

Abstract
Paroxysmal nocturnal hemoglobinuria (PNH) is a clonal, non-malignant, hematological disorder characterized by the expansion of hematopoietic stem cells and progeny mature blood cells which are deficient in some surface proteins, including the two complement regulators CD55 and CD59. PNH is the paradigm of diseases implying complement dysregulation as main pathogenic mechanism; in fact, PNH erythrocytes are uncapable to modulate on their surface physiologic complement activation, which eventually leads to the typical clinical hallmark of PNH – the chronic complement-mediated intravascular anemia. Indeed, due to the lack of CD55 complement is continuously activated on erythrocyte surface, which subsequently enables the terminal lytic complement because of the lack of CD59, finally resulting in erythrocyte lysis. The availability of eculizumab as the first complement inhibitor for clinical use renewed the interest for this rare hematological disease. Indeed, in the last decad the anti-C5 monoclonal antibody has proven effective for the treatment of PNH, resulting in a sustained control of complement-mediated intravascular hemolysis, with a remarkable clinical benefit. Anti-complement treatment allowed transfusion independence in at least half of PNH patients receiving eculizumab, with adequate control of all hemolysis-associated symptoms even in almost all remaining patients. In addition, the risk of thromboembolic events – an other clinical hallmark of PNH, which significantly affects prognosis and survival – seems substantially reduced on eculizumab treatment, apparently resulting in improved survival. Even with all these remarkable effects, eculizumab treatment does not result in hemoglobin normalization, and most patients remain anemic. It has been demonstrated that this is due to persistent activation of the early phases of complement activation (upstream the C5), leading to complement-mediated extravascular hemolysis. Ongoing researches are focusing on possible strategies to improve current anti-complement therapies, aiming to develop second-generation complement therapeutics. Here we review PNH and its complement-mediated pathophysiology, summarizing available data on anti-complement treatment; we’ll also discuss recent pathogenic insights which drive the development of novel strategies of complement inhibition.
Keywords: Paroxysmal Nocturnal Hemoglobinuria, complement alternative pathway, complement component 3, complement component 5, eculizumab, complement therapeutics, C3-targeted therapy
I. INTRODUCTION
PNH is a rare and puzzling hematological disorder clinically characterized by the triad of bone marrow failure, severe thrombophilia and complement-mediated intravascular hemolysis [1-4]; the most evident sign of this latter manifestation (namely the hemoglobinuria resulting from intravascular hemolysis) accounts for the picturesque name of the disease. A part from its clinical manifestations, PNH is a unique disease of the hematopoiesis characterized by the expansion of a few (or even one) mutated hematopoietic stem cells (and progeny mature blood cells) carrying the bizarre phenotype of the lack of several proteins from their surface [5-7].
All the proteins missing from the PNH cell surface share a unique biochemical feature [8]; in fact, they are not trans-membrane proteins, rather are linked to the cell membrane via a glycolipidic anchor – the glycosyl phosphatidyl-inositol (GPI)-moiety [9,10]. In the ‘90s, it has been documented that the reason accounting for this aberrant phenotype is a mutation in the X-linked phosphatidylinositol glycan class A (PIG-A) gene [11,12], which is necessary for the biosynthesis of the GPI-anchor. PNH is therefore an acquired genetic blood disorder, that cannot be transmitted to the progeny; however, a number of observations supports the concept that the mutation itself is not sufficient to cause PNH as a disease.
II. THE PATHOPHYSIOLOGY OF PNH
PNH as a disorder of hematopoiesis
It has been demonstrated that a few PNH-like cells carrying inactivating PIG-A mutations may be detected even in normal individuals (without any sign or symptom of PNH) [13]. On the other hand, the mutation does not reproduce the human disease in murine models; even if mice having a substantial proportion of PNH cells can be generated by using a complex technology (a conditional inactivation of the murine pig-a gene implemented using Cre recombinase specifically targeted to the hematopoietic stem cells [14], they do not really mimic the disease phenotype seen in humans, because PNH hematopoiesis tends to decrease over time [15]. This background raised the hypothesis of a dual pathophysiology for PNH (also known as the “relative advantage” [16] or “escape” theory [17]: the PIG-A mutation is not sufficient to cause the disease, and requires a second, independent event [18].
According to this view, a mutation in the PIG-A gene might be a fairly common phenomenon, with no major biological consequences, because in physiological conditions the mutated cell has no reason for expanding in the presence of a vast majority of normal cells. However, additional factors may alter this equilibrium, creating the conditions for the expansion of PNH clone(s); the most likely second event(s) is thought to be an (auto)-immune attack against hematopoiesis, as supported by the well-known clinical overlap between PNH and aplastic anemia (AA, which is in most cases immune-mediated) [19], as well as by direct demonstration of immune abnormalities in PNH patients [20].
It has been recently demonstrated that the GPI-anchor itself could be the target of such autoimmune attack, which would clearly spare PNH cells accounting for their relative expansion over normal hematopoiesis [21]. This pathogenic mechanisms accounts also for the one of typical manifestation of PNH – the moderate-to-severe bone marrow failure.
The other two typical features of PNH – intravascular hemolysis and thrombophilia – implies different specific pathogenic mechanisms. While the reasons for thrombophilia remain not fully understood and will not be discussed here (even if they are closely embedded with complement activation and hemolysis), the pathogenic meccanism accounting for hemolysis in PNH has been elucidated in details.
Complement dysregulation in PNH
The complement system is a key component of innate immunity evolved to recognize and to protect the host from both exogenous pathogenic microorganisms as well as injured self tissues. The complement system uses a number of plasma proteins , which may activate in the fluid phase along three distinct functional pathways – classical, alternative or lectin –, all finally merging into the a common final effector mechanism usually playing at a tissue level, the cytolytic membrane attack complex (MAC). Fluid-phase components (such as complement factor I [FI] and factor H [FH]) and membrane-bound proteins (such as complement receptor 1 [CR1], membrane cofactor protein [MCP], CD55 and CD59) have evolved as regulatory mechanisms tuning the complement system in specific conditions; it is now understood that possible derangements of these physiological regulatory mechanisms may lead to specific human diseases [22,23]. Historically, PNH is maybe the first disease in whom the complement cascade has been demonstrated as the key pathogenic mechanism.
As introduced above, among the several proteins lacking on PNH erythrocytes there are the two GPI-linked complement regulators CD55 and CD59. CD55, (also known as Decay Accelerating Factor, DAF; [6,24] is a 70-kd protein which inhibits the formation and the stability of C3 convertase (both C3bBb and C4b2a, alternative and classical pathways, respectively) [25]. Historically, CD55 was the first complement regulator found absent on PNH erythrocytes [6,26,27], leading to the hypothesis that its lack could account for the increased susceptibility of PNH erythrocytes to complement mediated lysis. However, further studies demonstrated that factors other than CD55 also were involved, likely acting downstream in the complement cascade [8,28]. Subsequently, CD59 (or Membrane Inhibitor of Reactive Lysis, MIRL; [29,30]) was identified as the key complement inhibitor which was found deficient on PNH cells [29]. CD59 interferes with the terminal effector complement, blocking the incorporation of C9 onto the C5b-C8 complex, thus preventing MAC formation [31].
The hierarchical contribution of CD55 and CD59 to hemolysis suggests that CD59 is the pivotal molecule which, if absent, results into lysis [32]. This is also supported by the observation that subject with isolated deficiency of CD55 (the so-called Inab phenotype) usually do not show any clinical or laboratory hemolysis in vivo (this may be due to redundant regulatory mechanisms, including CD59 itself) [33,34], whereas anecdotic cases of inherited CD59 deficiency harbor a clinical phenotype undistinguishable from PNH [35,36].
The in vitro susceptibility of PNH erythrocytes has been initially described by Dr. Ham in his seminal studies started almost a century ago. Dr. Ham demonstrated that erythrocytes from PNH patients lyse in autologous serum upon complement activation by acidification, using a hemolytic assay which take his own name (the Ham test, also known asthe acidified serum assay [37]). This feature of PNH erythrocytes was subsequently described more in depth by Dr. Rosse and Dr. Dacie, who showed that distinct phenotype of PNH erythrocytes may exist, according to their specific sensitivity to complement-mediated lysis in vitro [38,39]. In fact, in PNH one may find erythrocytes with a dramatic hypersensitivity to complement-mediated lysis (15-25 times the normal one), or just a moderate hypersentivity (3-5 times normal). These phenotypes are referred now as PNH type III and type II, respectively [38,39], which by flow cytometry correspond to a complete (type III) or partial (type II) deficiency of GPI-APs respectively [40]. While the in vitro susceptibility of PNH erythrocytes has been extensively elucidated, the actual mechanisms leading to complement activation in vivo and subsequent hemolysis have not been definitely demonstrated. However, it is conceivable that chronic hemolysis of PNH is due to a continuous steady-state complement activation coming from the low-grade spontaneous C3 tick over, with subsequent continous activation of the complement alternative pathway (CAP) on PNH erythrocyte surface [41,42]. Infections or inflammatory status usually result in hemolytic crises (the so-called paroxysms), eventually as a result of massive complement activation. At the moment, it is not clear which pathway accounts for complement activation in each of these specific conditions, even if it is conceivable that all the three pathways may co-operate, possibly with some hierarchical dominance of the CAP, which is specifically uncontrolled due to CD55 deficiency, and may amplify any initial complement activation.
III. ANTI-COMPLEMENT TREATMENT IN PNH
Clinical results of eculizumab in PNH
Eculizumab (h5G1.1-mAb) is a humanized monoclonal antibody (mAb) [43] derived from the murine anti-human C5 mAb; it binds to the complement component 5 (C5) and inhibits its further cleavage into C5a and C5b, disabling the progression to the terminal effector complement MAC [44]. Initial translation plans for eculizumab (Soliris®, Alexion Pharmaceuticals) started in autoimmune diseases; however, given its well-proven complement-mediated pathophysiology, PNH was subsequently identified as the best disorder for investigating this first complement inhibitor in humans. The prediction was that eculizumab in PNH, by preventing MAC assembly, could compensate for the absence of CD59 on PNH erythrocytes, preventing their intravascular lysis upon complement activation.
The initial phase II pilot study conducted on eleven heavily transfusion dependent PNH patients showed that eculizumab treatment was safe and well tolerated, and provided the proof-of-principle of effective blockade of intravascular hemolysis, as shown by substantial reduction of LDH level [45]. Eculizumab was administered intravenously dosed at 600 mg weekly for four weeks (loading phase), followed one week later by 900 mg fortnightly (maintenance phase); all patients were vaccinated against Neisseria Meningiditis at least two weeks before starting the treatment.
The subsequent study was a large double-blind, placebo-controlled, multinational randomized trial which enrolled 86 transfusion-dependent PNH patients [46]. Treatment with eculizumab resulted in a dramatic reduction of intravascular hemolysis, as measured by LDH, leading to hemoglobin stabilization and transfusion independence in about half of the patients. Control of intravascular hemolysis was found in all patients, and even cases not achieving transfusion independence showed a reduction of their transfusional needs and the disappearance of most clinical symptoms of hemolysis (painful abdominal crises, smooth muscle dystonia, erectile dysfunction). The effects of eculizumab on hemolysis were evident since the initial administration, and lasted for the whole study period. In comparison to placebo, eculizumab also resulted in a significant improvement in fatigue and quality of life, as measured by validated questionnaires. The study also showed that eculizumab treatment was extremely safe, with negligible side effects and incidence of adverse events comparable to that of the placebo.
These data were confirmed in another large open-label phase III study SHEPHERD, which enrolled a broader PNH population including patients with moderate marrow failure and minimal transfusion requirement [47]. Based on the 96 patients enrolled in the study, treatment with eculizumab resulted in a remarkable control of intravascular hemolysis, regardless of the pretreatment transfusion requirement; transfusion independence was achieved in about half of the patients, with significant improvement in fatigue and quality of life in all treated patients [47].
These two initial studies were continued in a common open-label Extension study, which included a total of 187 patients who have previously completed one of the parent clinical trials [48]. The Extension study confirmed the efficacy and the safety of eculizumab with a longer follow up, demonstating that the effects of eculizumab treatment on intravascular hemolysis were retained over time, and long-term safety remained excellent.
The Extension study also looked to additional clinically relevant endpoint, such as the incidence of thromboembolicevents. By comparing the rate of thrombosis between the pretreatment and treatment periods in the same patients, the Extension study showed a 85% reduction of the incidence of thrombosis in comparison to the pre-treatment period [48]. Whether eculizumab exerts its effect on thrombophilia of PNH directly (the pathophysiologyof thrombosis in PNH is still debated), or through the blockade of intravascular hemolysis (e.g., by reduction of nitric oxyde consumption or reduced release of procoagulant microvesicles), is still unknown. However, the effects of eculizumab on thrombophilia of PNH seem quite specific, given that distinct biomarkers of coagulation pathway activation, reactive fibrinolysis and endothelial cell activation decrease during eculizumab treatment [49]. Sustained safety and efficacy of eculizumab was confirmed again in a recent report which ruled out the risk of cumulative toxicity [50]. Given that nowadays the median survival of PNH patients not receiving specific treatment is estimated in 22 years, it is not yet time for a robust analysis assessing the effect of eculizumab on survival. However recent data in a limited cohort of patients provid evidence that continuous eculizumab treatment may result in an improvement of survival of PNH patients [51], at least as compared to historical data [52].
Unmet medical needs in current treatment for PNH
The availability of eculizumab represented a remarkable breakthrough in PNH therapy, which has dramatically impacted the natural history of the disease. Rarely in medicine a single agent has changed so quickly and so effectively the treatment of a disease, especially of such a rare disease; however, as always in medicine, there are still clinical needs which remain unmet in PNH regardless of eculizumab. In fact, even if eculizumab results in an adequate control of intravascular hemolysis in most (if not all) PNH patients for whom anti-hemolytic treatment is indicated (eculizumab is not indicated in PNH patients with bone marrow failure, where anemia is mostly due to impaired red cell production rather than to hemolysis), the majority of patients continue to harbor mild-to-moderate anemia. Eculizumab also represented a unique opportunity for a better understanding of PNH pathophysiology in absence and in presence of clinical anti-complement (anti-C5) inhibition, allowing the dissection of the pathogenic contribution of the different phases of complement cascade which remain uncontrolled in PNH. We have demonstrated that additional mechanisms of disease may account for residual anemia seen in PNH patients during eculizumab treatment. Indeed, PNH erythrocytes survive to complement activation because of the sustained inhibition of the terminal effector complement – C5 cleavage and subsequent MAC assembly; however, they may progressively become decorated on their surface by C3 fragments, which may eventually work as opsonins with subsequent entrapment of C3-bound PNH erythrocytes in the hepatosplenic macrophages. We have documented the phenomenon of C3 deposition by flow cytometry in all of our initial 41 PNH patients on eculizumab, whereas 15 untreated PNH patients do not show any C3+ erythrocyte [53].
The proportion of C3+ PNH erythrocytes is heterogeneous among patients, and in some way may correlate with the hematologic response during eculizumab treatment, as subsequently confirmed by another group [54]. In fact, we have also observed that most PNH patients on eculizumab remain anemic, regardless of an adequate control of intravascular hemolysis (as shown by normal or almost normal LDH levels), and all of them continue to have substantial reticulocytosis [53]. Thus, we came to the conclusion that eculizumab treatment may result in a complete control of the intravascular hemolysis typical of PNH, but in some patients may unmask a novel mechanism of disease, which is C3-mediated extravascular hemolysis. This working hypothesis was supported by the observation that these patients also have persistent reticulocytosis, hyperbilirubinemia and anemia, and it was definitely confirmed by an in vivo erythrocyte survival study by 51Cr labeling, which in paradigmatic cases showed reduced survival and hepatosplenic 51Cr uptake [53].
The observation of clinically remarkable C3-mediated extravascular hemolysis in PNH patients on eculizumab was for sure disappointing, but it is not necessarily detrimental to eculizumab. The reasons underlying this phenomenon are not related to eculizumab itself, rather they are linked to the absence of CD55 on PNH erythrocytes, accounting for persistent uncontrolled C3 convertase activation even regardless of the terminal complement inhibitor eculizumab (which acts downstream in the complement cascade). Indeed, it is only when eculizumab prevents MAC-mediated intravascular hemolysis that PNH erythrocytes survive longer enough to become susceptible to C3-fragment opsonization, and possible subsequent hemolysis through the C3-specific receptors expressed on reticuloendothelial cells (because even massive C3 activation does not lead to MAC assembly and lysis) [55,56].
It is not entirely clear why this mechanism of disease remains subclinical in some patients (who only have minimal residual anemia and persistent reticulocytosis) and led to a remarkable anemia in other cases; preliminary data suggest that genetic heterogeneity in other physiological modulators of complement activity (e.g., CR1) may account for higher risk of residual anemia during eculizumab treatment [57]. Nevertheless, it is conceivable that continuous uncontrolled complement activation through the alternative pathway is the ultimate cause of this phenomenon. In fact, CAP is physiologically in a state of continuous activation because of the C3 tick-over (a low-grade, spontaneous hydrolysis of the internal thioester bond of C3) generating a C3b-like molecule, C3(H2O); nascent C3(H2O) is able to recruit factor B in forming (in the fluid phase) an unstable pro-C3 convertase. Once cleaved by factor D (generating C3(H2O)Bb), this complex will in turn cleave additional C3 molecules to generate C3b, which binds predominantly to glycophorin A and activates (now in a membrane-bound phase on erythrocytes) the CAP amplification loop [22,58,59]. As stated above, this process is finely regulated by CD55, which is absent on PNH erythrocytes; thus, early complement activation is self-limiting on normal cells, but it eventually leads to progressive CAP-mediated amplification on PNH erythrocytes, regardless of the presence of eculizumab (which acts downstream C3).
The reasons why only a fraction of PNH erythrocytes has membrane-bound C3, and why the proportion varies among patients, are not fully understood, even if in vitro data support the concept that PNH erythrocytes are all susceptible to C3 deposition once exposed to conditions causing complement activation [60]. Our current understanding is that each individual erythrocyte has to reach a specific threshold needed to start CAP activation, and that in vivo only a small fraction of erythrocytes is exposed to such a threshold, possibly as a result of specific microenvironmental conditions generating in particular vascular districts (this may open a possible similarity with the site-specific risk of thrombotic events), or simply stochastically. However, even assuming as well-established the mechanism leading to C3 decoration on PNH erythrocytes, their subsequent clearance through the reticuloendothelial system remains to be elucidated. In fact, it is not clear why some patients do not show clinically relevant extravascular hemolysis regardless of a large proportion of C3-bound PNH erythrocytes [56]. Neverthless, residual anemia during eculizumab treatment is an emerging clinical need in PNH, which so far has no adequate treatment option. In fact, steroids are not useful and should be avoided [56], and splenectomy may represent an option [61], but it is not routinely recommended given its considerable medical risks, such as intra- or peri-operatory thrombotic complications and life-long infectious events [62]. Furthermore, long-lasting continuous extravascular hemolysis might lead to additional long-term complications such as gallstone formation or iron overload [63], possibly requiring further medical intervention.
IV. DEVELOPMENT OF NOVEL ANTI-COMPLEMENT STRATEGIES IN PNH
The observation that early phases of the complement cascade, and in particular C3 activation, represent a common biological event possibly resulting in clinical consequences remarks the need of novel strategies of complement modulation. Thus, investigators have started to think about alternative strategies to target early events of the complement cascade, possibly directly at the initial C3 activation (Figure 1).
Figure 1.
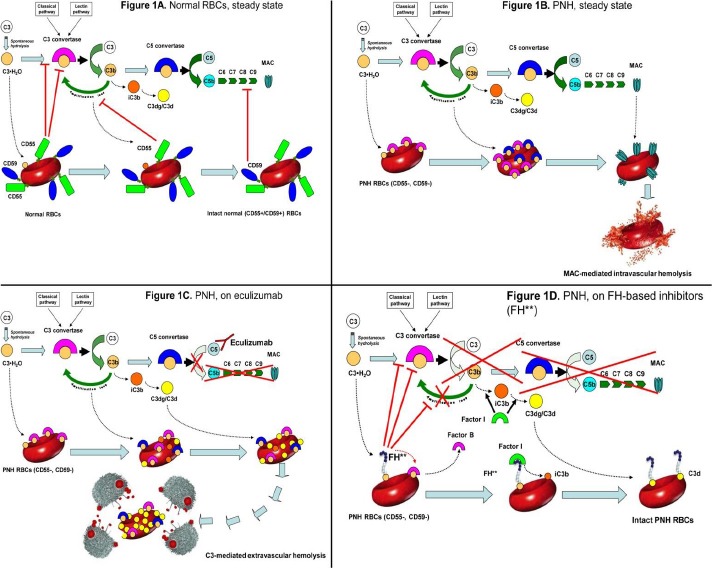
Complement cascade modulation on normal and PNH red blood cells (RBCs), with or without complement inhibitors.
A. Normal RBCs: steady state.
Normal RBCs (CD55+, CD59+) are protected from complement activation by CD55, which down-regulate the C3-convertase, and by CD59, which inhibits the MAC assembly. Thus, in steady state normal RBCs can withstand the challenge of complement activation.
B. PNH RBCs: steady state.
Given their deficiency in surface CD55, PNH RBCs are susceptible to complement activation through C3 tick-over and the alternative pathway, as well as through any other complement pathway. After initial C3 activation and subsequent C3b-binding to erythrocyte surface, the complement cascade may proceed toward the MAC formation, which is not inhibited due to the lack of CD59. As a result, PNH RBCs succumb because of MAC-mediated intravascular hemolysis.
C. PNH RBCs: effect of eculizumab.
Eculizumab binds to C5 in the fluid phase and prevents its cleavage to C5a and C5b. As a result, the progression of the complement cascade toward the MAC assembly is completely inhibited, and PNH RBCs result protected from MAC-mediated intravascular hemolysis. However, given the lack of CD55, initial CAP activation through the C3 tick-over and the CAP-mediated amplification loop remain uncontrolled; thus, early C3 activation continues, and because of their increased survival PNH RBCs progressively bind substantial amount of C3 fragments on their surface. Subsequently, C3-opsonized PNH RBCs become susceptible to C3-receptor mediated clearance by the hepato-splenic reticulo-endothelial system, eventually resulting in C3-mediated extravascular hemolysis.
D. PNH RBCs: effect of C3 inhibitors.
FH-based proteins (FH**), either TT30 or mini-FH, bind to C3b initially activated on PNH RBCs, and deliver their complement-modulatory effect via their FH domain. Thus, FH-based proteins disable the C3-convertase, and, as co-factor of factor I, they also promote the conversion of active C3b into iC3b and then C3dg/C3d, preventing all downstream events due to the complement cascade (as well as additional complement activation on the red cell membrane). As a result, irrespective of the blockade of C5, C3 inhibitors may confer to PNH RBCs a normal survival even in presence of complement activation. All other agents acting at the level of C3 convertase (e.g., compstatin analogs) may work in a similar way, inhibiting initial complement activation on PNH erythrocyte surface and preventing all the downstream events leading to either intravascular or extravascular hemolysis.
“This figure is modified from the research originally published in Blood. Risitano AM, Notaro R, Pascariello C, Sica M, Del Vecchio L, Horvath CJ, Fridkis-Hareli M, Selleri C, Lindorfer MA, Taylor RP, Luzzatto L, Holers VM. The complement receptor 2/factor H fusion protein TT30 protects paroxysmal nocturnalhemoglobinuria erythrocytes from complement-mediated hemolysis and C3 fragment opsonization. Blood. 2012;119(26):6307-16. © the American Society of Hematology.”
Neverthless, it has to be acknowledged that a broad C3 inhibition would carry substantial safety concerns, given that inherited C3 deficiency is associated with a remarkable risk of developing both severe infectioins and autoimmune diseases [64]. Ideally, one would like delivering a complete inhibition of the complement activation occurring on erythrocyte surface due to the lack of the complement inhibitors CD55 and CD59, possibly retaining at least part of the physiological role of the complement in the clearance of microorganisms and injured tissues. In keeping with the terrific clinical results of the anti-C5, different anti-C3 mAb have been initially considered, aiming to bind native C3 in the fluid phase preventing its activation along all the three pathways; however, this approach was abandoned because of the high plasmatic concentration of C3, which makes this approach quite problematic and expensive. In additional, it would carry several concerns because of the risk of possible infectious and autoimmune complications secondary to a complete disabling of the complement cascade.
Another antibody-based anti-C3 strategy was developed, aiming to target activated C3 (C3b/iC3b) rather than the native C3 in the fluid phase. Indeed, the anti-C3b/iC3b murine mAb 3E7 and its chimeric-deimmunized derivative H17 were shown to selectively inhibit the activity of C3 and C5 convertases of the CAP only, providing the opportunity for a selective inhibition of different complement pathway [65]. These antibodies were tested in vitro on PNH erythrocytes, and were shown effective in preventing complement-mediated hemolysis of CD55/CD59 deficient erythrocytes [65]. This finding was elegant and conceptually innovative, but likely still far from a clinical translation, because given that in PNH C3 and C5 convertases localized on erythrocyte surface, these antibodies would finally work as additional opsonins which should even increase PNH erythrocyte clearance by the reticuloendothelial cells, through both the C3- and Fc-specific macrophage receptors. Indeed, before being tested in the clinic these antibodies should be modified by removing their Fc moiety; however, according to available information, such engineered anti-C3 mAb (or Fab fragments) are not available yet.
More recently, we and others have developed a novel strategy of complement inhibition, which aims to deliver a selective (for the CAP) and targeted (on PNH erythrocytes) inhibition of early phases (C3 activation) of the complement cascade, while retaining intact the functioning of the other two complement pathways. This strategy is based on the use of an anti-complement activity deriving from the endogenous complement FH, modified by recombinant DNA technologies. FH is a physiological complement inhibitor that modulates the initial CAP activation in the fluid phase by preventing C3 convertase activity and by promoting C3b inactivation into iC3b [66]. Indeed, FH defuses the CAP amplification loop (which starts regardless the initial pathway of complement activation), and it has been demonstrated protective from lysis for PNH erythrocytes in vitro [67].
At least two different FH-derived agents have been developed. The first one has been designed by creating a recombinant fusion protein between complement FH and another complement-related protein, complement receptor 2 (CR2). In the aim to deliver FH activity locally at the site of complement activation, FH was fused with the iC3b/C3d-binding domain of CR2. Using C3 fragments as target seem quite appropriate given our observation that complement activation in PNH starts with surface C3-deposition, eventually leading to intravascular hemolysis or, in presence of eculizumab, to possible extravascular hemolysis. This novel compound named TT30 was recently investigated by our group in an in vitro model based on exposure of PNH erythrocytes to CAP-activated serum. We have demonstrated that TT30 completely inhibits complement-mediated hemolysis of PNH erythrocytes; in addition, it effectively prevents initial C3 activation and further C3 deposition on PNH erythrocytes [68]. We were able to demonstrate the presence of TT30 on PNH erythrocyte surface, as well as the reversion of the protective effect if anti-CR2 antibodies are added, consistent with the initial assumption that TT30 mainly work at the surface level, and cell membrane targeting is required for full inhibitory effect. Mechanistically, as a FH derivative, TT30 is able to act as a cofactor for FI to convert nascent C3b into iC3b, thus generating more of its own target and disabling the CAP amplification loop. At the same time, TT30 serves a CD55 surrogate on PNH erythrocytes, preventing the formation and promoting the decay of the CAP C3 convertase by displacing factor B and factor Bb, respectively, from the C3b-bound state. These in vitro data support the concept that TT30 in vivo could be useful to prevent both intravascular and extravascular hemolysis of PNH erythrocytes.
Based on this robust background, a phase I clinical trial has just started to enroll PNH patients in a first-in-human study [69]. This initial trial will also be useful to rule our possible concerns about this more intensive strategy of complement inhibition: the hope is that physiological immune-complex clearance and microorganism elimination may be preserved because of the CAP-selective and membrane-preferential action of TT30. If this anticipation will be confirmed, one may hypothesize that TT30 will be extensively investigated in PNH, given that based on the in vitro data it is very likely that the anti-hemolytic action on PNH erythrocytes should be extremely effective.
The second FH-derived recombinant protein follows the same idea of TT30, thus aiming to maximize FH activity at the sites of complement activation. Here the surface-recognition domain does not come from a different protein (e.g., CR2); rather, the FH domains SCR19-20 (which have a strong affinity for the opsonins C3b, iC3b and C3d) are utilized, in a recombinant protein which is called “mini-FH”. Mini-FH is an engineered 43kDa protein that retains both convertase decay acceleration and cofactor activities typical of endogenous human FH, combined with an increased surface-recognition activities, resulting in a potent and selective inhibition of activation and amplification of the complement alternative pathway, without affecting the classical and the mannose/lectin pathway. This compound too was investigated by our group in vitro: mini-FH showed a dose-dependent inhibition of hemolysis, with an IC50 of 0.05 µM and full inhibition at 0.1 µM. Mini-FH also prevented surface deposition of C3 fragments on PNH erythrocytes. on exposure of PNH erythrocytes to CAP-activated serum. Notably, mini-FH was far more potent of TT30, with full inhibition achieved at concentrations about 1 log lower than TT30 [70].
Thus, the spectrum of complement inhibitors moved from monoclonal antibodies to recombinant proteins, which yet may carry elevated production costs (that hopefully will not lead to treatment cose as outrageous as that of eculizumab).
However, C3-targeted complement inhibition also include strategies based on small peptide inhibitors, which potentially are more easy and more cheap to develop. Compstatin is a 13-residue disulphide-bridged peptide that selectively binds to C3 and its active fragment C3b [71]. Compstatin prevents the conversion of C3 to C3b and thus it impairs all initiation, amplification, and terminal pathways of the complement cascade [72]. As a result, compstatin and its derivatives are considered highly promising candidate drugs for treating different complement-mediated diseases [73]. We have recently investigated the in vitro effects of different compstatin analogs; preliminary data showed that, similarly to FH-based proteins, compstatin analogs inhibit complement activation on PNH erytrocytes preventing both hemolysis and C3-deposition on their surface [74]. Further preclinical investigations are currently ongoing to collect all the information needed to support translational plans for this class of agents in human diseases, including PNH. The list of candidate agents for novel complement therapeutics might be even longer, if one wish to include inhibitors of complement FB or FD, or of properdin, as well as other recombinant proteins able to tune complement activity (e.g., recombinant complement FI).
As for C3 inhibitors, all these candidate agents share the concept that targeting early events in the complement cascade may be a worthy option, especially in medical conditions where the terminal complement blockade by eculizumab may result in pharmacological leaks that leads to clinical consequences.
Of course the second generation complement inhibitors will require careful initial clinical investigations to assess whether such a more profound or broader complement inhibition will replicate the excellent safety profile seen with eculizumab. In fact, it will be only when all concerns about increased infectious and autoimmune risks will have been ruled out that the efficacy of this promising strategies could be extensively studied in PNH and other complement-mediated human diseases.
V. CONCLUSIONS
Therapeutic complement inhibition has led to remarkable clinical results in PNH in the last decade. As long as time passes, that the lesson from PNH becomes more clear: the current anti-C5 agent eculizumab is safe and effective, but residual complement activity resulting from upstream complement components may account for suboptimal clinical results. Complement inhibition targeting the C3 is an emerging option to develop novel complement therapeutics, and hopefully will be useful to handle current unmet clinical needs in PNH. Different strategies are currently under development, and a numeber of candidate agents are in preclinical or even clinical studies; the winner will result not only from objective data, but also on the efforts put by the investigators and on the investments put by the companies (treatment cost remains a remarkable issue in all Countries). A novel era for complement modulation has just started; the challenge is to establish targeted and specific treatment for PNH and other complement-mediated diseases.
REFERENCES
- [1].Dunn DE, Liu JM, Young NS. 2000. Paroxysmal nocturnal hemoglobinuria. Young NS.(ed) Bone Marrow Failure Syndromes. Philadelphia: W.B. Saunders Company; p99-121 [Google Scholar]
- [2].Parker CJ, Ware RE. 2003. Paroxysmal nocturnal hemoglobinuria. Greer J, et al.(ed) Wintrobe’s clinical hematology –11th ed Philadelphia: Lippincott Williams & Wilkins; p1203-1221 [Google Scholar]
- [3].Luzzatto L, Notaro R. 2003. Paroxysmal nocturnal hemoglobinuria. Handin RI, Lux SE, Stossel TP.(eds): Blood, principles and practice of hematology (2nd ed.). Philadelphia, PA (USA), Lippincot Williams & Wilkins; p319-34 [Google Scholar]
- [4].Risitano AM. Paroxysmal nocturnal hemoglobinuria. Silverberg DS, Anemia. Rijeka, Croatia: InTech; 2012:331-374 [Google Scholar]
- [5].Kunstling TR, Rosse WF. Erythrocyte acetylcholinesterase deficiency in paroxysmal nocturnal hemoglobinuria (PNH). A comparison of the complement-sensitive and insensitive populations. Blood 1969; 33:607-616 [PubMed] [Google Scholar]
- [6].Nicholson-Weller A, March JP, Rosenfeld SI, Austen KF. Affected erythrocytes of patients with paroxysmal nocturnal hemoglobinuria are deficient in the complement regulatory protein, decay accelerating factor. Proc Natl Acad Sci U S A 1983; 80:5066-5070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Selvaraj P, Rosse WF, Silber R, Springer TA. The major Fc receptor in blood has a phosphatidylinositol anchor and is deficient in paroxysmal nocturnal haemoglobinuria. Nature 1988; 333:565-567 [DOI] [PubMed] [Google Scholar]
- [8].Medof ME, Gottlieb A, Kinoshita T, Hall S, Silber R, Nussenzweig V, Rosse WF. Relationship between decay accelerating factor deficiency, diminished acetylcholinesterase activity, and defective terminal complement pathway restriction in paroxysmal nocturnal hemoglobinuria erythrocytes. J Clin Invest 1987; 80:165-174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Mahoney JF, Urakaze M, Hall S, DeGasperi R, Chang HM, Sugiyama E, Warren CD, Borowitz M, Nicholson-Weller A, Rosse WF. Defective glycosylphosphatidylinositol anchor synthesis in paroxysmal nocturnal hemoglobinuria granulocytes. Blood 1992;79:1400-1403 [PubMed] [Google Scholar]
- [10].Takahashi M, Takeda J, Hirose S, Hyman R, Inoue N, Miyata T, Ueda E, Kitani T, Medof ME, Kinoshita T. Deficient biosynthesis of N-acetylglucosaminyl-phosphatidylinositol, the first intermediate of glycosyl phosphatidylinositol anchor biosynthesis, in cell lines established from patients with paroxysmal nocturnal hemoglobinuria. J Exp Med 1993;177:517-521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Takeda J, Miyata T, Kawagoe K, Iida Y, Endo Y, Fujita T, Takahashi M, Kitani T, Kinoshita T. Deficiency of the GPI anchor caused by a somatic mutation of the PIG-A gene in paroxysmal nocturnal hemoglobinuria. Cell 1993;73:703-711 [DOI] [PubMed] [Google Scholar]
- [12].Miyata T, Takeda J, Iida Y, Yamada N, Inoue N, Takahashi M, Maeda K, Kitani T, Kinoshita T. The cloning of PIG-A, a component in the early step of GPI-anchor biosynthesis. Science 1993;259:1318-1320 [DOI] [PubMed] [Google Scholar]
- [13].Araten DJ, Nafa K, Pakdeesuwan K, Luzzatto L. Clonal populations of hematopoietic cells with paroxysmal nocturnal hemoglobinuria genotype and phenotype are present in normal individuals. Proc Natl Acad Sci U S A 1999;96:5209-5214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Keller P, Payne JL, Tremml G, Greer PA, Gaboli M, Pandolfi PP, Bessler M. FES-Cre targets phosphatidylinositol glycan class A (PIGA) inactivation to hematopoietic stem cells in the bone marrow. J Exp Med 2001;194:581-589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Jasinski M, Keller P, Fujiwara Y, Orkin SH, Bessler M. GATA1-Cre mediates Piga gene inactivation in the erythroid/megakaryocytic lineage and leads to circulating red cells with a partial deficiency in glycosyl phosphatidylinositol-linked proteins (paroxysmal nocturnal hemoglobinuria type II cells). Blood 2001; 98:2248-2255 [DOI] [PubMed] [Google Scholar]
- [16].Luzzatto L, Bessler M, Rotoli B. Somatic mutations in paroxysmal nocturnal hemoglobinuria: a blessing in disguise? Cell 1997; 88:1-4 [DOI] [PubMed] [Google Scholar]
- [17].Young NS, Maciejewski JP. Genetic and environmental effects in paroxysmal nocturnal hemoglobinuria: this little PIG-A goes "Why? Why? Why?". J Clin Invest 2000;106:637-641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Rotoli B, Luzzatto L. Paroxysmal nocturnal haemoglobinuria. Baillieres Clin Haematol 1989;2:113-138 [DOI] [PubMed] [Google Scholar]
- [19].Lewis SM, Dacie JV. The aplastic anaemia--paroxysmal nocturnal haemoglobinuria syndrome. Br J Haematol 1967;13:236-251 [DOI] [PubMed] [Google Scholar]
- [20].Karadimitris A, Manavalan JS, Thaler HT, Notaro R, Araten DJ, Nafa K, Roberts IA, Weksler ME, Luzzatto L. Abnormal T-cell repertoire is consistent with immune process underlying the pathogenesis of paroxysmal nocturnal hemoglobinuria. Blood 2001; 96:2613-2620 [PubMed] [Google Scholar]
- [21].Gargiulo L, Papaioannou M, Sica M, Talini G, Chaidos A, Richichi B, Nikolaev AV, Nativi C, Layton M, de la Fuente J, Roberts I, Luzzatto L, Notaro R, Karadimitris A. Glycosylphosphatidylinositol-specific, CD1d-restricted T cells in paroxysmal nocturnal hemoglobinuria. Blood 2013;121(14):2753-2761 [DOI] [PubMed] [Google Scholar]
- [22].Müller-Eberhard HJ. Molecular organization and function of the complement system. Annu Rev Biochem 1988;57:321-347 [DOI] [PubMed] [Google Scholar]
- [23].Holers VM. The spectrum of complement alternative pathway-mediated diseases. Immunol Rev 2008;223:300-316 [DOI] [PubMed] [Google Scholar]
- [24].Nicholson-Weller A, Burge J, Fearon DT, Weller PF, Austen KF. Isolation of a human erythrocyte membrane glycoprotein with decay-accelerating activity for C3 convertases of the complement system. J Immunol 1982; 129:184-189 [PubMed] [Google Scholar]
- [25].Nicholson-Weller A. Decay accelerating factor (CD55). Curr Top Microbiol Immunol 1992;178:7-30 [DOI] [PubMed] [Google Scholar]
- [26].Pangburn MK, Schreiber RD, Trombold JS, Müller-Eberhard HJ. Paroxysmal nocturnal hemoglobinuria: deficiency in factor H-like functions of the abnormal erythrocytes. J Exp Med 1983;157:1971-1980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Pangburn MK, Schreiber RD, Müller-Eberhard HJ. Deficiency of an erythrocyte membrane protein with complement regulatory activity in paroxysmal nocturnal hemoglobinuria. Proc Natl Acad Sci U S A 1983;80:5430-5434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Shin ML, Hänsch G, Hu VW, Nicholson-Weller A. Membrane factors responsible for homologous species restriction of complement-mediated lysis: evidence for a factor other than DAF operating at the stage of C8 and C9. J Immunol 1986;136:1777-1782 [PubMed] [Google Scholar]
- [29].Holguin MH, Fredrick LR, Bernshaw NJ, Wilcox LA, Parker CJ. Isolation and characterization of a membrane protein from normal human erythrocytes that inhibits reactive lysis of the erythrocytes of paroxysmal nocturnal hemoglobinuria. J Clin Invest 1989; 84:7-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Holguin MH, Wilcox LA, Bernshaw NJ, Rosse WF, Parker CJ. Relationship between the membrane inhibitor of reactive lysis and the erythrocyte phenotypes of paroxysmal nocturnal hemoglobinuria. J Clin Invest 1989;84:1387-1394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Meri S, Morgan BP, Davies A, Daniels RH, Olavesen MG, Waldmann H, Lachmann PJ. Human protectin (CD59), an 18,000-20,000 MW complement lysis restricting factor, inhibits C5b-8 catalysed insertion of C9 into lipid bilayers. Immunology. 1990; 71:1-9 [PMC free article] [PubMed] [Google Scholar]
- [32].Wilcox LA, Ezzell JL, Bernshaw NJ, Parker CJ. Molecular basis of the enhanced susceptibility of the erythrocytes of paroxysmal nocturnal hemoglobinuria to hemolysis in acidified serum. Blood 1991;78:820-829 [PubMed] [Google Scholar]
- [33].Holguin MH, Martin CB, Bernshaw NJ, Parker CJ. Analysis of the effects of activation of the alternative pathway of complement on erythrocytes with an isolated deficiency of decay accelerating factor. J Immunol. 1992; 148:498-502 [PubMed] [Google Scholar]
- [34].Merry AH, Rawlinson VI, Uchikawa M, Daha MR, Sim RB. Studies on the sensitivity to complement-mediated lysis of erythrocytes (Inab phenotype) with a deficiency of DAF (decay accelerating factor). Br J Haematol 1989;73:248-253 [DOI] [PubMed] [Google Scholar]
- [35].Yamashina M, Ueda E, Kinoshita T, Takami T, Ojima A, Ono H, Tanaka H, Kondo N, Orii T, Okada N, Okada H, Inoue K, Kitani T. Inherited complete deficiency of 20-kilodalton homologous restriction factor (CD59) as a cause of paroxysmal nocturnal hemoglobinuria. N Engl J Med 1990; 323:1184-1189 [DOI] [PubMed] [Google Scholar]
- [36].Motoyama N, Okada N, Yamashina M, Okada H. Paroxysmal nocturnal hemoglobinuria due to hereditary nucleotide deletion in the HRF20 (CD59) gene. Eur J Immunol. 1992; 22:2669-73 [DOI] [PubMed] [Google Scholar]
- [37].Ham TH, Dingle JH. Studies on the destruction of red blood cells. II. Chronic hemolytic anemia with paroxysmal nocturnal hemoglobinuria: certain immunological aspects of the hemolytic mechanism with special reference to serum complement. J Clin Invest 1939;18:657-672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Rosse WF, Dacie JV. Immune lysis of normal human and paroxysmal nocturnal hemoglobinuria (PNH) red blood cells. I. The sensitivity of PNH red cells to lysis by complement and specific antibody. J Clin Invest 1966; 45:736-748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Rosse WF. The life-span of complement-sensitive and –insensitive red cells in paroxysmal nocturnal hemoglobinuria. Blood 1971;37:556-562 [PubMed] [Google Scholar]
- [40].van der Schoot CE, Huizinga TW, van 't Veer-Korthof ET, et al. Deficiency of glycosyl-phosphatidylinositol-linked membrane glycoproteins of leukocytes in paroxysmal nocturnal hemoglobinuria, description of a new diagnostic cytofluorometric assay. Blood. 1990; 76:1853-1859 [PubMed] [Google Scholar]
- [41].Pangburn MK, Schreiber RD, Müller-Eberhard HJ. Formation of the initial C3 convertase of the alternative complement pathway. Acquisition of C3b-like activities by spontaneous hydrolysis of the putative thioester in native C3. J Exp Med. 1981;154:856–867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Pangburn MK, Müller-Eberhard HJ. Initiation of the alternative complement pathway due to spontaneous hydrolysis of the thioester of C3. Ann N Y Acad Sci 1983; 421:291–298 [DOI] [PubMed] [Google Scholar]
- [43].Rother RP, Rollins SA, Mojcik CF, Brodsky RA, Bell L. Discovery and development of the complement inhibitor eculizumab for the treatment of paroxysmal nocturnal hemoglobinuria. Nat Biotechnol 2007; 25:1256-1264 [DOI] [PubMed] [Google Scholar]
- [44].Matis LA, Rollins SA. Complement-specific antibodies: designing novel anti-inflammatories. Nat Med 1995;1:839-842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Hillmen P, Hall C, Marsh JC, Elebute M, Bombara MP, Petro BE, Cullen MJ, Richards SJ, Rollins SA, Mojcik CF, Rother RP. Effect of eculizumab on hemolysis and transfusion requirements in patients with paroxysmal nocturnal hemoglobinuria. N Engl J Med 2004; 350:552-559 [DOI] [PubMed] [Google Scholar]
- [46].Hillmen P, Young NS, Schubert J, Brodsky RA, Socié G, Muus P, Röth A, Szer J, Elebute MO, Nakamura R, Browne P, Risitano AM, Hill A, Schrezenmeier H, Fu CL, Maciejewski J, Rollins SA, Mojcik CF, Rother RP, Luzzatto L. The complement inhibitor eculizumab in paroxysmal nocturnal hemoglobinuria. N Engl J Med 2006; 355:1233-1243 [DOI] [PubMed] [Google Scholar]
- [47].Brodsky RA, Young NS, Antonioli E, Risitano AM, Schrezenmeier H, Schubert J, Gaya A, Coyle L, de Castro C, Fu CL, Maciejewski JP, Bessler M, Kroon HA, Rother RP, Hillmen P. Multicenter phase III study of the complement inhibitor eculizumab for the treatment of patients with paroxysmal nocturnal hemoglobinuria. Blood 2008;114:1840-1847 [DOI] [PubMed] [Google Scholar]
- [48].Hillmen P, Muus P, Dührsen U, Risitano AM, Schubert J, Luzzatto L, Schrezenmeier H, Szer J, Brodsky RA, Hill A, Socié G, Bessler M, Rollins SA, Bell L, Rother RP, Young NS. Effect of the complement inhibitor eculizumab on thromboembolism in patients with paroxysmal nocturnal hemoglobinuria. Blood 2007; 110:4123-4128 [DOI] [PubMed] [Google Scholar]
- [49].Helley D, de Latour RP, Porcher R, Rodrigues CA, Galy-Fauroux I, Matheron J, Duval A, Schved JF, Fischer AM, Socié G; French Society of Hematology Evaluation of hemostasis and endothelial function in patients with paroxysmal nocturnal hemoglobinuria receiving eculizumab. Haematologica. 2010; 95:574-581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Hillmen P, Muus P, Röth A, Elebute MO, Risitano AM, Schrezenmeier H, Szer J, Browne P, Maciejewski JP, Schubert J, Urbano-Ispizua A, de Castro C, Socié G, Brodsky RA. Long-term safety and efficacy of sustained eculizumab treatment in patients with paroxysmal nocturnal hemoglobinuria. Br J Haematol 2013; 162(1):62-73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Kelly RJ, Hill A, Arnold LM, Brooksbank GL, Richards SJ, Cullen M, Mitchell LD, Cohen DR, Gregory WM, Hillmen P. Long-term treatment with eculizumab in paroxysmal nocturnal hemoglobinuria: sustained efficacy and improved survival. Blood 2011;117:6786-6792 [DOI] [PubMed] [Google Scholar]
- [52].de Latour RP, Mary JY, Salanoubat C, Terriou L, Etienne G, Mohty M, Roth S, de Guibert S, Maury S, Cahn JY, Socié G; French Society of Hematology; French Association of Young Hematologists. Paroxysmal nocturnal hemoglobinuria: natural history of disease subcategories. Blood. 2008; 112:3099-106 [DOI] [PubMed] [Google Scholar]
- [53].Risitano AM, Notaro R, Marando L, Serio B, Ranaldi D, Seneca E, Ricci P, Alfinito F, Camera A, Gianfaldoni G, Amendola A, Boschetti C, Di Bona E, Fratellanza G, Barbano F, Rodeghiero F, Zanella A, Iori AP, Selleri C, Luzzatto L, Rotoli B. Complement fraction 3 binding on erythrocytes as additional mechanism of disease in paroxysmal nocturnal hemoglobinuria patients treated by eculizumab. Blood 2009;113:4094-4100 [DOI] [PubMed] [Google Scholar]
- [54].Hill A, Rother RP, Arnold L, Kelly R, Cullen MJ, Richards SJ, Hillmen P. Eculizumab prevents intravascular hemolysis in patients with paroxysmal nocturnal hemoglobinuria and unmasks low-level extravascular hemolysis occurring through C3 opsonization. Haematologica 2010; 95,567-573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Luzzatto L, Risitano AM, Notaro R. Paroxysmal nocturnal hemoglobinuria and eculizumab. Haematologica 2010; 95:523-526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Risitano AM, Notaro R, Luzzatto L, Hill A, Kelly R, Hillmen P. Paroxysmal Nocturnal Hemoglobinuria: Hemolysis Before and After Eculizumab. New Engl J Med 2010; 363:2270-2272 [DOI] [PubMed] [Google Scholar]
- [57].Rondelli T, Risitano AM, Peffault de Latour R, Peruzzi B, Ricci P, Barcellini W, Iori AP, Boschetti C, Valle V, Frémeaux-Bacchi V, De Angioletti M, Socie G, Luzzatto L, Notaro R. Genetic Polymorphism of the Complement Receptor-1 (CR1) Gene Correlates with the Clinical Response to Eculizumab of Patients with Paroxysmal Nocturnal Hemoglobinuria (PNH). Blood 2012; 120: 983.(abs) [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Pangburn MK, Schreiber RD, Müller-Eberhard HJ. C3b deposition during activation of the alternative complement pathway and the effect of deposition on the activating surface. J Immunol 1983;131:1930-1935 [PubMed] [Google Scholar]
- [59].Parker CJ, Baker PJ, Rosse WF. Increased enzymatic activity of the alternative pathway convertase when bound to the erythrocytes of paroxysmal nocturnal hemoglobinuria. J Clin Invest 1982;69:337-346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Sica M, Pascariello C, Rondelli T, et al. In vitro complement protein 5 (C5) blockade recapitulates the Complement protein 3 (C3) binding to GPI-negative erythrocytes observed in Paroxysmal Nocturnal Hemoglobinuria (PNH) patients on eculizumab. Haematologica. 2010; 95(s2):196.(abs) [Google Scholar]
- [61].Risitano AM, Marando L, Seneca E, Rotoli B. Hemoglobin normalization after splenectomy in a paroxysmal nocturnal hemoglobinuria patient treated by eculizumab. Blood 2008; 112:449-451 [DOI] [PubMed] [Google Scholar]
- [62].Brodsky RA. How I treat paroxysmal nocturnal hemoglobinuria. Blood 2009;113:6522-6527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Risitano AM, Imbriaco M, Marando L, Seneca E, Soscia E, Malcovati L, Iori AP, Pane F, Notaro R, Matarazzo M. From perpetual hemosiderinuria to possibile iron overload: iron redistribution in paroxysmal nocturnal haemoglobinuria on eculizumab by magnetic resonance imaging. Br J Haematol 2012;158(3):415-418 [DOI] [PubMed] [Google Scholar]
- [64].Botto M, Walport MJ. Hereditary deficiency of C3 in animals and humans. Int Rev Immunol 1993;10(1):37-50 [DOI] [PubMed] [Google Scholar]
- [65].Lindorfer MA, Pawluczkowycz AW, Peek EM, Hickman K, Taylor RP, Parker CJ. A novel approach to preventing the hemolysis of paroxysmal nocturnal hemoglobinuria: both complement-mediated cytolysis and C3 deposition are blocked by a monoclonal antibody specific for the alternative pathway of complement. Blood 2010;115:2283-2291 [DOI] [PubMed] [Google Scholar]
- [66].Ferreira VP, Pangburn MK. Factor H mediated cell surface protection from complement is critical for the survival of PNH erythrocytes. Blood 2007; 15;110:1290-1292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Whaley K, Ruddy S. Modulation of the alternative complement pathway by β1H globulin. J Exp Med 1976;144:1147-1163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Risitano AM, Notaro R, Pascariello C, Sica M, del Vecchio L, Horvath CJ, Fridkis-Hareli M, Selleri C, Lindorfer MA, Taylor RP, Luzzatto L, Holers VM. The complement receptor 2/factor H fusion protein TT30 protects paroxysmal nocturnal hemoglobinuria erythrocytes from complement-mediated hemolysis and C3 fragment opsonization. Blood 2012;119:6307-6316 [DOI] [PubMed] [Google Scholar]
- [69].ClinicalTrials.gov. A service of the U.S. National Institutes of Health. http://clinicaltrials.gov/ct2/show/NCT01335165
- [70].Schmidt CQ, Bai H, Lin Z, Risitano AM, Barlow PN, Ricklin D, Lambris JD. Rational engineering of a minimized immune inhibitor with unique triple-targeting properties. J Immunol 2013; 190(11):5712-5721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Sahu A, Kay BK, Lambris JD. Inhibition of human complement by a C3-binding peptide isolated from a phage-displayed random peptide library. J Immunol 1996;157:884–891 [PubMed] [Google Scholar]
- [72].Sahu A, Soulika AM, Morikis D, Spruce L, Moore WT, Lambris JD. Binding kinetics, structureactivity relationship, and biotransformation of the complement inhibitor compstatin. J Immunol 2000;165:2491–2499 [DOI] [PubMed] [Google Scholar]
- [73].Ricklin D, Lambris JD. Complement-targeted therapeutics. Nat Biotechnol 2007;25(11):1265-1275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Risitano AM, Ricci P, Pascariello C, Raia M, Schmidt CQ, Li Y, Reis ES, Sica M, Notaro R, Del Vecchio L, Pane F, Ricklin D, Lambris JD. Novel Complement Modulators for Paroxysmal Nocturnal Hemoglobinuria: Peptide and Protein Inhibitors of C3 Convertase Prevent Both Surface C3 Deposition and Subsequent Hemolysis of Affected Erythrocytes in Vitro. Blood 2012; 120:370 [Google Scholar]