

Abstract
Mature Large Granular lymphocytes (LGL) disorders include a spectrum of conditions, ranging from polyclonal to clonal indolent and/or overt leukemic LGL proliferations. Most cases are represented by clonal expansions of TCRα/β+ LGL displaying a CD8+ phenotype with expression of cytotoxic T-cell antigens (CD57, CD16, TIA-1, perforin and granzyme B). Proliferations of CD3-CD16+ NK cells with a restricted patter of NK receptors are less common, usually comprising 15% of the cases. Main features are cytopenias, splenomegaly and autoimmune phenomena. Morphology, immunophenotyping and molecular analyses are crucial to establish a correct diagnosis of disease. According to the 2008 WHO classification, two separate entities account for the majority of cases, T-LGL leukemia and Chronic Lymphoproliferative Disease of NK cell (this latter still provisional). Although these disorders are characterized by the expansion of different cells types i.e. T and NK cells, with specific genetic features and abnormalities, compelling evidence supports the hypothesis that a common pathogenic mechanism would be involved in both disorders. As a matter of fact, a foreign antigen driven clonal selection is considered the initial step in the mechanism ultimately leading to generation of both conditions. In this chapter we will discuss recent advances on the pathogenesis of chronic T and NK disorders of granular lymphocytes, challenging the current WHO classification on the opportunity to separate T and NK disorders, which are likely to represent two sides of the same coin.
Keywords: CTL, NK cells, T-LGL leukemia, CLPD-NK
I. INTRODUCTION
Large Granular lymphocytes (LGL) disorders include a spectrum of conditions, ranging from polyclonal to clonal indolent and/or overt leukemic LGL proliferations. Since its original description, large granular lymphocyte leukemia (LGLL) has represented a matter of controversy, as to whether we are dealing with a lymphoid malignancy or with an exaggerated reactive T-cell process [1-3]. LGLL is actually defined as a clonal chronic lymphoproliferative disorder that can be phenotypically subdivided into T-cell LGLL and natural killer LGLL. Although derived from distinct cell lineages, both subtypes are morphologically similar and are characterized by the accumulation of large granular lymphocytes that correspond to a mature cytotoxic effector type. Accordingly, the 2008 WHO considers NK and T lymphocytes disorders (i.e., NK-CLPD and T-LGLL) as two different and well separated entities [4,5]. However, the WHO classification relies on different lineage features of T and NK cells, not taking into account putative common pathogenic mechanisms and clinical features. As a matter of fact, chronic NK and T cell expansions display a similar morphology and, on clinical grounds, in both types of patients cytopenias and other comorbidities are often associated [6,7]. Furthermore, the treatment in T-LGLL and NK-CLPD patients is very similar, entailing an immunosuppressive regimen or careful observation of chronic lymphocytosis [8]. A series of recent new observations, in addition to the previously acquired evidence, point to the suggestion that chronic LGL disorders should be more properly included within only one category.
II. DIAGNOSIS
The evidence of a granular lymphocytosis greater than 2,000/μl lasting for more than 6 months has been generally accepted as the most relevant criteria for the diagnosis of disease [7], considering that normally circulating LGL counts are 0.25 x 109/L [8]. This figure obviously requires additional evaluation of immunophenotyping and molecular analyses. This is particularly relevant when only some of the above criteria might be present. On blood films, the LGL nucleus is typically round with condensed, mature chromatin; the cytoplasm is pale, where randomly distributed azurophilic granules are easily detectable [7,8]. BM involvement by chronic LGL disorders is often subtle and difficult to identify, even when apparent in the PB [9].
In most cases, flow cytometry immunophenotyping defines a CD3+ LGLL, which usually express the TCR α/β+, CD2+, CD4-, CD8+, CD16+, CD57+, CD45RA+, CD122+ (p75 IL-2R), CD25- (p55 IL-2R) phenotype. CD5 and CD7 are weakly expressed or negative; CD56 is rarely positive and it has been claimed to be associated with a more aggressive clinical course [7]. These cells usually display a cytotoxic phenotype corresponding to that of a fully differentiated mature cytotoxic T lymphocyte (CD45RA+, CD27-, CD28-, CD62L-, CCR7) [10]. Most cases represent expansions of T cells bearing the TCR αβ+ while only a minority is derived from TCRγδ+ cells bearing a Vγ9+/Cδ2+ or a Vγ9-/Cδ1 phenotypic profile with TCRγ monoclonal restriction [11]. The expression of the inhibitory complex CD94/NKG2A, belonging to NK receptor, in these cells seems to correlate with a less aggressive behavior of proliferation [12]. Flow cytometry analysis with MoAb against the various Vβ regions of the TCR allows to establish the clonality of the LGLs by showing the preferential use of one or two TCR-Vβ segments [13]. These techniques should be applied to all suspected cases and are useful in those patients in whom the absolute LGL count is not significantly increased.
Formal proof that we are dealing with a T-LGL is provided by the establishment of clonality through gene rearrangement studies [7-8], these techniques allowing to distinguish LGL proliferations as a neoplastic disease or reactive lymphocytosis. Methods used to determine TCR gene rearrangement are polymerase chain reaction (PCR) and Southern blotting. While Southern blotting has fewer false negative and false positive results, PCR is a more sensitive technique for detection. Also, PCR is considered more user friendly in that it is less labor-intensive and does not require high-molecular weight DNA, which allows for DNA to be used from paraffin-embedded tissues when fresh or frozen samples are not available [14].
CD3- LGLL are characterized by the CD16+CD56+CD45RA+CD122+CD25- phenotype. CD57 antigen is usually weakly detectable. CD56 antigen is normally expressed by LGL, although some negative cases are reported. No clinical correlation with this marker has been performed, although CD56 negative cases seem to be more frequently associated to cytopenia. CD94 antigen is found at high density on patients’ NK cells; this antigen is usually associated with the inhibitory subunit NKG2A, although in some cases the association CD94/NKG2C has been reported [13]. Patients’ NK cells characteristically express functional β and γ chains of IL-2/IL-15 receptor, which are strictly related to the role of these cytokines in the pathogenesis of disease [15]. Expression of NK receptors, mostly represented by KIR, is altered in patients with CLPD-NK. A restricted pattern of KIR expression has usually been reported in these patients, which is characterized either by a dominant expression of a relevant KIR, or by a lack of KIR expression [16,17]. NK-LGL cells are not equipped with the TCR and therefore clonality cannot be determined by the rearrangement of TCR genes. In these cases a KIR restricted pattern of expression has been considered as a marker of clonality [16]. In addition, the demonstration of somatic STAT3 mutations [18-20] may represent a useful tool in the diagnosis of disease, although not easily available in all labs (see below).
III. CLINICAL FEATURES
The disease usually affects older people (mean 60 years). It is more common in Eastern than in Western countries. The disease runs asymptomatic in nearly 30% of cases, with lymphocytosis representing the only observed hematological abnormality [1-3]. These asymptomatic patients may have other associated conditions; among these, RA is the most commonly reported co-morbidity condition [8, 21] but several other connective tissue diseases such as systemic lupus erythematosus have been reported to be associated with T-cell LGL leukemia. Hematological conditions are another well represented group including monoclonal gammopathies, multiple myeloma, myelodysplastic syndromes, myelofibrosis, Hodgkin’s and non Hodgkin’s lymphomas [8, 21]; non-hematological neoplasms have also been reported in association with LDGL. The term of T-cell clonopathy of unknown significance (TCUS) has been suggested to designate these asymptomatic patients [22]. Symptomatic patients frequently present with fever due to infections or mouth lesions, often related to neutropenia. Weakness due to anemia represents another relevant finding. B-related symptoms (fever, night sweats, weight loss) are observed in nearly 25% of cases. Another intriguing association has been found between LDGL and pulmonary hypertension, with documented infiltration of the lung by GL [23]. Up to a half of the patients have splenomegaly, around 20% of cases present skin lesions and only a minority have hepatomegaly; lymphadenopathy is rare.
Most CLPD-NK patients are asymptomatic, and the disease has a chronic indolent clinical course, similar to that reported for patients with T LGL leukemia [24]. Associated conditions are reported even for CLPD-NK, including pure red cell aplasia, vasculitic syndromes, solid and hematologic tumors, splenectomy, neuropathy and autoimmune disorders [8,24]. Recently in patients with chronic myeloid leukaemia an association between treatment with dasatinib and the development of the lymphoproliferative disease has been reported, the hypothesis having been suggested that the CLPD-NK might have a beneficial therapeutical effect on Ph+ leukemic cells [25]. Cytopenia (either neutropenia and anemia) are quite common. The leukocyte count may be normal or slightly elevated but in most patients there is an increase in circulating LGL, even without having an absolute lymphocytosis; some cases are lymphopenic. In discrete patients lymphocytosis develops after splenectomy. Lymphoadenopathy, hepatomegaly, splenomegaly and cutaneous lesions are uncommon [24]. Occasionally, patients present with a slowly progressive increase of peripheral blood NK cells and with organ involvement. In rare cases the disease transforms to aggressive NK cell leukemia and cases with EBV positive NK cells tend to evolve [25]. Several cases with a spontaneous complete remission have also been reported [24].
IV. THERAPY
Disease may run asymptomatic for many years in the majority of patients, whereas in other cases therapy is needed, usually for cytopenia-related manifestations. The percentage of patients who require therapy at some time during the disease ranges from 30 to 70%, according to different series [21]. In one of the largest published multicenter study including 151 cases, coordinated by our institution, mortality after 4 year follow-up was 20% and a median survival greater than 10 years [6]. Most deaths are due to sepsis and rarely occur due to disease progression.
Indications for treatment include severe and symptomatic neutropenia, transfusion dependent anemia or thrombocytopenia as far as progressive disease (i.e., appearance of organomegaly, B symptoms and rapidly LGL raising counts). Correction of cytopenias with therapy may be achieved without eradication of the clone, which often persists even after treatment. Immunomodulatory drugs, such as methotrexate (10 mg/m2/weekly), cyclosporine A (5-10 mg/kg/day) or low dose cyclophosphamide (50 to 100 mg/day) have been commonly used [27-30]. Corticosteroids may be useful as a part of the initial treatment to accelerate response and growth factors are often used. Adverse events are not severe and are more common with cyclosporine A, particularly in elderly patients. Splenectomy may be considered as an adjuvant in patients with relevant splenomegaly and refractory cytopenias [31]. Once patients with LGL leukemia begin treatment, the regimen should not be altered for a period of 4 months, and they must be closely observed through complete blood counts [8]. After this time point, a hematological complete response is considered achieved when blood counts reveal platelets >150 x109/L, ANC >1.5 x 109/L, lymphocytes <4 x 109/L, and hemoglobin >12 g/dL. Complete molecular remission is reached when the T-cell clone is no longer detectable through PCR analysis [8]. Partial response is considered when overall blood counts improve but the ANC has not achieved levels > 500 cells/μL, still leaving the patient at potential risk for secondary infections. Should an improvement not be achieved after 4 months of continued treatment, then one of the alternative therapies described above should be taken into account.
Chemotherapeutic agents currently being tested in cutaneous T-cell lymphoma (CTCL) and peripheral T-cell lymphoma (PTCL), such as gemcitabine, liposomal doxorubicin as well as the purine analogs, such as fludarabine, cladribine and nelarabine, represent possible new agents to be considered for symptomatic LGL disorders [8, 21]. Purine analogs have been only used in few patients with response rates of 40 to 67% for pentostatin, fludarabine alone or in combination. The use of these agents should be considered for young patients as they allow the achievement of good remissions, including the reduction of bone marrow infiltration.
Monoclonal antibodies anti-CD52 (Campath-1H), anti-CD122 and anti-CD2 are being incorporated in the therapeutic scenario [8,21]. Good responses have been obtained in patients with symptomatic T-LGL leukemia and CLPD-NK with the use of RAS farnesyltransferase inhibitor (FTI), tipifarnib (Zarnestra®, Johnson & Johnson), according to the findings of a constitutively active signaling of the Ras/MAPK/ERK pathway [32]. Recently, the efficacy of extracorporeal photopheresis has been evaluated in 5 refractory/relapsed patients with LGL leukemia, with 2 of 5 patients achieving a CR [33]. In addition, the proteasome inhibitor bortezomib has been reported to display anticancer activity against aggressive NK leukemia and extranodal NK/T cell lymphoma in vitro and in vivo, opening new therapeutical prespectives for these patients [34].
V. ARE T-LGLL AND CLPD-NK REALLY TWO DIFFERENT DISORDERS?
During infection exposure or antigen stimulation, LGLs undergo proliferation by many thousands of times upon priming by target cells, and at a later time after antigen clearance, are selectively eliminated by a process called activation induced cell death (AICD) [34]. The etiology of chronic LGL proliferations is largely unknown in most cases. This is consequent to the fact that not a single specific agent can establish the LGL proliferation, which instead is likely due to the expression of an erroneous management of a foreign agent. In other words, different agents are supposed to induce the disease through a common pathogenetic mechanism. Crucial cornerstones for disease development have been recently identified. A number of reports strongly support the role of a chronic/persistent antigenic stimulation by an auto- or foreign infective antigen as an initial event [36-38]. This would lead to the expansion of a fully differentiated effector/cytotoxic LGL, which is not eliminated due to an impairment on the apoptotic pathway [39]. Multiple cell survival pathways, including JAK2/STAT3, sphingolipid signaling, RAS/MEK/ERK, and SFK/PI3K/Akt, have been found to be constitutively activated in LGL leukemia patients (reviewed in [40]). Some of these altered pathways seem more correlated with T-type, others with NK cell type of proliferation, whereas others are common of both T and NK cell proliferation. A biology approach identified IL-15 and PDGF as master survival signaling switches that may have a profound effect on all known deregulations in T-LGL leukemia [39].
However, if both disorders share the feature of a persistent antigenic stimulation, it is reasonable to think that in the same patient both T and NK cell can be under antigenic pressure. Consequence of this finding rests on the fact that growth mechanisms shared by T and NK cells represent the cornerstone for the genesis of a LGL proliferation. Accordingly, some questions can be raised.
VI. IS THERE A COMMON ANTIGEN SHARED BETWEEN THE TWO DISORDERS?
It has been suggested that bone marrow, which is frequently, although minimally, involved in LGL proliferation patients, represents the setting where the putative inciting antigen could reside and dendritic cells (DC) have been indicated to play a role as presenting cells and, perhaps, as IL-15 producers in these patients [41]. Data pointing to a putative pathogenic role for some virus, in particular herpes virus [42, 43] and retroviral agents have been reported [44-46]. The evidence that sera from a series of patients from Europe and USA suffering from LGLL or CLPD-NK reacted with the recombinant human T lymphotropic virus (HTLV) env protein p21E suggests that exposure to a protein containing homology to BA21 may be important in the pathogenesis of these lymphoproliferative disorders [45-46]. Taken together, all these data point to a putative immune system’s inability to clear the proposed virus, favoring the survival of LGL leukemia cells [47].
VII. WHICH EVIDENCE OF ANTIGEN PERSISTENCE?
Two theories, not necessarily mutually exclusive, have polarized the investigations on the pathogenesis of chronic LGL disorders, possibly representing two different steps of the natural history of disease. The first hypothesis considers the proliferation and accumulation of a transformed T or NK cell originated upon acquired intrinsic molecular defect (for example as a mutation [18-20] or other inhibitory mechanisms [48]). Alternatively, they might represent the expression of a global dysregulation of cytotoxic T or NK cell repertoire homeostasis because of persistent antigenic drive in combination with immunogenetic factors favoring persistent cell expansions [49]. This latter theory is supported by the phenomenon of clonal drift, reported in nearly 50% of LGLL patients, which is characterized by the change of dominant clone during time [50]. Taken together, these results are consistent with the evidence that T-LGLL may involve multiple clones occurring either concurrently or even serially. Our group recently demonstrated the presence of monoclonal T cell populations in 48% of cases in a cohort of 42 patients with KIR restricted NK-CLPD. Even more intriguing is the finding that these monoclonal populations can be detected not only at the time of diagnosis, but in some cases they can occur during the natural history of disease, indicating that the association of T and NK proliferations is much more frequent that initially thought. The T cell clone can eventually became so relevant to be dominant, leading to the shift from NK-CLPD to T-LGLL [Gattazzo et al, submitted]. Both these observations indicate that cells are under antigenic pressure, suggesting that the putative antigen is likely to persist for many years and possibly for the lifelong of patients. This finding points to the relevance of a long lasting follow up of these patients and the possibility that a sudden worsening of clinical features even in indolent cases could never be ruled out, as reported in the literature [51].
VIII. IS STAT3 ACTIVATION A CENTRAL POINT?
The constitutive activation of JAK/STAT pathway has been claimed to be involved in the development of several human cancers, including hematologic neoplasms (acute myeloid leukemias, Sezary syndrome, multiple myeloma) [52-54]. In T-LGLL Epling-Burnette et al reported that LGL from patients constitutively express high levels of activated STAT3 as compared to PBMC of healthy individuals [55]. They also showed that STAT3 activation contributes to Fas resistance resulting in abnormal survival of leukemic LGL and is also responsible of the over-expression of the anti-apoptotic protein Mcl-1 [55]. We recently confirmed that over-expression and activation of STAT3 is a constitutive feature present in all LGLL patients and extend this observation to the comprehension of the mechanisms leading to the constitutive activation of STAT3 in these patients [48]. In fact, we provided evidence that the IL-6 produced in vivo by autologous monocytic cells represents a cytokine that is central for STAT3 upregulation. We also demonstrated that the lack of inhibitory signals counterbalancing STAT3 activation, mediated by epigenetic inhibition of SOCS3, plays a role in the persistence of this activation pathway [48]. Recently, in vivo activation of STAT3 has been reported also in patients with CLPD-NK ([19] and Zambello, unpublished data), indicating that STAT3 activation is a unifying mechanism working both in T and NK chronic LGL proliferations.
IX. HOW DOES DISEASE PROGRESS?
Recently, it has been observed that recurrent somatic missense mutations of STAT3 (more often Y640F and D661Y) might be demonstrated in one third of patients with LGLL [18]. The same Authors updated their case study in more than 100 LGLL patients confirming that STAT3 mutations were also detectable in nearly one third of patients with CLPD-NK [19]. These data have also been confirmed by other groups, although the percentage of mutated cases ranged from 22 to 77% [48, 56-57]. It has been suggested that mutated STAT3 still retains its physiologic ability to stimulate some target genes. Jerez et al. also suggested that STAT3 mutation can be helpful in discriminating true leukemic from possibly reactive conditions [19]. This conclusion however should be viewed with caution. As a matter of fact, whereas STAT3 activation is indeed a constitutive mechanism in all T-LGLL patients [48, 55], STAT3 mutations are reported only in a fraction of cases, despite the presence of monoclonal rearrangement of TCR was shown in all cases [18, 19]. In addition, considering each single patient, STAT3 mutated LGL have been shown to account for only a minority of the entire monoclonal LGL population [19]. Furthermore, in the STAT3 mutated patients, consistent with unmutated cases, we observed a high expression of P-STAT3 and IL-6, associated to SOCS3 down-regulation, ruling out any specific peculiarity for these patients [48]. Finally, by incubating pathological LGL with the downstream STAT3 selective inhibitor STA-21 (a novel synthetic inhibitor of STAT3 dimerization, DNA binding, and STAT3-dependent luciferase reporter activity) apoptosis was induced in both CLPD-NK or T-LGLL, irrespective of the mutational status [19].
According with the above quoted considerations, we suggest that the acquisition of STAT3 mutations might be an event occurring late during the natural history of disease, certainly later than the establishment of clonality. This event is likely to represent a marker of the progression of disease, defining in patients with T-LGLL and CLPD-NK a common specific clinical and pathogenic pattern driven by a shared genetic lesion, irrespective of the cell lineage they originate from.
X. CONCLUSION
Chronic LGL proliferations can arise from either cytotoxic T-cell lymphocytes or NK-cell lymphocytes. Individuals with either type of cell proliferations have a good prognosis and respond quite well to current available immunosuppressive therapies. To get insight into the characteristics of LGL disorders, it is mandatory to fully understand the development pathways of normal LGL. Until the distinctive features between cytotoxic T and NK cells are notcompletely elucidated, the shared altered pathways and clinical behavior strongly support the opportunity of merging both entities in one disease category, as we initially suggested under the definition of lymphoproliferative disease of granular lymphocytes [1]. DNA profiling and Next Generation Sequencing using purified normal and pathological T and NK cells will help to address this issue and would likely help in elucidating the differences of clinical behavior between cytotoxic T and NK derived proliferations.
Figure 1.
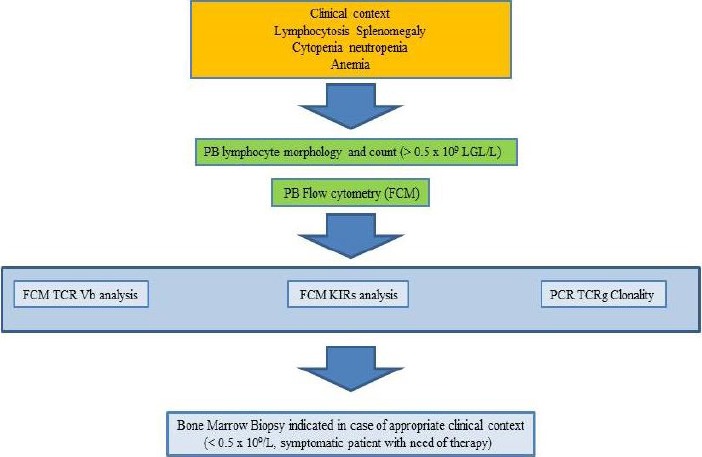
See text for more details.
Abbreviations. PB = peripheral blood; LGL = large granular lymphocytes; TCR = T cell receptor; KIRs = Killer Ig-like receptors.
TABLE I.
DIFFERENCES AND SHARED FEATURES IN T- LGL-LEUKEMIA AND CLPD-NK
| Types | Clinical features | Associated Diseases | Relevant Immunophenotype | Monoclonal T cell population | STAT3/5 Mutations | HTLV-BA21 seroreactivity | Treatment options |
|---|---|---|---|---|---|---|---|
| T-LGLL | Indolent clinical course | RA and autoimmune diseases, malignancy | CD3+ CD8+ CD16+ CD57+ TCR ab+ KIRs± CD94/NKG2C ± | 100 % | 37% (110/297)* 1.7% (3/173)** |
30% ° (21/70) |
Observation or immune suppressive regimen |
| CLPD-NK | Indolent clinical course | Malignancy, RA and autoimmune diseases | CD3- CD16+ CD56+ CD57+ KIRs± CD94/NKG2A± | 48% ^ (23/48) |
20% § (22/109) 2.6% §§ (1/38) |
73% °° (11/15) |
Observation or immuno suppressive regimen |
Abbreviations. RA: Rheumatoid Arthritis.
*Data summarize results reported in Koskela et al [18], Jerez et al [20], Fasan et al [56], Teramo et al [48], Sekiguchi et al [57];
** Data summarize results reported in Rajala et al [21];
§ Data summarize results reported in Jerez et al [20], Sekiguchi et al [57], Gattazzo et al, submitted;
§§ Data summarize results reported in Rajala et al [21];
^ Data summarize results reported in Gattazzo et al, submitted;
° Data summarize results reported in Loughran et al [45];
°°Data summarize results reported in Sokol et al [46].
Acknowledgment
This work was supported by Associazione Italiana per la Ricerca sul Cancro (AIRC), Cariparo, Cariverona, Ministero dell'Istruzione, dell’Università e della Ricerca Scientifica (MIUR) and Lions Club International. A.T. is a recipients of a fellowship from Associazione Italiana contro le Leucemie, Linfomi e Mieloma (AIL) and Lions Club, Padua.
REFERENCES
- [1].Semenzato G, Pandolfi F, Chisesi T, De Rossi G, Pizzolo G, Zambello R, et al. The lymphoproliferative disease of granular lymphocytes. A heterogeneous disorder ranging from indolent to aggressive conditions. Cancer 1987;60:2971-2978 [DOI] [PubMed] [Google Scholar]
- [2].Loughran TP., Jr Clonal diseases of large granular lymphocytes. Blood 1993;82:1-14 [PubMed] [Google Scholar]
- [3].Oshimi K, Yamada O, Kaneko T, Nishinarita S, Iizuka Y, Urabe A, Inamori T, Asano S, Takahashi S, Hattori M, et al. Laboratory findings and clinical courses of 33 patients with granular lymphocyte-proliferative disorders. Leukemia 1993;7(6):782-788 [PubMed] [Google Scholar]
- [4].Chan WC, Foucar KM, Morice WG, Catovsky D. T-cell large granular lymphocytic leukaemia. Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, et al. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon: IARC Press, 2008. p.272-273 [Google Scholar]
- [5].Villamor N, Morice WG, Chan WC, Foucar K. Chronic lymphoproliferative disorders of NK cells. Swerdlow SH, Campo E, Harris NL, Jaffe EJ, Pileri SA, Stein H, et al. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon: IARC Press, 2008:274-275 [Google Scholar]
- [6].Pandolfi F, Loughran TP, Jr, Starkebaum G, Chisesi T, Barbui T, Chan WC, Brouet JC, De Rossi G, McKenna RW, Salsano F, et al. Clinical course and prognosis of the lymphoproliferative disease of granular lymphocytes. A multicenter study. Cancer 1990;65(2):341-348 [DOI] [PubMed] [Google Scholar]
- [7].Semenzato G, Zambello R, Starkebaum G, Oshimi K, Loughran TP. The lymphoproliferative disease of granular lymphocytes: updated criteria for diagnosis. Blood 1997;89:256-260 [PubMed] [Google Scholar]
- [8].Lamy T, Loughran TP., Jr How I treat LGL leukemia. Blood 2011; 117: 2764-2774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Morice WG, Kurtin PJ, Tefferi A, Hanson CA. Distinct bone marrow findings in T-cell granular lymphocytic leukemia revealed by paraffin section immunoperoxidase stains for CD8, TIA-1, and granzyme B. Blood 2002;99:268-274 [DOI] [PubMed] [Google Scholar]
- [10].Baesso I, Pavan L, Boscaro E, Miorin M, Facco M, Trentin L, Agostini C, Zambello R, Semenzato G. T-cell type lymphoproliferative disease of granular lymphocytes (LDGL) is equipped with a phenotypic pattern typical of effector cytotoxic cells. Leuk Res 2007;31:371-377 [DOI] [PubMed] [Google Scholar]
- [11].Bourgault-Rouxel AS Loughran TP Jr Zambello R Epling-Burnette PK Semenzato G Donadieu J Amiot L Fest T Lamy T. Clinical spectrum of gammadelta+ T cell LGL leukemia: analysis of 20 cases. Leuk Res 2008;32:45-48 [DOI] [PubMed] [Google Scholar]
- [12].Angelini DF, Zambello R, Galandrini R, Diamantini A, Placido R, Micucci F, Poccia F, Semenzato G, Borsellino G, Santoni A, Battistini L. NKG2A inhibits NKG2C effector functions of γδ T cells: implications in health and disease. J Leukoc Biol 2011;89:75-84 [DOI] [PubMed] [Google Scholar]
- [13].Lima M, Almeida J, Santos AH, dos Anjos Teixeira M, Alguero MC, Queirós ML, Balanzategui A, Justiça B, Gonzalez M, San Miguel JF, Orfão A. Immunophenotypic analysis of the TCR-Vbeta repertoire in 98 persistent expansions of CD3(+)/TCR-alphabeta(+) large granular lymphocytes: utility in assessing clonality and insights into the pathogenesis of the disease. Am J Pathol 2001;159:1861-1868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Beck RC, Stahl S, O’ Keefe CL, Maciejewski JP, Theil KS, Hsi ED. Detection of mature T-cell leukemias by flow cytometry using anti-T-cell receptor V beta antibodies. Am J Clin Pathol 2003;120:785-794 [DOI] [PubMed] [Google Scholar]
- [15].Zambello R, Facco M, Trentin L, Sancetta R, Tassinari C, Perin A, Milani A, Pizzolo G, Rodeghiero F, Agostini C, Meazza R, Ferrini S, Semenzato G. Interleukin-15 triggers the proliferation and cytotoxicity of granular lymphocytes in patients with lymphoproliferative disease of granular lymphocytes. Blood 1997;89:201-211 [PubMed] [Google Scholar]
- [16].Zambello R, Falco M, Della Chiesa M, Trentin L, Carollo D, Castriconi R, Cannas G, Carlomagno S, Cabrelle A, Lamy T, Agostini C, Moretta A, Semenzato G, Vitale M. Expression and function of KIR and natural cytotoxicity receptors in NK-type lymphoproliferative disease of granular lymphocytes. Blood 2003;102:1797-1805 [DOI] [PubMed] [Google Scholar]
- [17].Epling-Burnette PK, Painter JS, Chaurasia P, Bai F, Wei S, Djeu JY, Loughran TP., Jr Dysregulation of NK receptor expression in patients with lymphoproliferative disease of granular lymphocytes. Blood 2004;103:3431-3439 [DOI] [PubMed] [Google Scholar]
- [18].Koskela HL, Eldfors S, Ellonen P, van Adrichem AJ, Kuusanmäki H, Andersson EI, Lagström S, Clemente MJ, Olson T, Jalkanen SE, Majumder MM, Almusa H, Edgren H, Lepistö M, Mattila P, Guinta K, Koistinen P, Kuittinen T, Penttinen K, Parsons A, Knowles J, Saarela J, Wennerberg K, Kallioniemi O, Porkka K, Loughran TP, Jr, Heckman CA, Maciejewski JP, Mustjoki S. Somatic STAT3 mutations in large granular lymphocytic leukemia. N Engl J Med 2012;366:1905-1913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Jerez A, Clemente MJ, Makishima H, Koskela H, Leblanc F, Peng Ng K, Olson T, Przychodzen B, Afable M, Gomez-Segui I, Guinta K, Durkin L, Hsi ED, McGraw K, Zhang D, Wlodarski MW, Porkka K, Sekeres MA, List A, Mustjoki S, Loughran TP, Maciejewski JP. STAT3 mutations unify the pathogenesis of chronic lymphoproliferative disorders of NK cells and T cell large granular lymphocyte leukemia. Blood 2012;120:3048-3057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Rajala HL, Eldfors S, Kuusanmäki H, van Adrichem AJ, Olson T, Lagström S, Andersson EI, Jerez A, Clemente MJ, Yan Y, Zhang D, Awwad A, Ellonen P, Kallioniemi O, Wennerberg K, Porkka K, Maciejewski JP, Loughran TP, Jr, Heckman C, Mustjoki S. Discovery of somatic STAT5b mutations in large granular lymphocytic leukemia. Blood 2013;121:4541-4550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Zambello R, Semenzato G. Large granular lymphocyte disorders: new etiopathogenetic clues as a rationale for innovative therapeutic approaches. Haematologica 2009;94:1341-1345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Dhodapkar MV, Li CY, Lust JA, Tefferi A, Phyliky RL. Clinical spectrum of clonal proliferations of T-large granular lymphocytes: a T-cell clonopathy of undetermined significance? Blood 1994;84:1620-1627 [PubMed] [Google Scholar]
- [23].Lamy T, Bauer FA, Liu JH, Li YX, Pillemer E, Shahidi H, Gregory SA, Zambello R, Marcolongo R, Semenzato G, Loughran TP. Clinicopathological features of aggressive large granular lymphocyte leukaemia resemble Fas ligand transgenic mice. Br J Haematol 2000;108:717-723 [DOI] [PubMed] [Google Scholar]
- [24].Semenzato G, Marino F, Zambello R. State of the art in natural killer cell malignancies. Int J Lab Hematol 2012;34:117-128 [DOI] [PubMed] [Google Scholar]
- [25].Mustjoki S, Ekblom M, Arstila TP, Dybedal I, Epling-Burnette PK, Guilhot F, Hjorth-Hansen H, Höglund M, Kovanen P, Laurinolli T, Liesveld J, Paquette R, Pinilla-Ibarz J, Rauhala A, Shah N, Simonsson B, Sinisalo M, Steegmann JL, Stenke L, Porkka K. Clonal expansion of T/NK-cells during tyrosine kinase inhibitor dasatinib therapy. Leukemia 2009;23:1398-1405 [DOI] [PubMed] [Google Scholar]
- [26].Kawa-Ha K, Ishihara S, Ninomiya T, Yumura-Yagi K, Hara J, Murayama F, Tawa A, Hirai K. CD3-negative lymphoproliferative disease of granular lymphocytes containing Epstein-Barr viral DNA. J Clin Invest 1989;84:51-55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Loughran TP., Jr Results of a prospective multicenter phase II study of initial treatment with methotrexate in LGL leukemia (ECOG protocol E5998) [abstract]. Blood (ASH Annual Meeting Abstracts) 2010;116(21):2595 [Google Scholar]
- [28].Gabor EP, Mishalani S, Lee S. Rapid response to cyclosporine therapy and sustained remission in large granular lymphocyte leukemia. Blood 1996;87:1199-1200 [PubMed] [Google Scholar]
- [29].Osuji N, Matutes E, Tjonnfjord G, Grech H, Del Giudice I, Wotherspoon A, Swansbury JG, Catovsky D. T-cell large granular lymphocyte leukemia: a report on the treatment of 29 patients and a review of the literature. Cancer 2006;107:570-578 [DOI] [PubMed] [Google Scholar]
- [30].Fujishima N, Sawada K, Hirokawa M, Oshimi K, Sugimoto K, Matsuda A, Teramura M, Karasawa M, Arai A, Yonemura Y, Nakao S, Urabe A, Omine M, Ozawa K; PRCA Collaborative Study Group. Long-term responses and outcomes following immunosuppressive therapy in large granular lymphocyte leukemia-associated pure red cell aplasia: a Nationwide Cohort Study in Japan for the PRCA Collaborative Study Group. Haematologica 2008;93(10):1555-1559 [DOI] [PubMed] [Google Scholar]
- [31].Subbiah V, Viny AD, Rosenblatt S, Pohlman B, Lichtin A, Maciejewski JP. Outcomes of splenectomy in T-cell largegranular lymphocyte leukemia with splenomegaly and cytopenias. Exp Hematol 2008;36:1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Epling-Burnette PK, Sokol L, Chen X, Bai F, Zhou J, Blaskovich MA, Zou J, Painter JS, Edwards TD, Moscinski L, Yoder JA, Djeu JY, Sebti S, Loughran TP, Jr, Wei S. Clinical improvementby farnesyltransferase inhibition in NK large granular lymphocyte leukemia associated with imbalanced NK receptor signaling. Blood 2008;112:4694-4698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Garban F, Carras S, Drillat P, Jacob MC, Fabre B, Callanan M, Courby S, Makowski C, Cahn JY, Gressin R. Extracorporeal photopheresisas a curative treatment strategy in non epidermotropic T-cell lymphoma and large granular lymphocyte leukemia. Ann Oncol 2012;23:2396-2390 [DOI] [PubMed] [Google Scholar]
- [34].Shen L, Au WY, Guo T, Wong KY, Wong ML, Tsuchiyama J, Yuen PW, Kwong YL, Liang RH, Srivastava G. Proteasome inhibitor bortezomib-induced apoptosis in natural killer(NK)-cell leukemia and lymphoma: an in vitro and in vivo preclinical evaluation. Blood 2007;110:469-470 [DOI] [PubMed] [Google Scholar]
- [35].Williams MA, Bevan MJ. Effector and memory CTL differentiation. Annu Rev Immunol 2007;25:171-192 [DOI] [PubMed] [Google Scholar]
- [36].Epling-Burnette PK, Loughran TP., Jr Survival signals in leukemic large granular lymphocytes. Semin Hematol 2003;40:213-220 [DOI] [PubMed] [Google Scholar]
- [37].Zambello R, Trentin L, Facco M, Cerutti A, Sancetta R, Milani A, Raimondi R, Tassinari C, Agostini C, Semenzato G. Analysis of the T cell receptor in the lymphoproliferative disease of granular lymphocytes: superantigen activation of clonal CD3-granular lymphocytes. Cancer Res 1995;55:6140-6145 [PubMed] [Google Scholar]
- [38].Wlodarski MW, Nearman Z, Jankowska A, Babel N, Powers J, Leahy P, Volk HD, Maciejewski JP. Phenotypic differences between healthy effector CTL and leukemic LGL cells support the notion of antigen-triggered clonal transformation in T-LGL leukemia. J Leukoc Biol 2008;83:589-601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Zhang R, Shah MV, Yang J, Nyland SB, Liu X, Yun JK, Albert R, Loughran TP., Jr Network model of survival signaling in large granular lymphocyte leukemia. Proc Natl Acad Sci U S A 2008;105:16308-16313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Zhang D, Loughran TP., Jr Large granular lymphocytic leukemia: molecular pathogenesis, clinical manifestations, and treatment. Hematology. Am Soc Hematol Educ Program 2012; pag.652-659 [DOI] [PubMed] [Google Scholar]
- [41].Zambello R, Berno T, Cannas G, Baesso I, Binotto G, Bonoldi E, Bevilacqua P, Miorin M, Facco M, Trentin L, Agostini C, Semenzato G. Phenotypic and functional analyses of dendritic cells in patients with lymphoproliferative disease of granular lymphocytes (LDGL). Blood 2003;102:1797-1805 [DOI] [PubMed] [Google Scholar]
- [42].Kawa-Ha K, Ishihara S, Ninomiya T, Yumura-Yagi K, Hara J, Murayama F, Tawa A, Hirai K. CD3-negative lymphoproliferative disease of granular lymphocytes containing Epstein-Barr viral DNA. J Clin Invest 1989;84:51-55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Zambello R, Loughran TP, Jr, Trentin L, Pontisso P, Battistella L, Raimondi R, Facco M, Sancetta R, Agostini C, Pizzolo G, et al. Serologic and molecular evidence for a possible pathogenetic role of viral infection in CD3-negative natural killer-type lymphoproliferative disease of granular lymphocytes. Leukemia 1995;9(7):1207-1211 [PubMed] [Google Scholar]
- [44].Hart DN, Baker BW, Inglis MJ, Nimmo JC, Starling GC, Deacon E, Rowe M, Beard ME. Epstein-Barr viral DNA in acute large granular lymphocyte (natural killer) leukemic cells. Blood 1992;79:2116-2123 [PubMed] [Google Scholar]
- [45].Loughran TP, Jr, Hadlock KG, Yang Q, Perzova R, Zambello R, Semenzato G, Foung SK, Poiesz BJ. Seroreactivity to an envelope protein of human T-cell leukemia/lymphoma virus in patients with CD3- (natural killer) lymphoproliferative disease of granular lymphocytes. Blood 1997;90:1977-1981 [PubMed] [Google Scholar]
- [46].Sokol L, Agrawal D, Loughran TP., Jr Characterization of HTLV envelope seroreactivity in large granular lymphocyte leukemia. Leuk Res 2005;29:381-387 [DOI] [PubMed] [Google Scholar]
- [47].Nearman ZP, Wlodarski M, Jankowska AM, Howe E, Narvaez Y, Ball E, Maciejewski JP. Immunogenetic factors determining the evolution of T-cell large granular lymphocyte leukaemia and associated cytopenias. Br J Haematol 2007;136:237-248 [DOI] [PubMed] [Google Scholar]
- [48].Teramo A, Gattazzo C, Passeri F, Lico A, Tasca G, Cabrelle A, Martini V, Frezzato F, Trimarco V, Ave E, Boscaro E, Piazza F, Facco M, Trentin L, Semenzato G, Zambello R. Intrinsic and extrinsic mechanisms contribute to maintain the JAK/STAT pathway aberrantly activated in T-type large granular lymphocyte leukemia. Blood 2013;121:3843-3854 [DOI] [PubMed] [Google Scholar]
- [49].Yang J, Epling-Burnette PK, Painter JS, Zou J, Bai F, Wei S, Loughran TP., Jr Antigen activation and impaired Fas-induced death inducing signaling complex formation in T-large granular lymphocyte leukemia. Blood 2008;111:1610-1616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Clemente MJ, Wlodarski MW, Makishima H, Viny AD, Bretschneider I, Shaik M, Bejanyan N, Lichtin AE, Hsi ED, Paquette RL, Loughran TP, Jr, Maciejewski JP. Clonal drift demonstrates unexpected dynamics of the T-cell repertoire in T-large granular lymphocyte leukemia. Blood 2011;118:4384-4393 Erratum in: Blood 2012;120:1963. His, Eric D [corrected to Hsi, Eric D] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Roullet MR, Cornfield DB. Large natural killer cell lymphoma arising from an indolent natural killer cell large granular lymphocyte proliferation. Arch Pathol Lab Med 2006;130:1712-1714 [DOI] [PubMed] [Google Scholar]
- [52].Schuringa JJ, Wierenga AT, Kruijer W, Vellenga E. Constitutive Stat3, Tyr705, and Ser727 phosphorylation in acute myeloid leukemia cells caused by the autocrine secretion of interleukin-6. Blood 2000;95:3765-3770 [PubMed] [Google Scholar]
- [53].Lin TS, Mahajan S, Frank DA. STAT signaling in the pathogenesis and treatment of leukemias. Oncogene 2000;19:2496-2504 [DOI] [PubMed] [Google Scholar]
- [54].Eriksen KW, Kaltoft K, Mikkelsen G, Nielsen M, Zhang Q, Geisler C, Nissen MH, Röpke C, Wasik MA, Odum N. Constitutive STAT3-activation in Sezary syndrome: tyrphostin AG490 inhibits STAT3-activation, interleukin-2 receptor expression and growth of leukemic Sezary cells. Leukemia 2001;15:787-793 [DOI] [PubMed] [Google Scholar]
- [55].Epling-Burnette PK, Liu JH, Catlett-Falcone R, Turkson J, Oshiro M, Kothapalli R, Li Y, Wang JM, Yang-Yen HF, Karras J, Jove R, Loughran TP., Jr Inhibition of STAT3 signaling leads to apoptosis of leukemic large granular lymphocytes and decreased Mcl-1 expression. J Clin Invest 2001;107:351-362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Fasan A, Kern W, Grossmann V, Haferlach C, Haferlach T, Schnittger S. STAT3 mutations are highly specific for large granular lymphocytic leukemia. Leukemia 2012Dec 4, “in press” [DOI] [PubMed] [Google Scholar]
- [57].Sekiguchi N, Matsuda K, Momose K, Makishima H, Ito T, Ishida F. STAT3 gene mutations and the association with pure red cell aplasia in large granular lymphocyte leukemia. Blood (ASH Annual Meeting Abstracts), Nov2012;120:2668. [DOI] [PMC free article] [PubMed] [Google Scholar]