

Abstract
The cellular process of autophagy (literally “self-eating”) is important for maintaining the homeostasis and bioenergetics of mammalian cells. Two of the best-studied mechanisms of autophagy are macroautophagy and chaperone-mediated autophagy (CMA). Changes in macroautophagy activity have been described in cancer cells and in solid tumors, and inhibition of macroautophagy promotes tumorigenesis. Because normal cells respond to inhibition of macroautophagy by up-regulation of the CMA pathway, we aimed to characterize the CMA status in different cancer cells and to determine the contribution of changes in CMA to tumorigenesis. Here, we show consistent up-regulation of CMA in different types of cancer cells regardless of the status of macroautophagy. We also demonstrate an increase in CMA components in human cancers of different types and origins. CMA is required for cancer cell proliferation in vitro because it contributes to the maintenance of the metabolic alterations characteristic of malignant cells. Using human lung cancer xenografts in mice, we confirmed the CMA dependence of cancer cells in vivo. Inhibition of CMA delays xenograft tumor growth, reduces the number of cancer metastases, and induces regression of existing human lung cancer xenografts in mice. The fact that similar manipulations of CMA also reduce tumor growth of two different melanoma cell lines suggests that targeting this autophagic pathway may have broad antitumorigenic potential.
INTRODUCTION
Autophagy is a cellular process that mediates the targeting and degradation of intracellular components—proteins and organelles—in lysosomes (1). This continuous turnover contributes to the maintenance of cellular homeostasis, quality control, defense against intra- and extracellular insults, and preservation of the cellular energetic balance. It does this by providing essential macromolecules (amino acids, free fatty acids, etc.) through the degradation of existing intracellular components (2, 3).
Of the different autophagic pathways that coexist in all cells, the two best characterized in mammalian cells are macroautophagy and chaperone-mediated autophagy (CMA) (4, 5). Macroautophagy delivers proteins and organelles to lysosomes for degradation upon sequestration in a double-membraned vesicle (autophagosome) (2). In contrast, protein substrates for CMA are selectively identified and targeted to the lysosomes through the interaction of a cytosolic chaperone protein (hsc70) (6), which has a pentapeptide motif in the substrate amino acid sequence (7). The targeted substrate binds to lysosomes through the cytosolic tail of the lysosome-associated membrane protein type 2A (LAMP-2A) (8), and after unfolding (9), chaperones on both sides of the membrane contribute to substrate internalization through a multimeric protein complex in the membrane (10–12). Both macroautophagy and CMA are up-regulated in response to stressors, including nutritional stress, but basal activity can be detected in most cells (13, 14).
Recent studies support the existence of a cross-talk between these two autophagic pathways that allows one pathway to compensate for the other (15, 16). Deficiency in either of the two autophagic pathways often becomes evident once the impaired cells are subjected to stress, but this compensatory up-regulation suffices under basal conditions (15, 16). This autophagic cross-talk has been shown to be beneficial in certain pathogenic conditions in which one of these pathways becomes primarily compromised (17, 18). Here, we investigate whether cancer cells also take advantage of this cross-talk to assure their own survival.
Macroautophagy has been shown to function as a tumor suppressor in part by contributing to the maintenance of genome stability (19, 20). Spontaneous tumorigenesis is common in mouse models with compromised autophagy because reduced macroautophagy facilitates cell cycle progression, inhibits the senescence program, and favors the accumulation of proteins involved in oncogenesis (21–24). Despite the low levels of macroautophagy observed in different types of cancer cells, these cells still need to handle the continuous stress that the proteome and organelles are exposed to in the context of high metabolic activity. It is thus likely that cancer cells may be able to switch between different states of macroautophagy activity and that, by the same token, they could have adopted mechanisms to activate different autophagic pathways to maintain homeostasis and accommodate specific metabolic requirements.
We have examined whether up-regulation of CMA is one such mechanism. We report that high basal CMA activity is a common feature among different types of cancer cells and human tumors. In contrast to normal cells, this up-regulation of CMA occurs independently of the status of macroautophagy in these cancer cells. Using two human lung cancer cell lines with very different macroautophagic activity, we show here that inhibition of CMA reduces cell proliferation and increases cell death. In contrast to nontumor cells, protein quality control upon CMA blockade in these cancer cells can be performed by up-regulation of the ubiquitin/proteasome system. In contrast, metabolic changes imposed by inhibiting CMA cannot be compensated for in these cancer cells, limiting their proliferative capability in vivo. We show that blockade of CMA delays tumor growth and induces regression of already formed human lung cancer xenografts in mice. The fact that similar manipulations of CMA also reduce tumor growth of two different melanoma cell lines suggests that targeting this autophagic pathway may have broad antitumorigenic potential.
RESULTS
Constitutive activation of CMA is a common feature of malignant cells
To gain insights into the macroautophagic activity of different types of cancer cells and their CMA status, we analyzed the autophagic profiles of several cancer cell lines that originated from lung (A549 and H460), breast (MCF7), liver (HuH-7), and epithelia (Saos-2 osteosarcoma). We compared their autophagic activity to that of non-oncogenic cell types used as controls: mouse embryonic fibroblasts (MEFs), mouse striatal neuronal cultures, immortalized mouse fibroblasts (NIH 3T3), human lung fibroblasts, and human breast epithelial cells (MCF12; often used as a control for MCF7 cells). In agreement with previous studies (24), measurement of macroautophagic activity in the form of protein degradation that is sensitive to the phosphatidylinositol 3-kinase inhibitor 3-methyladenine (fig. S1A) or autophagic flux of the autophagosome protein LC3 (fig. S1B) revealed a broad spectrum of macroautophagic activities in cancer cells compared to relatively uniform activity in control cells.
In contrast, CMA activity, measured by different approaches, was consistently higher in all cancer cell lines included in this study compared to untransformed control cells. We first monitored CMA using a photoconvertible reporter that behaves as a CMA substrate (25). The KFERQ-PA-mCherry1 reporter protein displays a cytosolic diffuse fluorescence pattern under resting conditions, but it binds to the lysosomal membrane when CMA is activated (such as during prolonged serum removal), highlighting lysosomes as red fluorescent puncta. Figure 1A shows examples of the fluorescence pattern of KFERQ-PA-mCherry1 in a nontumor cell line (NIH 3T3) maintained in the presence or absence of serum (low and high CMA, respectively), in human breast epithelial and cancer cell lines, and in two human lung cancer cell lines with different degrees of macroautophagic efficiency. Quantification of the fluorescent puncta revealed that CMA activity was significantly higher (about 2.8-fold increase; P < 0.05 to 0.01) in all cancer cell lines analyzed when compared with nontumor cells [Fig. 1A, right; removal of serum in nontumor NIH 3T3 cells (gray bar) is used as a positive control for up-regulation of CMA in normal cells]. In all studies, cell staining with the pH-sensitive fluorophore LysoTracker was used to confirm that the observed puncta coincided with lysosomes (fig. S1C).
Fig. 1.
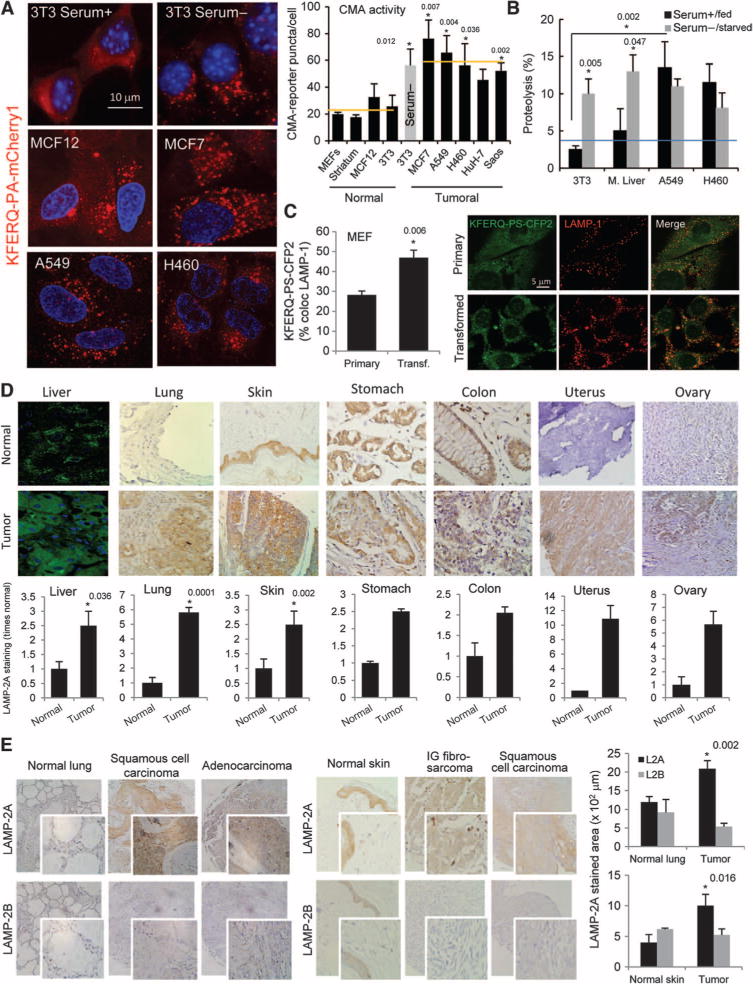
CMA is up-regulated in cancer cell lines and in human tumors. (A) CMA activity in nontumor and tumor cell lines transfected with a photoactivatable KFERQ-PA-mCherry1 fluorescent reporter. Left: standard pattern in resting or CMA up-regulated (serum−) NIH 3T3 cells and in cancer cells. Right: average number of fluorescent puncta per cell (n = 4 to 6; *P = 0.012, 0.007, 0.004, 0.0036, and 0.002, t test). Serum removal in cultured NIH 3T3 cells (gray bar) is used as a positive control of CMA induction in nontumor cells. (B) Uptake and degradation of radiolabeled proteins by lysosomes in two human lung cancer cell lines and in mouse fibroblasts (NIH 3T3) maintained in the presence or absence of serum and in liver cells from fed mice or mice starved for 48 hours. Values are percentage of proteolysis and are means ± SE (n = 3 to 6; *P = 0.005 and 0.0047 compared to serum+ and 0.002 compared to NIH 3T3 cells, t test). (C) CMA activity in MEFs untransfected (primary) or after transfection with SV40 T antigen determined using the photoswitchable KFERQ-PS-CFP2 reporter. Left: quantification of the percentage of lysosomes positive for the reporter (mean ± SE of >50 cells; *P = 0.006, t test). Right: representative fields. Individual channels for the reporter, LAMP-1, and the merge images are shown. (D) Immunostaining for LAMP-2A of human tumors from the indicated organs. Top: example of LAMP-2A staining in normal tissue (top) and tumor (bottom). Liver samples were observed by immunofluorescence and the rest of the samples by immunohistochemistry. Bottom: quantification of the total LAMP-2A–positive area in normal and tumor samples expressed as fold staining in normal tissue after correction by tissue area. Values are means ± SE of 3 to 15 different tumors (*P = 0.036, 0.0001, and 0.002, t test). (E) Immunostaining for LAMP-2A and LAMP-2B of skin and lung tumors and normal tissue. Left: examples of staining (inset shows higher magnification). Right: quantification of the staining for each protein. Values are means ± SE for 10 and 11 different skin and lung tumors, respectively (*P = 0.002 and 0.016, t test).
We then confirmed enhanced CMA activity in the two lung-derived cancer cell lines using a well-established system that allows reconstituting of CMA in vitro through the degradation of a radiolabeled pool of cytosolic CMA substrates by intact lysosomes (26). CMA activity of lysosomes isolated from the two lung cancer cell lines was 2.5- to 3-fold higher than in those isolated from different nontumor cells and rodent liver, where CMA has been most extensively characterized (Fig. 1B; P < 0.01). Under the conditions of our assay, the stability of the lysosomal membrane was comparable between control cells and cancer cells, ruling out enzyme leakage as the cause of the observed higher degradation rates. In addition, disruption of the lysosomal membrane to directly assess proteolysis independently of uptake eliminated differences between control and cancer cells (fig. S1D), confirming that the overall activity of the luminal hydrolases was not responsible for the observed higher degradation of cytosolic proteins in cancer cell lysosomes. In further support of constitutive CMA up-regulation in these cells, removal of serum did not result in an additional increase in CMA (Fig. 1B). In agreement with CMA up-regulation, we observed greater enrichment for LAMP-2A and hsc70—the two limiting components of the CMA pathway (8, 10)—in lysosomes from the two lung cancer cell lines when compared to controls (fig. S1E). As above, removal of serum did not further increase lysosomal concentrations of these two CMA effectors, suggesting that CMA was already maximally up-regulated in these cells. We observed a similar significant increase (35% increase, P = 0.006) in CMA activity upon simian virus 40 (SV40) T antigen transformation of primary mouse fibroblasts (Fig. 1C), further strengthening the possibility that CMA up-regulation may be a common characteristic of most transformed cells.
To determine whether CMA activity is also increased in tumors in vivo, we performed immunostaining for LAMP-2A in primary human tumors from different tissues (liver, lung, skin, stomach, colon, uterus, and ovary) (Fig. 1D and figs. S2 and S3). Although the intensity of LAMP-2A staining varied depending on the type of tumor, overall, the LAMP-2A signal was significantly higher in all tumor samples when compared to that in respective normal tissues (Fig. 1D shows representative examples and LAMP-2A average concentrations in tumor and control tissues, and fig. S2, A to C, shows examples of individual types of tumors from each organ). In some cases, there was a gradual increase in LAMP-2A staining when transitioning from normal regions to perineoplastic and tumoral regions (fig. S2A) and with the stage of malignancy for the same tumor type (fig. S2D). It is unlikely that the observed increase in LAMP-2A merely reflects an expansion of the lysosomal compartment in tumor cells, because control and tumor samples showed no difference upon staining for LAMP-2B, a lysosomal protein splice variant of the same lamp2 gene (27) with 95% sequence homology to LAMP-2A but no involvement in CMA (Fig. 1E and fig. S3).
Overall, these results support the theory that, independent of the status of macroautophagy, CMA is constitutively up-regulated in different cancer cell lines and in transformed cells and that concentrations of LAMP-2A, the limiting component for this autophagic pathway (8), are increased in human tumors.
Cell proliferation is reduced in human lung cancer cells with impaired CMA
To determine whether up-regulation of CMA in tumor cells is required for oncogenic behavior, we down-regulated CMA activity in the two human lung cancer cell lines using replication-deficient lentivirus-targeted short hairpin RNA (shRNA) against LAMP-2A. We chose these two lines because they displayed markedly different macroautophagic activity (fig. S1A), but CMA activity was consistently up-regulated in both of them (Fig. 1, A and B). Reducing the concentrations of LAMP-2A is the most efficient way to down-regulate CMA, because manipulation of the cellular levels of the other well-characterized components of this pathway (hsc70 and hsp90) yields results that are difficult to interpret because they are shared with many other cellular pathways (15). Immunoblot with an antibody against the cytosolic tail of LAMP-2A (Fig. 2A) and reverse transcription–polymerase chain reaction (RT-PCR) analysis confirmed that there was a 70 and 50% reduction in LAMP-2A concentrations in A549 and H460 cells, respectively, after knockdown of LAMP-2A [L2A(−)]. Analysis of the pattern of fluorescence of the CMA-photoconvertible reporter confirmed that levels of LAMP-2A knockdown were sufficient to almost fully suppress CMA activity in both cell types (Fig. 2B).
Fig. 2.
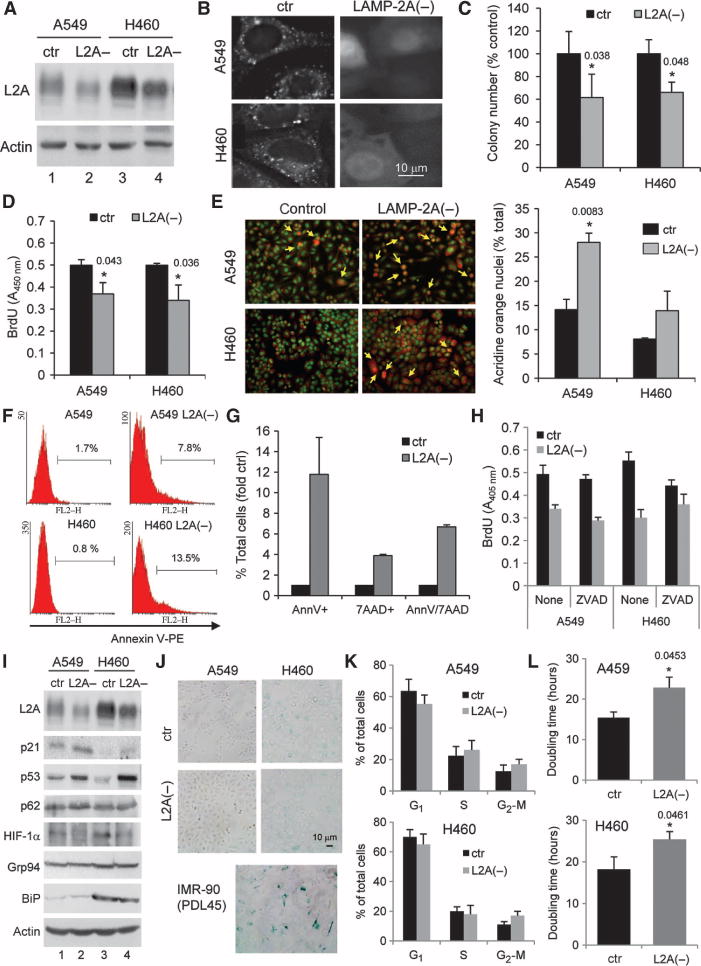
Blockade of CMA in lung cancer cells reduces cell proliferation and increases cell death. (A) Immunoblot for LAMP-2A (L2A) and actin of two human lung cancer cell lines either untreated [control (ctr)] or treated with RNAi to knock down LAMP-2A [L2A(−)]. (B) Fluorescent staining of the CMA reporter in the same cells. (C and D) Incorporation of BrdU (C) and clonogenicity (D) of human lung cancer cells with LAMP-2A knocked down or untreated [n = 6; *P = 0.038 and 0.048 in (C) and 0.043 and 0.036 in (D), t test]. (E) Staining with ethidium bromide/acridine orange of the same cells. Left: representative image. Right: quantification of the number of cells with orange nuclei (late apoptotic cells) per field. Values are means ± SE of four fields for three independent experiments (*P = 0.083, t test). (F and G) Staining for annexin V–PE or annexin V–7ADD in these cells quantified by FACS. Values in (F) indicate the percentage of cells positive for annexin V in each group, and in (G) the percentage of single- and double-stained cells (n = 3). (H) Incorporation of BrdU in the indicated cells treated with the pan-caspase inhibitor ZVAD or untreated (n = 4). (I) Immunoblots for the indicated stress-related proteins in human cancer cell lines: control (ctr) or after LAMP-2A knockdown [L2A(−)]. (J) β-Galactosidase staining of control and L2A(−) A549 and H460 cells. Staining in IMR-90 primary human lung fibroblasts after 45 population doublings (PDL45) is shown as a positive control. (K) Cell cycle distribution of cells labeled with PI and analyzed by FACS. Percentage of cells in each phase (n = 3). (L) Doubling time of control and L2A(−) A549 and H460 cells (n = 4; *P = 0.0453 and 0.0461, t test).
CMA inhibition significantly reduced the proliferative capability of both cancer cell lines, as determined by their ability to form colonies (Fig. 2C; P < 0.05) and incorporate 5-bromo-2′-deoxyuridine (BrdU) (Fig. 2D; P < 0.05). In addition, L2A(−) cancer cells also showed higher rates of cell death under basal culture conditions as demonstrated by ethidium bromide/acridine orange staining (Fig. 2E; P = 0.0083). Annexin V staining (Fig. 2F) and double staining for annexin V and 7-aminoactinomycin D (7AAD) (Fig. 2G) revealed not only higher rates of apoptosis in L2A(−) cells but also an increase in non-apoptotic cell death. Inhibition of apoptosis by treatment with the pan-caspase inhibitor ZVAD was not sufficient to correct the reduced BrdU incorporation in CMA-defective cells (Fig. 2H), supporting coexistence of both increased cell death and decreased proliferation in these cells.
Inhibition of CMA in human lung cancer cells induces metabolic changes
To gain insights into the mechanisms behind the reduced proliferative capability and increased cell death observed in the lung cancer cells after inhibition of CMA, we analyzed the changes in key molecular components of three major processes tightly related to cancer biology: senescence, resistance to stress, and metabolism.
Immunoblot of control cells and L2A(−) A549 and H460 cells, in which LAMP-2A had been knocked down, revealed marked increases in p53 and p21 in cells with compromised CMA activity (Fig. 2I). Up-regulation of p53 and p21 proteins has been linked to cell cycle arrest and to senescence. The percentage of cells positive for the senescent marker β-galactosidase did not change upon CMA blockade (Fig. 2J), suggesting that the observed increase in p53 and p21 did not lead to senescence. Flow cytometric analysis after propidium iodide (PI) labeling revealed discrete changes in the distribution of cells at different stages of the cell cycle (Fig. 2K), suggesting that the decrease in colony number may not be due to cell cycle arrest. In contrast, measurement of growth rate in culture revealed a consistent increase in cell doubling time (Fig. 2L; 35% and 18% increase in A459 and H460 cells, respectively; P < 0.05), which is likely responsible for the observed decrease in colony number.
We then compared resistance to stress and amounts of different cellular stress markers in both control and L2A(−) human lung cancer cells. In contrast to nontumor cells in which inhibition of CMA sensitizes cells to oxidative and endoplasmic reticulum (ER) stressors (15), cancer cells with impaired CMA did not show a further compromise of cellular viability than control cells upon treatment with the pro-oxidant paraquat or after inducing ER stress with thapsigargin (fig. S4, A and B). Blockade of macroautophagy exerts an antitumorigenic effect during metabolic stress through the accumulation of p62 and ER chaperones (19). We did not find significant accumulation of p62 or ER chaperones (Grp94 and BiP shown here) in CMA-deficient lung cancer cells (Fig. 2I). Also, in contrast to previous studies in nontumor cells (15), blockade of CMA in these cancer cells did not increase the cellular content of oxidized proteins (Fig. 3A) or of aggregated and soluble polyubiquitinated proteins (Fig. 3B), which were lower in L2A(−) A549 and H460 cells. To determine whether decreased protein damage resulted from a compensatory up-regulation of macroautophagy, we analyzed well-characterized macroautophagy regulators and effectors. As shown in Fig. 3C, we did not observe any differences in levels or phosphorylation of mammalian target of rapamycin (mTOR), a negative regulator of macroautophagy, beclin-1, the main macroautophagy activator, or the Atg5/12 complex, which forms upon initiation of macroautophagy to generate autophagosomes. In contrast to the constitutive up-regulation of macroautophagy observed in normal cells deficient in LAMP-2A (15), cancer cells with compromised CMA activity did not display a noticeable up-regulation of macroautophagy, determined by measurement of LC3-II clearance (Fig. 3D). These experiments also confirm that, as described in normal cells, most of the effect of LAMP-2A knockdown in cancer cells was directly on CMA, because RNA interference (RNAi) against LAMP-2A did not modify autophagosome content (levels of LC3-II without protease inhibitors) or their lysosomal clearance (measured as the difference in LC3-II levels in the presence and absence of inhibitors) (Fig. 3D).
Fig. 3.
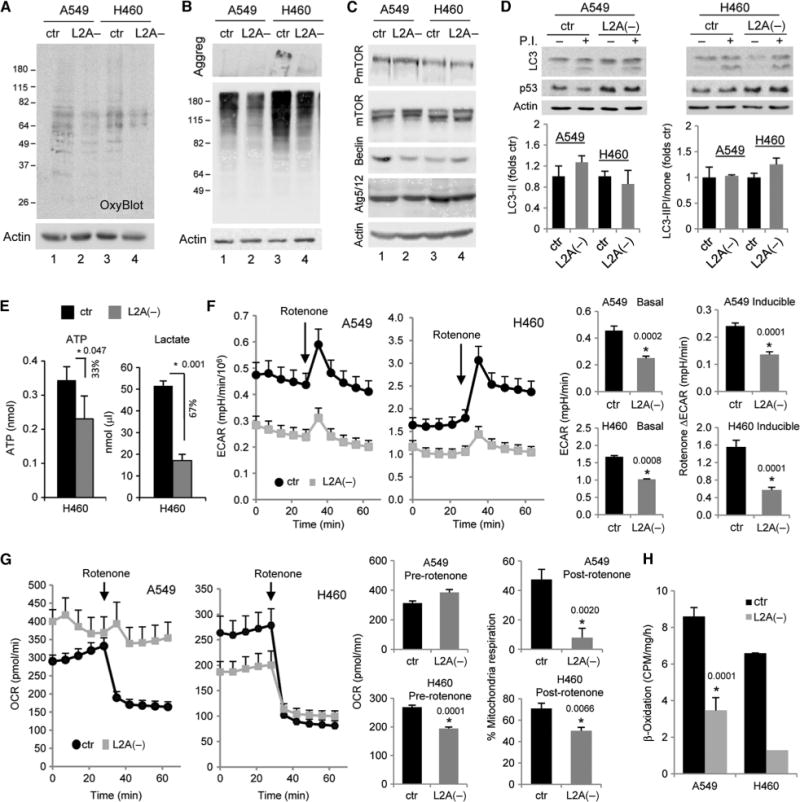
Blockade of CMA in human cancer cells induces changes in cellular metabolism that limit cell proliferation and induce cell death. (A) Concentrations of oxidized proteins in human lung cancer cell lines {control (ctr) or after LAMP-2A knockdown by RNAi [L2A(−)]} detected by the presence of carbonyl groups. (B) Pattern of polyubiquitinated proteins in the same cells. Stacking of the gel is shown to visualize possible protein aggregates that do not enter the gel. (C) Immunoblot for the indicated effectors and regulators of macroautophagy in the same cells. (D) Autophagic flux in the same cells was determined as the increase in LC3-II levels upon blockade of lysosomal hydrolysis with protease inhibitors (P.I.). Top: representative immunoblots. Increased levels of p53 in the knockdown cells are shown as a reference. Bottom: quantification of steady-state levels of LC3-II (left) and autophagic flux (right) defined as the ratio of LC3-II in cells treated with PI or untreated (n = 4). (E) Concentrations of intracellular ATP (left) and of lactate (right) released in the culture media of H460 human cancer cell lines, either control or cells in which LAMP-2A was knocked down by RNAi [L2A(−)] (n = 4; *P = 0.047 and 0.001, t test). (F) ECAR in A549 and H460 cells, control (ctr) or with LAMP-2A knocked down [L2A(−)]. Left: representative ECAR plots. Where indicated, rotenone was added to the culture media to block mitochondrial oxidative phosphorylation. Right: mean values of ECAR before rotenone addition (basal acidification, left) and average increase of ECAR values after rotenone addition (inducible acidification, right) (n = 6; *P = 0.0002, 0.0001, 0.00008, and 0.0001, t test). (G) OCR in the same cells. Left: representative OCR plots. Right: mean values of OCR before addition of rotenone, and percentage of OCR that is sensitive to rotenone (mitochondrial oxidative phosphorylation) (n = 6; *P = 0.0020 and 0.0066, t test). (H) Rates of β-oxidation of radiolabeled fatty acids in the same cells (n = 3; *P = 0.0001, t test).
It is likely that the lower concentrations of ubiquitinated proteins in L2A(−) cells are actually a consequence of their accelerated degradation through the proteasome system. After inhibition of the proteasome, polyubiquitinated proteins in L2A(−) A549 cells reached concentrations comparable to those in control A549 cells (fig. S4C). Up-regulation of the proteasome could also explain the lower levels of oxidized proteins in these cells, but it did not seem to be responsible for the observed reduction in cell proliferation, because treatment with lactacystin, a proteasome inhibitor, did not result in significant changes in cellular proliferation (fig. S4D). The enhanced proteasomal degradation in the CMA-incompetent cells was selective for a subset of proteins, because we found a reduction of 15 to 20% in the rates of proteasomal degradation of p53 in these cells (fig. S4C). Reduced degradation of p53 could thus contribute, at least in part, to the enhanced levels of this protein in cancer cells with compromised CMA activity. Further studies are necessary to determine the nature of the subset of ubiquitinated proteins that are more rapidly degraded, and how other proteins such as p53 escape increased proteasomal degradation.
Next, we investigated whether blockade of CMA in the human lung cancer cells affected their high basal metabolic rate, which is required to assure sustained proliferation. Concentrations of both adenosine 5′-triphosphate (ATP) and extracellular lactate, a metabolic by-product released into the culture medium, were lower in cells with impaired CMA (33 and 67% reduction in ATP and lactate, respectively; P < 0.05) (Fig. 3E). To gain a better understanding of the baseline bioenergetics of L2A(−) cancer cells, we used an extracellular flux analyzer to investigate the dynamics of lactic acid production, measured as ECAR (extracellular acidification rate), and cellular respiration, measured as OCR (oxygen consumption rate). Basal ECAR was lower in both A549 and H460 cells with compromised CMA activity (Fig. 3F, middle). In addition, when rotenone was administered to block mitochondrial respiration (traceable by a decrease in OCR, Fig. 3G), the typical increase in ECAR observed in control cells, indicative of a compensatory increase in glycolysis, was significantly reduced in L2A(−) cells (Fig. 3F, right).
In contrast to this uniform effect of CMA blockade on ECAR, changes in OCR differed between the two human lung cancer cell lines. We observed a slight but consistent elevation of OCR in L2A(−) A549 cells when compared to control cells, but significantly lower OCR in H460 cells upon LAMP-2A knockdown (Fig. 3G). However, analysis of OCR after rotenone treatment demonstrated that mitochondrial respiration (defined as the percentage of OCR sensitive to rotenone) was compromised in both cancer cell types after CMA blockage, although to a different extent (Fig. 3G, right; P < 0.01 to 0.001). Although further investigation is required, it is possible that the increase in rotenone-resistant OCR in the L2A(−) A549 cells could reflect a compensatory increase in nonmitochondrial respiration (mainly substrate oxidation and cell surface oxygen consumption) induced by the very low rates of mitochondrial respiration detected in these cells. Direct measurement of oxidation of radiolabeled fatty acids also confirmed a significant reduction in mitochondrial β-oxidation in the cancer cells with compromised CMA (Fig. 3H). Staining with MitoSOX, a fluorogenic dye that selectively detects superoxide generated as a by-product of oxidative phosphorylation, confirmed lower staining in L2A(−) cells (fig. S5A). The observed reduction in mitochondrial respiration in cancer cells with compromised CMA is unlikely to be a consequence of changes in total mitochondria mass, because amounts of specific mitochondrial proteins were comparable in control and L2A(−) cells (fig. S5B).
Although interesting, the changes in mitochondrial oxidative phosphorylation observed in L2A(−) cells may make only a small contribution to their overall energetic balance, because oxidative metabolism is already markedly reduced in cancer cells when compared with nontumor cells (fig. S5C shows rates of β-oxidation in the two human lung cancer cell lines compared to untransformed mouse fibroblasts). In contrast, persistent aerobic glycolysis (Warburg effect) is essential for survival of many cancer cells, which often uncouple glycolysis from respiration (28), and is a hallmark of advanced tumors. This prompted us to further investigate changes in glycolysis upon CMA blockade and the contribution of these changes to the reduced proliferative capability of these cells. First, we analyzed the origin of the reduced extracellular lactate, because most proliferating cells have two principal sources of lactate: glycolysis and glutaminolysis. Placement in media low in glucose reduced basal ECAR in all four cell groups and eliminated differences between control and L2A(−) tumor cells (Fig. 4A and fig. S6A), suggesting that differences in glucose metabolism were, to a large extent, behind the reduced ECAR after CMA blockade. Control A549 and H460 cells responded differently to glutamine depletion: ECAR was reduced by 20 to 30% in A549 cells but was consistently higher (50% increase compared to control) in H460 cells. However, ECAR was always lower in L2A(−) cells compared to control cells (about 60% decrease; P < 0.01 to 0.001) (Fig. 4A). In all cells, restriction of either glucose or glutamine almost eliminated the increase in ECAR observed upon blockade of mitochondrial oxidative phosphorylation (fig. S6B). Overall, these findings support the notion that glutaminolysis contributes to the generation of extracellular lactate in A549 cells and that, upon CMA blockade, this pathway was not as markedly affected as glycolysis. H460 cells use glutamine only in response to blockade in mitochondrial respiration but not under normal conditions (Fig. 4A and fig. S6B). Addition of glutamine after glutamine depletion was able to restore levels of ECAR in L2A(−) cells to those observed in complete media. However, even in the presence of an excess of glutamine (double molar concentration), ECAR rates did not reach those of control cells, not even after inhibition of mitochondrial phosphorylation (fig. S6C). Similar results were observed when cell proliferation was analyzed (fig. S6D).
Fig. 4.
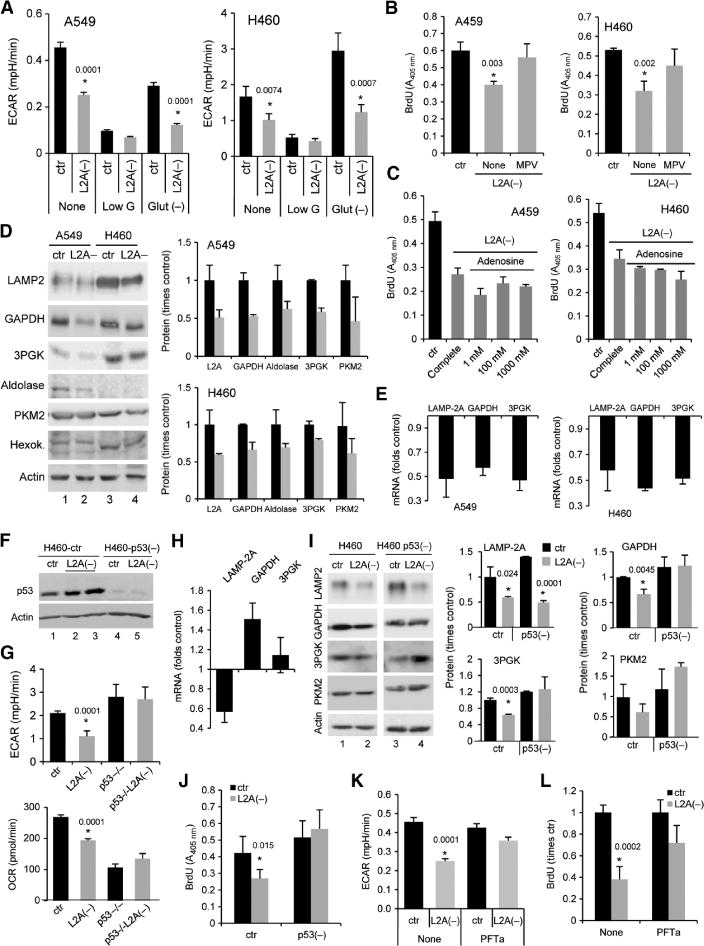
Abnormally increased p53 is responsible for reduced glycolytic metabolism and compromised proliferative capability after CMA blockade in cancer cells. (A) ECAR in A549 and H460 cells, control (ctr) or with LAMP-2A knocked down [L2A(−)], when maintained in complete media (None), media low in glucose (Low G), or depleted of glutamine [Glut(−)] (n = 4; *P = 0.0001, 0.0001, 0.0074, and 0.0007, t test). (B and C) BrdU incorporation in control and L2A(−) cells maintained in complete media alone (none) or supplemented with MPV (B) or the indicated concentrations of adenosine (C) (n = 3 to 5; *P = 0.003 and 0.002, t test). (D) Immunoblots for the indicated glycolytic enzymes in control and L2A(−) human lung cancer cell lines. Left: representative immunoblots. Right: protein concentrations expressed as fold increase over control cells (n = 3 to 5). (E) mRNA levels of the indicated glycolytic enzymes in the same cells. Values are expressed as fold increase over values in control cells after normalization to β-actin. LAMP-2A mRNA levels are shown as reference. (F) Immunoblot for p53 in control and L2A(−) H460 after RNAi knockdown of p53. (G) Mean basal ECAR (top) and OCR (bottom) in the same cells (n = 8; *P = 0.0001 and 0.0001, t test). (H) Changes in mRNA levels for the indicated proteins in L2A(−) cells after p53 knockdown (n = 3). (I) Immunoblot for the indicated proteins in control and L2A(−) cells, either control or with p53 knocked down. Left: representative immunoblot. Right: quantification of the changes in protein levels relative to those in control cells (n = 3). (J) BrdU incorporation in the same cells (n = 3; *P = 0.015, t test). (K) Mean ECAR values in control and L2A(−) A549 cells untreated (none) or treated with pifithrin α (PFTa) (n = 4; *P = 0.0001, t test). (L) BrdU incorporation in the same cells (n = 3; *P = 0.002, t test).
Supplementation with twice the normal concentration of glucose in the media failed to restore normal lactate production in L2A(−) cells, suggesting a primary defect in the ability of these cells to metabolize glucose (fig. S6E). Proliferative capability, however, could be at least partially restored in these cells if glycolysis was bypassed by directly supplementing them with its final product methylpyruvate (MPV) (Fig. 4B). Although MPV may improve cell proliferation in part by providing an alternative source of ATP downstream of glycolysis, titration with adenosine to increase intracellular ATP failed to restore proliferation (Fig. 4C), supporting the notion that MPV may be helping to overcome additional metabolic defects [that is, a possible primary defect in the tricarboxylic acid (TCA) cycle, as supported by the OCR changes in these cells]. Furthermore, these findings also support previous studies showing that the importance of high glycolytic metabolism in cancer cells is not merely due to ATP production but also to the generation of by-products and side reactions that favor cellular proliferation. Notably, we did not find significant changes in macroautophagy modulators and macroautophagy activity under the conditions that restored cellular proliferation (lack of effect of MPV supplementation on LC3-II, mTOR, and phosphorylated mTOR is shown in fig. S6, F and G).
The observed inability to restore normal lactate production upon supplementation with higher concentrations of glucose suggested that, among other possibilities, decreased glycolytic capability could be behind the reduced proliferation of cancer cells with compromised CMA. The fact that several key glycolytic enzymes are well-characterized CMA substrates led us to analyze the effect of blocking CMA on concentrations of these glycolytic enzymes in cancer cells. CMA substrates often accumulate in the cytosol of normal cells when CMA is compromised (15). In clear contrast with those previous findings, we observed a marked reduction (40 to 60%) in the cellular levels of glyceraldehyde-3-phosphate dehydrogenase (GAPDH), 3-phosphoglycerate kinase (3PGK), and aldolase (well-characterized CMA substrates), but not hexokinase, also a glycolytic enzyme previously shown not to undergo CMA (Fig. 4D). Using inhibitors of the lysosomal and proteasomal pathway, we confirmed that the reduced concentrations of these enzymes did not result from an increase in their degradation, because they were no longer degraded in lysosomes and we did not observe a switch in their degradation by the proteasome (fig. S4C). Instead, quantitative RT-PCR showed significant decreases (40 to 60% reduction) in the mRNA levels of these enzymes in both L2A(−) A549 and H460 cells (Fig. 4E).
The consistent increase in p53 observed in response to CMA blockade in the human lung cancer cells, the fact that p53 is not mutated in either of these two cancer cell lines, and the growing number of studies supporting a role for p53 in regulating different aspects of cellular metabolism [including down-regulation of glycolytic enzymes (29)] led us to determine whether the increase in p53 observed in L2A(−) cells mediated any of the metabolic differences in these cells. Genetic knockdown of p53 in H460 cells (Fig. 4F) was enough to (i) restore ECAR levels in L2A(−) cells to those observed in control cells (Fig. 4G), (ii) prevent the transcriptional down-regulation of key glycolytic enzymes (Fig. 4H), and (iii) restore these proteins to levels closer to those in control cells (Fig. 4I). Last, reducing p53 was sufficient to increase the proliferation rates of L2A(−) cells to those of control cells (Fig. 4J), likely through restoration of normal glycolytic activity. Reducing p53 resulted in a marked decrease in OCR in both control and L2A(−) cells (Fig. 4G, bottom), further supporting the notion that changes in oxidative phosphorylation are not behind the deficient cellular proliferation observed upon CMA blockade. Although we did not succeed in efficiently knocking down p53 in L2A(−) A549 cells, treatment of these cells with pifithrin α (a chemical inhibitor of p53) increased ECAR and the proliferation of these cells (30% increase compared to untreated cells; P = 0.001) (Fig. 4K). The marked reduction in extracellular lactate and in the concentrations of different glycolytic enzymes observed after LAMP-2A knockdown could not be reproduced by knocking down Atg7, an essential macroautophagy gene, or LAMP-2B, which is highly homologous to LAMP-2A and does not participate in CMA (fig. S7), providing further support that these changes were specific for CMA blockade.
In summary, our studies reveal that blockade of CMA in cancer cells may limit their proliferative capability and compromise their viability at least in part through changes in their glycolytic and oxidative metabolism. The glycolytic changes and the reduced cellular proliferation are consequences of the elevated levels of p53 observed upon CMA blockade.
Human lung cancer cells with compromised CMA are less tumorigenic
Next, we analyzed the effect of blocking CMA on the tumorigenic capability of the human lung cancer cells in vivo. Using A549 (Fig. 5A) and H460 (Fig. 5B) cells in which LAMP-2A was knocked down, we found that subcutaneous xenografts established in athymic nude mice grew markedly slower than xenografts from corresponding control cells. Analysis of the levels of LAMP-2A in individual tumors by either immunoblot or immunofluorescence confirmed that the efficiency of the knockdown was maintained in the tumor cells (Fig. 5, A and B, insets). Blockade of macroautophagy by atg7 knockdown did not have a pronounced effect on tumor growth in these cells (Atg7 was efficiently knocked down in the growing tumors and LC3-II lipidation was reduced in most of them, confirming efficient blockade of macroautophagy) (fig. S7D).
Fig. 5.
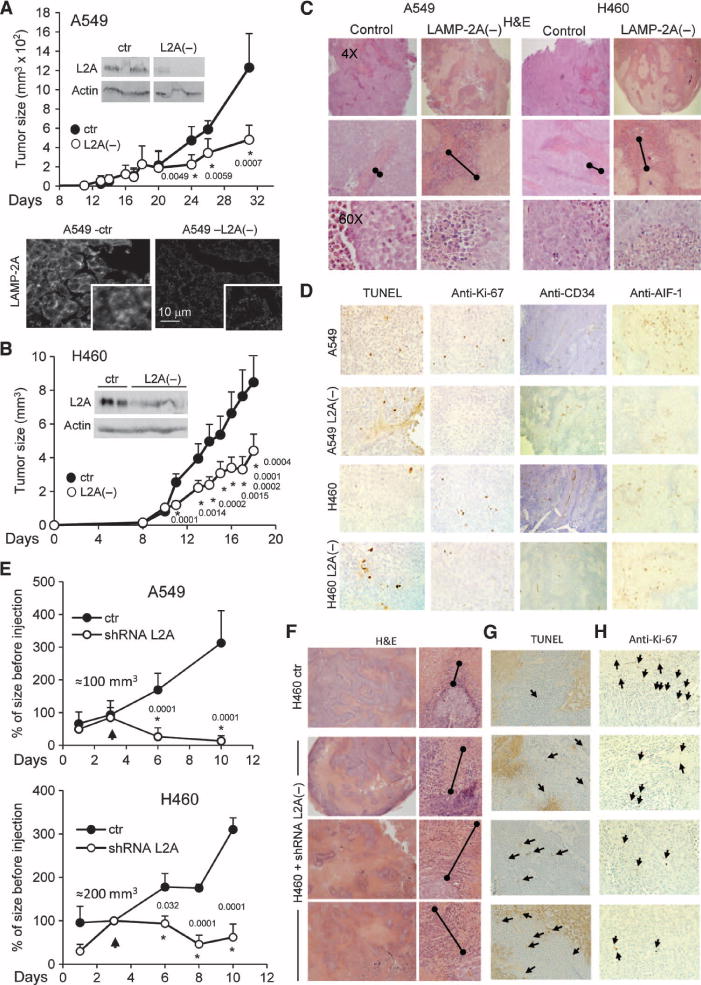
Blockade of CMA reduces tumor formation in xenograft mouse models and induces shrinkage of preexisting tumors. (A to D) Two human lung cancer cell lines, control (ctr) or with LAMP-2A knocked down [L2A(−)], were subcutaneously injected into nude mice, and their ability to form tumors was monitored. Rates of tumor growth of explants from A549 (A) and H460 (B) cell lines. Insets show immunoblots for LAMP-2A (L2A) in the tumors at the time of resection, and micrographs show examples of immunostaining for L2A in tumors from control cells and cells with LAMP-2A knocked down (n = 6 to 9 mice; *P = 0.0001 to 0.0059 as labeled, t test). Sections of individual tumors of each cell type were stained for H&E (C), TUNEL, Ki-67, CD34, and AIF-1 (D). Double-capped lines mark the area of debris from the border of the tumor. (E to H) Tumors grown from xenografts of human lung cancer cells of sizes up to 100 to 200 mm3 were directly injected with lentivirus carrying shRNA against LAMP-2A on 2 consecutive days. (E) Mean values of rates of tumor growth relative to the size of the tumor at first injection (n = 3 to 7 mice; *P = 0.0001 to 0.032 as labeled, t test). Arrows indicate time of injection. (F) H&E staining of sections of tumors from H460 human lung cancer cell xenografts unmodified (ctr) or after injection with the shRNA against LAMP-2A. Double-capped lines mark the area of debris from the border of the tumor. Staining for TUNEL (G) and immunostaining for Ki-67 (H) in the same tumors are shown. Arrows indicate positive cells.
Histological analysis of the tumors revealed a higher content of cellular debris at the periphery of tumor cells in which LAMP-2A had been knocked down [Fig. 5C; necrotic area was three times higher in L2A(−) xenografts]; these cells also showed discretely stronger TUNEL (terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick end labeling) staining (Fig. 5D) and lower reactivity for the cell proliferation marker Ki-67 (Fig. 5D). Staining for the endothelial cell marker CD34 (Fig. 5D) did not reveal differences in vascular supply between control tumors and tumors treated with the shRNA against LAMP-2A. Numbers of tumor-infiltrating inflammatory cells (Fig. 5C), including cells of the monocyte lineage expressing the allograft inflammatory factor–1 (AIF-1) (Fig. 5D), were also similar throughout the tumors. This argued against a stronger immune response being the reason for more cell death in these tumors. In summary, reduced proliferation and increased death of the human lung cancer cells seemed to be the main cause of the slower growth rates for tumors in which LAMP-2A has been knocked down. The antitumorigenic effect of LAMP-2A knockdown was not limited to human lung cancer cells, because we found similar effects on the growth of tumors from mouse melanoma cells (fig. S8). Knockdown of LAMP-2A by RNAi in the mouse melanoma cell line B16:F10 reduced cell proliferation and colony formation (fig. S8, A and B). Xenografts of this and a second melanoma line of lower metastatic potential (B16:F1) grew slower upon CMA blockade (fig. S8, C to F), whereas suppression of macroautophagy by knocking down Atg7 with RNAi did not affect their growth (fig. S8F).
Overall, our results suggest that blockade of CMA inhibited proliferation and stimulated apoptosis of lung and melanoma cancer cells both in culture and in vivo.
Blockade of LAMP-2A reduces the metastatic capacity of lung cancer cells
We next investigated whether the slower growth of tumors from lung cancer cells with reduced CMA activity could affect their ability to form metastases. Although the metastatic potential of the lung cancer cells used in this study is relatively low, fluorescence-activated cell sorting (FACS) analysis of the blood of mice with xenografts in their foot fat pad compared with control and LAMP-2A RNAi green fluorescent protein (GFP)–labeled A549 and H460 cells allowed us to detect a lower number of circulating cancer cells at any given time in animals injected with L2A(−) cancer cells [Fig. 6A; 72% and 75% decrease in circulating cells for L2A(−) A549 and H460 cells, respectively; P = 0.0001; values were corrected for the relative size of the primary tumor]. In agreement with these findings, histological analysis of the lungs of these animals showed a significant decrease in the amount of spontaneous metastasis in this tissue (Fig. 6A). Likewise, grafts of the mouse metastatic melanoma cell line B16:F10 also showed lower blood burden and fewer lung metastases after LAMP-2A knockdown (fig. S9A). Genetic blockade of macroautophagy reduced the number of circulating cells but did not result in a significant reduction in the number of lung metastases (fig. S9A), further reinforcing the different effects after blocking CMA and macroautophagy in these cells.
Fig. 6.
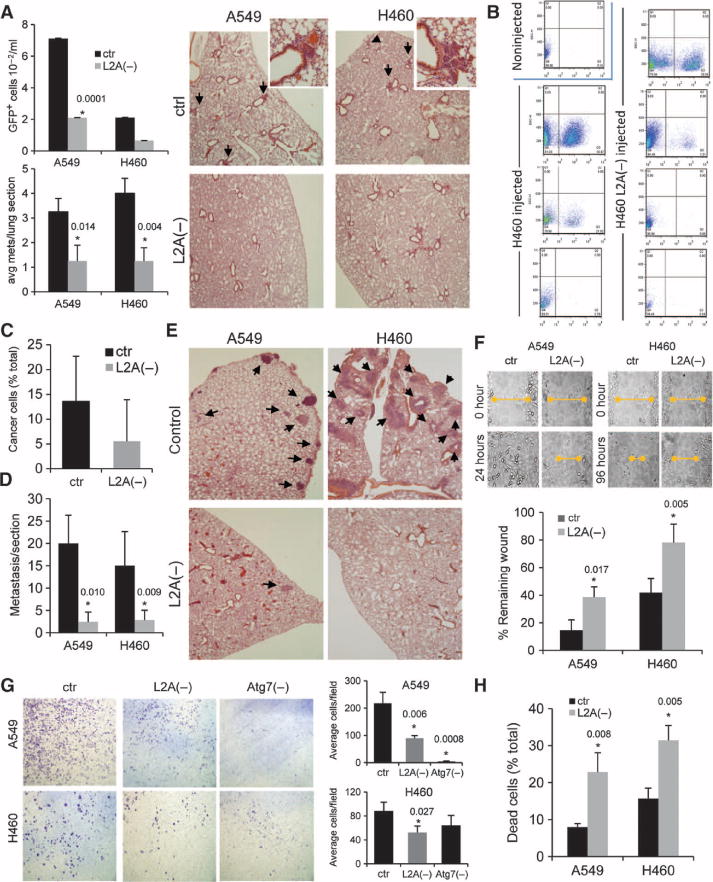
Inhibition of CMA reduces formation of human lung tumor metastases. (A) Nude mice were injected in the foot fat pad with A549 and H460 cells, either control or after LAMP-2A knockdown [L2A(−)]. The number of human cancer cells (detected by FACS analysis as GFP fluorescent cells) (top left) and the average number of metastases per lung section (bottom left) was quantified (n = 5 to 6 mice; *P = 0.0001, 0.014, and 0.004, t test). Right: representative H&E-stained sections of lungs. Arrows indicate cancer cell foci. (B to D) Nude mice were injected via the tail vein with H460 human lung cancer cells, either control or with LAMP-2A knocked down, and the lungs were subjected to treatment with collagenase to promote cell dissociation. The number of human cancer cells (detected as GFP fluorescent cells) was determined by FACS. Representative sorting plots of lungs from three to four different mice injected or uninjected with the different cell types (B) and mean values of the percentage of cancer cells in the total amount of cells sorted (C) (n = 3 to 5 animals). (D) Quantification of the number of lung metastases per mouse in sections (n = 3 to 5 mice; *P = 0.010 and 0.009, t test). (E) H&E-stained sections of the lungs from the same animals. Arrows mark areas of metastatic lesions. (F) Time course of wound closing in human lung cancer cells, either control or with LAMP-2A knocked down. Top: representative images of the size of the wound at different times. Bottom: quantification of the size of the wound at 24 hours. Values are expressed as percentage of the initial wound remaining (n = 3 to 4; *P = 0.017 to 0.005, t test). (G) Migration of human lung cancer cells, either control or with LAMP-2A and Atg7 knocked down, in the Transwell migration assay. Left: representative images of cells detected on the bottom side of the filter. Right: quantification of the number of cells detected per field (n = 3; *P = 0.006, 0.0008, and 0.027, t test). (H) Percentage of dead cells detected after preventing attachment of control and L2A(−) human lung cancer cells. Values are expressed as percentage of cells seeded (n = 3; *P = 0.008 to 0.005, t test).
To determine whether the decrease in the number of metastases after CMA inhibition resulted from changes in the primary tumor at the implantation site or when the cancer cells reached the bloodstream, we used an experimental lung metastasis model in which lung tumor cells were injected into the tail vein of mice. After injection of the same numbers of viable lung tumor cells, FACS analysis of lung cells dissociated by collagenase treatment revealed markedly lower numbers of human cancer cells in the lungs of mice injected with L2A(−) H460 cells (Fig. 6, B and C). In addition, histological analysis revealed a marked decrease in the number of metastatic foci in the lungs of mice injected with L2A(−) A549 and H460 cells (Fig. 6, D and E).
Of the multiple factors that could contribute to the reduction in metastasis number, we examined possible changes in cellular motility and resistance to cell death induced by detachment or anoikis. Cancer cells with a compromised CMA had a lower migratory capability as determined by slower closure of “wounds” made by scraping tumor cells from culture dishes (Fig. 6F and fig. S9, B and C). There was also a marked reduction in the number of cells crossing to the lower chamber in the Transwell migration assay in L2A(−) tumor cells compared to control tumor cells (Fig. 6G). Blockade of macroautophagy also reduced the migratory capability of both types of human lung cancer cells (Fig. 6G), whereas knockdown of LAMP-2B did not significantly affect cell motility (fig. S9D). Macroautophagy has been recently shown to protect against apoptosis after anoikis in breast cancer cells (30). Blockade of macroautophagy through knockdown of Atg7 did not increase death of human lung cancer cells maintained in suspension (to reproduce detachment conditions) over that observed in control cells (fig. S9E). However, knockdown of LAMP-2A, but not LAMP-2B, increased cell death by 2.5-fold (P < 0.01) (Fig. 6H and fig. S9E). These results suggest that CMA may help to protect some types of cancer cells against anoikis and could explain the lower blood burden and metastatic capacity of human lung cancer cells after CMA blockade.
Down-regulation of CMA reduces the size of preexisting lung tumors
We next investigated whether administration of replication-defective lentivirus carrying shRNA against LAMP-2A blocks the growth of preexisting xenograft-derived tumors in vivo. We proposed that the metabolic compromise induced by blockade of CMA in cancer cells may not only reduce their proliferation but could also compromise their ability to sustain cellular viability in the context of a solid tumor where nutrient and oxygen supply is limited. Both A549 and H460 tumors were established in subcutaneous sites in mice and were injected with the lentivirus (two injections on consecutive days) after they had reached 100 or 200 mm3, respectively. Tumors treated with LAMP-2A shRNA virus but not control virus showed rapid tumor regression with negative slopes of growth, and some tumors were no longer palpable 10 days after the viral injection (Fig. 5E and fig. S10). Histological analysis revealed the presence of extensive areas of necrosis in the LAMP-2A shRNA–injected tumors compared to tumors that originated from cells in which LAMP-2A was knocked down ex vivo (Fig. 5F). Tumor burden in blood remained undetectable after injection of tumors with shRNA, making it unlikely that increased extravasation (exit of cells from tissue to the bloodstream) was a cause of primary tumor shrinkage. The increase in TUNEL staining (Fig. 5G) and lower reactivity for Ki-67 (Fig. 5H) provide evidence that the reduction in tumor size upon CMA blockade is due to an increase in cell death and reduced rates of cancer cell proliferation.
Overall, our results suggest that blockade of CMA in lung cancer cells compromises their proliferative capacity, at least in part by interfering with glycolytic metabolism, and that the antitumorigenic effect of CMA inhibition has profound effects on tumor progression in vivo when this autophagic pathway is inhibited in tumors.
DISCUSSION
Here, using cancer cell lines of similar origin but with very different macroautophagic activities, we have investigated the role of a form of inducible autophagy, CMA, on cancer biology. On the basis of previous studies in normal cells, we anticipated that in cancer cells, CMA activity would change in a reverse direction relative to macroautophagy (15, 16). However, to our surprise, the seven cancer cell lines analyzed in this study displayed constitutive activation of CMA, despite the marked differences in macroautophagy. Induction of CMA was also observed after oncogenic transformation of primary cells, and the limiting CMA component LAMP-2A was up-regulated in 32 of the 41 different types of human tumor samples analyzed in this study. We demonstrate here that enhanced CMA activity is necessary to maintain the high proliferative capability of lung and melanoma cancer cells, because reduction in CMA activity through partial knockdown of an essential component of this pathway reduced cellular proliferation and tumor growth, and increased cellular death. In the context of lung cancer cells, CMA up-regulation is not as essential for the maintenance of quality control, but it is key for the support of aerobic glycolysis, which is characteristic of cancer cells (fig. S11).
Recent studies have shown that the antitumorigenic effect of macroautophagy is, to a large extent, dependent on the failure of macroautophagy-defective cells to eliminate damaged and stress-related molecules, such as p62 (19). The mechanism by which blockade of CMA prevents tumorigenesis, at least in lung cancer cells, is different, because we did not find accumulation of p62 or oxidized and aggregated proteins in these cells. Our results in cancer cells also differ compared with the effect of CMA blockade in normal cells, which respond to this blockade with the up-regulation of macroautophagy (15). In contrast, we did not find significant changes in macroautophagy upon inhibition of CMA in cancer cells. Cancer cells may have developed mechanisms to disrupt the autophagic cross-talk to be able to modulate macroautophagy without affecting the sustained constitutive activation of CMA, which seems to be required for proliferation. However, when CMA is compromised, cancer cells compensate for the loss of CMA-dependent quality control by up-regulating the proteasome system. Previous reports have documented common substrates for CMA and the proteasome (that is, IκBα, c-fos, aldolase, and α-synuclein) (31–33), and both participate in the removal of oxidized proteins (34–38). The compensatory proteasomal up-regulation in cancer cells with compromised CMA could thus be responsible for the lower concentrations of oxidized proteins in these cells. It would be of particular interest to further elucidate the molecular mediators of proteasome/CMA cross-talk in cancer cells, because they could become useful anti-oncogenic targets. In that respect, different catalytic and regulatory subunits of the proteasome can be degraded by CMA (39), making it possible that, under conditions of CMA blockade, stabilization of the proteasome subunits would lead to the observed increase in the activity of this system.
Blockade of CMA in cancer cells resulted in changes in different metabolic pathways. Because of the importance of glycolytic metabolism for cancer cells, we analyzed the mechanism behind the reduced glycolysis observed in CMA-compromised cancer cells. In agreement with the accepted idea that up-regulation of glycolysis in cancer cells is not just for ATP production but also contributes to the generation of by-products and metabolites for macromolecular biosynthesis that support cell growth, we found that an additional supply of energy in the form of adenosine was insufficient to sustain proliferation of CMA-compromised cancer cells. Our data support the notion that compromised glycolysis is a consequence of a reduction in concentrations of glycolytic enzymes and that the increased levels of p53 in these cells cause these changes. Because many glycolytic enzymes have been described as CMA substrates (14, 40), we expected that blockade of CMA would lead to an increase in their intracellular levels and enhanced glycolysis. However, we found the opposite: Levels of glycolytic enzymes, particularly those described as CMA substrates, decreased in cancer cells upon CMA blockade. Recent studies have described an inhibitory effect of p53 on glycolytic enzymes such as GAPDH through direct protein-protein interactions (41). Although we cannot ignore the possibility of a similar effect of p53 in the CMA-compromised cells, we have found that in these cells down-regulation of these enzymes occurs at least in part at the transcriptional level and in a p53-dependent manner. The exact mechanism by which p53 induces transcriptional down-regulation of glycolytic enzymes in these cells requires further investigation, but it is interesting that hypoxia-inducible factor 1α (HIF-1α), which was consistently reduced in these cells (Fig. 2I), is a strong transcriptional activator of these glycolytic enzymes (28). The decrease in levels of HIF-1α in these cells could underlie the transcriptional down-regulation of the glycolytic enzymes. Recent studies have shown that CMA contributes to the turnover of the glycolytic enzyme pyruvate kinase muscle isozyme 2 (PKM2), which is commonly expressed in cancer, and failure to degrade this enzyme by CMA favors cellular growth (42). It is possible that blockade of CMA in the human cancer cell lines used in this study could still reduce the turnover of PKM2 but that accumulation of this protein is not observed because of the overall p53-dependent transcriptional down-regulation of glycolytic enzymes in these cells. In agreement with the changes observed for the other glycolytic enzymes previously described as substrates for CMA, levels of PKM2 also decreased in human lung cancer cells upon CMA blockade (Fig. 4D) in a p53-dependent manner (Fig. 4I).
As described for other anti-oncogenic interventions, it is likely that more than one mechanism contributes to the antitumorigenic effect observed upon CMA blockade in preformed lung tumors. Both p53-dependent and p53-independent processes can contribute to the metabolic alterations, which in turn will reduce the proliferative capacity of the cancer cells, limiting tumor growth and increasing cell death rates in the aggressive tumor microenvironment, leading to tumor shrinkage (fig. S11). In this respect, it is noteworthy that CMA activity was up-regulated in cancer cell lines independent of their p53 status (A549, H460, MCF7, and B16:F10 cells express wild-type p53; HuH-7 cells express mutant p53; and Saos cells are null for p53). However, the cells used in the xenograft studies were all wild type for this tumor suppressor. Given the new roles of p53 in metabolism and the growing evidence supporting functions for this protein beyond the nucleus, it is possible that some of the metabolic effects of p53 are preserved or even enhanced in mutant forms of the protein, and that CMA would also be crucial for down-regulating p53 in these cancer cells.
Overall, our findings highlight a previously unknown dependence of several cancer cell types on CMA activation, which favors tumorigenesis. The marked effects of compromised CMA on tumor growth and, in particular, on the size of preexisting tumors support the feasibility of manipulating this autophagic pathway to prevent tumorigenesis and favor tumor regression. We propose that CMA could be a druggable target for anticancer therapy and that selective blockade of CMA in some types of cancer cells could be the basis for anticancer therapeutic intervention. Although blockade of CMA in tumor cells may not be sustainable for very prolonged periods of time, most nontumor cells respond to CMA blockade by up-regulating macroautophagy, which guarantees cellular survival under basal conditions. According to our findings, cancer cells are at a disadvantage when CMA is blocked, in part because they do not up-regulate macroautophagy under these conditions, and in part due to their higher dependence on glucose metabolism. Knockdown of LAMP-2A may be useful in those cases in which shRNAs can be efficiently delivered to the tumor in a local or targeted manner. Keeping in mind that a number of different human tumors were found to have up-regulated CMA, the future development of chemical modifiers capable of blocking CMA may be a broadly useful anti-oncogenic therapy.
MATERIALS AND METHODS
Animals, cells, and reagents
Nu/Nu athymic male mice and C57BL/6 male mice were used. Cancer cell lines were from the American Type Culture Collection. MEFs were prepared in our laboratory. Antibodies and reagents were as described previously (11, 15, 43). Sources of chemicals and antibodies were as described previously (11, 15, 43).
Autophagic measurements
Intracellular protein degradation was measured by metabolic labeling and pulse-chase experiments as described before (26). Autophagic flux was measured as changes in level of LC3-II upon inhibition of lysosomal hydrolases. CMA activity was determined with the photoactivatable KFERQ-PA-mCherry1 reporter that allows visualization of lysosomes as fluorescent puncta as CMA activity increases (25). An in vitro assay with isolated lysosomes was also used to determine CMA activity defined as the ability of intact lysosomes to take up and degrade a pool of radiolabeled cytosolic proteins as described before (26). Lysosomes were isolated from cultured cells by differential centrifugation and centrifugation in discontinuous metrizamide/Percoll gradients of a cell lysate obtained by rupture of the plasma membrane through nitrogen cavitation (44).
Lentivirus-mediated shRNA
Lentivirus-based constructs carrying shRNA against LAMP-2A, Atg7, LAMP-2B, and p53 were generated as described before and delivered to cultured cells upon packaging into replication-deficient lentiviral particles (15). Where indicated, the same virus was directly injected into 100- to 200-mm3 tumors grown from subcutaneous xenografts in mice.
Cellular proliferation and viability
Rates of cellular proliferation were determined as incorporation of BrdU 30 to 60 hours after plating and as their ability to form colonies after dilution in 96-well plates to 0.5 cell per well. Cellular death was determined by TUNEL, dual labeling with acridine orange/ethidium bromide, and staining for annexin V–phycoerythrin (PE) and FACScan analysis.
Metabolic measurements
Intracellular ATP and extracellular secretion of lactate were determined with the ATP Bioluminescence Assay Kit HS II (Roche Diagnostics) and the Lactate Assay Kit (MBL International), respectively. Measurement of ECAR, as well as OCR, was performed in XF96 plates with XF Extracellular Flux Analyzer (Seahorse Bioscience). Rates of fatty acid β-oxidation were determined by a modification of a previously used method (45).
Assessment of xenograft tumor growth, cell motility, and metastasis
Xenografts of human cancer cells in mice were generated by subcutaneous injection of 1 × 106 cells into their right flank or injection of 5 × 105 cells into their foot fat pad. Tumor growth was monitored by periodical caliper measurement of the tumor. Metastases were induced by direct injection of 1 × 106 cancer cells in the tail vein of nude mice. The number of spontaneous and induced metastatic cells in lungs was determined by histochemical analysis of their lungs or by FACS analysis of GFP-positive cells in the collagenized lungs. Cell motility was determined by the in vitro wound-healing and the Transwell invasion assays using the Boyden chambers.
Histological procedures
Tumor specimens from human and mouse were analyzed under a light microscope after hematoxylin and eosin (H&E) staining. Immunostaining of the paraffin-embedded sections was performed after dewaxing and antigen retrieval and incubation with the antibody of interest as previously described (46).
General methods
Protein concentration was determined by the Lowry method (47). β-Galactosidase expression levels were determined with the Senescence Kit (BioVision), and carbonyl groups were detected with the OxyBlot Oxidized Protein Detection Kit (Chemicon International). After SDS–polyacrylamide gel electrophoresis and immunoblot, gels were scanned in an Image Analyzer System (Innotech S-100). The ImageJ software (National Institutes of Health) was used for densitometric quantification of the scanned gels. Student’s t test for unpaired data and analysis of variance (ANOVA) followed by the Bonferroni post hoc test were used for statistical analysis.
Supplementary Material
Materials and Methods
Fig. S1. Autophagic/lysosomal system in human lung cancer cells.
Fig. S2. Levels of LAMP-2A in different types of human cancers.
Fig. S3. Immunostaining for LAMP-2A and LAMP-2B in different types of skin and lung human cancers.
Fig. S4. Effect of CMA blockage on stress resistance and intracellular proteolysis of human lung cancer cells.
Fig. S5. Effect of CMA blockage on mitochondrial activity and content of human lung cancer cells.
Fig. S6. Metabolic changes in CMA-compromised cancer cells in response to different supplementations.
Fig. S7. Effect of genetic blockage of macroautophagy in human lung cancer cells on metabolism and tumor growth.
Fig. S8. Effect of CMA blockage on proliferation and melanoma cell tumor growth.
Fig. S9. Effect of macroautophagy and CMA blockage on cancer cell motility and resistance to anoikis.
Fig. S10. Effect of CMA blockage on growth of preformed human lung cancer cell tumors.
Fig. S11. Hypothetical model of the consequences of CMA inhibition in human cancer cells.
References
Acknowledgments
We thank R. Sellers (Histopathology Core, Albert Einstein College of Medicine) for her assistance in the assessment of the stained tumor sections; the Stable Isotope and Metabolomics Core for their assistance with the metabolic analysis; A. Wolkoff and M. Olaywi for their help in accessing the human samples from the Clinical and Translational Science Award (CTSA) tissue repository of the Division of Gastroenterology at Einstein; E. Arias for providing the shRNA for tumor injection; B. T. Abe for all his guidance with different procedures; S. J. Orenstein for performing the mouse injections; R. Singh for his expert advice on the metabolic studies; and S. Kaushik and S. J. Orenstein for their critical evaluation of this manuscript.
Funding: Supported by NIH grants AG021904 (A.M.C.) and AG031782 (A.M.C. and F.M.), a Hirschl/Weill-Caulier Career Scientist Award (A.M.C.), a CTSA (UL1 RR025750), the Albert Einstein Cancer Center grant NCI PO1 13330, and TG32GM007288 (M.K.).
Footnotes
Author contributions: M.K. performed the experiments, analyzed the data, and contributed to writing the paper. R.K. performed the RT-PCR analysis and H.K. the studies on CMA in transformed cells. J.C. provided the human samples for the study. F.M. assisted with the proliferation, cell death, and cell cycle experiments, discussed experiments and interpretation, and revised the manuscript. L.V. provided reagents and helped with the design of experiments and revised the manuscript. A.M.C. designed the experiments, directed the study, and wrote the manuscript.
Competing interest: The authors declare that they have no competing interests.
REFERENCES AND NOTES
- 1.Yang Z, Klionsky DJ. Eaten alive: A history of macroautophagy. Nat Cell Biol. 2010;12:814–822. doi: 10.1038/ncb0910-814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–1075. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mizushima N. The pleiotropic role of autophagy: From protein metabolism to bactericide. Cell Death Differ. 2005;12(Suppl. 2):1535–1541. doi: 10.1038/sj.cdd.4401728. [DOI] [PubMed] [Google Scholar]
- 4.Yang Z, Klionsky DJ. Mammalian autophagy: Core molecular machinery and signaling regulation. Curr Opin Cell Biol. 2010;22:124–131. doi: 10.1016/j.ceb.2009.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cuervo AM. Chaperone-mediated autophagy: Selectivity pays off. Trends Endocrinol Metab. 2010;21:142–150. doi: 10.1016/j.tem.2009.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chiang HL, Terlecky SR, Plant CP, Dice JF. A role for a 70-kilodalton heat shock protein in lysosomal degradation of intracellular proteins. Science. 1989;246:382–385. doi: 10.1126/science.2799391. [DOI] [PubMed] [Google Scholar]
- 7.Dice JF. Peptide sequences that target cytosolic proteins for lysosomal proteolysis. Trends Biochem Sci. 1990;15:305–309. doi: 10.1016/0968-0004(90)90019-8. [DOI] [PubMed] [Google Scholar]
- 8.Cuervo AM, Dice JF. A receptor for the selective uptake and degradation of proteins by lysosomes. Science. 1996;273:501–503. doi: 10.1126/science.273.5274.501. [DOI] [PubMed] [Google Scholar]
- 9.Salvador N, Aguado C, Horst M, Knecht E. Import of a cytosolic protein into lysosomes by chaperone-mediated autophagy depends on its folding state. J Biol Chem. 2000;275:27447–27456. doi: 10.1074/jbc.M001394200. [DOI] [PubMed] [Google Scholar]
- 10.Agarraberes FA, Dice JF. A molecular chaperone complex at the lysosomal membrane is required for protein translocation. J Cell Sci. 2001;114(Pt. 13):2491–2499. doi: 10.1242/jcs.114.13.2491. [DOI] [PubMed] [Google Scholar]
- 11.Bandyopadhyay U, Sridhar S, Kaushik S, Kiffin R, Cuervo AM. Identification of regulators of chaperone-mediated autophagy. Mol Cell. 2010;39:535–547. doi: 10.1016/j.molcel.2010.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bandyopadhyay U, Kaushik S, Varticovski L, Cuervo AM. The chaperone-mediated autophagy receptor organizes in dynamic protein complexes at the lysosomal membrane. Mol Cell Biol. 2008;28:5747–5763. doi: 10.1128/MCB.02070-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell. 2004;15:1101–1111. doi: 10.1091/mbc.E03-09-0704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cuervo AM, Knecht E, Terlecky SR, Dice JF. Activation of a selective pathway of lysosomal proteolysis in rat liver by prolonged starvation. Am J Physiol. 1995;269:C1200–C1208. doi: 10.1152/ajpcell.1995.269.5.C1200. [DOI] [PubMed] [Google Scholar]
- 15.Massey AC, Kaushik S, Sovak G, Kiffin R, Cuervo AM. Consequences of the selective blockage of chaperone-mediated autophagy. Proc Natl Acad Sci USA. 2006;103:5805–5810. doi: 10.1073/pnas.0507436103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kaushik S, Massey AC, Mizushima N, Cuervo AM. Constitutive activation of chaperone-mediated autophagy in cells with impaired macroautophagy. Mol Biol Cell. 2008;19:2179–2192. doi: 10.1091/mbc.E07-11-1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang Y, Martinez-Vicente M, Krüger U, Kaushik S, Wong E, Mandelkow EM, Cuervo AM, Mandelkow E. Tau fragmentation, aggregation and clearance: The dual role of lysosomal processing. Hum Mol Genet. 2009;18:4153–4170. doi: 10.1093/hmg/ddp367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Martinez-Vicente M, Talloczy Z, Kaushik S, Massey AC, Mazzulli J, Mosharov EV, Hodara R, Fredenburg R, Wu DC, Follenzi A, Dauer W, Przedborski S, Ischiropoulos H, Lansbury PT, Sulzer D, Cuervo AM. Dopamine-modified α-synuclein blocks chaperone-mediated autophagy. J Clin Invest. 2008;118:777–788. doi: 10.1172/JCI32806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mathew R, Karp CM, Beaudoin B, Vuong N, Chen G, Chen HY, Bray K, Reddy A, Bhanot G, Gelinas C, Dipaola RS, Karantza-Wadsworth V, White E. Autophagy suppresses tumorigenesis through elimination of p62. Cell. 2009;137:1062–1075. doi: 10.1016/j.cell.2009.03.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Karantza-Wadsworth V, Patel S, Kravchuk O, Chen G, Mathew R, Jin S, White E. Autophagy mitigates metabolic stress and genome damage in mammary tumorigenesis. Genes Dev. 2007;21:1621–1635. doi: 10.1101/gad.1565707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Qu X, Yu J, Bhagat G, Furuya N, Hibshoosh H, Troxel A, Rosen J, Eskelinen EL, Mizushima N, Ohsumi Y, Cattoretti G, Levine B. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest. 2003;112:1809–1820. doi: 10.1172/JCI20039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Takahashi Y, Coppola D, Matsushita N, Cualing HD, Sun M, Sato Y, Liang C, Jung JU, Cheng JQ, Mulé JJ, Pledger WJ, Wang HG. Bif-1 interacts with Beclin 1 through UVRAG and regulates autophagy and tumorigenesis. Nat Cell Biol. 2007;9:1142–1151. doi: 10.1038/ncb1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ionov Y, Nowak N, Perucho M, Markowitz S, Cowell JK. Manipulation of nonsense mediated decay identifies gene mutations in colon cancer cells with microsatellite instability. Oncogene. 2004;23:639–645. doi: 10.1038/sj.onc.1207178. [DOI] [PubMed] [Google Scholar]
- 24.Wang RC, Levine B. Autophagy in cellular growth control. FEBS Lett. 2010;584:1417–1426. doi: 10.1016/j.febslet.2010.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Koga H, Martinez-Vicente M, Macian F, Verkhusha VV, Cuervo AM. A photoconvertible fluorescent reporter to track chaperone-mediated autophagy. Nat Commun. 2011;2:386. doi: 10.1038/ncomms1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kaushik S, Cuervo AM. Methods to monitor chaperone-mediated autophagy. Methods Enzymol. 2009;452:297–324. doi: 10.1016/S0076-6879(08)03619-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Eskelinen EL, Cuervo AM, Taylor MR, Nishino I, Blum JS, Dice JF, Sandoval IV, Lippincott-Schwartz J, August JT, Saftig P. Unifying nomenclature for the isoforms of the lysosomal membrane protein LAMP-2. Traffic. 2005;6:1058–1061. doi: 10.1111/j.1600-0854.2005.00337.x. [DOI] [PubMed] [Google Scholar]
- 28.Stubbs M, Griffiths J. The altered metabolism of tumors: HIF-1 and its role in the Warburg effect. Adv Enzyme Regul. 2010;50:44–55. doi: 10.1016/j.advenzreg.2009.10.027. [DOI] [PubMed] [Google Scholar]
- 29.Vousden KH, Ryan KM. p53 and metabolism. Nat Rev Cancer. 2009;9:691–700. doi: 10.1038/nrc2715. [DOI] [PubMed] [Google Scholar]
- 30.Fung C, Lock R, Gao S, Salas E, Debnath J. Induction of autophagy during extracellular matrix detachment promotes cell survival. Mol Biol Cell. 2008;19:797–806. doi: 10.1091/mbc.E07-10-1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cuervo AM, Hu W, Lim B, Dice JF. IκB is a substrate for a selective pathway of lysosomal proteolysis. Mol Biol Cell. 1998;9:1995–2010. doi: 10.1091/mbc.9.8.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Aniento F, Papavassiliou AG, Knecht E, Roche E. Selective uptake and degradation of c-Fos and v-Fos by rat liver lysosomes. FEBS Lett. 1996;390:47–52. doi: 10.1016/0014-5793(96)00625-4. [DOI] [PubMed] [Google Scholar]
- 33.Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D. Impaired degradation of mutant α-synuclein by chaperone-mediated autophagy. Science. 2004;305:1292–1295. doi: 10.1126/science.1101738. [DOI] [PubMed] [Google Scholar]
- 34.Kiffin R, Christian C, Knecht E, Cuervo AM. Activation of chaperone-mediated autophagy during oxidative stress. Mol Biol Cell. 2004;15:4829–4840. doi: 10.1091/mbc.E04-06-0477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shringarpure R, Grune T, Davies KJ. Protein oxidation and 20S proteasome-dependent proteolysis in mammalian cells. Cell Mol Life Sci. 2001;58:1442–1450. doi: 10.1007/PL00000787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Keller JN, Hanni KB, Markesbery WR. Possible involvement of proteasome inhibition in aging: Implications for oxidative stress. Mech Ageing Dev. 2000;113:61–70. doi: 10.1016/s0047-6374(99)00101-3. [DOI] [PubMed] [Google Scholar]
- 37.Goldberg AL, Akopian TN, Kisselev AF, Lee DH, Rohrwild M. New insights into the mechanisms and importance of the proteasome in intracellular protein degradation. Biol Chem. 1997;378:131–140. [PubMed] [Google Scholar]
- 38.Finn PF, Dice JF. Ketone bodies stimulate chaperone-mediated autophagy. J Biol Chem. 2005;280:25864–25870. doi: 10.1074/jbc.M502456200. [DOI] [PubMed] [Google Scholar]
- 39.Cuervo AM, Palmer A, Rivett AJ, Knecht E. Degradation of proteasomes by lysosomes in rat liver. Eur J Biochem. 1995;227:792–800. doi: 10.1111/j.1432-1033.1995.tb20203.x. [DOI] [PubMed] [Google Scholar]
- 40.Aniento F, Roche E, Cuervo AM, Knecht E. Uptake and degradation of glyceraldehyde-3-phosphate dehydrogenase by rat liver lysosomes. J Biol Chem. 1993;268:10463–10470. [PubMed] [Google Scholar]
- 41.Jiang P, Du W, Wang X, Mancuso A, Gao X, Wu M, Yang X. p53 regulates biosynthesis through direct inactivation of glucose-6-phosphate dehydrogenase. Nat Cell Biol. 2011;13:310–316. doi: 10.1038/ncb2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lv L, Li D, Zhao D, Lin R, Chu Y, Zhang H, Zha Z, Liu Y, Li Z, Xu Y, Wang G, Huang Y, Xiong Y, Guan KL, Lei QY. Acetylation targets the M2 isoform of pyruvate kinase for degradation through chaperone-mediated autophagy and promotes tumor growth. Mol Cell. 2011;42:719–730. doi: 10.1016/j.molcel.2011.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Martinez-Vicente M, Talloczy Z, Wong E, Tang G, Koga H, Kaushik S, de Vries R, Arias E, Harris S, Sulzer D, Cuervo AM. Cargo recognition failure is responsible for inefficient autophagy in Huntington’s disease. Nat Neurosci. 2010;13:567–576. doi: 10.1038/nn.2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Storrie B, Madden EA. Isolation of subcellular organelles. Methods Enzymol. 1990;182:203–225. doi: 10.1016/0076-6879(90)82018-w. [DOI] [PubMed] [Google Scholar]
- 45.Hoppel C, DiMarco JP, Tandler B. Riboflavin and rat hepatic cell structure and function. Mitochondrial oxidative metabolism in deficiency states. J Biol Chem. 1979;254:4164–4170. [PubMed] [Google Scholar]
- 46.Zhang C, Cuervo AM. Restoration of chaperone-mediated autophagy in aging liver improves cellular maintenance and hepatic function. Nat Med. 2008;14:959–965. doi: 10.1038/nm.1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Materials and Methods
Fig. S1. Autophagic/lysosomal system in human lung cancer cells.
Fig. S2. Levels of LAMP-2A in different types of human cancers.
Fig. S3. Immunostaining for LAMP-2A and LAMP-2B in different types of skin and lung human cancers.
Fig. S4. Effect of CMA blockage on stress resistance and intracellular proteolysis of human lung cancer cells.
Fig. S5. Effect of CMA blockage on mitochondrial activity and content of human lung cancer cells.
Fig. S6. Metabolic changes in CMA-compromised cancer cells in response to different supplementations.
Fig. S7. Effect of genetic blockage of macroautophagy in human lung cancer cells on metabolism and tumor growth.
Fig. S8. Effect of CMA blockage on proliferation and melanoma cell tumor growth.
Fig. S9. Effect of macroautophagy and CMA blockage on cancer cell motility and resistance to anoikis.
Fig. S10. Effect of CMA blockage on growth of preformed human lung cancer cell tumors.
Fig. S11. Hypothetical model of the consequences of CMA inhibition in human cancer cells.
References