

Abstract
Microcephaly-capillary malformation (MIC-CAP) syndrome exhibits severe microcephaly with progressive cortical atrophy, intractable epilepsy, profound developmental delay and multiple small capillary malformations on the skin. We employed whole-exome sequencing of five patients with MIC-CAP syndrome and identified novel recessive mutations in STAMBP, a gene encoding the deubiquitinating (DUB) isopeptidase STAMBP (STAM-binding protein)/AMSH (Associated Molecule with the SH3 domain of STAM), that plays a key role in cell surface receptor-mediated endocytosis and sorting. Patient cell lines showed reduced STAMBP expression associated with accumulation of ubiquitin-conjugated protein aggregates, elevated apoptosis and insensitive activation of the RAS-MAPK and PI3K-AKT-mTOR pathways. The latter cellular phenotype is significant considering the established connection between these pathways and their association with vascular and capillary malformations. Furthermore, our findings of a congenital human disorder caused by a defective DUB protein that functions in endocytosis, implicates ubiquitin-conjugate aggregation and elevated apoptosis as factors potentially influencing the progressive neuronal loss underlying MIC-CAP.
MIC-CAP was recently described in six children, including one brother-sister pair, who all presented with small scattered capillary malformations (CM), severe congenital microcephaly, early-onset intractable epilepsy, profound global developmental delay, spastic quadriparesis, hypoplastic distal phalanges, and poor growth1–4. CMs, sometimes referred to as “port-wine stains”, are non-regressing cutaneous vascular abnormalities5 that are seen in a growing number of congenital syndromes linked to dysregulated RAS-MAPK (RAS-mitogen activated protein kinase) function; these are collectively termed “RASopathies”. For example, mutations in RASA1, encoding p120-RasGAP, a negative regulator of the RAS pathway, were found in patients with capillary malformation-arteriovenous malformation syndrome (CM-AVM)6, while mutations in KRIT1, encoding a RAS-related protein 1A interactant, cause hyperkeratotic cutaneous capillary-venous malformations associated with cerebral capillary malformations7. Sequencing of RASA1 in two MIC-CAP patients was negative and sequencing of KRIT1 was not pursued7. Until now, the genetic mechanism responsible for this devastating disorder has been unknown.
We studied ten affected individuals from nine families with MIC-CAP syndrome (Fig. 1 and Table 1). Brain magnetic resonance imaging scans (MRI) of affected individuals showed enlarged extra-axial spaces and other changes suggesting prenatal onset cerebral atrophy with relative sparing of the cerebellum (Fig. 1a, b, and c). The gyral pattern was universally simplified, and associated with variable degrees of diffuse hypomyelination and hippocampal hypoplasia. All individuals with MIC-CAP were found to have intractable epilepsy, severe developmental delay and profound intellectual disability. Other distinguishing features of MIC-CAP include infantile spasms, hypoplasia of the distal phalanges characterized by variable degrees of nail and toe hypoplasia and CMs (Fig. 1d, e, f). The CMs are striking in appearance and visible at birth in all patients. They are generalized in distribution and tend to vary from small 2–3 mm to large 15–20 mm lesions. Interestingly, limited evidence suggests that the vascular anomalies are not restricted to skin CMs; one reported patient (P3.1 in this study) had a cerebellar angioma1 and another (Patient 9.1) had possible vascular malformations of the liver by ultrasound.
Figure 1.

Neuroimaging and clinical features of MIC-CAP in Patient 9.1. T1-weighted sagittal (a) and axial (b) and T2-weighted coronal (c) images of the brain of Patient 9.1 at 3 months of age. Note the low-sloping forehead, simplified gyral pattern, increased extra-axial space, diffuse hypomyelination, and hippocampal hypoplasia. Photos of Patient 9.1 at 3 weeks (d) and 18 months (e) showing generalized capillary malformations of variable sizes and hypoplastic toenails (f).
Table 1.
Clinical Characteristics and Molecular Findings of MIC-CAP Patientsa
| Microcephaly Capillary Malformation Syndrome | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Patient | P1.1 | P1.2 | P2.1 | P3.1 | P4.1 | P5.1 | P6.1 | P7.1 | P8.1 | P9.1 |
| Exome Sequencing |
+ | + | + | + | − | + | − | − | − | − |
| Validated Mutationsb (protein (cDNA)) |
p.Glu42Gly (c.125A>G) |
p.Glu42Gly (c.125A>G) |
p.Arg38Cys (c.112C>T) |
p.Phe100Tyr (c.299T>A) |
p.Arg424* (c.1270C>T) |
c.1005+358A>G | p.Lys378Asnfs*2 (c.1134_1138del ACTAA) |
p.Arg38Cys (c.112C>T) |
p.Arg38Cys (c.112C>T) |
p.Tyr63Cys (c.188A>G) |
| p.Arg178* (c.532C>T) |
p.Arg178* (c.532C>T) |
c.279+5G>T | p.Arg424* (c.1270C>T) |
p.Arg424* (c.1270C>T) |
c.1005+358A>G | p.Thr313Ile (c.938C>T) |
c.203+5G>A | p.Ile138Serfs*12 (c.411del C) |
p.Arg14Pro (c.41G>C) |
|
| Gender | F | M | M | M | M | F | M | F | F | M |
| Age at assessment |
2y | 9m | 12m | 2y | 22d | 5y4m | 2m | 28m | 8m | 15m |
| Ethnicity | African-American | African-American | European descent |
European descent |
European descent |
European descent |
European descent |
European descent |
Polynesian | European descent |
| Gestational Age (weeks) | 39 | 39 | 36+5 | 36 | 37 | Term | 36 | 37+2 | 37+6 | 35 |
| Clinical Features | ||||||||||
| Progressive congenital MIC |
+ | + | + | + | + | + | + | + | + | + |
| Small-for-gestational age |
+ | + | + | + | + | − | − | + | − | + |
| Birth OFC (SD) | −6 | −4 | −2 | −2 | −4 | −1.8 | −2 | −8 | −5 | −2 |
| Birth Weight (SD) | −1.5 | −1.5 | −1.5 | −2 | −2 | +1.8 | −1.5 | −4 | −1.5 | −1.5 |
| Birth Length (SD) | − 4 | − 2 | 0.5 | nd | − 1.5 | 0 (mean) | − 2 | − 4 | − 1 to − 2 | − 2 |
| Later age | 2.5y | 9m | 12m | 17m | 22d | 2y10m | 2m | 28m | 8m | 18 |
| Later OFC - SD (age) | − 8 | − 6 | − 6 | − 4 | − 3 | − 2.5 | − 6 | −8 | − 4 | − 4 |
| Later Weight (SD) | +1.5 to +2 | − 4 | +1 to +2 | − 1 to − 2 | −3 | +2 | − 2 | − 2.5 | +1 | − 3 |
| Later Length (SD) | nd | nd | −1 to −2 | − 2 | − 3 | 0 (mean) | − 3 | − 4 | nd | −3 to − 4 |
| Generalized Capillary Malformations |
+ | + | + | + | + | + | + | + | + | + |
| Early-onset intractable seizures |
+ | + | + | + | + | + | + | + | + | + |
| Infantile spasms |
+ | − | + | − | − | − | − | nd | + | + |
| Hypoplastic distal phalanges |
+ | + | − | + | + | + | + | + | + | + |
| Global DD | + | + | + | + | + | + | + | + | + | + |
| Spastic quadriparesis |
+ | + | + | + | + | − | − | + | + | + |
| Myoclonus/clonus | + | − | + | + | − | − | + | + | Intermittent dyskinetic/choreiform movements |
+ |
| Optic atrophy | + | + | + | + | + | nd | nd | + | − | − |
| Neuroimaging Features | ||||||||||
| Simplified gyral pattern |
+ | + | + | + | + | nd | + | + | + | + |
| Increased extra-axial space |
+ | + | + | + | + | nd | + | + | + | + |
| Hippocampal hypoplasia |
+ | + | + | nd | + | nd | − | + | + | nd |
| Hypomyelination | − | − | + | + | + | nd | nd | + | − | + |
| Published | +3 | +3 | +3 | +1 | +1 | +2 | − | − | − | − |
This table summarizes the clinical findings in study participants.
Numbering of mutations is relative to NM_006463 (gene) and NP_006454.1 (protein).
Abbreviations: F: female; M: male; y: years; m: months; OFC: Occipito-frontal circumference; SD: Standard deviations from the mean; nd: not determined.
To establish the genetic cause of MIC-CAP, we performed exome sequencing on DNA samples from five individuals (Table 1) diagnosed with the syndrome. The two affected children in Family 1, from non-consanguineous parents, suggested a recessive mode of inheritance for this disorder. Therefore, we focused on identifying genes in which a maximal number of patients had two rare protein-altering variants that were absent from dbSNP131, the 1000 Genomes project, and 159 in-house control exomes. In four (P1.1, P1.2, P2.1, P3.1) of the five patients studied by exome sequencing, including the two sibs, two variants in each individual were identified in STAMBP encoding STAM-binding protein (STAMBP/AMSH, hereafter referred to as STAMBP) (Fig. 2a; Supplementary Fig. 1a, 2 and Supplementary Table 1). Analysis of an additional three affected individuals (P6.1, P8.1, P9.1) by Sanger sequencing identified two coding STAMBP variants in each patient (Supplementary Table 2). Co-segregation analysis confirmed an autosomal recessive mode of inheritance in all families (Supplementary Fig. 3). Western blot analysis of whole cell extracts from patient-derived LCLs failed to detect STAMBP expression in Patient 1.2 (p.[Glu42Gly];[Arg178*]) (Fig. 2b). Patient 3.1 (p.[Phe100Tyr];[Arg424*]) showed a reduction of STAMBP expression compared to controls (Fig. 2b).
Figure 2.

Mutations in STAMBP cause MIC-CAP. (a) STAMBP (upper, chromosome 2, hg19: 74,056,114–74,094,295, RefSeq: NM_006463) and protein (lower, NP_006454.1) indicating MIC-CAP mutations. STAMBP contains a microtubule-interacting and transport (MIT) domain9,10, SH3 binding motif (SBM) (PX[V/I][D/N]RXXP)26, JAMM (JAB1/MPN/MOV34) motif12, nuclear localization signal (NLS)11 and the distal ubiquitin recognition site (DUR)27. For c.279+5G>T in P2.1 (tissue from patient not available), a computational splicing model predicted the inclusion of an extra codon in exon 4 (p=1.9e-9, sign test). We validated this model using the known mutation in P7.1 (p=1.9e-9, sign test) (Supplementary Fig. 1a). Five out of six missense mutations are located in the MIT domain; required for the interaction of STAMBP with CHMP3, an ESCRT-III subunit28. The sixth, Thr313Ile, located in the distal ubiquitin binding site within the JAMM domain, eliminates a hydrogen bond between the ubiquitin carbon backbone and STAMBP, likely decreasing ubiquitin binding to STAMBP (Supplementary Fig. 2). Two mutations were recurrent in multiple unrelated MIC-CAP families; Arg424* detected in Patients 3.1 and 4.1 and Arg38Cys detected in individuals P2.1, P7.1 and P8.1, suggestive of mutational hotspots in STAMBP. Within the ~5000 exomes in the NHLBI Exome variant server, only variant Arg38Cys was represented in 2 of 10756 alleles, suggesting a carrier frequency of approximately 1:5000 in a population of American/European ancestry, consistent with the prevalence of this very rare disorder. Western blot analysis of whole cell extracts (WCE) from LCLs P3.1, P5.1, P7.1, and P1.2, showing either equivocal (P3.1), reduced (P5.1) or absent STAMBP expression (P7.1, P1.2).
We identified one coding mutation in STAMBP in Patient 7.1. Analysis by Western blotting failed to detect STAMBP expression (Fig. 2b) and further sequencing of the gene revealed an intronic mutation (c.203+5G>A) believed to lead to an increase in skipping of the first coding exon (Table 1 and Supplementary Fig. 1b, 4a–d).
In Patient 5.1, no coding mutations were identified using exome sequencing. The depth of coverage across the exons of STAMBP did not suggest a deletion. However, analysis of SNP data from an Illumina Human Omni2.5 array, which contained 25 probes within STAMBP, suggested a 40Mb region of copy neutral homozygosity spanning STAMBP. Western blotting revealed a severe reduction in STAMBP expression (Fig. 2b), suggesting that P5.1 has MIC-CAP secondary to noncoding mutations in STAMBP. Sequencing of patient derived cDNA showed the presence of a 108bp pseudoexon containing a premature stop codon (Supplementary Fig.4e, 4f). Deep intronic sequencing identified a homozygous mutation (c.1005+358A>G). Application of a computational model of splicing regulation8 predicted that this mutation would activate a novel donor site as well as a cryptic AG acceptor site 114 bp upstream (p=8.7e-7, sign test). We believed that this mutation caused the leaky splicing of the full-length transcript and showed that patient cells have a 3-fold reduction of full-length transcript expression (Supplementary Fig. 1c and 4g).
Sanger sequencing in Patient 4.1 identified a homozygous stop mutation, p.Arg424*. Cosegregation analysis was not consistent with the suspected autosomal recessive mode of inheritance in this non-consanguineous family as only the mother was heterozygous for p.Arg424* mutation. We analyzed 10 microsatellite markers spanning chromosome 2 and found all markers to be homozygous; a diagnostic array performed using DNA extracted from whole-blood showed no evidence of copy number variation across chromosome 2. Therefore, we suspect that the mechanism of MIC-CAP in this patient is secondary to maternal isodisomy (Supplementary Fig. 5). In summary, we identified two mutations in STAMBP in a total of 10 patients; six missense variants, two nonsense mutations, two translational frameshift mutations predicted to cause a premature truncation of the STAMBP protein and three intronic mutations leading to alternative splicing of the STAMBP transcript (Fig. 2a).
STAMBP is a JAMM-family DUB containing a microtubule-interacting and transport (MIT) domain and a STAM-binding domain; both interact with the endosomal sorting and trafficking machinery (Fig. 2a and Supplementary Fig. 6a)9,10,11. STAMBP is recruited to the endosomal sorting complexes required for transport (ESCRTs), a group of distinct macromolecule assemblies that mediate the sorting and trafficking of ubiquitinated proteins from endosomes to lysosomes. STAMBP functions in regulating endosomal sorting of ESCRT machinery and ubiquitinated receptor ‘cargo’9,12,13–17. Endosomal sorting is a highly dynamic process fundamental to regulating protein homeostasis through the active regulation of receptor-mediated signal transduction and by enabling processes such as autophagy18,19 Impaired ESCRT-function is associated with the intracellular accumulation of ubiquitinated proteins. Brain lesions containing ubiquitinated protein aggregates have been noted in STAMBP −/− mice20, suggesting this to be a likely mechanism influencing microcephaly and its progression in MIC-CAP. Consistent with this, we observed elevated levels of conjugated-ubiquitin aggregates following siRNA mediated silencing of STAMBP in the human medullablastoma line T98G using indirect immunofluoresence (IF) with an antibody that specifically detects conjugated-ubiquitin (FK2) and not free ubiquitin (Fig. 3a. and Supplementary Fig. 6b). Strikingly, we also observed elevated levels of conjugated-ubiquitin aggregates in several STAMBP-patient LCLs, compared to wild-type (WT) controls following serum starvation (Fig. 3b). This phenotype was reversed following stable lentiviral transduction of patient-LCLs with STAMBP (Supplementary Fig. 6c, d) Furthermore, this was also associated with apoptosis induction, denoted by elevated levels of cleaved caspase-3 (Fig. 3c) and annexin V staining (Fig. 3d) in the STAMBP-patient LCLs, compared to WT. STAMBP functions with the ESCRT machinery to facilitate autophagy (ATG). Autophagic flux can be monitored by detection of the autophagosome-associated phosphatidylethanolamine-conjugated microtubule-associated light chain 3 (LC3-II isoform) expression in the presence of the ATG inhibitor, Bafilomycin A (BafA). Consistent with increased autophagic flux (i.e. increased levels of autophagosomes), elevated LC3-II levels were observed in STAMBP-patient LCLs compared to wild-type (WT) controls (Fig. 3e).
Figure 3.

Elevated ubiquitin protein aggregates, apoptosis and autophagic flux in MIC-CAP. (a) Elevated conjugated-ubiquitin protein aggregates were observed following siRNA mediated silencing of STAMBP. T98G human medullablastoma cells were either untransfected (Unt) or transfected with siRNA against STAMBP. 24hrs post-transfection cells were stained with anti-FK2 and ubiquitin aggregates visualised by indirect immunofluorescence (IF). The extent of STAMBP knockdown is shown in Supplementary Figure 6b. (b) STAMBP-patient LCLs exhibit elevated levels of conjugated-ubiquitin protein aggregates. IF using anti-FK2 showed elevated levels of ubiquitinated protein aggregates in LCLs from P7.1, P3.1 and P1.1, compared to WT following 24hrs serum starvation. Scale bar represents 10μm. (c) STAMBP-patient LCLs exhibit elevated levels of apoptosis following 24hrs serum starvation. Elevated levels of cleaved caspase 3 were observed in LCLs from P7.1, P1.2 and P3.1, compared to WT, following serum starvation (24hrs). (d) Under similar conditions to (c), elevated levels of annexin V were observed in MIC-CAP patient LCLs P7.1, P1.2 and P3.1, compared to WT. Unt; untreated, NS: no-serum. Mean of four seperate determinations + sd. (e) Elevated autophagic flux, as demonstrated by LC3-II expression, was seem in multiple MIC-CAP LCLs, following treatment with Bafilomycin A (BafA; 100nM 2hrs), compared to WT LCLs. These data are consistent with elevated levels of autophagosomes in STAMBP-mutant patient LCLs compared to WT. Mean of three separate determinations ± sd.
ESCRT-mediated endocytosis of activated cell surface receptors (e.g. activated receptor tyrosine kinases, G-protein coupled receptors) controls receptor distribution and coordinates signal transduction amplitude and duration17. Endocytosed ubiquitinated receptors are either recycled to the cell surface or targeted for degradation in the lysosome, leading to the proteolysis and termination of receptor signaling15. STAMBP interacts with key components of receptor signaling pathways such as Grb2 (Fig. 4a)10,22. Considering the known role of STAMBP in regulating receptor-mediated endocytosis, sorting and trafficking, we investigated aspects of the interconnected RAS-MAPK and PI3K-AKT-mTOR signal transduction pathways in our MIC-CAP LCLs, since mutations in components of these networks are associated with congenital capillary malformation disorders6,7,23. We found elevated levels of GTP-bound RAS (active RAS) in extracts from STAMP-patient LCLs compared with WT suggestive of elevated signaling through this pathway (Fig. 4b). Similarly, elevated levels of phosphorylated active phosphoinositol 3-kinase (PI3K) were observed in cell extracts from STAMBP-patient LCLs relative to WT cells even following serum starvation (Fig. 4c). Collectively, these data suggest elevated and insensitive active signal transduction in these interconnected pathways associated with defective STAMBP in patient LCLs.
Figure 4.

Elevated RAS-GTP (active RAS) and activated PI3-kinase in MIC-CAP. (a) Schematic overview of core components of the RAS-MAPK and PI3K-AKT-mTOR networks highlighting this inter-connectivity. As well as interacting with the ESCRT machinery and STAM, STAMBP has been shown to interact with other important components of these signal transduction pathways including the Grb2 adaptor and the class II PI3-kinase catalytic subunit. (b) GTP-bound active RAS levels were precipitated from whole cell extracts using recombinant RAF1-RBD (RAS binding domain) GST-beads followed by Western blotting for RAS. GDP was shown to effectively compete any interaction as expected. Elevated levels of RAS-GTP were pulled down from P7.1 and P.1 MIC-CAP patient cells compared to WT LCLs. Image J based quantification (a.u. arbitrary units) of active RAS-GTP from three separate experiments are represented (± sd) in the graph. (c) Serum starvation (24hrs) reduced PI3-kinase activation in WT LCLs as monitored by phosphorylation of of the PI3K subunit p55-Y199 and p85-Y458. Phospho-PI3K levels were found to be elevated in extracts from P7.1 and P1.1 MIC-CAP LCLs either endogenously or following serum starvation suggestive of hyperactive and insensitive PI3K activity.
To further characterize signaling abnormalities we examined the response of patient LCLs to serum starvation for both these pathways using a selection of substrates. Serum starvation induced a significant reduction in C-RAF phosphorylation on serine-338 in WT LCLs, consistent with inhibition of C-RAF activity under these conditions (Fig. 5a). STAMBP-patient LCLs maintained pS338-C-RAF levels in the absence of serum indicating persistent activation and insensitivity of this pathway. Further evidence suggesting insensitive signal transduction in the RAS-MAPK pathway in STAMBP-mutated LCLs is given by the relative insensitivity of these cells to the MEK1/2 inhibitor U0126. Active C-RAF phosphorylates and activates MEK1/2 kinase, which then phosphorylates and activates ERK1/2 (Fig. 4a). We repeatedly found elevated levels of phosphorylated-ERK1/2 in exponentially growing STAMBP-mutated LCLs compared to WT following a short treatment (1hr) with the MEK1/2 inhibitor U0126 (Fig. 5b and Supplementary Fig. 6e). The excess of phosphorylated-ERK1/2 in STAMBP-mutated LCLs under these robust inhibition conditions is further supportive of hyperactive and insensitive RAS-MAPK pathway in these cells. Analysis of several endpoints in the PI3K-AKT-mTOR pathway under identical conditions indicated a similar insensitive activation of this pathway. Serum starvation of WT LCLs reduced phosphorylation of AKT on T308, on the AKT-dependent T1462 of TSC2, and on S240/S244 of S6 protein. This is consistent with pathway inactivation under these conditions in WT LCLs (Fig. 5c and 5d). In contrast, STAMBP-patient LCLs maintained phosphorylation of all three proteins under these conditions (Fig. 5c and 5d). Furthermore, stable lentiviral transduction of patient LCLs with STAMBP resulted in reconstitution of a normal signaling response to serum starvation (Supplementary Fig. 6f).
Figure 5.
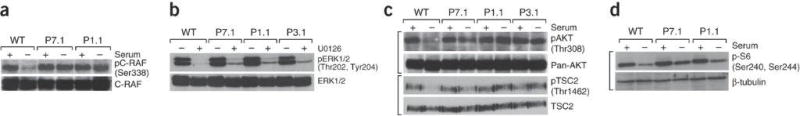
Elevated and insensitive RAS-MAPK and PI3K-AKT-mTOR signaling in MIC-CAP. (a) Serum starvation (24hrs) inhibits C-RAF activation (C-RAF-pS338) in wild-type (WT) LCLs in contrast to LCLs from P7.1 and P1.1. (b). LCLs were either treated (+) or untreated (-) with 10μM U0126, a specific MEK1/2 inhibitor for 1hr (Fig. 4a). Cells were harvested and WCEs were probed for phospho-ERK1/2 levels, which is mediated by MEK. Insensitivity to this treatment (as measured by relative phospho-ERK1/2 levels remaining after treatment with this MEK inhibitor), would reflect the magnitude/intensity of signal transduction from RAF to MEK to ERK (Fig. 4a). Residual pERK1/2 (pT202/pY204) signal (MEK-dependent phosphorylation) was seen in MIC-CAP LCLs, in contrast to WT. This phenotype is underscored following titration of U0126 in various MIC-CAP LCLs compared to WT (Supplementary Fig. 6e). Collectively, these data indicate a greater strength of MEK1/2 activity in MIC-CAP LCLs compared to WT cells. (c) Serum starvation (24hrs) reduces phosphorylation (activation) on AKT (T308) and on TSC2 at T1462, an AKT-dependent inhibitory TSC2 phosphorylation in WT LCLs in contrast to MIC-CAP LCLs from P7.1, P1.1 and P3.1. The TSC1/2 complex is the principal negative regulator of mTOR kinase complex (Fig. 4a). These data are consistent with active signal transduction from PI3K-AKT-mTOR in MIC-CAP cells under these conditions. (d) S6 protein is phosphorylated by S6-kinase in an mTOR-dependent fashion (Fig. 4a). Consistent with active signal transduction in this pathway under serum starvation conditions MIC-CAP LCLs from P7.1 and P1.1 maintained S6 phosphorylation on S240/S244 in contrast to WT LCLs.
The RAS-MAPK and PI3K-AKT-mTOR pathways regulate crucial cellular processes including cell growth, cell cycle progression and differentiation. Disorders characterized by hyperactivity of RAS-MAPK network including Noonan and Costello syndromes exhibit growth delay24. Considering the marked post-natal growth retardation and capillary abnormalities seen in MIC-CAP, hyperactive RAS-MAPK signaling may represent a significant biological consequence induced by impaired STAMBP function in humans suggesting that STAMBP-mutated MIC-CAP may have an overlapping pathomechanism with RASopathies. Furthermore, since the PI3K-AKT-mTOR pathway also plays a role in angiogenesis and vascularization, and considering the interconnectivity between these networks (Fig. 4a), it is possible that combined insensitive activation of both these networks may contribute to the MIC-CAP phenotype.
In summary, we identify mutations in STAMBP in MIC-CAP syndrome, a recently described severe developmental disorder. Analysis of MIC-CAP patient LCLs demonstrates elevated ubiquitin-conjugated protein aggregation and apoptosis activation. These data are consistent with elevated ubiquitin-conjugated protein aggregate-induced progressive apoptosis as a potential underlying mechanism for the microcephaly in this disorder. This is consistent with brain imaging and human pathological analysis of MIC-CAP1 and of the knockout mouse model of STAMBP/AMSH25. Furthermore, we document elevated autophagosome content and active and insensitive RAS-MAPK and PI3K-AKT-mTOR pathways as novel consequences of defective STAMBP, potentially contributing to the vasculature and growth characteristics of MIC-CAP syndrome. This work presents the first example of a human disorder caused by a congenitally defective DUB isopeptidase functioning in the endocytosis pathway; providing significant new insights into the pathophysiology of human microcephaly and capillary malformation.
ONLINE METHODS
Study Participants
All families provided written informed consent and this study was approved by the ethics review boards at the Children’s Hospital of Eastern Ontario, the University of Chicago, and Seattle Children’s Hospital. We studied a cohort of ten affected individuals from nine families with MIC-CAP syndrome. Genomic DNA was extracted from the whole blood of affected subjects and their family using standard techniques.
Sequencing Technology and Variant calling Pipeline
Using target capture with the Agilent SureSelect 50 Mb All Exon kit (Agilent Technologies, Santa Clara, CA) and sequencing of 100 bp paired end reads on Illumina Hiseq, we generated over 15 Gb of sequence for each sample such that approximately 90% of the coding bases of the exome defined by the consensus coding sequence (CCDS) project were covered by at least 20 reads. Reads were first quality trimmed from the 3′ end using the Fastx-toolkit and were then aligned to hg19 with BWA29. Duplicate reads were marked using Picard and excluded from downstream analyses. For each sample, single nucleotide variants (SNVs) and short insertions and deletions (indels) were called using samtools pileup and varFilter30 with the base alignment quality (BAQ) adjustment disabled, and were then quality filtered to require at least 20% of reads supporting the variant call. Coverage of the exome was determined using the Genome Analysis Toolkit (GATK). Variants were annotated using both Annovar31 and custom scripts to identify whether they affected protein coding sequence, and whether they had previously been seen in dbSNP131 or in the 1000 genomes pilot release (Nov. 2010).
Genetic analysis
To elucidate the molecular mechanism of MIC-CAP in Family 4, we PCR amplified 10 polymorphic microsatellite markers spanning the length of chromosome two, four on the short arm and six on the long arm. The amplification products were resolved using the IR2 DNA Analyzer and interpreted using the SAGA software (LI-COR). Analysis of the intronic mutations was performed using the computational model of splicing regulation first described by Barash Y. et al. (2010). Real-time PCR was performed using the Mastercycler Realplex (Eppendorf) in the presence of SYBR® Green PCR Mastermix reagents (Life Technologies, Applied Biosystems). Standard protocol was followed for the optimization of the real-time PCR primers, however reactions were scaled to 25μL per reaction. The PCR conditions were standard and all reagents, excluding the template and primers were provided in the SYBR® Green PCR Mastermix kit. Standard curves were generated using Beta-2 Microglobulin (NM_004048) as a control.
Functional Analysis
STAMBP expression in patient-derived lymphoblastoid cell lines (LCLs) (P1.1, P7.1 and P3.1) was assessed by Western blotting using anti-STAMBP (H-4) with an epitope directed to amino acids 131-270 of STAMBP (Santa Cruz Biotechnology, Santa Cruz, CA). The caspase 3 antibody was from Cell Signalling Technology (Beverly, MA). For annexin V-apoptosis assessment we used the Single Channel Annexin V Apoptosis Kit (Alexa Fluor-488 conjugated anti-Annexin V with SyTOX Green) from Life Technologies LTD (Paisley, UK), according to manufacturer’s instructions. The anti-LC3 was from Cell Signalling (D50G8 XP(R)) and the Bafilomycin A from SIGMA-ALDRICH (Poole, UK). Active RAS-GTP levels were determined using the RAS-activation assay kit (17–218) from Millipore, according to the manufacturer’s instructions.
For siRNA mediated silencing of STAMBP we used ONTARGET plus SMARTpool human STAMBP (L-012202-00-0005) from Dharmacon-Thermo Fisher Scientific (UK), and transfection using Metafectene-Pro from Canbio (Cambridge, UK), according to manufacturer’s instructions. Cells were analysed 24hrs post-transfection. The SMARTpool is a mixture of four oligonucleotides with distinct target sequences (Supplementary Table 3).
For the indirect immunofluorescence (IF), LCLs were pelleted, swollen in 75mM KCL (10mins), immobilised onto poly-lysine coated slides by cytospinning (CytoSpin. Shandon), permeabilised (0.1% triton X-100 in 5% BSA/PBS for 2mins) and blocked in 5% BSA/PBS (10mins), prior to sequential incubation with primary and secondary antibodies. Slides were counterstained with DAPI and preserved in anti-fade mounting media (Vectashield). Slides were analysed using the Zeiss AxioPlan platform and images captured using SimplePCI software at constant exposure times. Anti-conjugated ubiquitin mouse monoclonal clone FK2 was from Enzo Lifesciences UK LTD (Exeter, UK).
To interrogate RAS-MAPK pathway function, patient-derived LCLs were grown exponentially in complete medium in the presence or absence of foetal bovine serum for 24hrs. Antibodies, including phospho-specific antibodies against C-Raf (pS33) and MAPK/ERK1/2 (p42/44) (pT202/pY204), along with their corresponding native antibodies were from Cell Signalling Technology (Beverly, MA), The MEK1/2 inhibitor U0126 was used at 10μM for 1hr. Whole cell extracts were prepared by sonication in urea buffer (9M urea, 50mM Tris-HCl pH7.5, 10mM β-mercaptoethanol).
For lentiviral transduction of LCLs, high titre Precision LentiORF viral particles derived from the pLOC system were obtained from Thermo Scientific (Open Biosystems) and used according to the manufacturer’s instructions. Stable STAMBP-expressing clones were obtained following blasticidin S selection of transduced populations.
Supplementary Material
Acknowledgments
The authors would first like to thank the study patients and their families, without whose participation this work would not be possible. This work was funded by the Government of Canada through Genome Canada, the Canadian Institutes of Health Research (CIHR) and the Ontario Genomics Institute (OGI-049) (to K.M.B.). Additional funding was provided by Genome Quebec and Genome British Columbia (to K.M.B.), the US National Institutes of Health under NINDS grant NS058721 (to W.B.D.) as well as NICHD grant HD36657 and NIGMS grant 5-T32-GM08243 (to J.M.G. Jr.) and the Leukaemia Lymphoma Research (UK), Medical Research Council (UK) and Cancer Research UK (CR-UK) (to M.O’D). The authors wish to acknowledge the contribution of the high throughput sequencing platform of the McGill University and Génome Québec Innovation Centre, Montréal, Canada as well as Martin Moellers, (Pediatric Radiology, Evangelisches Krankenhaus Bielefeld, Germany). This work was selected for study by the FORGE Canada Steering Committee, consisting of K. Boycott (U. Ottawa), J. Friedman (U. British Columbia), J. Michaud (U. Montreal), F. Bernier (U. Calgary), M. Brudno (U. Toronto), B. Fernandez (Memorial U.), B. Knoppers (McGill U.), M. Samuels (U. de Montreal), and S. Scherer (U. Toronto). L.M.M is supported by a Frederick Banting Graduate Scholarship from CIHR. M.O’D is a CR-UK Senior Cancer Research Fellow. K.M.B. is supported by a Clinical Investigatorship Award from the CIHR Institute of Genetics.
Footnotes
URLs
NHLBI Exome variant server, http://evs.gs.washington.edu/EVS/ ; FASTX-Toolkit, http://hannonlab.cshl.edu/fastx_toolkit/ ; Picard tools, http://picard.sourceforge.net/ ; Samtools, http://samtools.sourceforge.net/
AUTHOR CONTRIBUTIONS
K.M.B., M.O’D., W.B.D. and D.E.B. directed the study. M.T.C., L.J.L., C.L.C., J.M.G., D.J.M., T.P., G.A., S.T., S.W., A.H., B.I., C.D.S., A.R.P., M.W., J.W., S.D., M.T.G., G.M.M., W.B.D. and K.M.B. provided clinical data. L.M.M. performed Sanger sequencing, genotyping studies and variant analysis supervised by K.M.B. and D.E.B.. D.A. performed the protein biochemistry and cell biology studies which were directed by M.O’D.. J.S. and J.M. performed exome variant calling analysis. The manuscript was written by L.M.M., G.M.M., M.O’D. and K.M.B.. K.M.B. is lead of the FORGE Canada Consortium and is assisted by C.L.B. and Janet.M. All authors reviewed the manuscript.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
References
- 1.Carter MT, et al. A new syndrome with multiple capillary malformations, intractable seizures, and brain and limb anomalies. Am J Med Genet A. 2011;155:301–306. doi: 10.1002/ajmg.a.33841. [DOI] [PubMed] [Google Scholar]
- 2.Isidor B, Barbarot S, Bénéteau C, Le Caignec C, David A. Multiple capillary skin malformations, epilepsy, microcephaly, mental retardation, hypoplasia of the distal phalanges: Report of a new case and further delineation of a new syndrome. Am J Med Genet A. 2011;155:1458–1460. doi: 10.1002/ajmg.a.34048. [DOI] [PubMed] [Google Scholar]
- 3.Mirzaa GM, et al. The microcephaly-capillary malformation syndrome. Am J Med Genet A. 2011;155A:2080–2087. doi: 10.1002/ajmg.a.34118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carter MT, Boycot KM. Microcephaly–capillary malformation syndrome: A story of rapid emergence of a new recognizable entity. Am J Med Genet A. 2011;155:2078–2079. doi: 10.1002/ajmg.a.34149. [DOI] [PubMed] [Google Scholar]
- 5.Jacobs AH, Walton RG. The incidence of birthmarks in the neonate. Pediatrics. 1976;58:218–222. [PubMed] [Google Scholar]
- 6.Eerola I, et al. Capillary Malformation-Arteriovenous Malformation, a New Clinical and Genetic Disorder Caused by RASA1 Mutations. Am J Hum Genet. 2003;73:1240–1249. doi: 10.1086/379793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Eerola I, et al. KRIT1 is mutated in hyperkeratotic cutaneous capillary-venous malformation associated with cerebral capillary malformation. Hum Mol Genet. 2000;9:1351–1355. doi: 10.1093/hmg/9.9.1351. [DOI] [PubMed] [Google Scholar]
- 8.Barash Y, et al. Deciphering the splicing code. Nature. 2010;465:53–59. doi: 10.1038/nature09000. [DOI] [PubMed] [Google Scholar]
- 9.Sierra MI, Wright MH, Nash PD. AMSH interacts with ESCRT-0 to regulate the stability and trafficking of CXCR4. J Biol Chem. 2010;285:13990–14004. doi: 10.1074/jbc.M109.061309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tsang HTH, et al. A systematic analysis of human CHMP protein interactions: Additional MIT domain-containing proteins bind to multiple components of the human ESCRT III complex. Genomics. 2006;88:333–346. doi: 10.1016/j.ygeno.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 11.Tanaka N, et al. Possible involvement of a novel STAM-associated molecule ‘AMSH’ in intracellular signal transduction mediated by cytokines. J Biol Chem. 1999;274:19129–19135. doi: 10.1074/jbc.274.27.19129. [DOI] [PubMed] [Google Scholar]
- 12.McCullough J, et al. Activation of the endosome-associated ubiquitin isopeptidase AMSH by STAM, a component of the multivesicular body-sorting machinery. Curr Biol. 2006;16:160–165. doi: 10.1016/j.cub.2005.11.073. [DOI] [PubMed] [Google Scholar]
- 13.Agromayor M, Martin-Serrano J. Interaction of AMSH with ESCRT-III and deubiquitination of endosomal cargo. J Biol Chem. 2006;281:23083–23091. doi: 10.1074/jbc.M513803200. [DOI] [PubMed] [Google Scholar]
- 14.Mizuno E, Kobayashi K, Yamamoto A, Kitamura N, Komada M. A deubiquitinating enzyme UBPY regulates the level of protein ubiquitination on endosomes. Traffic. 2006;7:1017–1031. doi: 10.1111/j.1600-0854.2006.00452.x. [DOI] [PubMed] [Google Scholar]
- 15.Kim MS, Kim J, Song HK, Jeon H. STAM-AMSH interaction facilitates the deubiquitination activity in the C-terminal AMSH. Biochem Biophys Res Commun. 2006;351:612–618. doi: 10.1016/j.bbrc.2006.10.068. [DOI] [PubMed] [Google Scholar]
- 16.Kyuuma M, et al. AMSH, an ESCRT-III associated enzyme, deubiquitinates cargo on MVB/late endosomes. Cell Struct Funct. 2006;31:159–162. doi: 10.1247/csf.06023. [DOI] [PubMed] [Google Scholar]
- 17.Raiborg C, Stenmark H. The ESCRT machinery in endosomal sorting of ubiquitylated membrane proteins. Nature. 2009;458:445–452. doi: 10.1038/nature07961. [DOI] [PubMed] [Google Scholar]
- 18.Komada M. Controlling receptor downregulation by ubiquitination and deubiquitination. Curr Drug Discovery Technol. 2008;5:78–84. doi: 10.2174/157016308783769469. [DOI] [PubMed] [Google Scholar]
- 19.Wright MH, Berlin I, Nash PD. Regulation of Endocytic Sorting by ESCRT-DUB-Mediated Deubiquitination. Cell Biochem Biophys. 2011;60:39–46. doi: 10.1007/s12013-011-9181-9. [DOI] [PubMed] [Google Scholar]
- 20.Suzuki S, et al. AMSH is required to degrade ubiquitinated proteins in the central nervous system. Biochem Biophys Res Commun. 2011;408:582–588. doi: 10.1016/j.bbrc.2011.04.065. [DOI] [PubMed] [Google Scholar]
- 21.Williams RL, Urbé S. The emerging shape of the ESCRT machinery. Nat Rev Mol Cell Biol. 2007;8:355–368. doi: 10.1038/nrm2162. [DOI] [PubMed] [Google Scholar]
- 22.Sowa ME, Bennett EJ, Gygi SP, Harper JW. Defining the Human Deubiquitinating Enzyme Interaction Landscape. Cell. 2009;138:389–403. doi: 10.1016/j.cell.2009.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Boon LM, Mulliken JB, Vikkula M. RASA1: Variable phenotype with capillary and arteriovenous malformations. Curr Opin Genet Dev. 2005;15:265–269. doi: 10.1016/j.gde.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 24.Tidyman WE, Rauen KA. The RASopathies: developmental syndromes of Ras/MAPK pathway dysregulation. Curr Opin Genet Dev. 2009;19:230–236. doi: 10.1016/j.gde.2009.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ishii N, et al. Loss of neurons in the hippocampus and cerebral cortex of AMSH-deficient mice. Mol Cell Biol. 2001;21:8626–8637. doi: 10.1128/MCB.21.24.8626-8637.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kato M, Miyazawa K, Kitamura N. A deubiquitinating enzyme UBPY interacts with the Src homology 3 domain of Hrs-binding protein via a novel binding motif PX(V/I)(D/N)RXXKP. J Biol Chem. 2000;275:37481–37487. doi: 10.1074/jbc.M007251200. [DOI] [PubMed] [Google Scholar]
- 27.Davies CW, Paul LN, Kim M, Das C. Structural and thermodynamic comparison of the catalytic domain of AMSH and AMSH-LP: Nearly identical fold but different stability. J Mol Biol. 2011;413:416–429. doi: 10.1016/j.jmb.2011.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yu MM, et al. Targeting of AMSH to endosomes is required for epidermal growth factor receptor degradation. J Biol Chem. 2007;282:9805–9812. doi: 10.1074/jbc.M611635200. [DOI] [PubMed] [Google Scholar]
- 29.Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26:589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li H, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.