

Abstract
In 2005, the discovery of Janus kinase 2 (JAK2) V617F mutation in approximately half of patients with myelofibrosis (MF) marked an important milestone in our understanding of the pathophysiology of MF. This has broadened our understanding of the disease pathogenesis and became the foundation for the development and subsequent clinical use of JAK inhibitors for MF. However, it is clear that other pathogenetic modifiers contribute to the disease diversity and phenotypic variability of MF. Novel genome scanning technologies were useful in the identification of recurrent molecular mutations in other genes including MPL, TET2, IDH1/2, DNMT3A, SH3B2 (LNK) and CBL in MF pointing out that other pathways might be important in addition to the JAK/STAT pathway. The biologic role and clinical implications of these molecular mutations in MF is currently under investigation. The main challenge is to understand the mechanisms whereby molecular mutations whether alone or in combination with other genetic and non-genetic events contribute to the pathogenesis of MF and eventually can explain the phenotypic variability among the MF patients. In the present review we will provide an overview of the molecular pathogenesis of MF describing past and recent discoveries in molecular markers and their possible relevance in disease phenotype.
Keywords: Myelofibrosis, pathogenesis, molecular mutations
I. INTRODUCTION: OVERVIEW OF PRIMARY MYELOFIBROSIS
Myelofibrosis (MF) is a Philadelphia chromosome negative myeloproliferative neoplasm (MPN) clinically characterized by disease related constitutional symptoms, extramedullary hematopoiesis (EMH) especially splenomegaly, leukoerythroblastosis and variable degrees of cytopenia. Pathologically, the bone marrow (BM) of MF patients demonstrates atypical megakaryocytes, megakaryocytic hyperplasia, thickening and distortion of bony trabeculae and fibrosis [1]. Clonal markers exemplified by cytogenetics can be helpful in diagnosis, prognosis and therapeutic stratification in myeloid malignancies. Key examples include t(9;22)(q34;q11) in chronic myeloid leukemia, 5q- in MDS with del5q abnormalities and inv(16)(p13;q22), t(16;16)(p13;q22), t(15;17)(q24;q21) and t(8;21)(q22;q22) in de novo acute myeloid leukemia. In MPN, the JAK2 V617F mutation provided one of the most important genetic findings in the non-Philadelphia chromosome myeloid malignancies. Many non-JAK2 clonal markers have been recently identified in MF, including genes like MPL (exon 10)[2], TET2 [3], ASXL1 [4], IDH1/2 [5], DNMT3A [6], EZH2 [7], CBL [8] and SH2B3 (LNK). Their exact role in the development of disease specific phenotypic features and clinical manifestations in MF is currently the subject of investigation but will likely provide new insight into disease specific mechanisms. For example, the ring sideroblast phenotype of MDS and even MF, mastocytosis and pure red cell aplasia patients can be explained by mutations in the SF3B1 gene [9]. Identification of phenotype-genotype associations specific to individual genetic mutations will hopefully translate to new therapeutic options for patients with MF that can lead to better clinical outcomes.
II. MOLECULAR MUTATIONS IN THE PATHOPHYSIOLOGY OF MYELOFIBROSIS
JAK2 (exon 14): the discovery of mutations in the Janus kinase 2 (JAK2) signalling pathway opened a new perspective in the pathophysiology and treatment of MF [10]. The frequency of JAK2V617F mutation ranges between 43-59% in MF. The most commonly detected mutation results in a guanine to thymine change at nucleotide 1849 [11]. JAK2 is a member of JAK family (JAK1, JAK3 and TYK2) proteins which are cytoplasmic non-receptor tyrosine kinases with a significant function in cell proliferation and survival of hematopoietic stem cells (HSC). The JAK2V617F mutation is essential for self-reprogramming properties of HSC and it has been associated with some phenotypes of MF. Experiences in transgenic mice showed that, a low level of JAK2V617F load can induce essential thrombocytosis (ET)-type features, while the higher levels of mutant alleles can cause polycythemia vera (PV) and MF-like phenotypes [12]. However, the presence of this mutation cannot consolidate all other MF related findings including cytopenia and the tendency of MF to transform to acute myeloid leukemia (AML) which is observed in 5-10% of patients with MF [13]. Determination of the molecular basis for the constitutive activation of JAK2V617F is crucial for understanding its clinical implication. It has been elucidated that the position at amino acid 617 it is important for protein-protein interaction. Mutations at position 617 induce autophosphorylation, gene transcription and in vitro kinase activity of JAK2 [14]. Recently, it was reported that activation of JAK2 can promoting the activation of three homodimeric myeloid receptors like EPO-receptor, MPL (TPO-receptor), and GM-CSF (Figure 1)[15].
Figure 1.
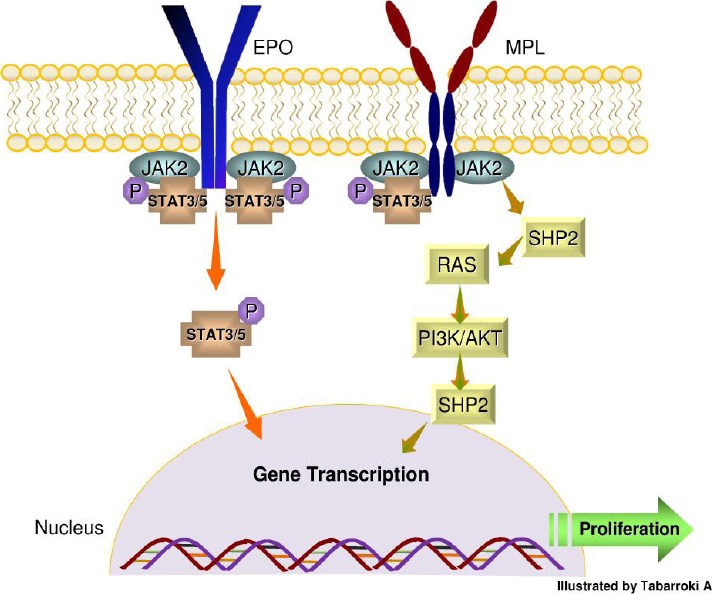
JAK2 receptor signaling and activation of STAT pathway. The binding of erythropoietin (EPO) to a receptor results in receptor dimerization of JAK2. This dimerization leads to phosphorylation of STAT3 and 5 and formation of stable homodimers and heterodimers which subsequently induce transcription of genes that regulate the cell proliferation and survival. The JAK2V617F mutation can cause the monotonous activation of STAT pathway in the absence of any other trigger. Furthermore, activation of MPL can initiate the downstream signaling cascade of JAK2 via recruitment of Src homology 2 (SH2) which can stimulate RAS protein and phosphoinositide-3 kinase (PI3K) leading to transcriptional activity of certain genes.
Beside its role in mediating the signalling pathways of EPO and MPL, JAK2 signal can induce the over-expression of several oncogenes like LIM domain only 2 (LMO2) by an epigenetic regulation. JAK2 signal can re-model the chromatin structure by phosphorylating the histone H3Y41 and consequently blocking the recruitment of the repressor heterochromatin 1α leading to over-expression of LMO2 [16].
JAK2 (exon 12): a gain-of-function mutation in exon 12 is detected in a small proportion (3-5%) of JAK2 wild type (WT) MF patients. Murine studies have shown that mutations in this exon impacts erythrocyte proliferation [17]. New alterations (deletions, insertions, substitutions) were found in positions 537 through 543. However, it is still unclear how these mutations can alter the function of JAK2 [17]. It has been elucidated that exon 12 links to SRC homology 2 (SH2) and JH2 domains of JAK2. Compared to V617F mutation, molecular alterations in exon 12 produce higher ligand-independent signaling and phosphorylation through JAK2 [17]. In humans, these mutations are associated with younger age, lower level of erythropoietin, and lower predominance of erythroid myeloproliferation [18]. Patients with polycythemia vera (PV) harboring mutations in exon 12 seem to be predisposed to transformation to MF.
MPL (Myeloproliferative Leukemia Virus): the absence of JAK2V617F in some MF patients suggests the presence of alternative mechanism(s) that can activate the JAK/STAT pathway (Figure 1)[19]. MPL W515L/K can be detected in 5-10% in MF patients and appears to be a gain-of-function mutation [20-21]. The MPL gene encodes a 635 amino acid protein called CD110. CD110 has 4 functional domains including a putative signal peptide, an extracellular domain, a transmembrane domain, and an intracellular domain [22]. The ligand, thrombopoietin (TPO) can lead to dimerization of CD110 and subsequent downstream activation of the JAK-STAT pathway. The CD110-TPO is important in renewal of HSC and in the development and proliferation of megakaryocytes and platelets [23]. The relationship between MPL W515L/K and the pathogenesis of MF are based on several important disease observations: 1) MPL W515L/K are found in the amphipathic KWQFP motif of CD110 which is important in preventing spontaneous activation of CD110, 2) Nude mice injected with MPL-transduced Ba/F3 cells developed subcutaneous tumors and splenomegaly [24], and 3) BM transplantation assays conducted by injecting murine cells with W515L mutation led to features of ET and tendency of rapid evolution to MF [25].
TET2 (Ten Eleven Translocation-2): TET2 is a tumor suppressor gene located in a small region (0.35 Mb) of chromosome 4 (4q24). Mutations of this gene are frequently detected in various myeloid malignancies. In MPN, TET2 has a frequency of 15-25% in PMF, 15-20% in post-PV MF, 10-15% in post-ET MF and 20-25% in MF that have progressed to AML [26]. This molecular mutation can occur in conjunction with JAK2V617F mutation leading to a bi-clonal model or can precede the JAK2 mutational event during disease formation [26-27]. TET2 mutations can lead to truncated proteins resulting in a total or partial loss-of-function. It has been postulated that molecular alterations in this gene may have been one of the first events in early hematopoiesis [27]. In addition, the epigenetic role of TET2 has been studied [28]. TET2 catalyzes the conversion of the 5-methylcytosine (5-MC) to 5-hydroxymethylcytosine (5-hMC) by oxidizing 5-MC. In patients carrying TET2 mutations, the level of 5-hMC is lower than WT with variability across diseases (lower in myelodysplastic syndromes (MDS) and higher in AML)[29]. In human hematopoiesis, knock-down of TET2 resulted in increased monocytes differentiation and decreased erythroid proliferation [16]. In a study conducted in our laboratory, TET2 mutations were not present in JAK2 WT MF patients in comparison to JAK2 mutant counterpart [30]. This observation was also reported by other investigators (Figure 2)[31].
Figure 2.
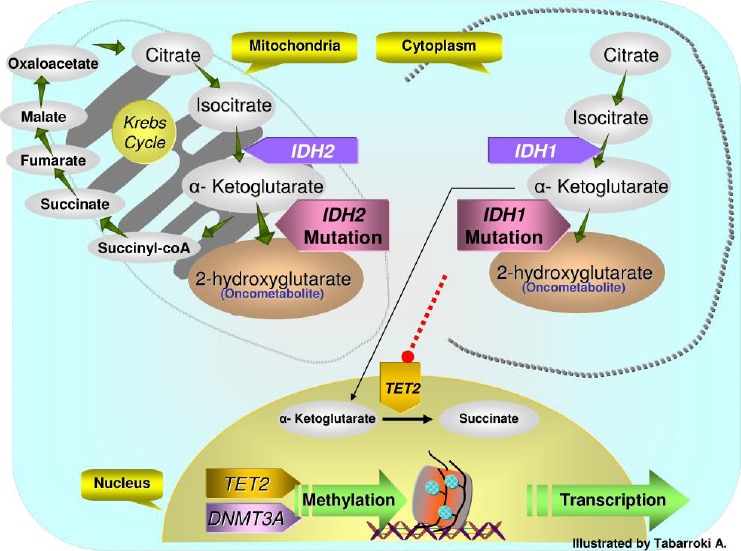
Krebs cycle to DNA methylation. IDH1/2 wild-type enzymes convert isocitrate to α-ketoglutarate (αKG), a TET2 substrate. IDH1/2 mutations result in neomorphic enzymes converting αKG to 2-hydroxyglutarate (2HG) which is an oncometabolite. 2HG can compromise the function of TET2. In addition, TET2 and DNMT3A genes are involved in epigenetic transcriptional regulation by maintaining the DNA methylation during DNA replication and cell division.
ASXL1 (Additional Sex Combs-like 1): ASXL1 is a polycomb gene required for long-term repression of the HOX genes. Genetically, in Drosophila, Asx gene behaves like an inducer of the trithorax complex. In general, polycomb genes are important in the manifestation of the bidirectional homeotic phenotypes suggesting their roles in the activation and silencing of HOX genes [32]. ASXL1, is a potential epigenetic modifier, and mutations of this gene cam cause the loss of polycomb repressive complex 2 (PRC2)-mediated histone H3 lysine 27 (H3K27) tri-methylation which results in the transcriptional repression in hematopoietic cells [33].
Loss of ASXL1 can lead to the depletion of PRC2 and H3K27 involvement in myeloid leukemic cells which can cause the perturbation of polycomb-mediated gene silencing. It was elucidated that dysregulation of this interaction can make the cells prone to leukemia transformation [33]. In addition, it has been shown to down-regulate the retinoic acid receptor signaling which can alter the transcription of particular genes [34]. The frequency of ASXL1 mutations in MF is about 15-20% albeit, in overlap syndromes particularly in chronic myelomonocytic leukemia (CMML) half of the patients acquire those mutations [35-36]. It was reported that ASXL1 mutations are usually mutually exclusive with JAK2V617F even though in one study ASXL1 mutations were noted in conjunction with JAK2V617F or TET2 mutations rather than IDH1 [35]. ASXL1 mutations are usually seen in exon 12 and are missense, frame-shifts, and duplications in the case of the controversial variant (p.Gly646TrpfsX12)[35].
CBL (Casitas B-cell Lymphoma): CBL gene is located in the long arm of chromosome 11 (11q23.3) and encodes for a cytosolic protein with dual functionality; 1) down regulates tyrosine kinase signaling trigged by E3 ubiquitin ligase which leads to internalization and lysosomal/ proteosomal degradation and 2) modulation of downstream signaling of JAK2 and MPL [37]. In cell culture studies, a CBL mutation induces oncogenic phenotype and proliferation with activating the RAS-pathway in the absence of growth factor stimulation [37]. CBL knockout mice manifested splenomegaly, HSC proliferation, and sensitivity to growth factors [38]. The mutations of this gene were first described in AML as MLL-CBL fusion (MLL, exon 6 and CBL, exon 8) resulted in induction of the FLT3 signaling. (38) CBL has been considered a tumor suppressor gene. The frequency of mutant CBL is about 15-20% and 3-6% in CMML and PMF, respectively [39]. The mutations are mainly missense or in-frame deletions and usually coexist with JAK2, TP53, FLT3 and RUNX1 mutations [40]. However, some studies have shown that at the time of disease progression, CBL mutations can occur after JAK2V617F mutation suggesting that two different cell lineages harbor these mutations [37].
IDH1/2 (Isocitrate Dehydrogenase 1/2): IDH1/2 are homodimeric NADP+ dependent enzymes involved in the conversion of isocitrate to α-ketoglutarate by oxidative decarboxylation prior to NADPH synthesis [41]. Heterozygous mutations of these genes were first described in gliomas and secondary glioblastomas and then in AML [42-43]. Mutations in IDH1/2 can lead to an increase in NADPH-dependent reaction of α-ketoglutarate resulting in over production of α-hydroxyglutarate which is a potential toxic substance and conversely have a negative effect on the function of the TET2 protein (Figure 2)[44]. IDH1/2 mutations usually involve the amino acid R132 and R172, respectively. The frequency of IDH1/2 mutation is about 3-5%, 1-2%, and 10-20% in PMF, post-PV/ET MF and in MF cases evolving to AML [45]. Furthermore, mutations in these genes are usually observed to be mutually exclusive from mutations of JAK2, TET2, and MPL [39]. In addition, in gliomas the presence of IDH1/2 mutations correlated to treatment response to chemotherapy. A study conducted in MPN has correlated the presence of a panel of molecular mutations including IDH1/2 with a failure to response to treatment with PEG-interferon-α [16]. Ultimately, IDH1/2 mutations have been associated with transformation to AML even though it is not clear at this time where it falls in the hierarchy of mutational events in clonal evolution [46].
DNMT3A (cytosine-5-methyltransferase 3-α): the DNMT3A heterozygous mutations were initially described in AML and MDS with a frequency of 5-22% [47]. However, in MF its frequency is lower (5 -12%)[48]. This gene encodes a DNA methyltransferase that is essential in de novo methylation. Acquisition of mutations can cause loss-of-function resulting in homo-dimerization and activation of the protein, reduction of the activity of methyltransferase, and consequent increased cell proliferation [48]. This gene can play a significant role in progression of MPN to AML in the presence of JAK2 and MPL mutations. It has been showed that the time of mutational event acquisition reflects the disease course [49]. In order to further dissect the role of this gene in MPN, a study described the isolation of different cellular lineages and the successive assessment of the cell type harboring high frequency of DNMT3A mutations. Mutations were identified with high frequency in CD14+ (monocytes) enriched fraction and with low frequency in CD3+ and CD19+ (T and B lymphocytes, respectively) suggesting that the aberrant clone does not occur in lymphoid lineages [49]. Indeed, DNMT3A mutations are associated with over expression of other relevant genes in advanced myeloid malignancies [48].
SH2B3 (LNK): the lymphocyte (LNK or SH2B3) adaptor protein is a member of the SH2B family and plays an important role in hematopoiesis and cytokine regulation. The protein encoded by this gene is a plasma adaptor component which binds to JAK2 and inhibits the JAK/STAT signal transduction pathway [50]. In addition, LNK negatively regulates the EPO receptor and MPL signaling resulting in the inhibition of the JAK-STAT pathway [51]. In JAK2V617F mutant patients, LNK expression may increase which contributes to the developing of myeloproliferative phenotype [14]. LNK-deficient mice showed a phenotype similar to MPN patients like splenomegaly, thrombocytosis, leukocytosis, abnormal megakaryocytes, and BM fibrosis [52]. The prevalence of LNK mutations is low (<5%) in MPN; however it is slightly higher (10-13%) in leukemic transformation of MF patients [53]. Concomitant mutations in JAK2V617F and LNK were associated with disease evolution [54]. In another study, LNK deficiency in mice increased cytokine independent- JAK-STAT signaling and also cooperates with JAK2 activation in the development of a MPN like phenotype [55].
EZH2 (enhancer of zeste homolog 2): this gene is located in chromosome 7 (7q36.1) and belongs to the complex 2 of Polycomb, a mediator of transcriptional silencing and a regulator of multiple cellular process like proliferation, maturation, aging and hematopoietic cell plasticity [56]. EZH2 over-expression is noted in a variety of solid tumors like prostate and breast cancer and it has been reported to contribute to tumor aggressiveness and poor cellular differentiation [57]. EZH2 mutations appear to be a gain-of-function genetic change acting as a repressor by methylating the histone H3 on lysine 27 (H3K27) and consequently inactivating the chromatin [58]. The frequency of EZH2 mutations is approximately 12% in MF; however, mutations are not exclusively found in MPN since they are more frequent in MDS (2-6%) and MDS/MPN (~15%). Clinically, EZH2 mutations have been associated with poor prognosis [7].
RNA Splicing Machinery: the recurrent mutations in genes encoding for different members of the RNA spliceosome machinery (SF3B1, SRSF2, U2AF1 and ZRSR2) have been reported in myeloid malignancies in relatively high frequency (MDS with ring sideroblast ~86%, MDS without ring sideroblast ~44%, and in CMML ~55%)[59]. All of the previously identified spliceosome genes are involved in the 3´-splice site recognition of a premature RNA. The U2 small nuclear RNA auxiliary factor 1 (U2AF1), establishes a substantial interaction with the serine/ argentine domain of another splicing factor called SRSF2 resulting in the exposure of the 3´-splice site and adjacent polypyrimidine tract. This synergism provides the possibility of actively recruiting also SF3B1 in the formation of the splicing A complex and in the formation of a mature RNA (Figure 3). Molecular alterations in these genes can cause defect in splicing of downstream targets. U2AF1 and SRSF2 have been associated with leukemogenesis [60]. The role of SRSF2 in genomic stability has also been studied. Indeed, perturbation of the function of this gene resulted in double-strand DNA breaks [60]. SF3B1 mutations have been associated with good clinical outcomes and ring sideroblast formation in MDS [9,61]. We reported the presence of an SF3B1 mutation (K700E) in a patient with MF whose BM erythroid precursors also showed occasional ring sideroblast further supporting the role of SF3B1 mutation on the pathogenesis of ring sideroblast formation even in non-MDS myeloid neoplasms [62]. The clinical relevance of ZRSR2 remains unclear due to the low frequency (3.1%) of mutations in MDS [63]. In our laboratory we have been actively working in defining the frequency of the three most prevalent spliceosome genes in MDS (SF3B1, U2AF1, SRSF2) in a cohort of 130 patients with MF, finding a frequency of SF3B1 (2%), U2AF1 (8%), and SRSF2 (17%)(Unpublished data). Table 1 summarizes the frequency of all the genes discussed above.
Figure 3.
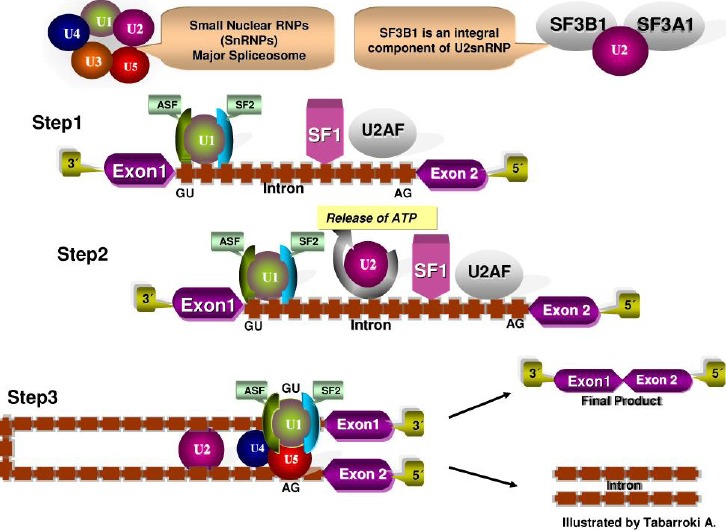
RNA Splicing machinery. RNA splicing is a mechanism in which non-coding regions (introns) are removed and coding regions (exons) strip to form a mature RNA. The GU dinucleotide at the 3’ and terminal AG dinucleotide at the 5’ ends serve as specific recognition sites. Several proteins forming complexes called: spliceosomes (U1, U2, SF1, U2AF) are involved in the removal of an intron lariat and assist in further steps in the formation of a mature messenger RNA.
TABLE I.
FREQUENCY OF MOLECULAR MUTATIONS AND CORRELATION WITH DISEASE PHENOTYPES IN MYELOFIBROSIS
| Gene Symbol | Frequency (%) | Associated Disease-Phenotype/ Impact on Survival Outcomes | Studies |
|---|---|---|---|
| JAK2 | 40-59 | Higher Hgb Leukocytosis BM Fibrosis |
James et al, 2005 Levine et al, 2005 Baxter et al, 2005 Kralovics et al, 2005 |
| MPL | 5-10 | Thrombocytosis Lower Hgb |
Beer et al, 2008 Boyd et al, 2010 Chaligne et al, 2008 |
| TET2 | 15-20 | Anemia | Vannucchi et al, 2013 Brecqueville et al, 2012 Delhommeau et al, 2009 |
| LNK | <5 | BM Fibrosis | Ha et al, 2011 St et al , 2010 Pardanani et al, 2010 |
| DNMT3A | 5-12 | Poor Prognosis | Vannucchi et al, 2013 Stegelmann et al, 2011 Abdel-Wahab et al, 2011 |
| IDH1/2 | <5 | Poor prognosis | Vannucchi et al, 2013 Brecqueville et al, 2012 Tefferi et al, 2010 |
| ASXL1 | 17-25 | BM Fibrosis/ Poor Prognosis | Brecqueville et al, 2012 Ricci et al, 2012 Stein et al, 2011 |
| CBL | <5 | BM Fibrosis | Schnittger et al, 2012 Brecqueville et al, 2012 Vainchenker et al, 2011 |
| EZH2 | 5-8 | Poor prognosis | Score et al, 2012 Guglielmelli et al, 2011 Ernst et al, 2010 |
| SF3B1 | 2-7 | Ring sideroblast/ Good prognosis | Visconte et al, 2012 |
| U2AF1 | 5-9 | Poor prognosis | Lasho et al, 2012 |
| SRSF2 | 15-25 | Anemia/ Poor prognosis | Lehmann et al, 2013 Vannucchi et al, 2013 Lasho et al, 2012 |
| ZRSR2 | <5§; 1.9† | Unknown | Thol et al, 2012 Yoshida et al, 2011 |
Table Legend: Summary of the frequencies, disease-phenotypes, and impact on survival outcomes of molecular mutations in Myelofibrosis.
§the frequency refers to Myelodysplastic syndromes.
†this frequency refers to a study conducted with whole exome sequencing
Abbreviations. Hgb, hemoglobin; BM, bone marrow.
III. THE IMPACT OF MOLECULAR MUTATIONS ON THE PHENOTYPES OF MYELOFIBROSIS
MF usually affects patients with advanced age (>65 years old) and usually experience constitutional symptoms (90%), splenomegaly (90%), fatigue (70%), anemia (60%), circulating blasts (50%), thrombocytopenia (30%), pulmonary hypertension (30-40%) and higher risk of thrombosis (10%)[64-65].
-Anemia and other hematologic abnormalities. There are many proposed mechanisms on the cause of anemia in MF patients and includes decrease bone marrow production due to decreased hematopoietic production sites in the BM due to fibrosis, splenic sequestration and consequent destruction of red cells in spleen, bleeding and autoimmune hemolysis. Yet, no distinctive molecular mutation has been proven to be the causative factor in the development of anemia in MF. Various studies that have looked at the clinical phenotypes of patients with MF with various molecular mutations have shown some some correlation between specific molecular subtypes and anemia. For example, patients with MPL and TET2 mutations have lower Hgb levels [26,66]. Conversely, observations from our laboratory showed that JAK2 mutant patients who also harbor spliceosome mutations have lower Hgb levels (8.98 vs 10.5 g/dL) and slightly higher leukocyte counts (27.5 vs 20.5 x109/L) compared to JAK2 WT cases without spliceosome mutations (Unpublished data). Leukocytosis and thrombocytosis are other hematologic features of MF patients, the higher allele burden of JAK2V617F mutation has been associated with leukocytosis, reticulocytosis and splenomegaly while [67] MPL mutations has been associated with thrombocytosis [68].
-Extramedullary hematopoiesis. Splenomegaly is the most common type of extramedullary hematopoiesis (EMH) in MF; however the definitive mechanisms involved in the development of splenomegaly remains elusive. The prevailing hypothesis for the development of splenomegaly includes cytokine stimulation of the spleen, deposition of BM erythroid precursors in the spleen and extramedullary hematopoietic compensation to sustain blood production. Little is known regarding the effects of molecular mutations in the causation of splenomegaly. Among various molecular mutations, patients with MF who carry JAK2 V617F and CBL mutations have enlarged spleens although the mechanisms whereby these mutations lead to EMH remain unclear [69].
-Bone Marrow Fibrosis. In MF, BM fibrosis is one of the important disease features and represents one of the major criteria for diagnosis. Fibrotic deposition can be detected using reticulin and/ or trichrome stains. It was alluded that fibrosis is the result of a reactive process of normal fibroblasts stimulated by aberrant cytokine activation resulting in the deposition of BM stromal fibers. Cytokines like transforming growth factor beta (TGF-β), platelet derived growth factor (PDGF,) and fibroblast growth factor (FGF) have been linked to the formation of fibrosis in BM [70-71]. Disruption of MPL function can cause over-activation of the thrombopoietin-receptor, that can over-stimulate megakaryocytes to release several cytokines that can then contributes to fibrosis [2].
IV. THE IMPACT OF MOLECULAR MUTATIONS ON PROGNOSIS OF MYELOFIBROSIS
A number of molecular mutations and genetic modifiers like single nucleotide polymorphisms have been described in MPN with some associated with poor prognosis and leukemia transformation [72]. For instance, it has been shown that the presence of the A3669G polymorphism in an MF patient with JAK2V617F mutation is associated with shorter overall survival and blast transformation free survival [73]. It has also been suggested that low JAK2V617F allele burden is associated with pancytopenia and higher susceptibility to infections which can result in lower overall survival [74].
Additionally, presence of mutations in molecular markers like IDH1/2, ASXL1, SRSF2 or EZH can independently predict shorter overall survival [75]. Some of these mutations (eg, IDH1/2 and SRSF2) have also been associated with inferior leukemia-free survival [75].
Despite the high frequency of JAK2V617F and TET2 mutations in MF, both genetic changes do not appear to affect survival outcomes [34,69]. Conversely, MPL mutations are not associated with inferior outcomes. The existence of EZH2 mutations in MDS was associated with poor overall survival and complex cytogenetic abnormalities. However, the same finding has not been established in MF [76].
In our studies, the presence of SRSF2 mutations contributed to a significant increase in the percentage of BM blasts and a higher incidence of progression to AML in MF patients (Unpublished data).
V. THE IMPACT OF MOLECULAR MARKERS ON RESPONSE TO TREATMENTS IN MYELOFIBROSIS
The presence of molecular mutations can alter the response to therapies in MDS, MDS/MPN, and secondary AML [77]. It was suggested that the presence of TET2 mutations could have negative impact on response to JAK inhibitors [78].
However, in other myeloid malignancies like MDS and AML, patients carrying TET2 and DNMT3A mutations responded better to therapies specifically hypomethylating (HMA) agents [79].
In MF, the presence of JAK2V617F mutation has been shown to be a predictor of better response to lenalidomide [80].
VI. CONCLUSIONS
In the JAK-inhibitor era, the management of some disease related features of MF has improved although there are still a lot of unmet needs including management of allele burden, treatment of cytopenias, and prevention of clonal evolution and therapy of patients who have evolved to AML. The discovery of molecular mutations has been expedited by next generation genomic technologies and this may lead to the identification of better therapeutic options for this disease similar to advancements being made in other hematologic malignancies [79,81]v (unpublished data). It may also allow us to identify patients who may respond to treatments and those that are likely to develop resistance or relapse from their therapies. The discovery of novel genetic signatures and alternative signaling pathways will hold the key to understanding the basic help pathophysiology of MF and will open new avenues in the management of MF and other MPNs that can improve patient management and survival.
ACKNOWLEDGMENT
The work was supported in part by the Cleveland Clinic Seed Support, Scott Hamilton CARES grant, and the American Cancer Society (RVT).
REFERENCES
- [1].Tabarroki A, Tiu RV. Immunomodulatory agents in myelofibrosis. Expert Opin Investig Drugs 2012;21(8):1141-1154 [DOI] [PubMed] [Google Scholar]
- [2].Pikman Y, Lee BH, Mercher T, McDowell E, Ebert BL, Gozo M, Cuker A, Wernig G, Moore S, Galinsky I, DeAngelo DJ, Clark JJ, Lee SJ, Golub TR, Wadleigh M, Gilliland DG, Levine RL. MPLW515L is a novel somatic activating mutation in myelofibrosis with myeloid metaplasia. PLoS Med 2006;3(7):e270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Pronier E, Delhommeau F. Role of TET2 mutations in myeloproliferative neoplasms. Curr Hematol Malig Rep 2012;7(1):57-64 [DOI] [PubMed] [Google Scholar]
- [4].Abdel-Wahab O Tefferi A Levine RL. Role of TET2 and ASXL1 mutations in the pathogenesis of myeloproliferative neoplasms. Hematol Oncol Clin North Am 2012;26(5):1053-1064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Green A, Beer P. Somatic mutations of IDH1 and IDH2 in the leukemic transformation of myeloproliferative neoplasms. N Engl J Med 2010;362(4):369-370 [DOI] [PubMed] [Google Scholar]
- [6].Shih AH, Abdel-Wahab O, Patel JP, Levine RL. The role of mutations in epigenetic regulators in myeloid malignancies. Nat Rev Cancer 2012;12(9):599-612 [DOI] [PubMed] [Google Scholar]
- [7].Ernst T, Chase AJ, Score J, Hidalgo-Curtis CE, Bryant C, Jones AV, Waghorn K, Zoi K, Ross FM, Reiter A, Hochhaus A, Drexler HG, Duncombe A, Cervantes F, Oscier D, Boultwood J, Grand FH, Cross NC. Inactivating mutations of the histone methyltransferase gene EZH2 in myeloid disorders. Nat Genet 2010;42(8):722-726 [DOI] [PubMed] [Google Scholar]
- [8].Schnittger S, Bacher U, Alpermann T, Reiter A, Ulke M, Dicker F, Eder C, Kohlmann A, Grossmann V, Kowarsch A, Kern W, Haferlach C, Haferlach T. Use of CBL exon 8 and 9 mutations in diagnosis of myeloproliferative neoplasms and myelodysplastic/myeloproliferative disorders: an analysis of 636 cases. Haematologica 2012;97(12):1890-1894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Visconte V, Rogers HJ, Singh J, Barnard J, Bupathi M, Traina F, McMahon J, Makishima H, Szpurka H, Jankowska A, Jerez A, Sekeres MA, Saunthararajah Y, Advani AS, Copelan E, Koseki H, Isono K, Padgett RA, Osman S, Koide K, O'Keefe C, Maciejewski JP, Tiu RV. SF3B1 haploinsufficiency leads to formation of ring sideroblasts in myelodysplastic syndromes. Blood 2012;120(16):3173-3186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Levine RL, Wadleigh M, Cools J, Ebert BL, Wernig G, Huntly BJ, Boggon TJ, Wlodarska I, Clark JJ, Moore S, Adelsperger J, Koo S, Lee JC, Gabriel S, Mercher T, D'Andrea A, Fröhling S, Döhner K, Marynen P, Vandenberghe P, Mesa RA, Tefferi A, Griffin JD, Eck MJ, Sellers WR, Meyerson M, Golub TR, Lee SJ, Gilliland DG. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell 2005;7(4):387-397 [DOI] [PubMed] [Google Scholar]
- [11].Baxter EJ, Scott LM, Campbell PJ, East C, Fourouclas N, Swanton S, Vassiliou GS, Bench AJ, Boyd EM, Curtin N, Scott MA, Erber WN, Green AR; Cancer Genome Project. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet 2005;365(9464):1054-1061 [DOI] [PubMed] [Google Scholar]
- [12].Scott LM, Scott MA, Campbell PJ, Green AR. Progenitors homozygous for the V617F mutation occur in most patients with polycythemia vera, but not essential thrombocythemia. Blood 2006;108(7):2435-2437 [DOI] [PubMed] [Google Scholar]
- [13].Huntly BJ, Shigematsu H, Deguchi K, Lee BH, Mizuno S, Duclos N, Rowan R, Amaral S, Curley D, Williams IR, Akashi K, Gilliland DG. MOZ-TIF2, but not BCR-ABL, confers properties of leukemic stem cells to committed murine hematopoietic progenitors. Cancer Cell 2004;6(6):587-596 [DOI] [PubMed] [Google Scholar]
- [14].Gnanasambandan K, Magis A, Sayeski PP. The constitutive activation of Jak2-V617F is mediated by a pi stacking mechanism involving phenylalanines 595 and 617. Biochemistry 2010;49(46):9972-9984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Oku S, Takenaka K, Kuriyama T, Shide K, Kumano T, Kikushige Y, Urata S, Yamauchi T, Iwamoto C, Shimoda HK, Miyamoto T, Nagafuji K, Kishimoto J, Shimoda K, Akashi K. JAK2 V617F uses distinct signalling pathways to induce cell proliferation and neutrophil activation. Br J Haematol 2010;150(3):334-344 [DOI] [PubMed] [Google Scholar]
- [16].Vainchenker W, Delhommeau F, Constantinescu SN, Bernard OA. New mutations and pathogenesis of myeloproliferative neoplasms. Blood 2011;118(7):1723-1735 [DOI] [PubMed] [Google Scholar]
- [17].Scott LM, Tong W, Levine RL, Scott MA, Beer PA, Stratton MR, Futreal PA, Erber WN, McMullin MF, Harrison CN, Warren AJ, Gilliland DG, Lodish HF, Green AR. JAK2 exon 12 mutations in polycythemia vera and idiopathic erythrocytosis. N Engl J Med 2007;356(5):459-468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Pietra D, Li S, Brisci A, Passamonti F, Rumi E, Theocharides A, Ferrari M, Gisslinger H, Kralovics R, Cremonesi L, Skoda R, Cazzola M. Somatic mutations of JAK2 exon 12 in patients with JAK2 (V617F)-negative myeloproliferative disorders. Blood 2008;111(3):1686-1689 [DOI] [PubMed] [Google Scholar]
- [19].Majka M, Ratajczak J, Villaire G, Kubiczek K, Marquez LA, Janowska-Wieczorek A, Ratajczak MZ. Thrombopoietin, but not cytokines binding to gp130 protein-coupled receptors, activates MAPKp42/44, AKT, and STAT proteins in normal human CD34+ cells, megakaryocytes, and platelets. Exp Hematol 2002;30(7):751-760 [DOI] [PubMed] [Google Scholar]
- [20].Pardanani AD, Levine RL, Lasho T, Pikman Y, Mesa RA, Wadleigh M, Steensma DP, Elliott MA, Wolanskyj AP, Hogan WJ, McClure RF, Litzow MR, Gilliland DG, Tefferi A. MPL515 mutations in myeloproliferative and other myeloid disorders: a study of 1182 patients. Blood 2006;108(10):3472-3476 [DOI] [PubMed] [Google Scholar]
- [21].Brecqueville M, Rey J, Bertucci F, Coppin E, Finetti P, Carbuccia N, Cervera N, Gelsi-Boyer V, Arnoulet C, Gisserot O, Verrot D, Slama B, Vey N, Mozziconacci MJ, Birnbaum D, Murati A. Mutation analysis of ASXL1, CBL, DNMT3A, IDH1, IDH2, JAK2, MPL, NF1, SF3B1, SUZ12, and TET2 in myeloproliferative neoplasms. Genes Chromosomes Cancer 2012;51(8):743-755 [DOI] [PubMed] [Google Scholar]
- [22].Mignotte V, Vigon I, Boucher de Crevecoeur E, Romeo PH, Lemarchandel V, Chretien S. Structure and transcription of the human c-mpl gene (MPL). Genomics 1994;20(1):5-12 [DOI] [PubMed] [Google Scholar]
- [23].de Sauvage FJ, Hass PE, Spencer SD, Malloy BE, Gurney AL, Spencer SA, Darbonne WC, Henzel WJ, Wong SC, Kuang WJ, Oles KJ, Hultgren B, Solberg LA, Jr, Goeddel DV, Eaton DL. Stimulation of megakaryocytopoiesis and thrombopoiesis by the c-Mpl ligand. Nature 1994;369(6481):533-538 [DOI] [PubMed] [Google Scholar]
- [24].Chaligné R, Tonetti C, Besancenot R, Roy L, Marty C, Mossuz P, Kiladjian JJ, Socié G, Bordessoule D, Le Bousse-Kerdilès MC, Vainchenker W, Giraudier S. New mutations of MPL in primitive myelofibrosis: only the MPL W515 mutations promote a G1/S-phase transition. Leukemia 2008;22(8):1557-1566 [DOI] [PubMed] [Google Scholar]
- [25].Kawamata N, Ogawa S, Yamamoto G, Lehmann S, Levine RL, Pikman Y, Nannya Y, Sanada M, Miller CW, Gilliland DG, Koeffler HP. Genetic profiling of myeloproliferative disorders by single-nucleotide polymorphism oligonucleotide microarray. Exp Hematol 2008;36(11):1471-1479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Tefferi A, Pardanani A, Lim KH, Abdel-Wahab O, Lasho TL, Patel J, Gangat N, Finke CM, Schwager S, Mullally A, Li CY, Hanson CA, Mesa R, Bernard O, Delhommeau F, Vainchenker W, Gilliland DG, Levine RL. TET2 mutations and their clinical correlates in polycythemia vera, essential thrombocythemia and myelofibrosis. Leukemia 2009;23(5):905-911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Swierczek SI, Yoon D, Bellanné-Chantelot C, Kim SJ, Saint-Martin C, Delhommeau F, Najman A, Prchal JT. Extent of hematopoietic involvement by TET2 mutations in JAK2V617F polycythemia vera. Haematologica 2011;96(5):775-778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, Agarwal S, Iyer LM, Liu DR, Aravind L, Rao A. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 2009;324(5929):930-935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Ko M, Huang Y, Jankowska AM, Pape UJ, Tahiliani M, Bandukwala HS, An J, Lamperti ED, Koh KP, Ganetzky R, Liu XS, Aravind L, Agarwal S, Maciejewski JP, Rao A. Impaired hydroxylation of 5-methylcytosine in myeloid cancers with mutant TET2. Nature 2010;468(7325):839-843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Tabarroki A RH, Visconte V, Hasrouni E, Advani AS, Sekeres MA, Duong HK, Kalaycio M, Copelan E, Stein BL, Tiu RV. The molecular and cytokine profile of Triple-Negative (JAK2 V617F, JAK2 exon 12, MPL negative) Myelofibrosis, a Myeloproliferative Neoplasm with distinct clinico-pathologic characteristics. ASH abstract 2012;54th annual meeting of American Society of Hematology (ASH) [Google Scholar]
- [31].Delhommeau F, Dupont S, Della Valle V, James C, Trannoy S, Massé A, Kosmider O, Le Couedic JP, Robert F, Alberdi A, Lécluse Y, Plo I, Dreyfus FJ, Marzac C, Casadevall N, Lacombe C, Romana SP, Dessen P, Soulier J, Viguié F, Fontenay M, Vainchenker W, Bernard OA. Mutation in TET2 in myeloid cancers. N Engl J Med 2009;360(22):2289-2301 [DOI] [PubMed] [Google Scholar]
- [32].Fisher CL, Lee I, Bloyer S, Bozza S, Chevalier J, Dahl A, Bodner C, Helgason CD, Hess JL, Humphries RK, Brock HW. Additional sex combs-like 1 belongs to the enhancer of trithorax and polycomb group and genetically interacts with Cbx2 in mice. Dev Biol 2010;337(1):9-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Abdel-Wahab O, Adli M, La Fave LM, Gao J, Hricik T, Shih AH, Pandey S, Patel JP, Chung YR, Koche R, Perna F, Zhao X, Taylor JE, Park CY, Carroll M, Melnick A, Nimer SD, Jaffe JD, Aifantis I, Bernstein BE, Levine RL. ASXL1 mutations promote myeloid transformation through loss of PRC2-mediated gene repression. Cancer Cell 2012;22(2):180-193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Ricci C, Spinelli O, Salmoiraghi S, Finazzi G, Carobbio A, Rambaldi A. ASXL1 mutations in primary and secondary myelofibrosis. Br J Haematol 2012;156(3):404-407 [DOI] [PubMed] [Google Scholar]
- [35].Carbuccia N, Murati A, Trouplin V, Brecqueville M, Adélaïde J, Rey J, Vainchenker W, Bernard OA, Chaffanet M, Vey N, Birnbaum D, Mozziconacci MJ. Mutations of ASXL1 gene in myeloproliferative neoplasms. Leukemia 2009;23(11):2183-2186 [DOI] [PubMed] [Google Scholar]
- [36].Gelsi-Boyer V, Trouplin V, Adélaïde J, Bonansea J, Cervera N, Carbuccia N, Lagarde A, Prebet T, Nezri M, Sainty D, Olschwang S, Xerri L, Chaffanet M, Mozziconacci MJ, Vey N, Birnbaum D. Mutations of polycomb-associated gene ASXL1 in myelodysplastic syndromes and chronic myelomonocytic leukaemia. Br J Haematol 2009;145(6):788-800 [DOI] [PubMed] [Google Scholar]
- [37].Grand FH, Hidalgo-Curtis CE, Ernst T, Zoi K, Zoi C, McGuire C, Kreil S, Jones A, Score J, Metzgeroth G, Oscier D, Hall A, Brandts C, Serve H, Reiter A, Chase AJ, Cross NC. Frequent CBL mutations associated with 11q acquired uniparental disomy in myeloproliferative neoplasms. Blood 2009;113(24):6182-6192 [DOI] [PubMed] [Google Scholar]
- [38].Beer PA, Delhommeau F, LeCouédic JP, Dawson MA, Chen E, Bareford D, Kusec R, McMullin MF, Harrison CN, Vannucchi AM, Vainchenker W, Green AR. Two routes to leukemic transformation after a JAK2 mutation-positive myeloproliferative neoplasm. Blood 2010;115(14):2891-2900 [DOI] [PubMed] [Google Scholar]
- [39].Tefferi A, Vainchenker W. Myeloproliferative neoplasms: molecular pathophysiology, essential clinical understanding, and treatment strategies. J Clin Oncol 2011;29(5):573-582 [DOI] [PubMed] [Google Scholar]
- [40].Martinez-Aviles L, Besses C, Alvarez-Larran A, Torres E, Serrano S, Bellosillo B. TET2, ASXL1, IDH1, IDH2, and c-CBL genes in JAK2- and MPL-negative myeloproliferative neoplasms. Ann Hematol 2012;91(4):533-541 [DOI] [PubMed] [Google Scholar]
- [41].Dang L, Jin S, Su SM. IDH mutations in glioma and acute myeloid leukemia. Trends Mol Med 2010;16(9):387-397 [DOI] [PubMed] [Google Scholar]
- [42].Okita Y, Narita Y, Miyakita Y, Ohno M, Matsushita Y, Fukushima S, Sumi M, Ichimura K, Kayama T, Shibui S. IDH1/2 mutation is a prognostic marker for survival and predicts response to chemotherapy for grade II gliomas concomitantly treated with radiation therapy. Int J Oncol 2012;41(4):1325-1336 [DOI] [PubMed] [Google Scholar]
- [43].Abbas S, Lugthart S, Kavelaars FG, Schelen A, Koenders JE, Zeilemaker A, van Putten WJ, Rijneveld AW, Löwenberg B, Valk PJ. Acquired mutations in the genes encoding IDH1 and IDH2 both are recurrent aberrations in acute myeloid leukemia: prevalence and prognostic value. Blood 2010;116(12):2122-2126 [DOI] [PubMed] [Google Scholar]
- [44].Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, Li Y, Bhagwat N, Vasanthakumar A, Fernandez HF, Tallman MS, Sun Z, Wolniak K, Peeters JK, Liu W, Choe SE, Fantin VR, Paietta E, Löwenberg B, Licht JD, Godley LA, Delwel R, Valk PJ, Thompson CB, Levine RL, Melnick A. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 2010;18(6):553-567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Pardanani A, Lasho TL, Finke CM, Mai M, McClure RF, Tefferi A. IDH1 and IDH2 mutation analysis in chronic- and blast-phase myeloproliferative neoplasms. Leukemia 2010;24(6):1146-1151 [DOI] [PubMed] [Google Scholar]
- [46].Marcucci G, Maharry K, Wu YZ, Radmacher MD, Mrózek K, Margeson D, Holland KB, Whitman SP, Becker H, Schwind S, Metzeler KH, Powell BL, Carter TH, Kolitz JE, Wetzler M, Carroll AJ, Baer MR, Caligiuri MA, Larson RA, Bloomfield CD. IDH1 and IDH2 gene mutations identify novel molecular subsets within de novo cytogenetically normal acute myeloid leukemia: a Cancer and Leukemia Group B study. J Clin Oncol 2010;28(14):2348-2355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Lin J, Yao DM, Qian J, Chen Q, Qian W, Li Y, Yang J, Wang CZ, Chai HY, Qian Z, Xiao GF, Xu WR. Recurrent DNMT3A R882 mutations in Chinese patients with acute myeloid leukemia and myelodysplastic syndrome. PLoS One 2011;6(10):e26906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Stegelmann F, Bullinger L, Schlenk RF, Paschka P, Griesshammer M, Blersch C, Kuhn S, Schauer S, Döhner H, Döhner K. DNMT3A mutations in myeloproliferative neoplasms. Leukemia 2011;25(7):1217-1219 [DOI] [PubMed] [Google Scholar]
- [49].Rao N, Butcher CM, Lewis ID, Ross DM, Melo JV, Scott HS, Bardy PG, D'Andrea RJ. Clonal and lineage analysis of somatic DNMT3A and JAK2 mutations in a chronic phase polycythemia vera patient. Br J Haematol 2012;156(2):268-270 [DOI] [PubMed] [Google Scholar]
- [50].Mullighan CG, Miller CB, Radtke I, Phillips LA, Dalton J, Ma J, White D, Hughes TP, Le Beau MM, Pui CH, Relling MV, Shurtleff SA, Downing JR. BCR-ABL1 lymphoblastic leukaemia is characterized by the deletion of Ikaros. Nature. 2008;453(7191):110-114 [DOI] [PubMed] [Google Scholar]
- [51].Tong W, Zhang J, Lodish HF. Lnk inhibits erythropoiesis and Epo-dependent JAK2 activation and downstream signaling pathways. Blood 2005;105(12):4604-4612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Velazquez L, Cheng AM, Fleming HE, Furlonger C, Vesely S, Bernstein A, Paige CJ, Pawson T. Cytokine signaling and hematopoietic homeostasis are disrupted in Lnk-deficient mice. J Exp Med 2002;195(12):1599-1611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Lasho TL, Tefferi A, Finke C, Pardanani A. Clonal hierarchy and allelic mutation segregation in a myelofibrosis patient with two distinct LNK mutations. Leukemia 2011;25(6):1056-1058 [DOI] [PubMed] [Google Scholar]
- [54].Baran-Marszak F, Magdoud H, Desterke C, Alvarado A, Roger C, Harel S, Mazoyer E, Cassinat B, Chevret S, Tonetti C, Giraudier S, Fenaux P, Cymbalista F, Varin-Blank N, Le Bousse-Kerdilès MC, Kiladjian JJ, Velazquez L. Expression level and differential JAK2-V617F-binding of the adaptor protein Lnk regulates JAK2-mediated signals in myeloproliferative neoplasms. Blood 2010;116(26):5961-5971 [DOI] [PubMed] [Google Scholar]
- [55].Bersenev A, Wu C, Balcerek J, Jing J, Kundu M, Blobel GA, Chikwava KR, Tong W. Lnk constrains myeloproliferative diseases in mice. J Clin Invest 2010;120(6):2058-2069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Shen X, Liu Y, Hsu YJ, Fujiwara Y, Kim J, Mao X, Yuan GC, Orkin SH. EZH1 mediates methylation on histone H3 lysine 27 and complements EZH2 in maintaining stem cell identity and executing pluripotency. Mol Cell 2008;32(4):491-502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].He LR, Liu MZ, Li BK, Jia WH, Zhang Y, Liao YJ, Chen YC, Zhang LJ, Guan XY, Zeng YX, Kung HF, Xie D. High expression of EZH2 is associated with tumor aggressiveness and poor prognosis in patients with esophageal squamous cell carcinoma treated with definitive chemoradiotherapy. Int J Cancer 2010;127(1):138-147 [DOI] [PubMed] [Google Scholar]
- [58].Cao R, Wang L, Wang H, Xia L, Erdjument-Bromage H, Tempst P, Jones RS, Zhang Y. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science 2002;298(5595):1039-1043 [DOI] [PubMed] [Google Scholar]
- [59].Lasho TL, Jimma T, Finke CM, Patnaik M, Hanson CA, Ketterling RP, Pardanani A, Tefferi A. SRSF2 mutations in primary myelofibrosis: significant clustering with IDH mutations and independent association with inferior overall and leukemia-free survival. Blood 2012;120(20):4168-4171 [DOI] [PubMed] [Google Scholar]
- [60].Makishima H, Visconte V, Sakaguchi H, Jankowska AM, Abu Kar S, Jerez A, Przychodzen B, Bupathi M, Guinta K, Afable MG, Sekeres MA, Padgett RA, Tiu RV, Maciejewski JP. Mutations in the spliceosome machinery, a novel and ubiquitous pathway in leukemogenesis. Blood 2012;119(14):3203-3210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Papaemmanuil E, Cazzola M, Boultwood J, Malcovati L, Vyas P, Bowen D, Pellagatti A, Wainscoat JS, Hellstrom-Lindberg E, Gambacorti-Passerini C, Godfrey AL, Rapado I, Cvejic A, Rance R, McGee C, Ellis P, Mudie LJ, Stephens PJ, McLaren S, Massie CE, Tarpey PS, Varela I, Nik-Zainal S, Davies HR, Shlien A, Jones D, Raine K, Hinton J, Butler AP, Teague JW, Baxter EJ, Score J, Galli A, Della Porta MG, Travaglino E, Groves M, Tauro S, Munshi NC, Anderson KC, El-Naggar A, Fischer A, Mustonen V, Warren AJ, Cross NC, Green AR, Futreal PA, Stratton MR, Campbell PJ; Chronic Myeloid Disorders Working Group of the International Cancer Genome Consortium. Somatic SF3B1 mutation in myelodysplasia with ring sideroblasts. N Engl J Med 2011;365(15):1384-1395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Visconte V, Makishima H, Maciejewski JP, Tiu RV. Emerging roles of the spliceosomal machinery in myelodysplastic syndromes and other hematological disorders. Leukemia 2012;26(12):2447-2454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Thol F, Kade S, Schlarmann C, Löffeld P, Morgan M, Krauter J, Wlodarski MW, Kölking B, Wichmann M, Görlich K, Göhring G, Bug G, Ottmann O, Niemeyer CM, Hofmann WK, Schlegelberger B, Ganser A, Heuser M. Frequency and prognostic impact of mutations in SRSF2, U2AF1, and ZRSR2 in patients with myelodysplastic syndromes. Blood 2012;119(15):3578-3584 [DOI] [PubMed] [Google Scholar]
- [64].Barbui T, Carobbio A, Cervantes F, Vannucchi AM, Guglielmelli P, Antonioli E, Alvarez-Larrán A, Rambaldi A, Finazzi G, Barosi G. Thrombosis in primary myelofibrosis: incidence and risk factors. Blood 2010;115(4):778-782 [DOI] [PubMed] [Google Scholar]
- [65].Dingli D, Utz JP, Krowka MJ, Oberg AL, Tefferi A. Unexplained pulmonary hypertension in chronic myeloproliferative disorders. Chest 2001;120(3):801-808 [DOI] [PubMed] [Google Scholar]
- [66].Guglielmelli P, Pancrazzi A, Bergamaschi G, Rosti V, Villani L, Antonioli E, Bosi A, Barosi G, Vannucchi AM; GIMEMA--Italian Registry of Myelofibrosis; MPD Research Consortium. Anaemia characterises patients with myelofibrosis harbouring Mpl mutation. Br J Haematol 2007;137(3):244-247 [DOI] [PubMed] [Google Scholar]
- [67].Vannucchi AM, Antonioli E, Guglielmelli P, Pardanani A, Tefferi A. Clinical correlates of JAK2V617F presence or allele burden in myeloproliferative neoplasms: a critical reappraisal. Leukemia 2008;22(7):1299-1307 [DOI] [PubMed] [Google Scholar]
- [68].Pardanani A, Guglielmelli P, Lasho TL, Pancrazzi A, Finke CM, Vannucchi AM, Tefferi A. Primary myelofibrosis with or without mutant MPL: comparison of survival and clinical features involving 603 patients. Leukemia 2011;25(12):1834-1839 [DOI] [PubMed] [Google Scholar]
- [69].Tefferi A. Novel mutations and their functional and clinical relevance in myeloproliferative neoplasms: JAK2, MPL, TET2, ASXL1, CBL, IDH and IKZF1. Leukemia 2010;24(6):1128-1138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Kuter DJ, Bain B, Mufti G, Bagg A, Hasserjian RP. Bone marrow fibrosis: pathophysiology and clinical significance of increased bone marrow stromal fibres. Br J Haematol 2007;139(3):351-362 [DOI] [PubMed] [Google Scholar]
- [71].Bedekovics J, Kiss A, Beke L, Karolyi K, Mehes G. Platelet derived growth factor receptor-beta (PDGFRbeta) expression is limited to activated stromal cells in the bone marrow and shows a strong correlation with the grade of myelofibrosis. Virchows Arch 2013;463(1):57-65 [DOI] [PubMed] [Google Scholar]
- [72].Susini MC, Guglielmelli P, Spolverini A, Biamonte F, Mannarelli C, Barosi G, Zoi K, Reiter A, Duncombe A, Cervantes F, Cazzola M, Cross N, Vannucchi AM; Associazione Italiana per la Ricerca sul Cancro Gruppo Italiano Malattie Mieloproliferative (AGIMM) Investigators. The ERCC2 Gln/Gln polymorphism at codon 751 is not associated with leukaemic transformation in primary myelofibrosis. Br J Haematol 2013;162(3):424-427 [DOI] [PubMed] [Google Scholar]
- [73].Poletto V, Rosti V, Villani L, Catarsi P, Carolei A, Campanelli R, Massa M, Martinetti M, Viarengo G, Malovini A, Migliaccio AR, Barosi G. A3669G polymorphism of glucocorticoid receptor is a susceptibility allele for primary myelofibrosis and contributes to phenotypic diversity and blast transformation. Blood 2012;120(15):3112-3117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Guglielmelli P, Barosi G, Specchia G, Rambaldi A, Lo Coco F, Antonioli E, Pieri L, Pancrazzi A, Ponziani V, Delaini F, Longo G, Ammatuna E, Liso V, Bosi A, Barbui T, Vannucchi AM. Identification of patients with poorer survival in primary myelofibrosis based on the burden of JAK2V617F mutated allele. Blood 2009;114(8):1477-1483 [DOI] [PubMed] [Google Scholar]
- [75].Vannucchi AM, Lasho TL, Guglielmelli P, Biamonte F, Pardanani A, Pereira A, Finke C, Score J, Gangat N, Mannarelli C, Ketterling RP, Rotunno G, Knudson RA, Susini MC, Laborde RR, Spolverini A, Pancrazzi A, Pieri L, Manfredini R, Tagliafico E, Zini R, Jones A, Zoi K, Reiter A, Duncombe A, Pietra D, Rumi E, Cervantes F, Barosi G, Cazzola M, Cross NC, Tefferi A. Mutations and prognosis in primary myelofibrosis. Leukemia 2013;27(9):1861-1869 [DOI] [PubMed] [Google Scholar]
- [76].Nikoloski G, Langemeijer SM, Kuiper RP, Knops R, Massop M, Tönnissen ER, van der Heijden A, Scheele TN, Vandenberghe P, de Witte T, van der Reijden BA, Jansen JH. Somatic mutations of the histone methyltransferase gene EZH2 in myelodysplastic syndromes. Nat Genet 2010;42(8):665-667 [DOI] [PubMed] [Google Scholar]
- [77].Itzykson R, Kosmider O, Cluzeau T, Mansat-De Mas V, Dreyfus F, Beyne-Rauzy O, Quesnel B, Vey N, Gelsi-Boyer V, Raynaud S, Preudhomme C, Adès L, Fenaux P, Fontenay M; Groupe Francophone des Myelodysplasies (GFM). Impact of TET2 mutations on response rate to azacitidine in myelodysplastic syndromes and low blast count acute myeloid leukemias. Leukemia 2011;25(7):1147-1152 [DOI] [PubMed] [Google Scholar]
- [78].Santos FP, Kantarjian HM, Jain N, Manshouri T, Thomas DA, Garcia-Manero G, Kennedy D, Estrov Z, Cortes J, Verstovsek S. Phase 2 study of CEP-701, an orally available JAK2 inhibitor, in patients with primary or post-polycythemia vera/essential thrombocythemia myelofibrosis. Blood 2010;115(6):1131-1136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Traina F VV, Elson P, Tabarroki A. Impact of Molecular Mutations on Treatment Response to DNMT Inhibitors in Myelodysplasia and Related Neoplasms Leukemia 2013, "in press" [DOI] [PubMed] [Google Scholar]
- [80].Quintás-Cardama A, Kantarjian HM, Manshouri T, Thomas D, Cortes J, Ravandi F, Garcia-Manero G, Ferrajoli A, Bueso-Ramos C, Verstovsek S. Lenalidomide plus prednisone results in durable clinical, histopathologic, and molecular responses in patients with myelofibrosis. J Clin Oncol 2009;27(28):4760-4766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Patel JP, Gönen M, Figueroa ME, Fernandez H, Sun Z, Racevskis J, Van Vlierberghe P, Dolgalev I, Thomas S, Aminova O, Huberman K, Cheng J, Viale A, Socci ND, Heguy A, Cherry A, Vance G, Higgins RR, Ketterling RP, Gallagher RE, Litzow M, van den Brink MR, Lazarus HM, Rowe JM, Luger S, Ferrando A, Paietta E, Tallman MS, Melnick A, Abdel-Wahab O, Levine RL. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med 2012;366(12):1079-1089 [DOI] [PMC free article] [PubMed] [Google Scholar]