

Abstract
Various substrates, catalysts, and assay methods are currently used to screen inhibitors for their effect on the proteolytic activity of botulinum neurotoxin. As a result, significant variation exists in the reported results. Recently, we found that one source of variation was the use of various catalysts, and have therefore evaluated its three forms. In this paper, we characterize three substrates under near uniform reaction conditions using the most active catalytic form of the toxin. Bovine serum albumin at varying optimum concentrations stimulated enzymatic activity with all three substrates. Sodium chloride had a stimulating effect on the full length synaptosomal-associated protein of 25 kDa (SNAP25) and its 66-mer substrates but had an inhibitory effect on the 17-mer substrate. We found that under optimum conditions, full length SNAP25 was a better substrate than its shorter 66-mer or 17-mer forms both in terms of kcat, Km, and catalytic efficiency kcat/Km. Assay times greater than 15 min introduced large variations and significantly reduced the catalytic efficiency. In addition to characterizing the three substrates, our results identify potential sources of variations in previous published results, and underscore the importance of using well-defined reaction components and assay conditions.
Introduction
The “anthrax letter” scare in the fall of 2001 [1] generated renewed interest in finding remedies to real and perceived bio threat agents. These interests include development of sensitive detection methods for environmental samples as well as dependable activity assay methods to screen large compound databases for potential drug candidates against these bio threat agents. One such bio threat agent is botulinum neurotoxin (BoNT), the most toxic compound known to humans [2]. Its ultimate toxicity relies on a zinc-endopeptidase activity on a neuronal protein. For more than a decade, this protease activity has been the intense focus as a target of inhibitor screening for small molecule drug development. Inhibitory properties of a handful of available inhibitors obtained from these efforts often are not in agreement to each other [3]–[5] because of variations in the particular assay methods employing various catalysts, substrates, and reaction components. An investigation into the effects that these variations have upon enzyme activity is extremely important for adoption of a balanced and appropriate assay conditions in drug discovery [6].
BoNT is a secretory protein produced primarily by several strains of Clostridium botulinum [2], [7]. Depending on the bacterial strain producing the BoNT, it has traditionally been classified into seven distinct serotypes, designated as BoNT/A through BoNT/G. Of these, BoNT/A afflicts humans most, followed by BoNT/B and BoNT/E. Although serologically different, all have very similar primary, secondary, tertiary, and quaternary structures. They are produced as single polypeptides of ∼1300 amino acid residues. A posttranslational cleavage, either in the bacterial or host environment cleaves this polypeptide approximately 440 amino acids from the N-terminus resulting in a large C-terminal heavy chain (HC) binding domain, and a small N-terminal light chain (Lc) catalytic domain. Lc is a zinc-endopeptidase in which zinc, bound at the active site, is essential for its enzymatic activity. HC and Lc however remains linked through a conserved disulfide bond. HC is again divided into two subdomains of N-terminal heavy chain (HcN) and C-terminal heavy chain (HcC).
A number of laboratories have synthesized scores of peptides and peptidomimetics [4], [8]–[15], or have synthesized and screened tens of thousands of compounds in small molecule libraries [3], [16]–[22] as inhibitors of BoNT catalytic activity. Most of these efforts however employed a unique combination of BoNT catalyst, its cognate substrate, and a particular assay method. Results from these assays, however, are not always in agreement [3]–[5]. The disagreement of published results poses a major impediment for selecting a useful scaffold that would provide a relevant model for screening small molecules as inhibitors of Lc activity.
Two major sources of discrepancies are (a) the choice of the Lc catalyst and (b) the choice of a substrate (23,24) (Table 1). A third potential source of discrepancy results from the (c) variations in the reaction composition. Other apparent reasons that often remains ignored are (d) reaction time and (e) the analytical technique used to follow the reaction. A recent review has addressed these aspects in more detail [23].
Table 1. Substrates, LcA catalytic activity assay methods and results.
| Substrate | Assay Method | Km (µM) | Kcat (Sec−1) | Reference |
| Live mice | Lethality | - | - | [50] |
| 66-mer SNAP25 | SDS-PAGE | 25 | 11 | [29] |
| 16 | 60 | [30] | ||
| HPLC | 27 | <3 | [31] | |
| SNAP25 | SDS-PAGE | 41 | 2 | [51] |
| GST-SNAP25 | 14 (64*) | 5 (1*) | [52] | |
| GST-SNAP25 | SDS-PAGE | 10 | 17 | [53] |
| HPLC | 51* (106*) | 10* (4*) | [54] | |
| ELISA | [55] | |||
| SNAP25 (in phrenic hemidiaphraghm) | Electrophysio-logical | [56] | ||
| 17-mer SNAP25 | HPLC | 1000 | 23 | [26], [27] |
| HPLC | 3000–5000 | 12 | [28] | |
| HPLC | 3400 | 9 | [25] | |
| UPLC | 1000 | 24 | [12] | |
| UPLC | 1513 | 28 | This study | |
| SNAPtide™ | ALISSA | [57] | ||
| SNRTRIDEAN[dnpK]RA[daciaC]RML | Fluorescence | 10 | 7 | [26] |
| (FITC)-TDRIDQANQRATK(DABCYL)nL-amide | Fluorescence | [58] | ||
| (FITC)-ATDRIDQANQRATK(DABCYL)nL-amide | Fluorescence | 19 | [32] | |
| SNAPtide520 | Fluorescence | [33] | ||
| SNAPtide521 (FITC/DABCYL) | 55 | [52] | ||
| -SNAPtide522 | ||||
| CFP-SNAP25141–206 –YFP | Fluorescence | 0.7 | 4.1 | [59] |
*The catalyst used in these cases was whole BoNT/A toxin instead of only LcA in all other cases.
It was not until the availability of comparative data on catalytic constants with and without several types of inhibitors towards four different versions of the catalytic BoNT/A Lc [5], [24], [25] that a clear picture of the reason for discrepancy emerged. Inhibition constant ki or the extent of inhibition depended on which of the several C-terminally truncated BoNT/A Lc was used [5]. In the past, we have demonstrated that a full length Lc free from rest of the BoNT/A molecule is the most catalytically active species [25]. In light of the inhibitor development problems, we extended that study to include two C-terminally truncated LcA and demonstrated that a full length BoNT/A Lc containing 1–448 residues has the highest catalytic activity because its C-terminal appeared to play a product removal role from the active site of LcA [24]. There was little variation in the substrate Km catalyzed by these Lcs and by various BoNT/A forms [25].
The cellular target for BoNT/A or its LcA is the 206-residue SNAP25. For convenience, investigators have often used two versions of SNAP25 [12], [25]–[28]: a truncated 66-mer [29]–[31], or a shorter 17-mer version [12], [25]–[28], in addition to a modified Forster resonance energy transfer (FRET) version of the 17mer [3], [32], [33] as the substrate. Data compiled in Table 1 using various forms of the substrate, show that the Km and kcat values vary considerably, even if the same substrate is used. This is probably due to major differences in the buffer, reaction component, or the particular analytical tool used. However, except for the 17-mer, and cyan fluorescent protein (CFP) and yellow fluorescent protein (YFP)-tagged CFP-SNAP25141–206 –YFP substrates, properties of the rest have not been fully characterized. It is often argued that the 66-mer is a more reasonable counterpart of the full length SNAP25 substrate for LcA. However, no systematic investigation comparing these substrates under a standard set of conditions has been done so far.
Depending upon the concentration, addition of zinc and dithiothreitol (DTT) to the LcA reaction mixtures can be both stimulating and inhibitory [34]. Similarly, both Km and kcat of the 17-mer substrate are dramatically affected by increasing concentrations of bovine serum albumin [35]. Salts and buffer components like NaCl, Na-phosphate, tris.HCl, and ethylenediaminetetraacetate (EDTA) are inhibitory to BoNT/A activity [34], [36]. Presence of these components in the LcA or substrate preparations or in the reaction mixtures can potentially give misleading activities and false inhibitory results. Thus, it is very important that activity of one standard LcA catalyst be determined using several of the currently used substrates, so that the effects of various additives on the rate of the reaction can be evaluated. Results obtained from such a study will allow a direct comparison of the properties of LcA and the substrates for a more realistic evaluation of inhibitor screening.
In this backdrop, the current investigation compares the substrate properties of the 17-mer, the 66-mer, and the full length SNAP25 with the most active BoNT/A catalyst under near identical assay conditions. We also examined the effects of several additives that have been in use in each of these assays. Our results provide a direct comparison of these effects demonstrating for the first time that reaction components, particularly NaCl, exert completely different effects depending upon which substrate is used. Additionally, we show that the reaction time has a profound effect on the enzyme constants, and the full length SNAP25 is by far the best substrate that yields the lowest Km and highest kcat values.
Results
17-Mer substrate
Previously, we reported that a LcA preparation solubilized from inclusion bodies behaved very similar to that of whole BoNT/A toxin when assayed with the synthetic 17-mer substrate [34]. These similarities included activity stimulation by BSA [27], and in Km and kca t values [34], [35]. We had also reported [34] that the solubilized LcA activity was inhibited by including 5 mM DTT that could be neutralized by the addition of ZnCl2. Conversely, in the absence of DTT or β-mercaptoethanol, the reaction was inhibited by increasing concentrations of ZnCl2. We had concluded that our LcA preparation, having stoichiometric amounts of zinc, behaved similar to other Zn-metalloproteases [37]. We repeated these experiments with the new LcA preparation purified from soluble extracts and found essentially same behavior was displayed by adding BSA, ZnCl2, and DTT (data not shown). Therefore, in routine assays we included 0.2 mg/ml BSA but omitted ZnCl2 and DTT. Under these conditions and employing a 17-mer substrate concentration of 0.13, 0.27, 0.33, 0.44,0.67, and 1.17 mM, we calculated its Km as 1.5 mM and a Vmax of 33.6 mM/min/mg (kcat of 28/sec) (Table 1–3). In terms of Vmax using the 17-mer substrate, this LcA preparation has the highest activity reported in the literature [28], [34], [35].
Table 3. Steady state kinetic constants for LcA reactions utilizing various SNAP25 substrates.
| SNAP25 Substrate | Km (µM)a | kcat (Sec−1)a | kcat/Km (µM/Sec) |
| 17-merb | 1513±281.8 | 28±3.5 | 0.019 |
| 66-mer | 32.43±0.9281 | 23±0.22 | 0.72 |
| Full Length | 33.34±4.592 | 76±2.2 | 2.3 |
The values are averages of results from 5, 10, and 15-min reactions reported in Table 2.
10 min incubation data. Kinetic parameters were obtained by hyperbolic Michaelis-Menten curve fitting (R2 = 0.99345) of experimental 5 replicate data.
Because LcA is now routinely used to screen for BoNT/A protease inhibitors using a variety of reactions such as in high throughput formats, the purity, concentration, time of incubation, and its temperature dependent stability needs to be carefully established. LcA used in this study is a highly pure protein showing only one stained band in sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and western blot [38]. Figure 1A shows that 51 nM LcA rapidly consumes the 17-mer substrate and that substrate conversion-time linearity is clearly lost after 5 min, the first time point at which the reaction measurement can be recorded accurately. By lowering the LcA concentration to 0.4–1.6 nM, linearity was observed for 60 min although detection of product at earlier time points, particularly with 0.8 and 0.4 nM LcA yielded results with high standard deviation. Yet the results in Figure 1A inset indicate that in the presence of 0.2 mg/ml BSA, LcA at concentration as low as 0.4 nM remains stable at 37°C for at least 1 hour. By plotting the amount of substrate converted (or product formed) in 5 min as a function of LcA concentration, we found a fairly linear relationship between LcA concentration and extent of substrate conversion up to 36%. In addition to characterizing the LcA preparation as a stable protein under the specified conditions, results in these two figures allow us to select a combination of LcA concentration, reaction time, and sensitivity of detection that is most suitable for a particular assay including high throughput screening platforms using this substrate.
Figure 1.

A, Time course of reaction of LcA utilizing the 17-mer peptide (0.5 mM) as a substrate in the presence of 0.2 mg/ml BSA in 50 mM NA-HEPES, pH 7.4 at 37°C. LcA concentrations used was 51 nM (purple circle), and in the inset, 1.6 nM (gold circle), 0.8 nM (blue square), and 0.4 nM (green diamond). Each data point is an average of 5 assays. B, 5-Min data for each of the LcA concentrations used in A and others not shown are plotted as a function of LcA concentration. Bars in both panels represent standard deviations. These results show that (a) at high enough LcA concentration, the time course becomes nonlinear (due to substrate depletion), (b) LcA concentration as low as 0.4 nM remains stable (in the presence of 0.2 mg/ml BSA) at 37°C for at least 1 hour, and (c) if enough substrate is available, the reaction rate is linear to LcA concentration between 0.4 and 51 nM.
Behavior of LcA towards the full length SNAP25 and its synthetic 66-mer peptide substrate
The commercially-obtained full length SNAP25 is a reasonably pure protein of 206 residues. The protein concentration was verified by complete digestion with LcA (data not shown). SNAP25 is supplied by the vendor in 25 mM tris.HCl buffer containing 2 mM EDTA. We [34] and others [39] have demonstrated that tris.HCl is inhibitory to BoNT activity and that EDTA, in addition to being a metal chelator, is also an inhibitor of LcA activity. Therefore the full length SNAP25 was extensively dialyzed against 50 mM Na-HEPES, pH 7.4 before using in the assays.
Incubation of various LcA concentrations with a fixed, 11.3 µM concentration of SNAP25 yielded results as depicted in Figure 2. At the highest LcA concentration, more than 90% of the substrate was converted into products while at the lowest LcA concentration, only ∼20% substrate was consumed in 60 min. Please note that the lowest concentration (0.04 nM) of LcA used in this experiment is an order of magnitude lower than the lowest concentration used in previous experiments (Figure 1), because the substrate SNAP25 concentration was 45-fold lower than that used with the 17-mer peptide substrate due to almost 50-fold lower Km (see later). Even at this low concentration of 0.04 nM LcA, incubation at 37°C for 1 hour did not denature the enzyme, as was evidenced by the fact that time-dependent product formation maintained a linear relationship during the incubation (Figure 2A). The major difference observed in these experiments versus results shown in Figure 1A was that the SNAP25 substrate did not have a time dependent linearity with LcA concentration above 0.08 nM having incubations longer than 10 min (Figure 2B). This loss of linearity of the reaction must be due to depletion of substrate because the lowest LcA concentration (0.04 nM) yielded a straight line for 60 min (Figure 2A). Although a linear relationship between the % product formed in 5 and 10 min at 0.04–0.4 nM LcA concentrations (Figure 2B) was obtained, there was a nonlinear tendency at the 20-min incubations that was due to substrate depletion as noted before. Implications of these results are more thoroughly addressed later in Table 2.
Figure 2.
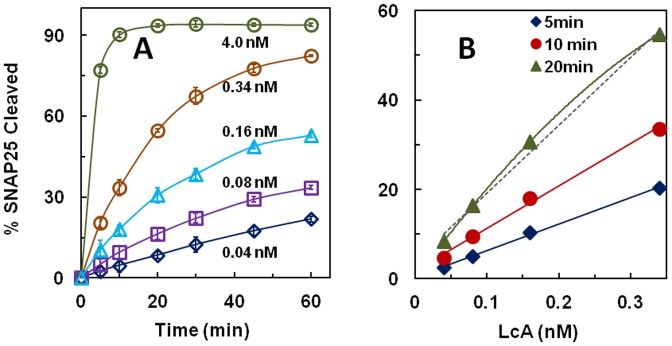
A, Time course of reaction of LcA utilizing the full length SNAP25 (11.3 µM) as a substrate in the presence of 0.2 mg/ml BSA, 0.25 mM ZnCl2 and 5 mM DTT in 50 mM NA-HEPES, pH 7.4 at 37°C. LcA concentrations ranging from 0.04 nM to 4.0 nM were as indicated. Each data point is an average of 5 assays. Bars represent standard deviations. B, 5, 10 and 20-min data for four of the LcA concentrations used in A are plotted as a function of LcA concentration. The dotted line is the best fit (y = mx + c) of the 20-min data points while the curved lines connect the actual data points for each of 5-min, 10-min and 20-min data points. These results show that (a) above 0.04 nM LcA concentration, the time course becomes nonlinear (due to substrate depletion), (b) LcA concentration as low as 0.04 nM remains stable at 37°C for at least 1 hour, and (c) if enough substrate is available, the reaction rate should be linear between 0.04 and 4.0 nM LcA concentration.
Table 2. Dependence of Km, kcat and kcat/Km for full length SNAP25 substrate on the LcA reaction time.
| Time (min) | Km (µM) | kcat (Sec−1) | kcat/Km (µM/Sec) |
| 5 | 29.8±4.02 | 73.9±5.32 | 2.47 |
| 10 | 31.6±4.35 | 76.8±5.63 | 2.43 |
| 15 | 38.5±1.19 | 78.2±1.38 | 2.03 |
| 30 | 67.4±1.61 | 88.4±1.14 | 1.31 |
| 45 | 180±18.8 | 159±14.9 | 0.883 |
| 60 | 189±14.6 | 134±8.75 | 0.709 |
The values were calculated by hyperbolic curve fitting by Michaelis-Menten equation of the data presented in Figure 5B.
These results underscore the importance of choosing the right LcA concentration, and the time of enzymatic reaction in devising a standard assay protocol. The LcA concentration-dependent reaction linearity shown in Figures 1B and 2B also show that the LcA preparation does not contain contaminants that would complicate the steady-state kinetics and data described below.
Data presented in Figure 3 show that increasing the NaCl concentration to 100 mM in the reaction mixture containing SNAP25 and LcA, dramatically increased LcA activity more than sevenfold. Higher concentrations of NaCl caused a decrease in LcA activity. The same was also observed when the 66-mer peptide was used as a substrate. These stimulating effects of NaCl towards the full length and 66-mer versions of SNAP25 are in quite contrast to its inhibitory effect towards the 17-mer substrate as shown in Figure 3 and previously noted elsewhere [34]. The most logical explanation for the stimulating effect of NaCl is that it disrupts nonspecific, inter- and intra-molecular protein-protein interactions between the large molecules of SNAP25, the 66-mer, and LcA. Such interactions will limit the availability of monomeric forms of the molecules needed to act as substrates. The 17-mer peptide being much smaller than the full length SNAP25, is most likely free from such interactions.
Figure 3. Substrate-dependent effects of sodium chloride on the catalytic activity of LcA.
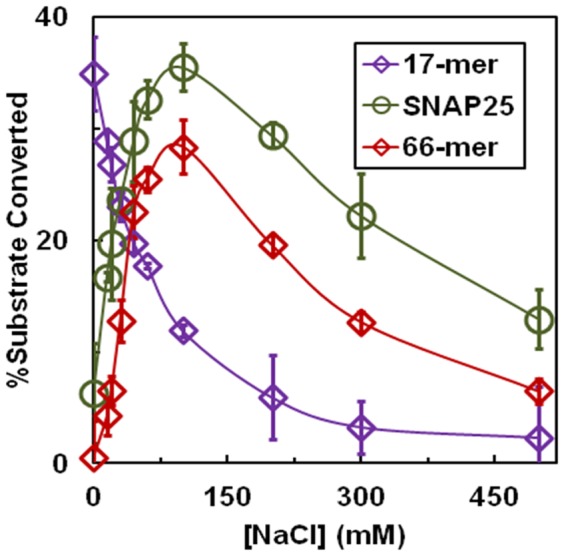
The 30-µl reaction mixtures containing 0.2 mg/ml BSA, 12 µM SNAP25 or 66-mer substrate +1.0 nM LcA, or 0.5 mM 17-mer substrate +50 nM LcA in 50 mM Na-HEPES, pH 7.4. The choice of 50-fold difference in LcA concentration was based on the fact that the SNAP25 substrate concentration used was 42-fold lower than the 17-mer substrate because of a ∼45-fold difference in the Km of SNAP25 or its 66-mer with the 17-mer (see later in Table 3). The SNAP25 and 66-mer substrate reaction mixtures also contained 5 mM DTT, and 250 µM ZnCl2, and the reactions were allowed to proceed for 10 min, while the 17-mer reaction was allowed to proceed for 5 min. Addition of 5 mM DTT and 250 µM ZnCl2 to the 17-mer substrate reactions yielded results identical to those without them as shown in this figure.
Effects of DTT, BSA, and Zn on LcA activity with SNAP25 and the 66-mer peptide
Unlike the whole BoNT/A toxin which contains an inter chain disulfide bond, LcA does not contain a disulfide bond. However SNAP25 containing four cysteine residues located near the middle of the molecule, have a propensity to form mixed disulfide bonds [40], [41] and other oxidation products that lead to insolubility and precipitation. The same would be expected with the 66-mer peptide because it contains the same four cysteine residues. Therefore 5 mM DTT was included in all LcA reactions using these two substrates. Reaction mixtures having no additive such as DTT, ZnCl2, BSA, or the 57-mer peptide were treated as controls in the respective experiments. DTT has a propensity to form complexes with divalent metal ions [42] such as the Zn++ bound at the LcA active site and could potentially inhibit LcA activity. Therefore, we measured LcA activity in the presence of 5 mM DTT and increasing concentrations of ZnCl2. There was a slight stimulation of the activity up to 100 µM ZnCl2 after which the activity declined with increasing concentrations although at 250 µM the activity remained above the control 100% (Figure 4A). Similarly, at a fixed 250 µM concentration of ZnCl2, slight stimulation of LcA activity was observed when 1–8 mM DTT was added to the reaction mixture (Figure 4B). Although optimal activity was obtained at 0.1 mM ZnCl2 and 4 mM DTT, there was little difference with the activities at 0.25 mM ZnCl2 (Figure 4A) and 5 mM DTT (Figure 4B), the optimal concentrations reported [34] and used earlier [4], [12], [14], [24], [26], [27], [34], [43]–[45].
Figure 4. Effects of ZnCl2 (A), DTT (B), and BSA (C) on the catalytic activity of LcA using SNAP25 as a substrate.
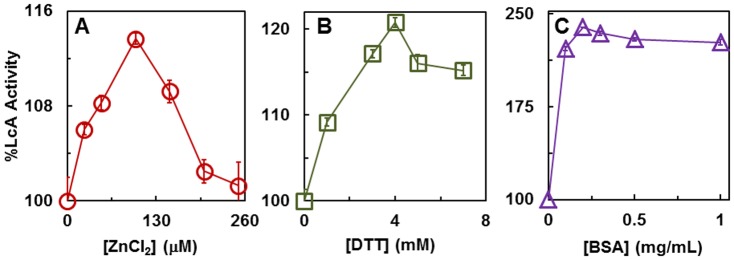
The 30-µl reaction mixtures containing 12 µM SNAP25, 0.34 nM LcA in 50 mM Na-HEPES, pH 7.4 were incubated at 37°C for 10 min. In addition, the reaction mixtures contained 5 mM DTT in A, 250 µM ZnCl2 in B, and 5 mM DTT and 250 µM ZnCl2 in C.
The stimulating effect of BSA on LcA activity was more evident than either DTT or ZnCl2 as the activity was more than doubled by the addition of 0.1 mg/ml BSA (Figure 4C). This level of stimulation did not change with up to 1 mg/ml BSA. We demonstrated earlier that LcA is prone to precipitation but BSA provided a stabilizing effect by keeping it more soluble [46]. Saturation of SNAP25 cleavage activity stimulation by the addition of low BSA concentrations (0.1∼0.2 mg/ml) (Figure 4C) compared to ∼2 mg/ml BSA needed for optimum cleavage of the17-mer substrate [27], may be related to the fact that the large SNAP25 substrate itself provides the stabilizing effect on LcA.
Results obtained in these experiments led us to formulate a standard reaction mixture recipe that contains 0.2 mg/ml BSA, 5 mM DTT and 0.25 mM ZnCl2 to use with SNAP25 or its 66-mer as substrates. In routine assays, the latter concentrations are more convenient to prepare from concentrated stocks.
Steady state kinetic constants using the three different substrates
The Michaelis constant Km for various forms of the 66-mer or the full length SNAP25 has been reported to range from 14 µM to 64 µM by various laboratories (Table 1), reflecting a fourfold to fivefold difference. A much more pronounced difference in the values of kcat (2–60/sec) using these substrates were also reported. Additionally, substrate differences, source and/or quality of LcA, time of the enzymatic reaction (5 min to 2 hours), and the analytic methods used for quantification varied as well. Another potential reason for the differences in the kinetic constants in Table 1 may be due to different methods of computation used. It occurred to us that because sub-saturating concentrations of the substrate (maximum fivefold above Km) was used in all cases in Table 1, and the reaction incubation time differed, time-dependent rapid depletion of substrate could account for the large variations in the reported values of Km and kcat. We therefore followed the LcA reaction from five to 60 minutes using full length SNAP25, and the 66-mer SNAP25 peptide substrates. Figure 5A shows a representative time course experiment using seven different concentrations ranging from 5 µM to 50 µM of the full length SNAP25. Longer the time of incubation, more products are formed (less substrate is remaining) such that most of the substrate was consumed over 60 min. As a result, except for the 10 min incubation, the kobs progressively decreased when calculated from the increasing incubation time data, as depicted in Figure 5B. These observations are reflected in the derived values of kcat and Km that increased with time of incubation (Table 2). Although both parameters increased with time of reaction, the catalytic efficiency, calculated as kcat/Km progressively decreased almost fourfold from 10 min to 60 min incubation (Table 2).
Figure 5. Time course of LcA reaction using various concentrations of SNAP25 (A), and linear plots of reaction rate (kobs) calculated at various time points versus SNAP25 concentrations (B).
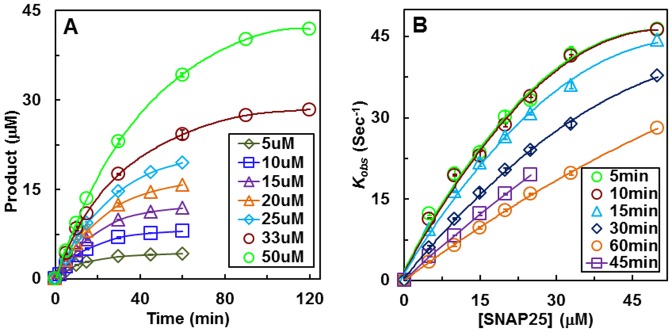
Each reaction mixture contained 0.2/ml BSA, 5 mM DTT, and 250 µM ZnCl2, 0.34 nM LcA, and 50 mM Na-HEPES, pH 7.4. The bars inside symbols represent standard deviation of three assays. The lines connecting the data points in B were generated by curve fitting using the Michaelis-Menten equation kobs = (kcat×[S])/(Km+[S]) in a KaleidaGraph software package. Km and kcat values derived from these curves are reported in Table 2.
For routine use SNAP25 stock solution can be obtained at a concentration of no more than 40 µM. So there is a practical limit of its concentration in the assays. With this limitation of using low concentrations of the substrate in routine assays, a 5–15 min reaction incubation time appears to provide a reasonably accurate Km (33.34±4.592 µM) and kcat (76±2.2/sec) values for full length SNAP25 (Table 3). Essentially identical experiments using the 66-mer as a substrate yielded an almost identical Km of 32.43±0.9281 µM but almost threefold reduced kcat of 23±0.22/sec (Table 3). Thus, the 66-mer peptide appears to be a poor substrate when compared with the full length SNAP25. At the same time, the 66-mer substrate with a very similar kcat but almost 50-fold lower Km is a better substrate than the 17-mer peptide (Table 3). In terms of Km, and kcat/Km, the 17-mer peptide is the poorest of all the three substrates followed by the 66-mer substrate The full length SNAP25 having the highest kcat/Km value, appeared to be the best substrate. Nonetheless, kcat with the 17-mer is comparable to that with the full-length and the 66-mer substrates as opposed to its very high Km. Because most of the active and catalytic site interactions of LcA is contained in this 17-mer substrate [15], its low cost and high solubility [27] should make the 17-mer a preferred choice in routine screening for inhibitors of LcA catalytic activity.
Inhibition of LcA activity by a product of its reaction with substrate
Cleavage of the 66-mer substrate with LcA results in a 57-mer N-terminal product and a 9-mer C-terminal product. To investigate if the nonlinearity of the time course of reaction observed in Figure 5A could be partly due to inhibition of the reaction by the formed product, we incubated LcA with SNAP25 in the presence of 50 µM 9-residue C-terminal product or 50 µM 57-residue N-terminal product. Only the 57-mer product affected the LcA activity. Figure 6 shows that irrespective of the two large substrates used, activity of LcA is inhibited by the 57-mer product. The inhibition appears to be more pronounced with the 66-mer substrate than with the full length SNAP25 substrate. In contrast to the inhibition by the 57-mer product, the reaction of LcA with the 17-mer substrate was not inhibited by either its N-terminal 11-mer or the C-terminal 6-mer or peptide products [24]. Only 5–10% inhibition of these larger substrate cleavage reactions at 10 µM peptide (Figure 6) suggest that the nonlinearity of the time course in Figure 5A may not be due to inhibition by the formed N-terminal product. However, we cannot rule out the possibility that the off rate of the formed product might be slower than the on rate of our extraneously added 57-mer product. Recently, we provided evidence for a LcA C-terminus-mediated N-terminal product removal step in the LcA catalysis of the 17-mer substrate reaction [24]. Our unpublished results indicate this might be true with the full length and 66-mer SNAP25 substrate too. In that respect, the decreased kobs values with increasing substrate concentration observed in Figure 5B may be partly due to inhibition of the reaction by the 57-mer product.
Figure 6. Inhibition of LcA activity by the 57-mer N-terminal product of the enzymatic reaction of LcA on SNAP25 and a 66-mer peptide substrate.
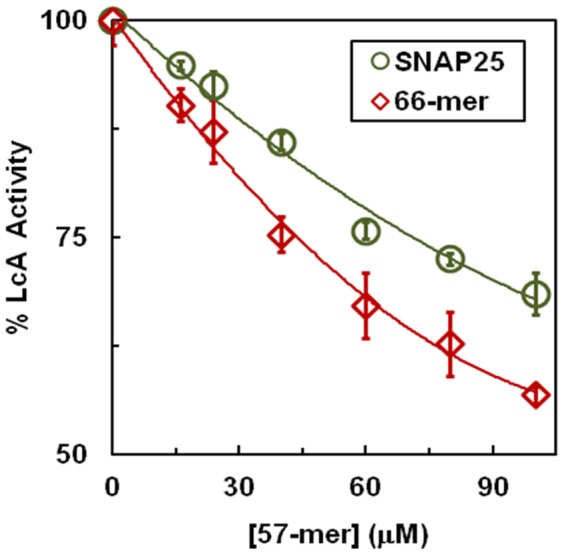
Reaction mixtures (30 µl) containing 10 µM substrate, 1.01 nM LcA, 0.2 mg/ml BSA, 5 mM DTT, 0.25 µM ZnCl2 and the indicated concentrations of the 57-mer product peptide in 50 mM Na-HEPES, pH 7.4 were incubated at 37°C for 5 min.
Discussion
To identify inhibitors of BoNT endoproteolytic activity, several quantitative methods and reagents are used by various investigators [12], [25]–[31]. As a result, there is significant variation in the data obtained which creates confusion in the interpretation of the results [3]–[5]. In many cases, the substrates and reaction components have not been characterized using uniform assay conditions and analytical methods. With the aim to fill this gap, the present investigation compared the behavior of three BoNT/A substrates and several additives that are commonly used in activity measurement assays. We have shown that sodium chloride, a common additive in many enzyme reactions, differently affects the rates of reactions using alternate substrates. We have also shown that incubation time and substrate concentrations affect the calculated values of the reaction rate (kobs). Based on these experimental demonstrations, we have formulated an assay protocol for each of the 3 substrates (Table 4). Because these substrates approximate the natural SNAP25 substrates more than the modified, FRET substrates, the assay formulations described below should provide a standard reference when comparing activity results using various substrates and methods.
Table 4. Standardized recipe for enzymatic activity assays of LcA using three different substrates.
| Reaction Component | Substrate | ||
| 17-mer | 66-mer | Full length | |
| LcA | 25 nM | 0.3 nM | 0.3 nM |
| BSA | 0.2 mg/ml | 0.2 mg/ml | 0.2 mg/ml |
| DTT | - | 5 mM | 5 mM |
| ZnCl2 | - | 250 µM | 250 µM |
| NaCl | - | 100 mM | 100 mM |
| Na-HEPES | 50 mM | 50 mM | 50 mM |
| pH | 7.4 | 7.4 | 7.4 |
| Substrate | 0.5 mM | 10 µM | 10 µM |
| Incubation temp | 37°C | 37°C | 37°C |
| Incubation time | 5 min | 5–15 min | 5–15 min |
Because both the substrate and enzyme in BoNT assays are proteins, their purification buffers often contain EDTA, NaCl, and phosphate, all of which inhibits BoNT activities [34], [36], [47]. In devising a standard assay protocol, one needs to avoid the presence of these or any known inhibitory buffer or additive in the reaction mixture. If an initial experiment shows substoichiometric zinc bound to the BoNT or its Lc preparation, low concentrations (not to exceed 250 µM) of zinc chloride should be added to the reaction mixture. Including 5 mM DTT helps to overcome the inhibitory effect of zinc [34]. Inclusion of zinc and DTT is more important when the catalyst is whole BoNT toxin or when the substrate is SNAP25 or its 66-mer, all of which contain one or multiple cysteine residues. BSA should be added to protect LcA at low concentration from denaturation before or during the reaction time. NaCl should not be used with the 17-mer or similar substrates, but should be included with full-length or the 66-mer SNAP25 substrate.
Usually the multi-well plate incubation chambers are set at temperatures lower than 37°C. Simultaneous reactions of large number of samples in plates with 24-, 96- or higher number wells will also require longer times of preparation. Therefore in adapting these reactions in multi-well plates, temperature and incubation times may need to be modified in order to provide a robust assay. Lower incubation temperatures will require higher LcA concentrations, but longer incubation times will require lower LcA concentration. For example, we observed that a 30 min incubation at 22°C was required to convert 40% of the substrate into products using 25 nM LcA with 0.5 mM 17-mer substrate or 0.34 nM LcA with 11 µM full length SNAP25 substrate. Similarly, it took 60 min under the same conditions for 60–65% substrate conversions. For comparison, almost all of the substrates were converted into products in 30 min at 37°C.
Although highly desirable, in routine assays it is not possible to use saturating concentration of full length soluble NSF attachment protein receptor (SNARE) or their peptide, or FRET substrates because of (a) solubility, (b) availability, (c) detection limitation of the instrument, and (d) cost. Therefore a compromised highest concentration of substrate near its Km value (Table 4) should be chosen.
Formulation of the three recipe in Table 4 is based on the consideration that no more than 25% of the substrate will be converted into products. The experiments described in this paper added 5 µl LcA to 25 µl substrate-additive master mix (as described in Table 4) to initiate the reaction. Identical results can also be obtained by adding 5 µl substrate to 25 µl LcA-additive master mix to initiate the reaction. Screening of inhibitors can be accomplished by adding such compounds to the master mix prior to addition of LcA. However, inhibitor libraries may contain compounds that are slow binders of an enzyme. In such a situation, a preincubation of the inhibitor candidates with LcA in the master mix followed by addition of substrate may be desirable and more convenient than adding LcA at the end. In either case, a short vortex for 1 sec immediately after the final addition must be done to ensure complete mixing of this small 30 µl volume.
In our standard practice, 25 µl master mix in a 1.8 ml screw capped eppendorf tube is preincubated for 5 min at 37°C. 5 µl LcA or substrate at ambient temperature is added to the eppendorf tube and immediately vortexed followed by incubation of the capped tube at 37°C. The reaction is stopped by acidifying the mixture with 90 µl of 1% TFA. A brief spin at 12,000 g for 2 min helps to precipitate any formed particulate material ensuring better chromatographic column performance. Fifty to 110 µl of the supernatant is transferred into UPLC™ or HPLC vials for product analyses.
Materials and Methods
Materials
Recombinant BoNT light chain of serotype A (LcA) was purified as described [38], [48], [49]. Human SNAP25 sequence-derived substrates and products were as follows: 66-mer substrate peptide (141-ARENEMDENLEQVSGIIGNLRHMALDMGNEIDTQNRQIDRIMEKADSNKTRIDEANQRATKMLGSG-206), 57-mer product peptide (141-ARENEMDENLEQVSGIIGNLRHMALDMGNEIDTQNRQIDRIMEKADSNKTRIDEANQ-197), 17-mer substrate peptide (SNKTRIDEANQ-RATKML), 11-mer product peptide (SNKTRIDEANQ), 9-mer product peptide: (198-RATKMLGSG-206), and the 6-mer product peptide (RATKML). All substrate peptides having N-terminal acetylated and C-terminal amidated, were custom-synthesized and purified to >95% by Peptide2.0, (Chantilly, VA 20153). The products of LcA reaction on the 17-mer, N-acetylated SNKTRIDEANQ (not C-amidated) and C-amidated RATKML (not N-acetylated) were also from the same vendor.
Full length recombinant human SNAP25 (1 mg/ml or ∼40 µM), purchased from GeneWay Biotech Inc. (Santa Clara, CA) was extensively dialyzed against 50 mM HEPES pH 7.4, and saved as small aliquots at −20°C until use. If a higher concentration of the protein was required, it was concentrated on a Centricon-10 (Amicon) ultrafiltration unit.
Enzymatic activity assays
Concentration of LcA was determined from its A1% ( = 10) at 280 nm in an Agilent 8453 diode array spectrophotometer [34]. Activity assays were based on ultra performance liquid chromatography (UPLC™) or high performance liquid chromatography (HPLC) separation and measurement of the cleaved products from the SNAP25 substrate. A master reaction mixture lacking the LcA was prepared and its aliquots were stored at −20°C. Stocks of 0.05–0.07 mg/ml LcA in 50 mM Na-HEPES, pH 7.4 containing 0.05% Tween-20 were also stored at −20°C. Before assay, a Lc stock was thawed and diluted further in 50 mM NA-HEPES, pH 7.4, containing bovine serum albumin (BSA). At the time of assay, 5 µl of diluted LcA was added to 25 µl of the thawed master mix to initiate the enzymatic reaction. Components and final concentration in this 30-µl reaction mixture were 0.005–1.5 mM substrate, 0.2 mg/ml BSA, 0.4–50 nM (or 0.04–4 nM as indicated) LcA, 0.25 mM ZnCl2, 5 mM dithiothreitol (DTT), and 50 mM N-2-ydroxyethylpiperazine-N-2-ethanesulfonic acid whose pH was adjusted with NaOH (Na-HEPES) pH 7.4. Because DTT and ZnCl2 had no effect on the kinetic constants [43] or specific activity [12] of LcA employed in this study using the 17-mer substrate, these reagents were omitted from the 17-mer substrate reactions. After 5 or 10 min (depending on the particular experiment) incubation (37°C), reactions were stopped by adding 90 µl of 1% trifluoroacetic acid (TFA). Unless and otherwise mentioned, LcA concentration and the time of incubation were adjusted so that no more than 20% of the substrate was converted into products.
The amounts of uncleaved 17-mer substrate and the products were measured after separation using a Waters Acquity UPLC™ system employing a reverse-phase C18 column (2.1×50 mm, 1.7-µm particle size) with 0.1% TFA in H2O as solvent A and 70% acetonitrile-0.1% TFA as solvent B. The peptides were eluted at a flow rate of 0.5 ml/min with a linear gradient of 10%B to 42%B over one min , 0.5 min after injection of 5 µl sample [27]. Column regeneration was for 0.7 min [27]. The 17-mer substrate, its N-terminal product, and its C-terminal product were eluted at 1.75, 1.3, and 0.9 min, respectively, and were completely separated [27]. When analyzed in the Waters HPLC column (see later), the reaction mixtures yielded identical results. However because of rapid analysis of minute sample volumes using little solvent, UPLC™ [27] was used in analyzing the 17-mer substrate reactions. The UPLC™ system was not suitable to resolve the products from the full length and the 66-mer substrates. Therefore, these reaction mixtures were analyzed on a Waters C18 (Synergy) 4.6×75 mm (3–5 µm particle size) HPLC column with a linear gradient of 10%B to 90%B over 12.5 min, one minute after injection of 100 µl sample, followed by a jump to 100%B that was held for 3 min. The smaller C-terminal products from these substrates eluted at 5.6 min while the larger N-terminal product and the substrates eluting between 11–12 min were not completely separated. Therefore, we used the area under the C-terminal products for accurate quantification of the reaction.
The UPLC™ was equipped with Waters Acquity™ Sample Manager (autosampler), a Waters Acquity™ photodiode array detector and Empower Pro software, while the Waters HPLC was equipped with Waters 717plus autosampler, a Waters 996 photodiode array detector, and Empower Pro software. Quantification of peptides was based on the area under their 210 nm absorbance peaks. The limit of detection in 5 µl UPLC™-injected sample for the 17-mer substrate and its products was 5 µM [27]. The limit of detection in 100 µl HPLC-injected sample of the C-terminal product from SNAP25 and its 66-mer substrates was 0.1 µM.
Insolubility of the substrates limited our ability to use saturating substrate concentration in the kinetic experiments. Therefore we used several concentrations of each substrate around their previously reported Km values. Kinetic parameter calculations used a “one substrate, two product” model (E + S ↔ ES → E + P1 +P2) ignoring the on and off rates of the substrates and products since they were not determined. Km and kcat were determined by hyperbolic curve-fitting of the experimental data at various substrate concentrations using the Michaelis-Menten equation, kobs = (kcat×[S])/(Km+[S]), in a KaleidaGraph (Synergy Software, Reading, PA) software package.
Acknowledgments
We thank Drs. Martha Hale and Chris Kane for careful reading and criticisms. We thank Dr. Sarah L. Norris for the statistical Analyses.
Funding Statement
This project received support from the Defense Threat Reduction Agency-Joint Science and Technology Office for Chemical and Biological Defense (Grant #JSTOCBD3.10012_06_RD_B and CB3849). Opinions, interpretations, conclusions, and recommendations are those of the author and are not necessarily endorsed by the U.S. Army. The authors also acknowledge National Research Council (NRC) for their support to RMM through their research associateship program. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Borio L, Frank D, Mani V, Chiriboga C, Pollanen M, et al. (2001) Death due to bioterrorism-related inhalational anthrax: report of 2 patients. JAMA 286: 2554–2559. [DOI] [PubMed] [Google Scholar]
- 2. Montecucco C, Schiavo G (1995) Structure and function of tetanus and botulinum neurotoxins. Q Rev Biophys 28: 423–472. [DOI] [PubMed] [Google Scholar]
- 3. Boldt GE, Kennedy JP, Janda KD (2006) Identification of a potent botulinum neurotoxin a protease inhibitor using in situ lead identification chemistry. Org Lett 8: 1729–1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zuniga JE, Schmidt JJ, Fenn T, Burnett JC, Arac D, et al. (2008) A Potent Peptidomimetic Inhibitor of Botulinum Neurotoxin Serotype A Has a Very Different Conformation than SNAP-25 Substrate. Structure 16: 1588–1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Molles BE, Collins EK, Levit MB, Sweeny RE, Zottola MA, et al.. (2010) Towards a sub-nanomolar affinity BoNT/A light chain protease inhibitor: clues from structure-activity studies of peptide inhibitors. In: Maslanka SE, editor. 47th Inter-institute Botulinum Research Coordinating Committee Meeting. Atlanta, GA: CDC. [Google Scholar]
- 6.Copeland RA (2005) Evaluation of enzyme inhibitors in drug discovery: A guide to chemists and pharmacologists. Hoboken, New Jersey, USA: Wiley-Interscience. 271 p. [PubMed] [Google Scholar]
- 7. Simpson LL (2004) Identification of the major steps in botulinum toxin action. Annu Rev Pharmacol Toxicol 44: 167–193. [DOI] [PubMed] [Google Scholar]
- 8. Schmidt JJ, Stafford RG (2005) Botulinum neurotoxin serotype F: identification of substrate recognition requirements and development of inhibitors with low nanomolar affinity. Biochemistry 44: 4067–4073. [DOI] [PubMed] [Google Scholar]
- 9. Schmidt JJ, Stafford RG (2002) A high-affinity competitive inhibitor of type A botulinum neurotoxin protease activity. FEBS Lett 532: 423–426. [DOI] [PubMed] [Google Scholar]
- 10. Schmidt JJ, Stafford RG, Bostian KA (1998) Type A botulinum neurotoxin proteolytic activity: development of competitive inhibitors and implications for substrate specificity at the S1′ binding subsite. FEBS Lett 435: 61–64. [DOI] [PubMed] [Google Scholar]
- 11. Kumar G, Kumaran D, Ahmed SA, Swaminathan S (2012) Peptide inhibitors of botulinum neurotoxin serotype A: design, inhibition, cocrystal structures, structure-activity relationship and pharmacophore modeling. Acta Crystallogr D Biol Crystallogr 68: 511–520. [DOI] [PubMed] [Google Scholar]
- 12. Hale M, Oyler G, Swaminathan S, Ahmed SA (2011) Basic tetrapeptides as potent intracellular inhibitors of type A botulinum neurotoxin protease activity. J Biol Chem 286: 1802–1811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ludivico M, Smith LA, Ahmed SA (2009) Structure-Based Design of Peptide Inhibitors of Botulinum Neurotoxin Serotypes A Proteolytic Activity. The Botulinum Journal 1: 297–308. [Google Scholar]
- 14. Kumaran D, Rawat R, Ludivico ML, Ahmed SA, Swaminathan S (2008) Structure- and substrate-based inhibitor design for Clostridium botulinum neurotoxin serotype A. J Biol Chem 283: 18883–18891. [DOI] [PubMed] [Google Scholar]
- 15. Kumaran D, Rawat R, Ahmed SA, Swaminathan S (2008) Substrate binding mode and its implication on drug design for botulinum neurotoxin A. PLoS Pathog 4: e1000165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Roxas-Duncan V, Enyedy I, Montgomery VA, Eccard VS, Carrington MA, et al. (2009) Identification and biochemical characterization of small-molecule inhibitors of Clostridium botulinum neurotoxin serotype A. Antimicrob Agents Chemother 53: 3478–3486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hermone AR, Burnett JC, Nuss JE, Tressler LE, Nguyen TL, et al. (2008) Three-dimensional database mining identifies a unique chemotype that unites structurally diverse botulinum neurotoxin serotype A inhibitors in a three-zone pharmacophore. ChemMedChem 3: 1905–1912. [DOI] [PubMed] [Google Scholar]
- 18. Eubanks LM, Hixon MS, Jin W, Hong S, Clancy CM, et al. (2007) An in vitro and in vivo disconnect uncovered through high-throughput identification of botulinum neurotoxin A antagonists. Proc Natl Acad Sci U S A 104: 2602–2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dickerson TJ, Janda KD (2007) The use of small molecules to investigate molecular mechanisms and therapeutic targets for treatment of botulinum neurotoxin A intoxication. ACS Chem Biol 2: 359–369. [DOI] [PubMed] [Google Scholar]
- 20. Capkova K, Yoneda Y, Dickerson TJ, Janda KD (2007) Synthesis and structure-activity relationships of second-generation hydroxamate botulinum neurotoxin A protease inhibitors. Bioorg Med Chem Lett 17: 6463–6466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ma H, Zhou B, Kim Y, Janda KD (2006) A cyclic peptide-polymer probe for the detection of Clostridium botulinum neurotoxin serotype A. Toxicon 47: 901–908. [DOI] [PubMed] [Google Scholar]
- 22. Mizanur RM, Gorbet J, Swaminathan S, Ahmed SA (2012) Inhibition of catalytic activities of botulinum neurotoxin light chains of serotypes A, B and E by acetate, sulfate and calcium. Int J Biochem Mol Biol 3: 313–321. [PMC free article] [PubMed] [Google Scholar]
- 23.Ahmed SA (2013) Recent advances in the botulinum neurotoxin catalytic activity assays. In: Pandalai SG, editor. Recent Research Developments in Analytical Biochemistry. 1st ed. Trivandrum, Kerala, India: Transworld Research Network. pp. 43. [Google Scholar]
- 24. Mizanur RM, Frasca V, Swaminathan S, Bavari S, Webb R, et al. (2013) The C terminus of the catalytic domain of type a botulinum neurotoxin may facilitate product release from the active site. J Biol Chem 288: 24223–24233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gul N, Smith LA, Ahmed SA (2010) Light chain separated from the rest of the type a botulinum neurotoxin molecule is the most catalytically active form. PLoS ONE 5: e12872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Schmidt JJ, Stafford RG (2003) Fluorigenic substrates for the protease activities of botulinum neurotoxins, serotypes A, B, and F. . Appl Environ Microbiol 69: 297–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rowe B, Schmidt JJ, Smith LA, Ahmed SA (2010) Rapid product analysis and increased sensitivity for quantitative determinations of botulinum neurotoxin proteolytic activity. Anal Biochem 396: 188–193. [DOI] [PubMed] [Google Scholar]
- 28. Ahmed SA, Olson MA, Ludivico ML, Gilsdorf J, Smith LA (2008) Identification of Residues Surrounding the Active Site of Type A Botulinum Neurotoxin Important for Substrate Recognition and Catalytic Activity. Protein J 27: 151–162. [DOI] [PubMed] [Google Scholar]
- 29. Henkel JS, Jacobson M, Tepp W, Pier C, Johnson EA, et al. (2009) Catalytic properties of botulinum neurotoxin subtypes A3 and A4. Biochemistry 48: 2522–2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chen S, Barbieri JT (2006) Unique substrate recognition by botulinum neurotoxins serotypes A and E. J Biol Chem 281: 10906–10911. [DOI] [PubMed] [Google Scholar]
- 31. Breidenbach MA, Brunger AT (2004) Substrate recognition strategy for botulinum neurotoxin serotype A. Nature 432: 925–929. [DOI] [PubMed] [Google Scholar]
- 32. Cai S, Lindo P, Park JB, Vasa K, Singh BR (2010) The identification and biochemical characterization of drug-like compounds that inhibit botulinum neurotoxin serotype A endopeptidase activity. Toxicon 55: 818–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.(2012) LIST Biological Laboratories Inc. Available: http://www.listlabs.com/.
- 34. Ahmed SA, Smith LA (2000) Light chain of botulinum A neurotoxin expressed as an inclusion body from a synthetic gene is catalytically and functionally active. J Protein Chem 19: 475–487. [DOI] [PubMed] [Google Scholar]
- 35. Schmidt JJ, Bostian KA (1997) Endoproteinase activity of type A botulinum neurotoxin: substrate requirements and activation by serum albumin. J Protein Chem 16: 19–26. [DOI] [PubMed] [Google Scholar]
- 36. Shone CC, Roberts AK (1994) Peptide substrate specificity and properties of the zinc-endopeptidase activity of botulinum type B neurotoxin. Eur J Biochem 225: 263–270. [DOI] [PubMed] [Google Scholar]
- 37. Auld DS (1995) Removal and replacement of metal ions in metallopeptidases. Methods Enzymol 248: 228–242. [DOI] [PubMed] [Google Scholar]
- 38. Jensen MJ, Smith TJ, Ahmed SA, Smith LA (2003) Expression, purification, and efficacy of the type A botulinum neurotoxin catalytic domain fused to two translocation domain variants. Toxicon 41: 691–701. [DOI] [PubMed] [Google Scholar]
- 39. Ekong TA, Feavers IM, Sesardic D (1997) Recombinant SNAP-25 is an effective substrate for Clostridium botulinum type A toxin endopeptidase activity in vitro. Microbiology 143 Pt 10: 3337–3347. [DOI] [PubMed] [Google Scholar]
- 40. Sadoul K, Berger A, Niemann H, Regazzi R, Catsicas S, et al. (1997) SNAP-25 can self-associate to form a disulfide-linked complex. Biol Chem 378: 1171–1176. [PubMed] [Google Scholar]
- 41. Glass PJ, Zeng CQ, Estes MK (2003) Two nonoverlapping domains on the Norwalk virus open reading frame 3 (ORF3) protein are involved in the formation of the phosphorylated 35K protein and in ORF3-capsid protein interactions. J Virol 77: 3569–3577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Creighton TE (1984) Proteins Structures and Molecular Properties. New York: W.H. Freeman and Company. 515 p. [Google Scholar]
- 43. Ahmed SA, Olson MA, Ludivico ML, Gilsdorf J, Smith LA (2008) Identification of residues surrounding the active site of type A botulinum neurotoxin important for substrate recognition and catalytic activity. Protein J 27: 151–162. [DOI] [PubMed] [Google Scholar]
- 44. Schmidt JJ, Bostian KA (1995) Proteolysis of synthetic peptides by type A botulinum neurotoxin. J Protein Chem 14: 703–708. [DOI] [PubMed] [Google Scholar]
- 45. Toth S, Brueggmann EE, Oyler GA, Smith LA, Hines HB, et al. (2012) Tyrosine phosphorylation of botulinum neurotoxin protease domains. Front Pharmacol 3: 102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Toth SI, Smith LA, Ahmed SA (2009) Extreme sensitivity of botulinum neurotoxin domains towards mild agitation. J Pharm Sci 98: 3302–3311. [DOI] [PubMed] [Google Scholar]
- 47. Foran P, Lawrence G, Dolly JO (1995) Blockade by botulinum neurotoxin B of catecholamine release from adrenochromaffin cells correlates with its cleavage of synaptobrevin and a homologue present on the granules. Biochemistry 34: 5494–5503. [DOI] [PubMed] [Google Scholar]
- 48. Ahmed SA, McPhie P, Smith LA (2003) Autocatalytically fragmented light chain of botulinum a neurotoxin is enzymatically active. Biochemistry 42: 12539–12549. [DOI] [PubMed] [Google Scholar]
- 49. Gilsdorf J, Gul N, Smith LA (2006) Expression, purification, and characterization of Clostridium botulinum type B light chain. Protein Expr Purif 46: 256–267. [DOI] [PubMed] [Google Scholar]
- 50.Hatheway CL (1988) Botulism. In: (eds), vol 1., New York,. In:Balows A, W.H.J. H, M. O, A. T, editors. Laboratory Diagnosis of Infectious Diseases: Principles and Practice. New York: Springer. pp. 112–133. [Google Scholar]
- 51. Li L, Singh BR (2000) Role of zinc binding in type A botulinum neurotoxin light chain's toxic structure. Biochemistry 39: 10581–10586. [DOI] [PubMed] [Google Scholar]
- 52. Baldwin MR, Bradshaw M, Johnson EA, Barbieri JT (2004) The C-terminus of botulinum neurotoxin type A light chain contributes to solubility, catalysis, and stability. Protein Expr Purif 37: 187–195. [DOI] [PubMed] [Google Scholar]
- 53. Binz T, Bade S, Rummel A, Kollewe A, Alves J (2002) Arg(362) and Tyr(365) of the botulinum neurotoxin type a light chain are involved in transition state stabilization. Biochemistry 41: 1717–1723. [DOI] [PubMed] [Google Scholar]
- 54. Cai S, Singh BR (2001) A correlation between differential structural features and the degree of endopeptidase activity of type A botulinum neurotoxin in aqueous solution. Biochemistry 40: 4693–4702. [DOI] [PubMed] [Google Scholar]
- 55. Sharma SK, Singh BR (2004) Enhancement of the endopeptidase activity of purified botulinum neurotoxins A and E by an isolated component of the native neurotoxin associated proteins. Biochemistry 43: 4791–4798. [DOI] [PubMed] [Google Scholar]
- 56. Adler M, Capacio B, Deshpande SS (2000) Antagonism of botulinum toxin A-mediated muscle paralysis by 3, 4-diaminopyridine delivered via osmotic minipumps. Toxicon 38: 1381–1388. [DOI] [PubMed] [Google Scholar]
- 57. Bagramyan K, Barash JR, Arnon SS, Kalkum M (2008) Attomolar detection of botulinum toxin type A in complex biological matrices. PLoS One 3: e2041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Rasooly R, Do PM (2008) Development of an in vitro activity assay as an alternative to the mouse bioassay for Clostridium botulinum neurotoxin type A. Appl Environ Microbiol 74: 4309–4313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ruge DR, Dunning FM, Piazza TM, Molles BE, Adler M, et al. (2011) Detection of six serotypes of botulinum neurotoxin using fluorogenic reporters. Anal Biochem 411: 200–209. [DOI] [PubMed] [Google Scholar]