

Abstract
The conversion of UDP-α-d-gdlucuronic acid to UDP-α-d-xylose and UDP-α-d-apiose by a bifunctional potato enzyme UDP-apiose/UDP-xylose synthase was studied using real-time nuclear magnetic resonance (NMR) spectroscopy. UDP-α-d-glucuronic acid is converted via the intermediate uridine 5′-β-l-threo-pentapyranosyl-4″-ulose diphosphate to UDP-α-d-apiose and simultaneously to UDP-α-d-xylose. The UDP-α-d-apiose that is formed is unstable and is converted to α-d-apio-furanosyl-1,2-cyclic phosphate and UMP. High-resolution real-time NMR spectroscopy is a powerful tool for the direct and quantitative characterization of previously undetected transient and labile components formed during a complex enzyme-catalyzed reaction.
Keywords: Apiose, UDP-apiose, UDP-xylose synthase, NMR spectroscopy, Stocsy
1. Introduction
The branched sugar 3-C-(hydroxymethyl)-d-glycero-tetrose or d-apiose (see Fig. 1) is a component of several plant flavonoids1–4 and is present in the plant cell-wall pectic polysaccharides5, apiogalacturonan6,7, and rhamnogalacturonan II (RG-II)8,9 Twoof the apiosyl residues present in separate RG-II molecules are crosslinked together by a borate diester that results in the formation of a dimer of RG-II. The formation of this dimer is believedtohave an important role in regulating the chemical and physical properties of the primary cell wall8 and in normal plant growth and development.10
Figure 1.

Proposed mechanism for the conversion of UDP-GlcA to UDP-xylose and UDP-apiose catalyzed by UAXS. The UDP-4-keto-xylose structure is depicted in the keto form, but it is most likely in equilibrium with the hydrated gem-diol form. The numbering schemes of the UDP-GlcA and UDP-apiose are independent. Carbon 4 of UDP-GlcA (marked with an asterisk) becomes carbon 3 of UDP-apiose. Note that the ring protons have been left off for clarity.
Plants are believed to use uridine 5′-diphosphate- α-d-apiofuranose (UDP-apiose, see Fig. 1) to donate apiosyl residues in the biosynthesis of polysaccharides and flavonoids. Previous work has proposed that UDP-apiose is formed from UDP-α-d-glucuronic acid (UDP-GlcA) by the β-NAD+-dependent, bifunctional enzyme UDP-d-apiose/UDP-d-xylose synthase (abbreviated herein as UAXS).11–15 Despite clear evidence of a biosynthetic route from UDP-GlcA to apiose, it has only been inferred that UDP-apiose is the direct product of UAXS, and by extension, the activated sugar donor for subsequent pathways.16 In fact it was never observed or isolated, but rather detected as α-d-apio-furanosyl-1,2-cyclic phosphate (apiofuranosyl-1,2-cyclic phosphate). Biochemically synthesized UDP-apiose was shown to be unstable, and it spontaneously converts to apiofuranosyl-1,2-cyclic phosphate,17 which can also be made chemically.18
Characterization of some of the proposed products formed by UAXS has led to the suggestion that uridine 5′-β-l-threo-pentopyr-anosyl-4″-ulose diphosphate (UDP-4-keto-xylose) is an intermediate in the reaction, and to a proposal of a mechanism for the rearrangement of the ring carbons19,20 as outlined in Fig. 1. However, unambiguous structural evidence supporting the formation of the intermediate is lacking.20
NMR spectroscopy is a powerful technique for monitoring chemical21 and enzymatic reactions.22–26 At high magnetic fields there is usually sufficient frequency resolution to follow the fate of each molecular species, even if they have closely related or iso-meric structures. Moreover, the linear relationship between resonance intensity and molar quantities of each component of the reaction makes measuring the time course of enzyme-catalyzed conversions relatively straightforward.
Here, we describe the use of real-time 1H NMR spectroscopy to examine the formation of products and intermediate when a cloned recombinant potato UAXS is reacted with UDP-GlcA. We show that enzyme kinetics and the conversion of substrate into products, including those that are labile and thus difficult to isolate and characterize, can be continuously monitored during the course of the enzyme-catalyzed reaction.
2. Results and discussion
2.1. Functional identification of UAXS and initial enzymatic characterization
Enzymes from diverse organisms are known to convert UDP-glucuronic acid (UDP-GlcA) into different products.27–29 We isolated a potato (Solanum tuberosum) cDNA that encodes a protein with 28% amino acid identity (see Supplemental Fig. 2) to a fungal UDP-GlcA decarboxylase (UXs) that converts UDP-GlcA to UDP-xy-lose28, and with 34% amino acid identity to a bacterial NAD+-dependent decarboxylase, ArnA29, that converts UDP-GlcA to UDP-4-keto-xylose. Motif analysis of the putative potato protein (GenBank ABC75032) revealed a conserved NAD+-binding motif, GxxGxxG (Gly21 to Gly27), and a conserved catalytic motif, YxxxK (Tyr182 to Lys186), associated with interconversion of nucleotide sugars. Thus, we reasoned that the potato protein (referred to as UAXS) may have UDP-xylose and/or UDP-apiose synthase activity.
To obtain potato UAXS in amounts sufficient for kinetic and product formation studies the protein was expressed in E. coli. A unique protein band was detected in the crude protein extract of the UAXS-expressing cells (Fig. 2A, lane A, marked by arrow), but it was absent in cells expressing an empty vector (lane B). UAXS was column purified yielding a distinct 43 kDa protein (Fig 2A lane C) that is not produced by E. coli cells expressing an empty vector as control (lane D). The amino acid sequence of the purified recombinant protein was confirmed by MS/MS analyses of its trypsin digestion products (data not shown).
Figure 2.
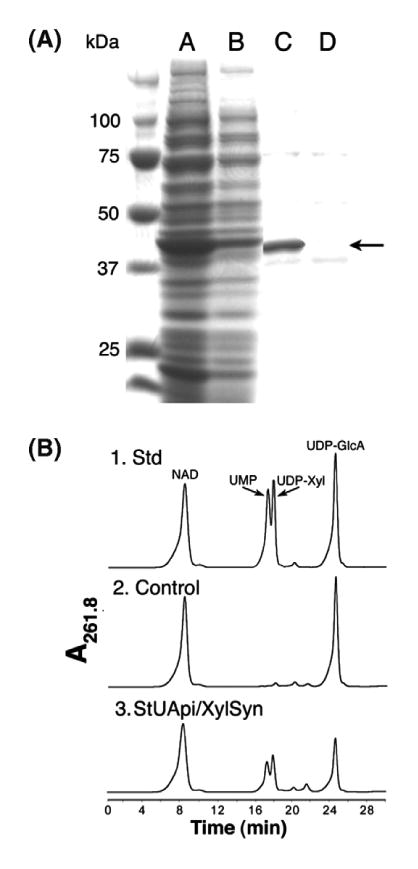
Protein expression of UAXS and analysis of enzymatic reaction using HPLC. Panel A: SDS–PAGE of soluble protein isolated from E. coli cell expressing recombinant UAXS (lane A) or control empty vector (lane B). Column fractionation of UAXS (lane C) compared to an equivalent fraction from cell containing the empty vector (lane D). The band indicated by an arrow is the over-expressed recombinant UAXS. Panel B: (1) HPLC analysis of standard NAD+, UMP, UDP-xylose, and UDPGlcA, (2) reaction products when UDP-GlcA and NAD+ are reacted with protein from control empty vector expression, and (3) reaction products when UDP-GlcA and NAD+ are reacted with the recombinant UAXS.
Recombinant potato UAXS is active only in the presence of NAD+ (data not shown); therefore, all activity assays contained UDP-GlcA and NAD+ as substrates. The enzymatic products of UAXS were analyzed by an anion-exchange column with UV detection (Fig. 2B panel 3) that showed the presence of two new products with retention times characteristic of UMP and UDP-xylose. These components were isolated, and their structures were confirmed by 1H NMR spectroscopy and by analyses of authentic standards (data not shown). No UDP-apiose was detected, which is consistent with previous work.14
To determine whether UMP was a specific product of UAXS or formed by degradation of UDP-apiose, anion-exchange chromatography was performed with a CAD-Corona detector (a charged aerosol HPLC detector). A new peak with elution time of 12.9 min (Fig. 3A, arrow) was detected, collected, and shown to be apiofuranosyl-1,2-cyclic phosphate (Fig. 3B) by 1D- and 2D-1H NMR spectroscopy (Fig. 3C). The chemical shift data (Supplemental Table 1) are comparable to those of a previous report of the NMR spectra of apiofuranosyl phosphates.18 The signals for H1 and H2 are shifted downfield relative to apiose30 or its methyl glucoside31, and show scalar couplings to 31P as expected for phosphate substitution at those positions.18 The COSY spectrum (Fig. 3C) shows four-bond scalar couplings between H2, H3, and H4. Small molecules with appropriate geometry will have measurable couplings over multiple bonds.32 In this case there is a relatively strong cross-peak between H2 (cA2) and one of the H4 protons (cA4), most likely due to a favorable ‘W’ orientation.33 A similar argument can be made for the cross-peak between one of the H3s (cA3) and the other H4.
Figure 3.
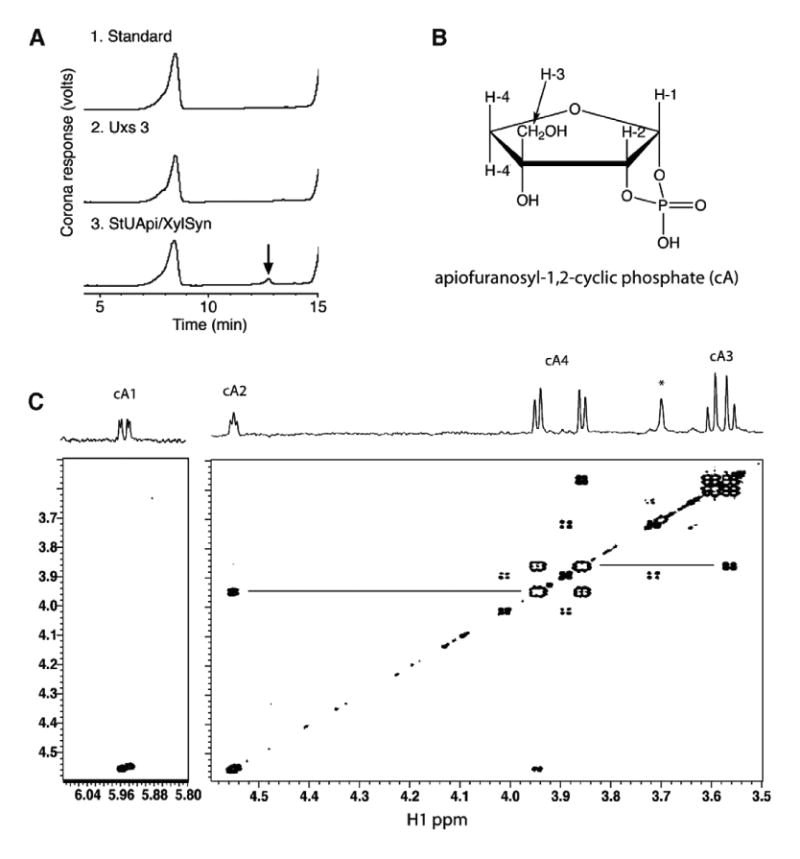
Identification of apiofuranosyl-1,2-cyclic phosphate (cA). Panel A: Ion-exchange HPLC chromatogram of products formed when UDP-GlcA and NAD+ were reacted with the recombinant UAXS. A peak (marked by arrow) was detected when the reaction mixture contained UAXS but was absent in the control reactions. Panel B: Chemical structure: in this numbering scheme the exocyclic methylene protons are labeled as H3, and ring methylene protons are labeled as H4. (C) COSY NMR spectrum: Four-bond couplings between H2 and H4, and H3 and H4, are indicated by horizontal lines. The signals are labeled as cA1, etc. to conform to subsequent figures.
2.2. Real-time 1H NMR spectroscopy establishes that UAXS converts UDP-GlcA to UDP-apiose
No evidence for the formation of UDP-apiose was obtained by HPLC analyses of the products formed by UAXS. Thus, we sought to further investigate the nature of these products in real time using 1H NMR spectroscopy. Preliminary studies indicated that enzymatic reactions preformed in 80% D2O gave an acceptable NMR signal-to-noise ratio and that D2O buffer exchange was not required after recombinant protein was column purified.
The spectra shown in Figure 4 cover the full time course for the reaction at pH 7.8 (upper set 4A) and at pH 6.5 (lower set 4B). The data for the reaction at pH 7.8 show the disappearance of UDP-GlcA, and the formation of products, some of which are transient. A comparison of spectra collected at the beginning and the end of the time course confirms that UDP-GlcA (peaks labeled G1–G5) has been consumed, and that at least two products, UDP-xy-lose (peaks X1–X5) and apiofuranosyl-1,2-cyclic phosphate (peaks cA1, cA3, and cA4) have been formed. The signals were assigned based on comparison to authentic samples of apiofuranosyl-1,2-cyclic phosphate and UDP-xylose.34 Signals labeled ‘U’ belong to the two H5 protons of ribose in UMP, which is generated when UDP-apiose is converted to the cyclic phosphate. Signals labeled A1–A4 that increase in amplitude from the earliest time point up to ∼60 min and then decay, were assigned to UDP-apiose. The triplet signal at δ 5.75 (A1) is consistent with an anomeric proton in a 1-phosphate linkage and has an H1-H2 coupling of 4.5 Hz and an H1-P coupling of 5.6 Hz.
Figure 4.
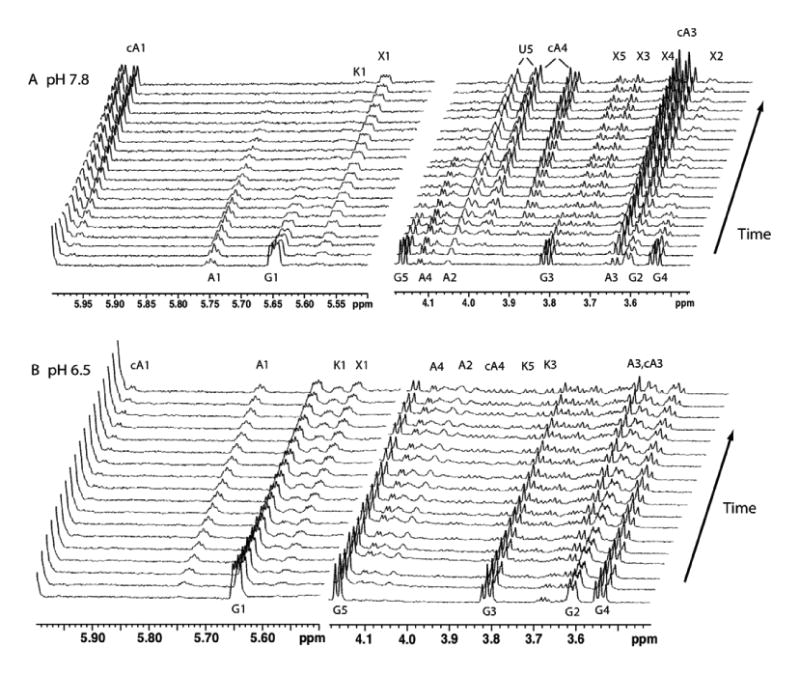
Real-time 1H NMR analysis of the products formed when recombinant UAXS is reacted at pH 7.8 in 80% D2O-20% H2O–phosphate buffer at 35 °C with UDP-GlcA and NAD+ (Panel A, upper set). Regions of the 800-MHz proton spectra are plotted at 25-min intervals starting at the bottom, from 15 min after the addition of UAXS. Signals are labeled as originating from UDP-GlcA (G), UDP-apiose (A), apiofuranosyl-1,2-cyclic phosphate (cA), UDP-xylose (X), UDP-4-keto-xylose (K), and the ribose of UMP (U). Panel B, lower set: UAXS activity with UDP-GlcA and NAD+ when reactions were carried out at pH 6.5 in 80% D2O–20% H2O–phosphate buffer at 35 °C Spectra are plotted at 48-min intervals starting at the bottom (15 min). Signals are labeled as in Panel A.
When UAXS was reacted at pH 6.5, the rate of product formation was slower, and the increased stability of UDP-apiose produced a different ratio of reactants and products.
We obtained a COSY dataset of the pH 6.5 UAXS reaction products and used this information to confirm the signal assignments of UDP-xylose, apiofuranosyl-1,2-cyclic phosphate, and UDP-GlcA (Supplementary data, Fig. S3). The coupling between H1 and H2 of UDP-apiose was observed, and a second resonance at δ 4.05 (A2) was therefore assigned to H2. Unlike the COSY of the isolated apiofuranosyl-1,2-cyclic phosphate, no long-range couplings between H2 and H3 or H4 in UDP-apiose were observed here. However, by examining 1D spectra corresponding to the time points when apiofuranosyl-1,2-cyclic phosphate has not yet formed (Fig. 5), we were able to assign signals to the H3 and H4 protons of UDP-apiose based on their chemical shifts, which are comparable to those observed in other apiofuranosides.30,31 Note that the spectrum shown in Figure 5 was collected in 90% H2O in order to minimize any proton–deuteron exchange at the alpha position to the ketone. Other spectra taken in 80% D2O show a minor change in the peak shape and intensity of the H4 of UDP-apiose attributed to this exchange.
Figure 5.
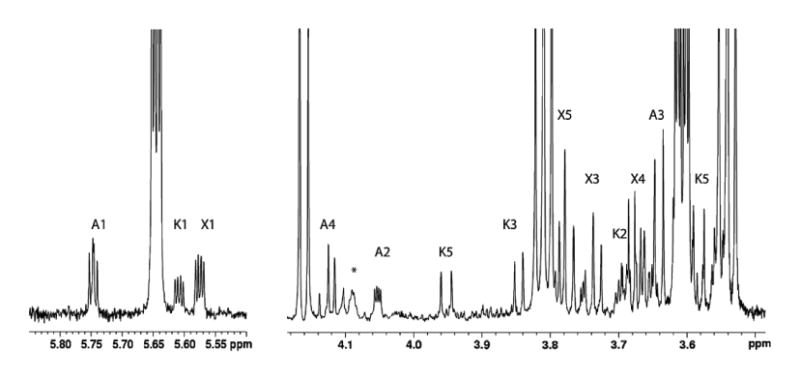
Regions of a 900-MHz 1H NMR spectrum of the reaction mixture after 2 h at pH 6.5 in 90% H2O buffer at 33 °C. Signals are labeled as UDP-apiose (A), UDP-xylose (X), and UDP-4-keto-xylose (K). No detectable amounts of apiofuranosyl-1,2-cyclic phosphate have been formed at this stage. The signal labeled A4 shows the expected AB quartet pattern of methylene protons, and the signal labeled A2 shows scalar coupling to A1 and long-range coupling to 31P. The broad signal at 4.09 ppm indicated with an asterisk is correlated to UDP-apiose and is tentatively assigned as a ribose H5 proton.
Additional evidence that the assigned signals originate from UDP-apiose was obtained by processing of the spectra shown in Figure 4 in the form of a covariance matrix which was then displayed as a ‘statistical TOCSY’ or STOCSY (Fig. 6B).35 Such analysis can identify sets of signals that are likely to originate from the same compound. For example, peaks whose amplitudes are strongly correlated over the reaction time course will give either strong positive or negative cross-peaks depending on whether they belong to the same substrate, or are a product derived from that substrate. In cases of multiple substrates and similar amplitude profiles, discrimination can be achieved by analyzing chemical shifts. Figure 6C shows a row of this covariance matrix taken through the diagonal peak corresponding to the A1 proton of UDP-apiose. For comparison, Figure 6A shows the corresponding segment from a 1D spectrum of the reaction mixture showing all species present. The peaks assigned to UDP-apiose (A1–A4) are positively correlated as expected. The reactant, UDP-GlcA (peaks G1–G5), also shows some positive correlation with A1, but can be eliminated based on its chemical shift signature. In contrast, products of the reaction, apiofuranosyl-1,2-cyclic phosphate (peaks cA1–cA4) and UDP-Xyl (peak X1–X5) are negatively correlated with A1. For example, the peaks corresponding to cA3 and A3 are almost completely overlapped in Figure 6A due to similar chemical shifts, but can be differentiated in the correlation trace 6C.
Figure 6.
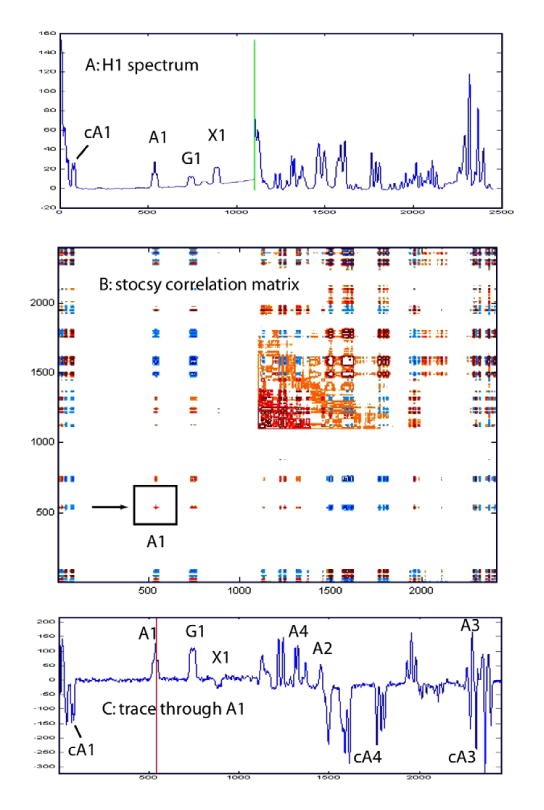
Covariance matrix representation (STOCSY) of the spectra shown in Figure 4A corresponding to the time interval from 90 to 490 min. During this period, the UDP-apiose converts to apiofuranosyl-1,2-cyclic phosphate. Top spectrum A shows the corresponding regions from a 1D spectrum of the reaction mixture. Bottom spectrum C shows the peaks that are correlated to UDP-apiose resonance A1, indicated by box in panel C. See text for further discussion.
2.3. Observation of the UDP-4-keto-xylose intermediate at pH 6.5
UAXS assays performed at pH 7.8 show that both UDP-apiose and UDP-xylose are formed within 15 min with approximately 20% of the UDP-GlcA consumed. At this time point, the ratio of UDP-apiose and UDP-xylose is 2:1 (Fig. 4A). After 6 h only apiofuranosyl-1,2-cyclic phosphate and UDP-xylose remain in a ratio of approximately 3:2, together with small amounts (<5%) of UDP-4-keto-xylose.
When the UAXS reaction is carried out at pH 6.5 (Figs. 4B and 5), the rate of the reaction is reduced and the ratio of products is altered such that after 115 min equal amounts of UDP-xylose and UDP-apiose are present, with ∼50% of the UDP-GlcA consumed. UDP-4-keto-xylose is present at half the levels of the UDP-xylose and UDP-apiose, but there are no detectable amounts of apiofuranosyl-1,2-cyclic phosphate.
The assignment of UDP-4-keto-xylose is based on chemical shifts29 and connectivities observed in the COSY spectrum (Supplementary data, Fig. 3). The proton–proton coupling constants (J1,2 3.8 Hz, J2,3 9.8 Hz, and J3,4 9.8 Hz) are consistent with an α-pyranose in the 4C1 chair form. At the final reaction time point, 70% of the UDP-glucuronic acid has been consumed, but the ratios of UDP-xylose, UDP-4-keto-xylose, and apiose, now in the form of both UDP-apiose and apiose-CP, remain approximately the same.
The increase of UDP-apiose at the lower pH is at least partially due to the reduced rate of conversion of UDP-apiose to apiose-CP. This conversion likely occurs via an SN2 displacement of UMP by attack of the 2-hydroxyl group at the phosphorous bonded to the anomeric hydroxyl oxygen. Breaking the phosphate–phosphate bond would be easier at higher pH where both phosphates are negatively charged. The increase in the UPD-4-keto-xylose inter-mediate by NAD+-assisted decarboxylation as depicted in Figure 1 produces a proton, and the reduction of the same intermediate by NADH consumes a proton. Therefore, one might expect the decarboxylation to proceed more slowly and the reduction to proceed more rapidly at a lower pH. Since our data suggest the opposite behavior, the observed change must be due to as yet unidentified aspects of the catalytic mechanism.
Real-time NMR data confirm that UAXS does convert UDP-GlcA directly to UDP-apiose and simultaneously to UDP-xylose. The data also show the transient nature of UDP-apiose formation under these reaction conditions, and production of a stable intermediate, UDP-4-keto-xylose. The application of NMR spectroscopy was key to observing and identifying signals from all the products, whether they were unstable or previously proposed as intermediates. The production of the UDP-4-keto-xylose and the dependence of the ratio of products on pH and temperature have implications for the mechanism and role of UAXS in other synthetic pathways. Whether the produced UDP-4-keto-xylose can be further modified or interconverted to other nucleotide sugars remains to be investigated.
3. Experimental
3.1. Cloning, expression, and purification of UAXS
The gene encoding the putative UDP-Api/UDP-Xyl synthase (UAXS) was identified by querying the NCBI-translated database for amino acid sequences similar to UDP-xylose synthase from Cryptococcus neoformans.28
A partial sequence of clone bf460135 from a cDNA library of potato tubers shared some amino acid sequence identity to UXSA. A 50-μL PCR was used to amplify the coding sequence of potato UAXS with one unit of high-fidelity proofreading Platinum DNA polymerase (Gibco-BRL), 0.2 μM of each forward and reverse primers, BamHI/Ncobf153387#1S 5′ -GGATCCATGGCAGGGAGGGTAGATCTAGACG, and bf153387#2ASTGA/NotI 5′GCGGCCGCTCAATTTGCCGTTGTCTTTGAC, respectively, a plasmid consisting of the potato full-length cDNA clone (pBS-SK+:31bf153387), 0.2 mM each dNTP, and buffer containing 60 mM Tris–SO4, pH 8.9, 18 mM NH4SO4, and 1 mM MgSO4. The amplified DNA fragment was cloned into pCR2.1 (Topo-TA vector, Invitrogen), and DNA sequencing of the resulting plasmid, pCR2.1:31.2, confirmed UASX identity (GenBank DQ345778). Subsequently, the Nco I—Not I fragment (1172 bp) containing the open reading frame of UAXS was cloned into E. coli expression vector to generate pET24d:31.2#7.
E. coli cells harboring pET24d:31.2#7 or an empty vector control were grown for 16 h at 37 °C in 20 mL of LB liquid medium supplemented with 50 μg/mL kanamycin and 34 μg/mL chloramphenicol. A portion (7 mL) of the cultured cells was transferred into 250 mL of fresh LB liquid medium supplemented with the same antibiotics, and the cells were grown at 37 °C at 200 rpm until cell density reached OD600 = 0.65. The culture was transferred to a 30 °C shaker, and gene expression was induced by adding isopropyl β-d-thiogalactoside (1 mM). After a 4-h incubation, cells were collected by centrifugation, suspended in 10 mL of buffer (50 mM Tris–HCl, pH 7.5, 10% glycerol, 1 mM EDTA) supplemented with 5 mM DTT and 0.5 mM phenylmethylsulfonyl fluoride, and were then lysed by six sonication intervals (each 10 s pulse followed by 20 s rest) using a Fisher 550 sonicator (Fisher Scientific, Loughborough, Leicestershire, UK) equipped with microtip probe. The lysed cells were centrifuged at 4 °C for 30 min at 20,000g, and the supernatant (termed s20) was recovered and stored at −20 °C.
UAXS was purified on Source-Q15 anion-exchange column (5 × 50 mm, Amersham Biosciences) using a Waters 626 LC HPLC system equipped with a photo diode-array detector (PDA 996). Chromatography was controlled by a Waters Millennium32 workstation, and spectra were analyzed with the PDA software. The column was equilibrated with 50 mM sodium phosphate at pH 7.6 (buffer A) at a flow rate of 1 mL min−1. The s20 supernatant (1 mL) was injected into the column, and the column was washed for 30 min with buffer A. Proteins were then eluted with a linear gradient of 0–0.5 M NaCl over 30 min in buffer A at a flow rate of 0.5 mL min−1. Eluted proteins were detected by monitoring the UV absorption at 280 nm, and fractions were collected for enzyme analysis. Size-exclusion chromatography was used to estimate the native molecular weight of recombinant UAXS. A total of 0.5 mL of protein sample of UAXS or a mixture of standard markers [10 mg each of alcohol dehydrogenase (150 kDa), ovalbumin (48.9 kDa), ribonuclease A (15.6 kDa), and cytochrome C (12.4 kDa)] was loaded at a flow rate of 1 mL min−1 onto a Superdex75 column (8 mm id × 900 mm long, Amersham) equilibrated with 0.1 M sodium phosphate, pH 7.6, containing 0.1 M NaCl. Protein elution was monitored at A280 nm, and fractions were collected every 20 s. Active and purified UAXS eluted as dimers (data not shown) and were stored at −20 °C. The purified recombinant protein was fragmented with trypsin, and the resulting peptides were sequenced by electrospray MS/MS analyses28 to confirm the UAXS amino acid sequence (data not shown).
3.2. HPLC analysis of enzymatic reactions
Enzymatic reactions were performed in 0.1 M sodium phosphate, pH 7.8, (50 μL) containing 1 mM β-NAD+, 1 mM UDP-GlcA, and recombinant UAXS unless otherwise specified. Reactions were kept at 37 °C for up to 2 h, and the reactions were then terminated by adding an equal volume of chloroform. After 30 s vortexing and 5 min centrifugation (12,000 rpm at room temperature), the upper aqueous phase was collected, and enzymatic reactants were chromatographed on an anion-exchange column (Q15-Amersham, 4.6 mm id × 250 mm) using an Agilent Series 1100 HPLC system equipped with an autosampler, diode-array detector, and a Corona Charged Aerosol Detector (ESA, Chelmsford, MA) as described.36 Chromatography was controlled by ChemStation software (Agilent), and the HPLC ChemStation Spectral Module was used for the collection and analyses of UV and Corona (ESA) spectrum. The effect of various nucleotides (2 mM) on UAXS activity is summarized in Supplementary data Figure S1.
3.3. NMR analysis of enzymatic reactions
All spectra were collected on Varian Inova 800-MHz or 900-MHz spectrometer equipped with cryogenic probes. The HPLC-isolated enzymatic products UMP and apiofuranosyl-1,2-cyclic phosphate were dissolved in 99.96% D2O at neutral pD. COSY experiments were performed using a standard Varian gCOSY pulse program and data were acquired for 16 h and 12 h for purified apiofuranosyl-1,2-cyclic phosphate and the UAXS reaction mixture having pH of 6.5, respectively. Data were collected as a 2048 × 512 matrix with spectral width of 8000 Hz and were processed with the Varian spectrometer software using a squared-sinebell function and zero-filling in both dimensions.
Most real-time NMR reactions (500 μL) were performed in D2O/H2O (4:1 v/v or 9:1 v/v) mixtures containing sodium phosphate (0.1 M, pH 7.8 or 6.5), β-NAD+ (0.5 mM), UDP-GlcA (0.5 mM), and recombinant UAXS (100 μL). In some cases, the D2O/H2O ratio was changed to 1:9. Immediately upon addition of protein, the reaction mixture was transferred to a 5-mm NMR tube and analyzed. Due to requirements of the NMR set-up, data acquisition was not recorded until 10–20 min into the enzymatic reaction. Consecutive individual 1D proton spectra were each acquired for five min and were stored over the course of the reaction (6–24 h). All spectra were referenced externally to the water resonance at 4.765 ppm downfield of 2,2-dimethyl-2-silapentane-5-sulfonate DSS). Processing of the data as covariance matrices was done with Matlab (The Mathworks, Inc.).
Supplementary Material
Acknowledgments
We thank Dr. Karen Welinder (Aalborg, Denmark) for providing the clone bf460135 on 12/2000. We also thank Professor J. Prestegard and M. O'Neill of the CCRC for their constructive comments on the manuscript. This research was supported by NSF: IOB-0453664 (MB-P); in part by U.S. Dept. of Agriculture grant 2002-35318-12620 (MB-P); and in part by BESC, the BioEnergy Science Center, that is supported by the Office of Biological and Environmental Research in the DOE Office of Science.
Footnotes
Supplementary data: Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.carres.2009.03.026.
References
- 1.Harborne JB, Williams CA. Nat Prod Rep. 2001;18:310–333. doi: 10.1039/b006257j. [DOI] [PubMed] [Google Scholar]
- 2.Su YF, Koike K, Guo D, Satou T, Liu JS, Zheng JH, Nikaido T. Tetrahedron. 2001;57:6721–6726. [Google Scholar]
- 3.El-Sayed NH, Wojcinska M, Drost-Karbowska K, Matlawska I, Williams J, Mabry TJ. Phytochemistry. 2002;60:835–838. doi: 10.1016/s0031-9422(02)00133-4. [DOI] [PubMed] [Google Scholar]
- 4.Siciliano T, De Tommasi N, Morelli I, Braca A. J Agric Food Chem. 2004;52:6510–6515. doi: 10.1021/jf040214q. [DOI] [PubMed] [Google Scholar]
- 5.Darvill AG, O'Neill MA, Albersheim P. Plant Physiol. 1978;62:418–422. doi: 10.1104/pp.62.3.418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Beck EZ. Pflanzenphysiol. 1967;57:444–461. [Google Scholar]
- 7.Longland JM, Fry SC, Trewavas AJ. Plant Physiol. 1989;90:972–976. doi: 10.1104/pp.90.3.972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.O'Neill MA, Ishii T, Albersheim P, Darvill AG. Annu Rev Plant Biol. 2004;55:109–139. doi: 10.1146/annurev.arplant.55.031903.141750. [DOI] [PubMed] [Google Scholar]
- 9.Ishii T, Matsunaga T, Pellerin P, O'Neill MA, Darvill A, Albersheim P. J Biol Chem. 1999;274:13098–13104. doi: 10.1074/jbc.274.19.13098. [DOI] [PubMed] [Google Scholar]
- 10.O'Neill MA, Eberhard S, Albersheim P, Darvill AG. Science. 2001;294:846–849. doi: 10.1126/science.1062319. [DOI] [PubMed] [Google Scholar]
- 11.Grisebach H. In: The Biochemistry of Plants: A Comprehensive Treatise. Stumpf PK, Conn EE, editors. Academic Press; New York: 1980. pp. 171–206. [Google Scholar]
- 12.Mendicino J, Abouissa H. Biochim Biophys Acta. 1974;364:159–172. doi: 10.1016/0005-2744(74)90143-0. [DOI] [PubMed] [Google Scholar]
- 13.Picken JM, Mendicino J. J Biol Chem. 1967;242:1629–1634. [PubMed] [Google Scholar]
- 14.Molhoj M, Verma R, Reiter WD. Plant J. 2003;35:693–703. doi: 10.1046/j.1365-313x.2003.01841.x. [DOI] [PubMed] [Google Scholar]
- 15.Pan YT, Kindel PK. Arch Biochem Biophys. 1977;181:131–138. doi: 10.1016/0003-9861(77)90427-1. [DOI] [PubMed] [Google Scholar]
- 16.Ortmann R, Sandermann H, Grisebach H. FEBS Lett. 1970;7:164–166. doi: 10.1016/0014-5793(70)80146-6. [DOI] [PubMed] [Google Scholar]
- 17.Gustine DL, Kindel PK. J Biol Chem. 1969;244:1382. [PubMed] [Google Scholar]
- 18.Mendicino J, Hanna R. J Biol Chem. 1970;245:6113–6124. [PubMed] [Google Scholar]
- 19.Kelleher WJ, Grisebach H, Ortmann R, Baron D. FEBS Lett. 1972;22:203–204. doi: 10.1016/0014-5793(72)80045-0. [DOI] [PubMed] [Google Scholar]
- 20.Gebb C, Baron D, Grisebach H. Eur J Biochem. 1975;54:493–498. doi: 10.1111/j.1432-1033.1975.tb04161.x. [DOI] [PubMed] [Google Scholar]
- 21.Gal M, Mishkovsky M, Frydman L. J Am Chem Soc. 2006;128:951–956. doi: 10.1021/ja0564158. [DOI] [PubMed] [Google Scholar]
- 22.Ishiyama N, Creuzenet C, Miller WL, Demendi M, Anderson EM, Harauz G, Lam JS, Berghuis AM. J Biol Chem. 2006;281:24489–24495. doi: 10.1074/jbc.M602393200. [DOI] [PubMed] [Google Scholar]
- 23.Schmid F, Stone BA, Brownlee RTC, McDougall BM, Seviour RJ. Carbohydr Res. 2006;341:365–373. doi: 10.1016/j.carres.2005.10.013. [DOI] [PubMed] [Google Scholar]
- 24.McNally DJ, Schoenhofen IC, Mulrooney EF, Whitfield DM, Vinogradov E, Lam JS, Logan SM, Brisson JR. ChemBioChem. 2006;7:1865–1868. doi: 10.1002/cbic.200600298. [DOI] [PubMed] [Google Scholar]
- 25.Barlow JN, Blanchard JS. Carbohydr Res. 2000;328:473–480. doi: 10.1016/s0008-6215(00)00135-x. [DOI] [PubMed] [Google Scholar]
- 26.Petersen BO, Krah M, Duus JO, Thomsen KK. Eur J Biochem. 2000;267:361–369. doi: 10.1046/j.1432-1327.2000.01008.x. [DOI] [PubMed] [Google Scholar]
- 27.Baron D, Wellmann E, Grisebac H. Biochim Biophys Acta. 1972;258:310–318. doi: 10.1016/0005-2744(72)90988-6. [DOI] [PubMed] [Google Scholar]
- 28.Bar-Peled M, Griffith CL, Doering TL. Proc Natl Acad Sci U S A. 2001;98:12003–12008. doi: 10.1073/pnas.211229198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Breazeale SD, Ribeiro AA, Raetz CRH. J Biol Chem. 2002;277:2886–2896. doi: 10.1074/jbc.M109377200. [DOI] [PubMed] [Google Scholar]
- 30.Snyder JR, Serianni AS. Carbohydr Res. 1987;166:85–99. doi: 10.1016/0008-6215(87)80046-0. [DOI] [PubMed] [Google Scholar]
- 31.Ishii T, Yanagisawa M. Carbohydr Res. 1998;313:189–192. [Google Scholar]
- 32.Otter A, Bundle DR. J Magn Reson., Ser B. 1995;109:194–201. doi: 10.1006/jmrb.1995.0009. [DOI] [PubMed] [Google Scholar]
- 33.Barfield M, Dean AM, Fallick CJ, Spear RJ, Sternhell S, Westerman PW. J Am Chem Soc. 1975;97:1482–1492. [Google Scholar]
- 34.Harper AD, Bar-Peled M. Plant Physiol. 2002;130:2188–2198. doi: 10.1104/pp.009654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cloarec O, Dumas ME, Craig A, Barton RH, Trygg J, Hudson J, Blancher C, Gauguier D, Lindon JC, Holmes E, Nicholson J. Anal Chem. 2005;77:1282–1289. doi: 10.1021/ac048630x. [DOI] [PubMed] [Google Scholar]
- 36.Gu XG, Bar-Peled M. Plant Physiol. 2004;136:4256–4264. doi: 10.1104/pp.104.052365. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.