128.7, 129.2, 134.6, 137.6, 166.2, 170.2, 170.9 ppm.
(2S,3 R)-tert-Butyl 2-(3-(benzyl(methyl)carbamoyl)benzamido)-3-methylpentanoate (16): According to general procedure B, reaction of derivative 5 afforded 16 as a white semi-solid (165 mg, 75 %): 1H NMR (CDCl3): δ=0.90–1.00 (m, 6 H), 1.18–1.32 (m, 1 H), 1.48 (s, 9 H), 1.46–1.58 (m, 1 H), 1.93–2.04 (m, 1 H), 2.84–3.08 (m, 3 H), 4.43–4.73 (m, 2 H), 4.76 (s br, 1 H), 6.63–6.78 (m, 1 H), 7.13–7.22 (m, 1 H), 7.27–7.32 (m, 1 H), 7.35 (s br, 3 H), 7.41–7.53 (m, 1 H), 7.58 (d, J=7.5, 1 H), 7.82–7.87 (m, 1 H), 7.89 ppm (s br, 1 H); 13C NMR (CDCl3): δ=11.8, 15.4, 25.5, 28.1, 33.3, 37.0, 38.4, 50.8, 55.1, 57.1, 82.3, 125.7, 126.6, 127.6, 128.0, 128.2, 128.4, 128.7, 128.9, 130.0, 134.8, 136.8, 138.8, 166.1, 168.3, 171.0 ppm.
(2S,3 R)-tert-Butyl 2-(3-(dibenzylcarbamoyl)benzamido)-3-methylpentanoate (19): According to general procedure B, reaction of derivative 5 afforded 19 as a white semi-solid (72 mg, 14 %): 1H NMR (CDCl3): δ=0.94 (d, J=7.0 Hz, 3 H), 0.95–1.00 (m, 3 H), 1.17–1.30 (m, 1 H), 1.46–1.58 (m, 1 H), 1.49 (s, 9 H), 1.92–2.02 (m, 1 H), 4.39 (s br, 2 H), 4.67 (dd, J=8.3 Hz, 4.5 Hz, 1 H), 4.68–4.82 (m, 2 H), 6.67 (d, J=8.2 Hz, 1 H), 7.08–7.18 (m, 2 H), 7.26–7.39 (m, 8 H), 7.45 (t, J=7.8 Hz, 1 H), 7.59–7.64 (m, 1 H), 7.83 (ddd, J=7.8 Hz, 1.7 Hz, 1.2 Hz, 1 H), 7.92 ppm (t, J=1.5 Hz, 1 H); 13C NMR (CDCl3): δ=11.8, 15.4, 25.5, 28.1, 38.4, 47.1, 51.5, 57.1, 82.3, 125.4, 126.9, 127.7, 128.2, 128.4, 128.7, 128.9, 129.6, 134.8, 136.1, 136.7, 166.0, 170.9, 171.3 ppm.
(2S,3 R)-tert-Butyl 2-(3-(benzyl(ethyl)carbamoyl)benzamido)-3-methylpentanoate (20): According to general procedure B, reaction of derivative 5 afforded 20 as a white semi-solid (97 mg, 43 %): 1H NMR (CDCl3): δ=0.92–1.00 (m, 6 H), 1.02–1.15 (m, 1 H), 1.15–1.27 (m, 3 H), 1.44–1.56 (m, 1 H), 1.47 (s, 9 H), 1.91–2.01 (m, 1 H), 3.18 (s br, 1 H), 3.51 (s br, 1 H), 4.46 (s, 1 H), 4.66 (s br, 1 H), 4.76 (s, 1 H), 6.70 (d, J=33.1 Hz, 1 H), 7.10–7.38 (m, 5 H), 7.50–7.38 (m, 1 H), 7.54 (s, 1 H), 7.79–7.89 ppm (m, 2 H); 13C NMR (CDCl3): δ=11.7, 12.1, 13.6, 15.3, 25.5, 28.0, 38.4, 40.0, 42.9, 46.9, 52.1, 57.1, 82.2, 125.1, 126.6, 127.4, 127.9, 128.1, 128.4, 128.6, 128.8, 129.4, 133.1, 134.7, 137.1, 162.5, 166.1, 170.9 ppm.
(2S,3 R)-tert-Butyl 2-(3-(diethylcarbamoyl)-4-methylbenzamido)-3-methylpentanoate (25): According to general procedure B, reaction of derivative 6 afforded 25 as a white semi-solid (43 mg, 22 %): 1H NMR (CDCl3): δ=0.94 (d, J=6.8 Hz, 3 H), 0.95 (t, J=7.2 Hz, 3 H), 1.02 (t, J=7.1 Hz, 3 H), 1.17–1.23 (m, 1 H), 1.25 (t, J=7.1 Hz, 3 H), 1.47 (s, 9 H), 1.49–1.56 (m, 1 H), 1.96 (ddt, J=9.2 Hz, 6.8 Hz, 4.6 Hz, 1 H), 2.32 (s, 3 H), 3.06–3.14 (m, 2 H), 3.22–3.81 (m, 2 H), 4.66 (dd, J=8.3 Hz, 4.5 Hz, 1 H), 6.65 (d, J=8.2 Hz, 1 H), 7.26 (dd, J=8.0 Hz, 0.4 Hz, 1 H), 7.59 (d, J=1.7 Hz, 1 H), 7.68 ppm (dd, J=8.0 Hz, 1.1 Hz, 1 H); 13C NMR (CDCl3): δ=11.7, 12.8, 14.0, 15.3, 18.9, 25.5, 28.0, 38.4, 38.8, 42.7, 57.0, 82.2, 124.2, 127.1, 130.6, 132.1, 137.4, 138.0, 166.2, 169.8, 171.0 ppm.
(2S,3 R)-tert-Butyl 2-(3-(benzyl(methyl)carbamoyl)-4-methylbenzamido)-3-methylpentanoate (27): According to general procedure B, reaction of derivative 6 afforded 27 as a white semi-solid (102 mg, 45 %): 1H NMR (CDCl3): δ=0.91–1.00 (m, 6 H), 1.18–1.34 (m, 1 H), 1.49 (s, 9 H), 1.51–1.58 (m, 1 H), 1.92–2.03 (m, 1 H), 2.36 (s, 3 H), 2.70 (s, 2 H), 3.08 (s, 1 H), 4.35 (s, 1 H), 4.68 (dd, J=8.3 Hz, 4.5 Hz, 1 H), 4.78 (s, 1 H), 6.67 (d, J=8.3 Hz, 1 H), 7.09 (d, J=7.6 Hz, 1 H), 7.25–7.36 (m, 3 H), 7.36–7.39 (m, 2 H), 7.66 (d, J=1.7 Hz, 1 H), 7.71 ppm (t, J=7.2 Hz, 1 H); 13C NMR (CDCl3): δ=11.8, 15.4, 19.0, 25.5, 28.1, 32.6, 35.7, 38.5, 50.2, 54.6, 57.0, 82.3, 124.6, 127.0, 127.6, 128.4, 128.7, 128.8, 130.9, 132.3, 136.0, 136.7, 138.0, 166.2, 170.5, 171.1 ppm.
(2S,3 R)-tert-Butyl 2-(3-(dibenzylcarbamoyl)-4-methylbenzamido)-3-methylpentanoate (30): According to general procedure B, reaction of derivative 6 afforded 30 as a white semi-solid (115 mg, 22 %): 1H NMR (CDCl3): δ=0.93 (d, J=6.9 Hz, 3 H), 0.97 (t, J=7.4 Hz, 3 H), 1.16–1.29 (m, 1 H), 1.49 (s, 9 H), 1.50–1.56 (m, 1 H), 1.91–1.99 (m, 1 H), 2.36 (s, 3 H), 4.22 (s, 2 H), 4.29–4.57 (m, 1 H), 4.64 (dd, J=8.3 Hz, 4.5 Hz, 1 H), 4.87–5.34 (m, 1 H), 6.59 (d, J=8.2 Hz, 1 H), 7.05–7.10 (m, 2 H), 7.26–7.39 (m, 9 H), 7.67–7.71 ppm (m, 2 H); 13C NMR (CDCl3): δ=11.8, 15.3, 19.2, 25.5, 28.1, 38.4, 46.7, 50.9, 57.0, 82.2, 124.6, 127.2, 127.5, 127.7, 127.8, 128.7, 128.8, 128.8, 130.9, 132.1, 135.8, 136.4, 136.7, 138.5, 166.0, 170.96, 170.99 ppm.
(2S,3 R)-tert-Butyl 2-(3-(benzyl(ethyl)carbamoyl)-4-methylbenzamido)-3-methylpentanoate (31): According to general procedure B, reaction of derivative 6 afforded 31 as a white semi-solid (101 mg; 45 %): 1H NMR (CDCl3): δ=0.90–1.00 (m, 7 H), 1.12–1.33 (m, 3 H), 1.48 (s, 9 H), 1.50–1.58 (m, 1 H), 1.92–2.02 (m, 1 H), 2.36 (s, 3 H), 3.01–3.10 (m, J=7.0 Hz, 2 H), 4.31–4.55 (m, 2 H), 4.68 (dd, J=8.2 Hz, 4.4 Hz, 1 H), 6.67 (d, J=8.2 Hz, 1 H), 7.10 (d, J=7.9 Hz, 1 H), 7.21–7.41 (m, 5 H), 7.60–7.70 ppm (m, 2 H); 13C NMR (CDCl3): δ=11.7, 12.2, 13.4, 15.3, 19.1, 25.5, 28.0, 38.4, 42.1, 51.5, 57.0, 82.2, 124.3, 127.0, 127.4, 127.7, 128.1, 128.6, 128.7, 130.6, 132.2, 136.3, 137.2, 138.1, 162.5, 166.0, 170.4 ppm.
(2S,3 R)-tert-Butyl 2-(3-(diethylcarbamoyl)-4-ethylbenzamido)-3-methylpentanoate (33): According to general procedure B, reaction of derivative 7 afforded 33 as a white semi-solid (100 mg, 48 %): 1H NMR (CDCl3): δ=0.99–0.93 (m, 6 H), 1.04 (t, J=7.1 Hz, 3 H), 1.29–1.20 (m, 7 H), 1.48 (s, 9 H), 1.59–1.49 (m, 1 H), 2.02–1.92 (m, 1 H), 2.65 (q, J=7.6 Hz, 2 H), 3.11 (q, J=6.7 Hz, 2 H), 3.83–3.29 (m, 2 H), 4.67 (dd, J=8.3 Hz, 4.5 Hz, 1 H), 6.64 (d, J=8.2 Hz, 1 H), 7.33 (d, J=8.1 Hz, 1 H), 7.58 (s, 1 H), 7.73 ppm (d, J=6.7 Hz, 1 H); 13C NMR (CDCl3): δ=11.8, 12.8, 14.0, 14.8, 15.4, 25.5, 25.9, 28.1, 38.5, 38.8, 42.9, 57.0, 82.2, 124.0, 127.0, 129.0, 132.1, 136.9, 144.1, 166.2, 169.8, 171.0 ppm.
(2S,3 R)-tert-Butyl 2-(3-(benzyl(methyl)carbamoyl)-4-ethylbenzamido)-3-methylpentanoate (34): According to general procedure B, reaction of derivative 7 afforded 34 as a white semi-solid (155 mg, 66 %): 1H NMR (CDCl3): δ=0.89–0.99 (m, 6 H), 1.16–1.28 (m, 4 H), 1.47 (s, 9 H), 1.49–1.56 (m, 1 H), 1.89–2.02 (m, 1 H), 2.65 (q, J=7.5 Hz, 3 H), 2.68 (s, 2 H), 3.06 (s, 1 H), 4.20–4.53 (m, 1 H), 4.67 (dd, J=8.2 Hz, 4.5 Hz, 1 H), 4.71–4.88 (m, 1 H), 6.69 (d, J=7.8 Hz, 1 H), 7.09 (d, J=7.9 Hz, 1 H), 7.32 (m, 5 H), 7.63 (d, J=1.8 Hz, 1 H), 7.70–7.78 ppm (m, 1 H); 13C NMR (CDCl3): δ=11.7, 14.8, 15.3, 25.5, 25.9, 28.0, 32.6, 35.9, 38.4, 50.2, 54.7, 57.0, 82.2, 124.4, 126.9, 127.6, 127.7, 128.3, 128.6, 128.8, 129.0, 132.2, 136.0, 136.7, 144.2, 166.0, 170.4, 171.0 ppm.
(2S,3 R)-tert-Butyl 2-(3-(benzyl(ethyl)carbamoyl)-4-ethylbenzamido)-3-methylpentanoate (35): According to general procedure B, reaction of derivative 7 afforded 35 as a white semi-solid (120 mg, 50 %): 1H NMR (CDCl3): δ=0.93–1.04 (m, 9 H), 1.19–1.33 (m, 4 H), 1.49 (s, 9 H), 1.54 (ddd, J=13.0 Hz, 7.5 Hz, 5.1 Hz, 1 H), 1.93–2.02 (m, 1 H), 2.68 (q, J=7.5 Hz, 2 H), 3.05 (q, J=7.1 Hz, 2 H), 4.32 (d, J=7.9 Hz, 2 H), 4.68 (dd, J=8.3 Hz, 4.5 Hz, 1 H), 6.67 (d, J=8.3 Hz, 1 H), 7.11 (d, J=7.3 Hz, 1 H), 7.21–7.41 (m, 7 H), 7.59–7.67 (m, 1 H), 7.75 ppm (d, J=7.8 Hz, 1 H); 13C NMR (CDCl3): δ=11.8, 13.3, 15.0, 15.4, 25.5, 26.0, 28.1, 38.4, 42.2, 51.7, 57.0, 82.2, 124.2, 127.0, 127.5, 127.7, 128.3, 128.6, 128.8, 129.1, 132.1, 136.3, 137.3, 144.2, 166.0, 170.4, 171.0 ppm.
(2S,3 R)-tert-Butyl 2-(3-(dibenzylcarbamoyl)-4-ethylbenzamido)-3-methylpentanoate (36): According to general procedure B, reaction of derivative 7 afforded 36 as a white semi-solid (339 mg, 62 %): 1H NMR (CDCl3): δ=0.89–0.94 (m, 3 H), 0.96 (t, J=7.4 Hz, 3 H), 1.15–1.27 (m, 1 H), 1.23 (t, J=7.6 Hz, 3 H), 1.47–1.55 (m, 1 H), 1.49 (s, 9 H), 1.94 (s, 1 H), 2.61–2.75 (m, 2 H), 4.10–4.27 (m, 2 H), 4.27–4.45 (m, 1 H), 4.64 (dd, J=8.3 Hz, 4.5 Hz, 1 H), 5.04–5.27 (m, 1 H), 6.57 (d, J=8.0 Hz, 1 H), 7.07–7.11 (m, 2 H), 7.24–7.39 (m, 9 H), 7.68 (d, J=1.9 Hz, 1 H), 7.75 ppm (dd, J=8.0 Hz, 1.9 Hz, 1 H); 13C NMR (CDCl3): δ=11.8, 15.0, 15.4, 25.5, 26.1, 28.1, 38.4, 46.6, 51.0, 57.0, 82.2, 124.5, 127.1, 127.7, 127.8, 128.7, 128.8, 128.9, 129.2, 132.1, 135.8, 136.0, 136.7, 144.5, 166.0, 170.9, 171.0 ppm.
(2S,3 R)-tert-Butyl 2-(4-(diethylcarbamoyl)benzamido)-3-methylpentanoate (65): According to general procedure B, reaction of derivative 8 afforded 65 as a white semi-solid (133 mg, 68 %): 1H NMR (CDCl3): δ=0.94–0.99 (m, 6 H), 1.05–1.14 (m, 3 H), 1.22–1.27 (m, 3 H), 1.28–1.35 (m, 1 H), 1.48 (s, 9 H), 1.49–1.61 (m, 1 H), 1.94–2.02 (m, 1 H), 3.14–3.26 (m, 2 H), 3.49–3.58 (m, 2 H), 4.69 (dd, J=8.2 Hz, 4.4 Hz, 1 H), 6.71 (d, J=7.6 Hz, 1 H), 7.40–7.46 (m, 2 H), 7.80–7.85 ppm (m, 2 H); 13C NMR (CDCl3): δ=11.8, 14.2, 15.4, 25.5, 28.1, 38.5, 39.4, 43.2, 57.1, 82.4, 126.5, 127.2, 134.9, 140.4, 166.3, 170.3, 171.1 ppm.
(2S,3 R)-tert-Butyl 2-(4-(benzyl(methyl)carbamoyl)benzamido)-3-methylpentanoate (66): According to general procedure B, reaction of derivative 8 afforded 66 as a white semi-solid (160 mg, 73 %): 1H NMR (CDCl3): δ=0.94 (s br, 6 H), 1.18–1.32 (m, 1 H), 1.46 (s, 9 H), 1.49–1.63 (m, 1 H), 1.91–2.02 (m, 1 H), 2.80 (s, 2 H), 3.02 (s, 1 H), 4.44 (s, 1 H), 4.68 (s br, 1 H), 4.73 (s, 1 H), 6.78 (s br, 1 H), 7.12 (d, J=6.6 Hz, 1 H), 7.37–7.25 (m, 4 H), 7.48 (t, J=6.4 Hz, 2 H), 7.74–7.85 ppm (m, 2 H); 13C NMR (CDCl3): δ=11.7, 15.3, 25.5, 28.0, 33.2, 36.8, 38.4, 50.7, 54.9, 57.1, 82.3, 126.5, 126.9, 127.2, 127.7, 128.2, 128.7, 128.8, 135.3, 136.1, 139.1, 166.1, 170.5, 171.0 ppm.
(2S,3 R)-tert-Butyl 2-(4-(dibenzylcarbamoyl)benzamido)-3-methylpentanoate (67): According to general procedure B, reaction of derivative 8 afforded 67 as a white semi-solid (40 mg, 16 %): 1H NMR (CDCl3): δ=0.96 (d, J=7.0 Hz, 3 H), 0.97 (t, J=7.6 Hz, 3 H), 1.21–1.30 (m, 1 H), 1.48 (s, 9 H), 1.50–1.58 (m, 1 H), 1.98 (ddt, J=9.2 Hz, 4.7 Hz, 2.4 Hz, 1 H), 4.72 (s, 2 H), 4.36 (s, 2 H), 4.68 (dd, J=8.2 Hz, 4.4 Hz, 1 H), 6.71 (d, J=8.2 Hz, 1 H), 7.12 (d, J=6.6 Hz, 2 H), 7.27–7.39 (m, 8 H), 7.53–7.57 (m, 2 H), 7.79–7.83 ppm (m, 2 H); 13C NMR (CDCl3): δ=11.8, 15.4, 25.5, 28.1, 38.5, 47.0, 51.4, 57.1, 82.4, 126.8, 126.9, 127.3, 127.7, 127.8, 128.5, 128.7, 128.9, 135.4, 136.0, 136.7, 139.2, 166.0, 171.1, 171.3 ppm.
General procedure C: Hydrolysis of tert-butyl esters: The ester (1 equiv, 0.08–0.6 mmol) was dissolved in CH2Cl2 (15 μL/μmol). Trifluoroacetic acid (TFA; 7.7 μL/μmol) added and the mixture was stirred at RT for 3 h. The solvent was evaporated, and the crude mixture was purified using HPLC (MeCN/H2O, 0–100%). The pure products were isolated in 46–96 % yield.
(2S,3 R)-2-(3-(Diethylcarbamoyl)benzamido)-3-methylpentanoic acid (42): According to general procedure C, reaction of ester 14 gave 42 as a white semi-solid (31 mg; 73 %): [α]=+10.3; 1H NMR (CD3OD): δ=0.97 (t, J=7.4 Hz, 3 H), 1.03 (d, J=6.8 Hz, 3 H), 1.14 (t, J=6.4 Hz, 3 H), 1.27 (t, J=6.4 Hz, 3 H), 1.30–1.41 (m, 1 H), 1.56–1.68 (m, 1 H), 1.98–2.10 (m, 1 H), 3.25–3.37 (m, 2 H), 3.57 (d, J=6.8 Hz, 2 H), 4.57 (d, J=6.3 Hz, 1 H), 7.56 (dt, J=5.8 Hz, 3.2 Hz, 2 H), 7.86 (s, 1 H), 7.91–7.97 ppm (m, 1 H); 13C NMR (CD3OD): δ=11.7, 13.1, 14.4, 16.1, 26.6, 38.1, 41.0, 45.0, 59.0, 126.5, 129.6, 130.0, 130.4, 135.9, 138.3, 169.4, 172.6, 174.9 ppm; HRMS (ESI): m/z [M+H]+ calcd for C18H26N2O4: 335.1971, found: 335.1972.
(2S,3 R)-2-(3-(Benzyl(methyl)carbamoyl)benzamido)-3-methylpentanoic acid (44): According to general procedure C, reaction of ester 16 gave 44 as a white semi-solid (51 mg, 65 %): [α]=+8.0; 1H NMR (CD3OD): δ=0.96 (t, J=7.3 Hz, 3 H), 1.03 (d, J=6.9 Hz, 3 H), 1.25–1.40 (m, 1 H), 1.54–1.68 (m, 1 H), 1.97–2.08 (m, 1 H), 2.89 (s, 2 H), 3.02 (s, 1 H), 4.52 (s br, 1 H), 4.57 (d, J=6.0 Hz, 1 H), 4.76 (s br, 1 H), 7.17 (d, J=6.8 Hz, 1 H), 7.25–7.42 (m, 4 H), 7.49–7.59 (m, 1 H), 7.62 (d, J=6.5 Hz, 1 H), 7.89–7.98 ppm (m, 2 H); 13C NMR (CD3OD): δ=11.8, 16.3, 26.7, 33.9, 37.7, 38.3, 52.0, 56.3, 59.1, 127.2, 128.1, 128.8, 129.3, 129.8, 130.0, 130.1, 131.1, 136.1, 137.8, 138.2, 169.6, 173.0, 175.0 ppm; HRMS (ESI): m/z [M+H]+ calcd for C22H26N2O4: 383.1971, found: 383.1974.
(2S,3 R)-2-(3-(Dibenzylcarbamoyl)benzamido)-3-methylpentanoic acid (47): According to general procedure C, reaction of ester 19 gave 47 as a white semi-solid (37 mg, 96 %): [α]=+5.5; 1H NMR (CD3OD): δ=0.95 (t, J=7.4 Hz, 3 H), 1.00 (d, J=6.9 Hz, 3 H), 1.23–1.37 (m, 1 H), 1.59 (dqd, J=14.9 Hz, 7.5 Hz, 4.3 Hz, 1 H), 1.95-2.08 (m, 1 H), 4.44 (s br, 2 H), 4.55 (d, J=6.3 Hz, 1 H), 4.63–4.77 (m, 2 H), 7.14 (s, 2 H), 7.29–7.44 (m, 8 H), 7.52 (t, J=7.7 Hz, 1 H), 7.64 (dt, J=7.7 Hz, 1.4 Hz, 1 H), 7.89–7.93 ppm (m, 1 H), 7.94 (t, J=1.5 Hz, 1 H); 13C NMR (CD3OD): δ=11.8, 16.3, 26.7, 38.3, 48.7, 53.3, 59.1, 127.0, 128.3, 128.9, 129.0, 129.5, 130.0, 130.1, 130.16, 130.21, 130.8, 136.2, 137.5, 137.7, 138.1, 169.6, 173.8, 175.0 ppm; HRMS (ESI): m/z [M+H]+ calcd for C28H30N2O4: 459.2284; found: 459.2287.
(2S,3 R)-2-(3-(Benzyl(ethyl)carbamoyl)benzamido)-3-methylpentanoic acid (48): According to general procedure C, reaction of ester 20 gave 48 as a white semi-solid (47 mg, 64 %): [α]=+6.4; 1H NMR (CD3OD): δ=0.97 (t, J=7.4 Hz, 3 H), 1.03 (d, J=6.7 Hz, 3 H), 1.06–1.25 (m, 2 H), 1.27–1.39 (m, 1 H), 1.54–1.69 (m, 1 H), 1.97–2.09 (m, 1 H), 3.22–3.29 (m, 1 H), 3.45–3.60 (m, 1 H), 4.54 (s br, 1 H), 4.56 (d, J=6.4 Hz, 1 H), 4.80 (s, 1 H), 7.16–7.43 (m, 5 H), 7.49–7.65 (m, 2 H), 7.88–7.98 ppm (m, 2 H); 13C NMR (CD3OD): δ=11.8, 12.7, 14.1, 16.3, 26.8, 38.3, 41.8, 44.9, 48.9, 53.7, 59.2, 126.7, 128.2, 128.7, 128.9, 129.2, 129.9, 130.1, 130.2, 130.6, 136.2, 138.1, 138.7, 169.6, 173.4, 175.1 ppm; HRMS (ESI): m/z [M+H]+ calcd for C23H28N2O4: 397.2122, found: 397.2122.
(2S,3 R)-2-(3-(Diethylcarbamoyl)-4-methylbenzamido)-3-methylpentanoic acid (53): According to general procedure C, reaction of ester 25 gave 53 as a white semi-solid (30 mg, 72 %): [α]=+9.7; 1H NMR (CD3OD): δ=0.96 (t, J=7.4 Hz, 3 H), 1.02 (d, J=6.8 Hz, 3 H), 1.07 (t, J=7.1 Hz, 3 H), 1.29 (t, J=7.1 Hz, 3 H), 1.31–1.39 (m, 1 H), 1.56–1.68 (m, 1 H), 1.08–2.08 (m, 1 H), 2.34 (s, 3 H), 3.08–3.28 (m, 2 H), 3.44–3.77 (m, 2 H), 4.55 (d, J=6.5 Hz, 1 H), 7.39 (d, J=8.0 Hz, 1 H), 7.70 (s, 1 H), 7.83 ppm (dd, J=8.0 Hz, 1.8 Hz, 1 H); 13C NMR (CD3OD): δ=11.8, 13.2, 14.3, 16.3, 19.1, 26.8, 38.2, 40.7, 44.7, 59.0, 126.0, 129.3, 131.9, 133.3, 138.2, 139.5, 169.5, 172.4, 175.1 ppm; HRMS (ESI): m/z [M+H]+ calcd for C19H28N2O4: 349.2127, found: 349.2136.
(2S,3 R)-2-(3-(Benzyl(methyl)carbamoyl)-4-methylbenzamido)-3-methylpentanoic acid (55): According to general procedure C, reaction of ester 27 gave 55 as a white semi-solid (46 mg, 68 %): [α]=+7.3; 1H NMR (CD3OD): δ=0.96 (t, J=7.4 Hz, 3 H), 1.02 (d, J=6.9 Hz, 3 H), 1.24–1.39 (m, 1 H), 1.54–1.67 (m, 1 H), 1.97–2.07 (m, 1 H), 2.32 (s, 3 H), 2.75 (s, 2 H), 3.08 (s, 1 H), 4.39 (s br, 1 H), 4.56 (d, J=6.7 Hz, 1 H), 4.66–4.87 (m, 1 H), 7.12 (d, J=7.1 Hz, 1 H), 7.23–7.35 (m, 2 H), 7.35–7.44 (m, 3 H), 7.71–7.78 (m, 1 H), 7.80–7.86 ppm (m, 1 H); 13C NMR (CD3OD): δ=11.8, 16.3, 19.2, 26.8, 33.5, 36.6, 38.2, 51.4, 55.8, 59.1, 126.2, 128.5, 129.0, 129.4, 129.5, 130.0, 130.1, 131.9, 133.6, 137.6, 138.2, 139.5, 169.5, 173.0, 175.1 ppm; HRMS (ESI): m/z [M+H]+ calcd for C23H28N2O4: 397.2127; found: 397.2117.
(2S,3 R)-2-(3-(Dibenzylcarbamoyl)-4-methylbenzamido)-3-methylpentanoic acid (58): According to general procedure C, reaction of ester 30 gave 58 as a white semi-solid (67 mg, 82 %): [α]=+5.5; 1H NMR (CD3OD): δ=0.95 (t, J=7.4 Hz, 3 H), 0.99 (d, J=6.8 Hz, 3 H), 1.23–1.35 (m, 1 H), 1.58 (ddt, J=11.7 Hz, 7.5 Hz, 4.3 Hz, 1 H), 1.96–2.05 (m, 1 H), 2.29 (s, 3 H), 4.28 (s, 2 H), 4.54 (d, J=6.4 Hz, 1 H), 4.92 (s br, 2 H), 7.04–7.07 (m, 2 H), 7.20–7.44 (m, 9 H), 7.78 (s, 1 H), 7.81 ppm (dd, J=7.9 Hz, 1.9 Hz, 1 H); 13C NMR (CD3OD): δ=11.8, 16.3, 19.4, 26.7, 38.3, 48.8, 52.8, 59.0, 126.4, 128.7, 129.0, 129.1, 129.5, 129.8, 129.95, 130.02, 132.1, 133.3, 137.2, 137.5, 138.1, 134.0, 169.3, 173.5, 175.0 ppm; HRMS (ESI): m/z [M+H]+ calcd for C29H32N2O4: 473.2440; found: 473.2445.
(2S,3 R)-2-(3-(Benzyl(ethyl)carbamoyl)-4-methylbenzamido)-3-methylpentanoic acid (59): According to general procedure C, reaction of ester 31 gave 59 as a white semi-solid (48 mg, 71 %): [α]=+6.4; 1H NMR (CD3OD): δ=0.96 (t, J=7.4 Hz, 3 H), 0.99 (d, J=6.9 Hz, 3 H), 1.01–1.26 (m, 3 H), 1.26–1.39 (m, 1 H), 1.54–1.67 (m, 1 H), 1.95–2.07 (m, 1 H), 4.40 (s br, 1 H), 4.55 (d, J=6.5 Hz, 1 H), 2.34 (s, 3 H), 3.07–3.40 (m, 2 H), 4.69–4.96 (m, 1 H), 7.11–7.16 (m, 1 H), 7.25–7.46 (m, 5 H), 7.69–7.77 (m, 1 H), 7.82 ppm (ddd, J=13.8 Hz, 8.0 Hz, 1.9 Hz, 1 H); 13C NMR (CD3OD): δ=11.8, 12.7, 13.8, 16.3, 19.3, 26.8, 38.4, 41.4, 44.3, 48.3, 53.2, 59.3, 126.1, 128.6, 128.9, 129.0, 129.3, 129.5, 129.9, 130.0, 132.0, 133.5, 137.9, 138.8, 139.7, 169.3, 173.0, 175.3 ppm; HRMS (ESI): m/z [M+H]+ calcd for C24H30N2O4: 411.2278, found: 411.2289.
(2S,3 R)-2-(3-(Diethylcarbamoyl)-4-ethylbenzamido)-3-methylpentanoic acid (61): According to general procedure C, reaction of ester 33 gave 61 as a white semi-solid (31 mg, 58 %): [α]=+9.7; 1H NMR (CD3OD): δ=0.96 (t, J=7.4 Hz, 3 H), 1.02 (d, J=6.9 Hz, 3 H), 1.08 (t, J=7.1 Hz, 3 H), 1.25 (t, J=7.6 Hz, 3 H), 1.28 (t, J=7.1 Hz, 3 H), 1.31–1.39 (m, 1 H), 1.62 (ddq, J=14.9 Hz, 7.5 Hz, 4.3 Hz, 1 H), 1.97–2.08 (m, 1 H), 2.61–2.72 (m, 2 H), 3.09–3.27 (m, 2 H), 3.39–3.56 (m, 1 H), 3.71 (s br, 1 H), 4.56 (d, J=6.5 Hz, 1 H), 7.45 (d, J=8.1 Hz, 1 H), 7.69 (s, 1 H), 7.88 ppm (dd, J=8.1 Hz, 2.0 Hz, 1 H); 13C NMR (CD3OD): δ=11.8, 13.1, 14.3, 15.4, 16.3, 26.8, 27.1, 38.3, 40.6, 44.8, 59.1, 126.1, 129.5, 130.4, 133.3, 137.7, 145.6, 169.4, 172.4, 175.0 ppm; HRMS (ESI): m/z [M+H]+ calcd for C20H30N2O4: 363.2278, found: 363.2278.
(2S,3 R)-2-(3-(Benzyl(methyl)carbamoyl)-4-ethylbenzamido)-3-methylpentanoic acid (62): According to general procedure C, reaction of ester 34 gave 62 as a white semi-solid (45 mg, 49 %): [α]=+4.2; 1H NMR (CD3OD): δ=0.96 (t, J=7.4 Hz, 3 H), 1.02 (d, J=6.9 Hz, 3 H), 1.18–1.28 (m, 3 H), 1.28–1.39 (m, 1 H), 1.54–1.67 (m, 1 H), 1.97–2.08 (m, 1 H), 2.60–2.73 (m, 2 H), 2.76 (s, 2 H), 3.08 (s, 1 H), 4.38 (s, 1 H), 4.55 (d, J=6.5 Hz, 1 H), 4.60–4.80 (m, 1 H), 7.11–7.16 (m, 1 H), 7.23–7.36 (m, 2 H), 7.37–7.47 (m, 3 H), 7.72 (t, J=7.5 Hz, 1 H), 7.87 ppm (dt, J=8.1 Hz, 1.8 Hz, 1 H); 13C NMR (CD3OD): δ=11.8, 15.5, 16.3, 26.8, 27.1, 33.5, 37.0, 38.2, 51.5, 56.0, 59.1, 126.4, 128.5, 129.0, 129.6, 129.8, 130.0, 130.1, 130.5, 133.6, 137.4, 138.2, 145.7, 169.6, 173.0, 175.1 ppm; HRMS (ESI): m/z [M+H]+ calcd for C24H30N2O4: 411.2284; found: 411.2276.
(2S,3 R)-2-(3-(Benzyl(ethyl)carbamoyl)-4-ethylbenzamido)-3-methylpentanoic acid (63): According to general procedure C, reaction of ester 35 gave 63 as a white semi-solid (46 mg, 52 %): [α]=+5.7; 1H NMR (CD3OD): δ=0.95 (t, J=7.4 Hz, 3 H), 1.02 (t, J=7.1 Hz, 3 H), 1.20 (t, J=7.6 Hz, 3 H), 1.24 (t, J=7.6 Hz, 3 H), 1.27–1.38 (m, 1 H), 1.51–1.66 (m, 1 H), 1.93–2.08 (m, 1 H), 2.59–2.70 (m, 2 H), 3.04–3.17 (m, 1 H), 3.61–3.86 (m, 1 H), 4.36 (d, J=4.6 Hz, 1 H), 4.54 (d, J=6.5 Hz, 1 H), 4.57–5.03 (m, 1 H), 7.10–7.16 (m, 1 H), 7.21–7.33 (m, 2 H), 7.34–7.45 (m, 3 H), 7.67–7.75 (m, 1 H), 7.86 ppm (dd, J=8.1 Hz, 2.0 Hz, 1 H); 13C NMR (CD3OD): δ=11.8, 12.6, 15.5, 16.3, 26.8, 27.2, 38.3, 41.3, 44.4, 48.1, 53.4, 59.1, 126.2, 128.6, 128.9, 129.0, 129.5, 129.9, 130.0, 130.5, 133.4, 137.4, 138.8, 145.7, 169.3, 173.0, 175.1 ppm; HRMS (ESI): m/z [M+H]+ calcd for C25H32N2O4: 425.2440; found: 425.2442.
(2S,3 R)-2-(3-(Dibenzylcarbamoyl)-4-ethylbenzamido)-3-methylpentanoic acid (64): According to general procedure C, reaction of ester 36 gave 64 as a white semi-solid (138 mg, 46 %): [α]=+5.5; 1H NMR (CD3OD): δ=0.96 (t, J=7.4 Hz, 3 H), 0.98–1.03 (m, 3 H), 1.20 (t, J=7.6 Hz, 3 H), 1.23–1.36 (m, 1 H), 1.53–1.65 (m, 1 H), 1.95–2.06 (m, 1 H), 2.63 (q, J=7.4 Hz, 2 H), 4.29 (s, 2 H), 4.33–4.49 (m, 1 H), 4.52 (d, J=6.3 Hz, 1 H), 4.90–5.22 (m, 1 H), 7.10 (d, J=7.2 Hz, 2 H), 7.22–7.41 (m, 8 H), 7.43 (d, J=8.1 Hz, 1 H), 7.76 (s br, 1 H), 7.85 ppm (dd, J=8.1 Hz, 1.9 Hz, 1 H); 13C NMR (CD3OD): δ=11.9, 15.5, 16.3, 26.7, 27.2, 38.5, 48.7, 53.0, 59.3, 126.2, 128.7, 129.05, 129.09, 129.7, 129.9, 130.0, 130.1, 130.6, 133.5, 137.0, 137.2, 138.2, 146.0, 169.2, 173.5, 175.5 ppm; HRMS (ESI): m/z [M+H]+ calcd for C30H34N2O4: 487.2591, found: 487.2606.
(2S,3 R)-2-(4-(Diethylcarbamoyl)benzamido)-3-methylpentanoic acid (68): According to general procedure C, reaction of ester 65 gave 68 as a white semi-solid (70 mg, 74 %): [α]=+15.4; 1H NMR (CD3OD): δ=0.97 (t, J=7.4 Hz, 3 H), 1.04 (d, J=6.9 Hz, 3 H), 1.12 (t, J=7.0 Hz, 3 H), 1.26 (t, J=7.0 Hz, 3 H), 1.30–1.41 (m, 1 H), 1.63 (ddq, J=14.9 Hz, 7.5 Hz, 4.3 Hz, 1 H), 1.98–2.11 (m, 1 H), 3.27 (q, J=7.0 Hz, 2 H), 3.56 (q, J=6.9 Hz, 2 H), 4.58 (d, J=6.4 Hz, 1 H), 7.44–7.49 (m, 2 H), 7.90–7.94 ppm (m, 2 H); 13C NMR (CD3OD): δ=11.9, 13.2, 14.5, 16.3, 26.7, 38.3, 41.0, 45.0, 59.0, 127.5, 129.1, 136.6, 141.2, 169.7, 172.7, 174.9 ppm; HRMS (ESI): m/z [M+H]+ calcd for C18H26N2O4: 335.1965, found: 335.1967.
(2S,3 R)-2-(4-(Benzyl(methyl)carbamoyl)benzamido)-3-methylpentanoic acid (69): According to general procedure C, reaction of ester 66 gave 69 as a white semi-solid (42 mg, 88 %): [α]=+13.2; 1H NMR (CD3OD): δ=0.90–0.98 (m, 3 H), 0.98–1.05 (m, 3 H), 1.25–1.38 (m, 1 H), 1.60 (dt, J=12.4 Hz, 7.5 Hz, 1 H), 1.96–2.06 (m, 1 H), 2.87 (s, 2 H), 3.02 (s, 1 H), 4.49–4.53 (m, 1 H), 4.55 (d, J=6.5 Hz, 1 H), 4.75 (s br, 1 H), 7.16 (d, J=7.4 Hz, 1 H), 7.24–7.39 (m, 4 H), 7.53 (d, J=8.1 Hz, 2 H), 7.86 (d, J=7.9 Hz, 1 H), 7.92 ppm (d, J=8.1 Hz, 1 H); 13C NMR (CD3OD): δ=11.8, 16.3, 26.7, 33.8, 37.7, 38.3, 51.9, 56.2, 59.1, 128.1, 128.2, 128.9, 129.0, 129.1, 129.3, 130.0, 130.1, 137.0, 137.7, 138.2, 140.5, 169.8, 173.0, 175.0 ppm; HRMS (ESI): m/z [M+H]+ calcd for C22H26N2O4: 383.1965, found: 383.1968.
(2S,3 R)-2-(4-(Dibenzylcarbamoyl)benzamido)-3-methylpentanoic acid (70): According to general procedure C, reaction of ester 67 gave 70 as a white semi-solid (20 mg, 57 %): [α]=+11.9; 1H NMR (CD3OD): δ=0.95 (t, J=7.4 Hz, 3 H), 1.01 (d, J=6.9 Hz, 3 H), 1.25–1.38 (m, 1 H), 1.60 (ddq, J=14.9 Hz, 7.5 Hz, 4.3 Hz, 1 H), 1.95–2.07 (m, 1 H), 4.43 (s br, 2 H), 4.54 (d, J=6.3 Hz, 1 H), 4.71 (s br, 2 H), 7.14 (d, J=6.9 Hz, 2 H), 7.26–7.40 (m, 8 H), 7.54–7.59 (m, 2 H), 7.86–7.91 ppm (m, 2 H); 13C NMR (CD3OD): δ=11.9, 16.3, 26.7, 38.4, 48.7, 53.2, 59.2, 127.9, 128.3, 128.9, 129.2, 129.4, 130.0, 130.1, 137.1, 137.5, 138.1, 140.4, 169.7, 173.8, 175.2 ppm; HRMS (ESI): m/z [M+H]+ calcd for C28H30N2O4: 459.2278, found: 459.2289.
Biology
Cell cultures: The human embryonic kidney 293 (HEK-293) cell line stably expressing human Flag-AT1 receptor was a generous gift from Dr. Richard Leduc (Department of Pharmacology, Université de Sherbrooke, Sherbrooke, Quebec, Canada) and prepared as previously described.[23] The native 293/FRT cell line (HEK-293 cell line with single genome-integrated Flp recombinase target site (FRT)) was maintained in high-glucose DMEM with 7 % FBS, 2 mm GlutaMAX and 100 μg mL−1 zeocin.
The stable cell line stably expressing the human AT2 receptor was established as described previously.[24] First, the forward primer containing a HindIII restriction site and a Myc epitope (ttaaacttaagcttaccatggaacaaaaactcatctcagaagaggatctgatgaagggcaactccacc) was used with a reverse primer containing a SalI site (agcaagcaagacacatgtcgacttaagacacaaaggtctcc) with the Expand High FidelityPLUS PCR System (Roche) to create a HindIII-Myc-AT2 receptor-SalI fragment, which was cloned into pcDNA5/FRT between its HindIII and XhoI sites, thus creating the pcDNA5/FRT/Myc-AT2 R vector. Then, the cell line stably expressing Myc-human AT2 receptor (293/FRT/Myc-hAT2R) was generated by Flp recombinase-mediated homologous recombination system (Flp-In™) and was maintained with 100 μg mL−1 hygromycin B.[24]
Binding experiments
First radioligand binding assay: This study was performed at Cerep (France) according to literature.[16–17] The assays were performed in HEK-293 cells transfected with recombinant human AT2R or AT1R and relying on the displacement of [125I]CGP-42112 A (AT2R)[16] and [125I][Sar1, Ile8]Ang II (AT1R)[17] with radiolabeled Ang II as reference compound in the AT2R assay and radiolabeled saralasin [Sar1, Val5, Ile8]Ang II in the AT1R assay. Unlabeled Ang II was used for nonspecific binding in both the AT2R (1 μm) and AT1R (10 μm) assay. In the initial screen the % inhibition of [125I]CGP-42112 A (AT2R) or [125I][Sar1, Ile8]Ang II (AT1R) binding was measured at 1 and 10 μm of the compounds. The Ki values were determined from at least six data points with test concentrations ranging from 30 pm to 1 mm. The concentration range was adjusted to be appropriate for the expected Ki values.
Second radioligand binding assay: Binding studies were conducted in HEK-293 transfected cells with human AT1R or AT2R and were performed as recently described.[8e] Briefly, the analogue [Sar1, Ile8]Ang II was iodinated by the Iodogen method, and binding assays were performed on cultured cells. The hormone binding reaction was initiated by addition of 0.1 nm of [125I][Sar1, Ile8]Ang II (1000 Ci mmol−1) to each Petri dish (1.0×106 cells/Petri dish) either alone (total binding) or in the presence of increasing concentrations of Ang II or the ligands under investigation, including 10 μm Ang II for nonspecific binding (which represents less than 10 % of total binding). Incubations were performed in duplicate for 30 min at RT (22 °C). After incubation, cells were rapidly detached from the substratum with a rubber policeman; cells and media were filtered through Whatman GF/C filters (presoaked overnight in 2 % BSA), rinsed three times and counted in a Beckman γ-counter. The endogenous ligand Ang II, the selective nonpeptide AT1 antagonist losartan, the selective AT2 agonist CGP42112 A, and the selective nonpeptide AT2 antagonist PD 123,319 were used as reference compounds for the binding studies. In radioligand binding experiments, IC50 values were obtained by fitting radioligand competition data to a sigmoidal function by use of a nonlinear least-squares program (GraphPad Software Inc., San Diego, CA). Ki values were determined using the Cheng–Prusoff equation: Ki=IC50/(1+H/KD) where H is the radioligand concentration and KD is the KD value for the radioligand.
Acknowledgments
We gratefully acknowledge the financial support from the Swedish Research Council. The authors wish to thank Lucie Chouinard and Sandra Pinard (Université de Sherbrooke, Sherbrooke, Quebec, Canada) for experimental assistance and for stimulating discussions. We sincerely thank Dr. Richard Leduc and Dr. Emmanuel Escher (Department of Pharmacology, Université de Sherbrooke) for the respective gifts of the AT1 and AT2 receptor cDNA and of [125I][Sar1, Ile8]-Ang II. We also thank Simon Roy (PhD student) for generating the AT1-and AT2-receptor-transfected HEK cells. Work from the Sherbrooke team was supported by grants from the Canadian Diabetes Association (grant no. OG-3–10–3021-NG) and by the Alzheimer Society of Canada (#1136). Finally, we would like to acknowledge the Beijer Laboratory (Uppsala, Sweden) where this research was conducted.
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/open.201300040.
References
- [1].de Gasparo M, Catt KJ, Inagami T, Wright JW, Unger T. Pharmacol. Rev. 2000;52:415. [PubMed] [Google Scholar]
- [2a)].Carey RM, Padia SH. Trends Endocrinol. Metab. 2008;19:84. doi: 10.1016/j.tem.2008.01.003. [DOI] [PubMed] [Google Scholar]
- [2b)].Steckelings UM, Rompe F, Kaschina E, Namsolleck P, Grzesiak A, Funke-Kaiser H, Bader M, Unger T. J. Renin Angiotensin Aldosterone Syst. 2010;11:67. doi: 10.1177/1470320309347791. [DOI] [PubMed] [Google Scholar]
- [3].Yu L, Zheng M, Wang W, Rozanski GJ, Zucker IH, Gao L. J. Renin Angiotensin Aldosterone Syst. 2010;11:214. doi: 10.1177/1470320310379065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4a)].Steckelings UM, Kaschina E, Unger T. Peptides. 2005;26:1401. doi: 10.1016/j.peptides.2005.03.010. [DOI] [PubMed] [Google Scholar]
- [4b)].Padia S, Carey R. Pflugers Arch.-Eur. J. Physiol. 2013;465:99. doi: 10.1007/s00424-012-1146-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5a)].Tamargo J, López-Sendón J. Nat. Rev. Drug Discovery. 2011;10:536. doi: 10.1038/nrd3431. [DOI] [PubMed] [Google Scholar]
- [5b)].Lauer D, Slavic S, Sommerfeld M, Thöne-Reineke C, Sharkovska Y, Hallberg A, Dahlöf B, Kintscher U, Unger T, Steckelings UM, Kaschina E. Hypertension. 2014;63:e60. doi: 10.1161/HYPERTENSIONAHA.113.02522. [DOI] [PubMed] [Google Scholar]
- [6a)].Rosenström U, Sköld C, Lindeberg G, Botros M, Nyberg F, Karlén A, Hallberg A. J. Med. Chem. 2004;47:859. doi: 10.1021/jm030921v. [DOI] [PubMed] [Google Scholar]
- [6b)].Rosenström U, Sköld C, Plouffe B, Lindeberg G, Botros M, Nyberg F, Wolf G, Karlén A, Gallo-Payet N, Hallberg A. J. Med. Chem. 2005;48:4009. doi: 10.1021/jm0491492. [DOI] [PubMed] [Google Scholar]
- [6c)].Georgsson J, Sköld C, Plouffe B, Lindeberg G, Botros M, Larhed M, Nyberg F, Gallo-Payet N, Gogoll A, Karlen A, Hallberg A. J. Med. Chem. 2005;48:6620. doi: 10.1021/jm050280z. [DOI] [PubMed] [Google Scholar]
- [6d)].Rosenström U, Sköld C, Lindeberg G, Botros M, Nyberg F, Karlén A, Hallberg A. J. Med. Chem. 2006;49:6133. doi: 10.1021/jm051222g. [DOI] [PubMed] [Google Scholar]
- [6e)].Georgsson J, Rosenström U, Wallinder C, Beaudry H, Plouffe B, Lindeberg G, Botros M, Nyberg F, Karlen A, Gallo-Payet N, Hallberg A. Bioorg. Med. Chem. 2006;14:5963. doi: 10.1016/j.bmc.2006.05.019. [DOI] [PubMed] [Google Scholar]
- [6f)].Georgsson J, Skold C, Botros M, Lindeberg G, Nyberg F, Karlen A, Hallberg A, Larhed M. J. Med. Chem. 2007;50:1711. doi: 10.1021/jm0613469. [DOI] [PubMed] [Google Scholar]
- [7a)].Perlman S, Schambye HT, Rivero RA, Greenlee WJ, Hjorth SA, Schwartz TW. J. Biol. Chem. 1995;270:1493. doi: 10.1074/jbc.270.4.1493. [DOI] [PubMed] [Google Scholar]
- [7b)].Kivlighn SD, Huckle WR, Zingaro GJ, Rivero RA, Lotti VJ, L. Chang RS, Schorn TW, Kevin N, Johnson Jr RG. Am. J. Physiol. 1995;268:R820. doi: 10.1152/ajpregu.1995.268.3.R820. [DOI] [PubMed] [Google Scholar]
- [8a)].Wan Y, Wallinder C, Johansson B, Holm M, Mahalingam AK, Wu X, Botros M, Karlen A, Pettersson A, Nyberg F, Faendriks L, Hallberg A, Alterman M. J. Med. Chem. 2004;47:1536. doi: 10.1021/jm031031i. [DOI] [PubMed] [Google Scholar]
- [8b)].Wan Y, Wallinder C, Plouffe B, Beaudry H, Mahalingam AK, Wu X, Johansson B, Holm M, Botros M, Karlen A, Pettersson A, Nyberg F, Faendriks L, Gallo-Payet N, Hallberg A, Alterman M. J. Med. Chem. 2004;47:5995. doi: 10.1021/jm049715t. [DOI] [PubMed] [Google Scholar]
- [8c)].Steckelings UM, Larhed M, Hallberg A, Widdop RE, Jones ES, Wallinder C, Namsolleck P, Dahlof B, Unger T. Curr. Opin. Pharmacol. 2011;11:187. doi: 10.1016/j.coph.2010.11.002. [DOI] [PubMed] [Google Scholar]
- [8d)].Steckelings UM, Paulis L, Namsolleck P, Unger T. Curr. Opin. Nephrol. Hypertens. 2012;21:142. doi: 10.1097/MNH.0b013e328350261b. [DOI] [PubMed] [Google Scholar]
- [8e)].Guimond M-O, Wallinder C, Alterman M, Hallberg A, Gallo-Payet N. Eur. J. Pharmacol. 2013;699:160. doi: 10.1016/j.ejphar.2012.11.032. [DOI] [PubMed] [Google Scholar]
- [8f)].McCarthy C, Widdop R, Denton K, Jones E. Curr Hypertens Rep. 2013;15:25. doi: 10.1007/s11906-012-0321-4. [DOI] [PubMed] [Google Scholar]
- [9a)].Murugaiah AMS, Wallinder C, Mahalingam AK, Wu X, Wan Y, Plouffe B, Botros M, Karlén A, Hallberg M, Gallo-Payet N, Alterman M. Bioorg. Med. Chem. 2007;15:7166. doi: 10.1016/j.bmc.2007.07.026. [DOI] [PubMed] [Google Scholar]
- [9b)].Wallinder C, Botros M, Rosenström U, Guimond M-O, Beaudry H, Nyberg F, Gallo-Payet N, Hallberg A, Alterman M. Bioorg. Med. Chem. 2008;16:6841. doi: 10.1016/j.bmc.2008.05.066. [DOI] [PubMed] [Google Scholar]
- [9c)].Mahalingam AK, Wan Y, Murugaiah AMS, Wallinder C, Wu X, Plouffe B, Botros M, Nyberg F, Hallberg A, Gallo-Payet N, Alterman M. Bioorg. Med. Chem. 2010;18:4570. doi: 10.1016/j.bmc.2010.03.064. [DOI] [PubMed] [Google Scholar]
- [10a)].Kaiser N-FK, Hallberg A, Larhed M. J. Comb. Chem. 2002;4:109. doi: 10.1021/cc010085f. [DOI] [PubMed] [Google Scholar]
- [10b)].Wu XY, Larhed M. Org. Lett. 2005;7:3327. doi: 10.1021/ol0512031. [DOI] [PubMed] [Google Scholar]
- [10c)].Wannberg J, Dallinger D, Kappe CO, Larhed M. J. Comb. Chem. 2005;7:574. doi: 10.1021/cc049816c. [DOI] [PubMed] [Google Scholar]
- [10d)].Lagerlund O, Larhed M. J. Comb. Chem. 2006;8:4. doi: 10.1021/cc050102r. [DOI] [PubMed] [Google Scholar]
- [11a)].Wannberg J, Kaiser N-FK, Vrang L, Samuelsson B, Larhed M, Hallberg A. J. Comb. Chem. 2005;7:611. doi: 10.1021/cc050016r. [DOI] [PubMed] [Google Scholar]
- [11b)].Wu X, Ekegren JK, Larhed M. Organometallics. 2006;25:1434. [Google Scholar]
- [12a)].Cho CH, Neuenswander B, Larock RC. J. Comb. Chem. 2010;12:278. doi: 10.1021/cc900172u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12b)].Wu X-F, Neumann H, Beller M. Chem. Asian J. 2010;5:2168. doi: 10.1002/asia.201000418. [DOI] [PubMed] [Google Scholar]
- [12c)].Pizzetti M, Russo A, Petricci E. Chem. Eur. J. 2011;17:4523. doi: 10.1002/chem.201100447. [DOI] [PubMed] [Google Scholar]
- [13a)].Hermange P, Lindhardt AT, Taaning RH, Bjerglund K, Lupp D, Skrydstrup T. J. Am. Chem. Soc. 2011;133:6061. doi: 10.1021/ja200818w. [DOI] [PubMed] [Google Scholar]
- [13b)].Wan Y, Alterman M, Larhed M, Hallberg A. J. Comb. Chem. 2003;5:82. doi: 10.1021/cc0200843. [DOI] [PubMed] [Google Scholar]
- [13c)].Friis SD, Taaning RH, Lindhardt AT, Skrydstrup T. J. Am. Chem. Soc. 2011;133:18114. doi: 10.1021/ja208652n. [DOI] [PubMed] [Google Scholar]
- [14a)].Letavic MA, Ly KS. Tetrahedron Lett. 2007;48:2339. [Google Scholar]
- [14b)].Begouin A, Queiroz MJRP. Eur. J. Org. Chem. 2009:2820. [Google Scholar]
- [14c)].Lagerlund O, Mantel MLH, Larhed M. Tetrahedron. 2009;65:7646. [Google Scholar]
- [14d)].Wiéckowska A, Fransson R, Odell LR, Larhed M. J. Org. Chem. 2011;76:978. doi: 10.1021/jo102151u. [DOI] [PubMed] [Google Scholar]
- [14e)].Odell L, Russo RF, Larhed M. Synlett. 2012:685. [Google Scholar]
- [15a)].Brechler V, Jones PW, Levens NR, de Gasparo M, Bottari SP. Regul. Pept. 1993;44:207. doi: 10.1016/0167-0115(93)90244-3. [DOI] [PubMed] [Google Scholar]
- [15b)].Buisson B, Bottari SP, de Gasparo M, Gallo-Payet N, Payet MD. FEBS Lett. 1992;309:161. doi: 10.1016/0014-5793(92)81086-2. [DOI] [PubMed] [Google Scholar]
- [16].Tsuzuki S, Ichiki T, Nakakubo H, Kitami Y, Guo DF, Shirai H, Inagami T. Biochem. Biophys. Res. Commun. 1994;200:1449. doi: 10.1006/bbrc.1994.1613. [DOI] [PubMed] [Google Scholar]
- [17].Le MT, De Backer J-P, Hunyady L, Vanderheyden PML, Vauquelin G. Eur. J. Pharmacol. 2005;513:35. doi: 10.1016/j.ejphar.2005.02.029. [DOI] [PubMed] [Google Scholar]
- [18].Pals DT, Masucci FD, Denning GS, Sipos F, Fessler DC. Circ. Res. 1971;29:673. doi: 10.1161/01.res.29.6.673. [DOI] [PubMed] [Google Scholar]
- [19].Bosnyak S, Jones E, Christopoulos SA, Aguilar M-I, Thomas W, Widdop GR. Clin. Sci. 2011;121:297. doi: 10.1042/CS20110036. [DOI] [PubMed] [Google Scholar]
- [20].Consortium TU. Nucleic Acids Res. 2012;40:D71. doi: 10.1093/nar/gkr981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21a)].Mogi M, Iwai M, Horiuchi M. Arterioscler. Thromb. Vasc. Biol. 2007;27:2532. doi: 10.1161/ATVBAHA.107.144154. [DOI] [PubMed] [Google Scholar]
- [21b)].Funke-Kaiser H, Reinemund J, Steckelings U, Unger MT. J. Renin Angiotensin Aldosterone Syst. 2010;11:7. doi: 10.1177/1470320309343652. [DOI] [PubMed] [Google Scholar]
- [22].Feng Y-H, Zhou L, Sun Y, Douglas JG. Kidney Int. 2005;67:1731. doi: 10.1111/j.1523-1755.2005.00270.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Servant G, Laporte SA, Leduc R, Escher E, Guillemette G. J. Biol. Chem. 1997;272:8653. doi: 10.1074/jbc.272.13.8653. [DOI] [PubMed] [Google Scholar]
- [24].Roy S, Rached M, Gallo-Payet N. Mol. Endocrinol. 2007;21:1656. doi: 10.1210/me.2007-0041. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.