

Abstract
Mastocytosis is a heterogeneous group of disorders characterized by a clonal proliferation and accumulation of mast cells in one or more organ, primarily in the skin and bone marrow. The clinical spectrum of the disease varies from relatively benign forms with isolated skin lesions to very aggressive variants with extensive systemic involvement and poor prognosis. The growth and proliferation of clonal mast cells is caused by an activating mutation of the tyrosine kinase receptor Kit for Stem Cell Factor, the main growth factor for mast cells. Clinical symptoms are related to mast-cell mediator release, to the tissue mast cell infiltration or both. The degree of infiltration and cell activation determines the highly variable clinical and morphological features. Current treatment of mastocytosis includes symptomatic, antimediator drugs and cytoreductive targeted therapies.
Keywords: mastocytosis
I. INTRODUCTION
In 1869, Nettleship and Tay described the first case of a “rare form of urticaria that result in a brownish discoloration” [1]. In 1878 Paul Ehrlich first described the mast cells “metachromasia”, along with the tendency for mast cells to be associated with blood vessels, glandular ducts, and nerves. Further in 1949, Ellis detailed an autopsy report of a 1-year-old child where mast cell infiltration was found in numerous organs (bone marrow, lymph nodes, spleen and liver) [2].
In 1957 mast cell leukemia was reported for the first time. In 1991 Metcalfe proposed the first classification of mastocytosis. In 2001 a WHO (World Health Organization) classification was published (Table I). The current WHO classification discriminates cutaneous mastocytosis (CM) and systemic mastocytosis (SM) [3]. The CM is a benign disease confined to the skin, while SM is a clonal hematological disease and is classified in indolent (ISM), aggressive (ASM) and SM with an associated clonal hematologic non-mast cell lineage disease (SM-AHNMD).
TABLE I.
CLASSIFICATION OF MASTOCYTOSIS (WHO)
| Variants | Prevalence | Sub-variants |
|---|---|---|
| Cutaneous mastocytosis (CM) | ~ 85% |
|
| Indolent systemic mastocytosis (ISM) | ~ 10% |
|
| Systemic mastocytosis with an associated clonal haematological non- mast cell disease(SM-AHNMD) | ~ 1% |
|
| Aggressive systemic mastocytosis (ASM) | ~ 5% |
|
| Mast cell leukaemia (MCL) | < 1% |
|
| Mast cell sarcoma | < 1% | |
| Extracutaneous mastocytoma | < 1% |
Abbreviations. SM-AML: systemic mastocytosis with acute myelogenous leukemia. SM-MDS: systemic mastocytosis with myelodysplastic syndrome. SM-MPS: systemic mastocytosis with myeloproliferative syndrome. SM-CEL OR SM-HES: systemic mastocytosis with hypereosinophilic syndrome. SM-CMML: systemic mastocytosis with chronic myelomonocytic leukemia. SM-NHL: systemic mastocytosis with non-Hodgkin lymphoma.
II. MAST CELLS
Mast cells are an important effectors cells of the immune system and are found in all vascularized tissue, especially in the skin and mucous membranes of the respiratory and gastrointestinal tracts. These cells release a variety of inflammatory mediators whose biological effects lead the heterogeneous symptoms in patients with mastocytosis [4-10].
Mast cells derive from CD34+ hematopoietic stem cells of the bone marrow and differentiate into mast cell precursors that are distributed via blood to tissues. Growth, differentiation and proliferation of mast cells are controlled by the tyrosine kinase receptor c-Kit (CD117) and its ligand stem cell factor (SCF), also of key importance in the development of mastocytosis. Mast cells can be identified by cell surface markers FcεR1, CD13, CD34, CD68, and KIT (CD117) [11].
A wide array of proinflammatory mediators is secreted by mast cells after IgE-receptor cross-linking by allergens or other stimuli. The preformed vasoactive and immunoregolatory mediators, contained within mast cells granules, include histamine, heparine, serotonin, neutral proteases (tryptase, chymase, carboxypeptidase A, cathepsin G), major basic protein and phospholipases. Tryptase and chymase are the most abundant protein components of mast cell granules. After cell activation, mast cells are also able to synthesize protein and lipid mediators, including lipoxigenase and cyclooxygenase metabolites of arachidonic acid. These include leukotrienes (mainly LTC4), prostaglandins (mainly PGD2) and platelet-activating factor (PAF). Mast cells also produce cytokines, growth factors including IL-5, IL-6, IL-13, IL-16, SCF, GMCSF, NGF, VEGF and various chemokines and possess numerous membrane-bound receptors [12].
In the bone marrow mast cells have four distinct morphological stages of maturation: the non-granulated blast cell (tryptase+), the matachromatic blast cell, the promastocyte (also called atypical mast cell type II), and the mature mast cell [13]. The stages of differentiation are of particular interest in that immature forms of mast cells are often seen in the more severe forms of systemic mastocytosis.
III. EPIDEMIOLOGY OF MASTOCYTOSIS
Mastocytosis is a rare disease. In various studies an incidence of 5 to 10 new cases per one million population per year was calculated. The prevalence (evaluated by epidemiologic study conducted in Europe and in United States) is 1/60.000.
The most frequent variants are cutaneous and indolent systemic mastocytosis, the rarest is probably mast cell leukemia. While children almost exclusively have cutaneous mastocytosis forms, in adults urticaria pigmentosa and indolent systemic mastocytosis, is much more likely. In adults mastocytosis often manifests between age 20 and 40 years, sometimes even later. On the other hand, as signs and symptoms of mastocytosis are unspecific and overlap with many other diseases, the correct diagnosis may be overlooked and there is an unusually long latency period between the first symptoms and the correct diagnosis.
IV. PATHOGENESIS OF MASTOCYTOSIS
The clonal nature of mastocytosis can be established through the demonstration of gain-of-function mutations involving the tyrosin kinase domain of kit receptor in skin and/or bone marrow cells. KIT (CD117) is a type III tyrosine kinase (TK) receptor that is characterized by an extracellular domain with five immunoglobulin-like loops, a transmembrane domain, an juxtamembrane autoinhibitory domain and a TK domain. The first three immunoglobulin (Ig)-like loops of the extracellular domain form the binding site for stem cell factor (SCF) or KIT ligand, while the fourth and fifth loops play a role in stabilizing the SCF-induced KIT dimer. The autoinhibitory juxtamembrane domain is essential for the downregulation of tyrosine phosporylation. The kinase portion of KIT is composed of two domains which are separated by a kinase insert: the TK1 domain is constituted by the small N-terminal lobe that expands from amino acids 582–684 and contains the ATP binding site, and the TK2 domain is formed by the large C-terminal lobe containing the phosphotransferase site and the activation loop (amino acids: 810–839). The interaction between KIT and its ligand, SCF, plays a key role in regulating mast cell proliferation, maturation, adhesion, chemotaxis, and survival.
In over 90 % of adults with mastocytosis the somatic activating kit point mutation on exon 17 at codon 816 (substitution of aspartate by valine in the tyrosine kinase domain of the kit receptor or Asp-816-Val or kitD816V) can be detected. This mutation leads to autophosphorylation of the receptor resulting in endogenous-autonomous mast cell proliferation [14-18]. However, have been reported other forms of kit mutations, not only on exon 17: these mutations affect several different domains of the receptor, such as the extracellular domain, transmembrane domain, juxtamembrane domain and activation loop domain [19]. In children mutagenesis is much less homogeneous as in adults. In a recent study, Bodemer et al. analyzed cutaneous biopsies of 50 children with mastocytosis (aged 0-16 years): the kit mutation at codon 816 on exon 17 was detected in 42 % of cases, while in 44% in of cases mutations were observed in the extracellular domain of kit on exon 8 and 9 [20]. All mutations were somatic and led to kit activation.
Other oncogenic mutations recently identified in mastocytosis patients include TET2 (TET oncogene family member 2) and N-RAS [21]. These mutation are not specific of mastocytosis and their pathogenetic role and/or prognostic impact is currently uncertain. TET2 is a putative tumor suppressor gene. In one study, the frequency of TET2 mutations in SM was 29% and its presence was associated with monocytosis. Further, TET2 mutations cosegregated with KITD816V but did not appear to affect survival in SM.
V. CLASSIFICATION OF MASTOCYTOSIS
Mastocytosis is a rare disease characterized by a clonal proliferation of morphologically and immunophenotypically abnormal mast cells, that accumulate in one or more organ systems (skin, bone marrow, lymph node, gastrointestinal tracts, liver, spleen, etc.) [1-8]. The clinical presentation of mastocytosis is heterogeneous, ranging from skin-limited disease (cutaneous mastocytosis, CM), particularly in childhood with spontaneous regression of skin lesions before adolescence [22] to more aggressive forms in adult patients with extracutaneous involvement (systemic mastocytosis, SM) that maybe associated with multiorgan failure [23].
Cutaneous mastocytosis
The most common clinical sign of mastocytosis (both cutaneous and systemic disease) is the presence of typical skin lesions of urticaria pigmentosa, which appear as fixed, dark red-brown macules or papules. These lesions exhibit Darier’s sign that is considered pathognomonic of mastocytosis and it consists in urticarial swelling induced by scratching of skin lesions [24-26]. Cutaneous mastocytosis is defined as an accumulation of mast cells limited to the skin. Clinically, various subtypes are described: urticaria pigmentosa, diffuse CM, and mastocytoma of the skin.
- Maculopapular cutaneous mastocytosis/Urticaria pigmentosa (MPCM/UP) is the most common variant of CM, that it manifests as 0,5- 1 cm yellowish tan to red-brown macules or slightly raised papules (Figure 1). The affected areas include the trunk and extremities, while the face, scalp, palms, and soles tend to be free of lesions.
Figure 1.
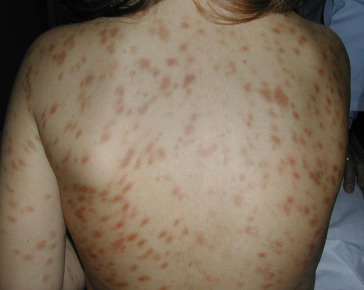
Urticaria Pigmentosa
Much rarer are the bullous variant (often in the first weeks of life), the plaque form, the nodular form and the Teleangectasia macularis-eruptiva perstans (TMEP) with multiple hyperpigmented maculae as well as erythema with telangiectases. This form is associated with either SM or with various hematological abnormalities, such as myelodysplasia, myeloproliferative disorders, acute myeloid leukaemia, and lymphoproliferative disease [27]. These variants do not constitute independent forms in the WHO classification.
- Diffuse cutaneous mastocytosis (DCM) can involve extensive areas of the skin. It is a rare (1-3% of the cases of CM) severe variant of CM, that occurs predominantly in children. DCM can appear at birth (congenital and neonatal) or in early infancy. Blistering and bullae may be the presenting symptoms and the blisters can be hemorrhagic. The skin may be leathery and thickened. Hyperpigmentation may persist into adulthood and dermographism may be prominent.
- Mastocytoma of skin is a single lesion of the skin, appearing as yellowish to reddish nodular lesions, in part with blistering. It manifests predominantly in childhood and in most cases heals spontaneously (Fig. 2).
Figure 2.
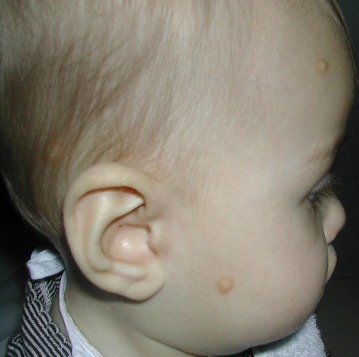
Mastocytomas of skin in child
Cutaneous mastocytosis has a highly favorable clinical prognosis. The majority of children with CM show a good prognosis with gradual resolution of both symptoms and skin lesions [28].
Systemic mastocytosis
Systemic Mastocytosis is highly heterogeneous diseases with by definition involvement of at least one extracutaneous tissue (skin involvement is frequent but not obligatory). Since the bone marrow is almost always involved, bone marrow biopsy is the method of choice to confirm the diagnosis of systemic mastocytosis. Patients with systemic mastocytosis have mediator related symptoms and/or signs and symptoms caused by mast cells infiltration [3,5]. Organ infiltration by MC may lead not only to organomegaly, but also to impairment or even loss of organ function and in severe (high grade) disease, bone marrow failure with anemia, thrombocytopenia, and/or recurrent infections (due to neutropenia) may occur. Typical clinical findings (B and C-findings) were established by WHO to determinate the type of systemic mastocytosis (Table II). B-findings referred to organ involvement without organ failure, while C-findings referred to organ involvement with organ dysfunction. The presence of a C- finding is sufficient to diagnose advanced SM and the patient is a candidate for chemotherapy.
TABLE II.
B-FINDINGS AND C-FINDINGS
| B-findings* | C-findings** Impairment of organ function |
Progressive C-findings |
|---|---|---|
|
-
|
-
|
* When two or three B-findings are recorded, the final diagnosis is SM smouldering
**Organ failure caused by infiltration of neoplastic mast cells: a single C-findings is sufficient to diagnose ASM
Variants and sub-variants of systemic mastocytosis
- Indolent systemic mastocytosis (ISM). ISM is the most common form of SM, accounting for about two thirds of all cases. Commonly, ISM involves both the skin and bone marrow, with urticaria pigmentosa seen in more than 90% of cases. Usually ISM manifests in adulthood. Bone marrow is affected in almost 100% of cases and usually contains multifocal mast cell infiltrates, but there are not associated hematologic disorders or organ dysfunction. In ISM “C findings” are not found; if “C findings” are present, the disease has progressed to ASM.
Two subvariants of ISM are Isolated Bone Marrow Mastocytosis (BMM) and Smoldering Systemic Mastocytosis (SSM). In the BMM the mast cell infiltration is only found in bone marrow, but not in the skin. The rare smoldering SM is differentiated from the more common indolent variety by massive mast cell proliferation and tryptase levels over 200 μg/l. Even in smoldering SM no signs of bone marrow insufficiency should be detectable (important differentiation from aggressive SM).
- Systemic mastocytosis with an associated clonal hematological non-mast cell disease (SM-AHNMD). Systemic mastocytosis with an associated clonal hematological non-mast cell disease (SM-AHNMD) is the second most frequent subtype of systemic mastocytosis, comprising one fourth to one third of the cases of SM. SM and AHNMD are usually diagnosed simultaneously in bone marrow biopsy. All subtypes of hematologic neoplasia have been reported within the context of SM-AHNMD. Myeloid disorders comprise 80-90% of the AHNMDs, while lymphoid disorders comprise the other 10-20 %. The prognosis of SM-AHNMD usually depends on the AHNMD and not on SM [29].
- Aggressive systemic mastocytosis (ASM). Aggressive systemic mastocytosis is a rare form (represents only about 5% of all patients with SM), characterized by the extensive infiltration of tissue depending on the involved organ leads to highly different symptoms as a result of dysfunction with signs of insufficiency being considered as C-findings in the WHO criteria. These C-findings different clinical sign of organ dysfunction: hepatopathy with hypoproteinemia, ascites, pleural and/or pericardical effusion, prolongation of prothrombin time, and bleeding disorders; bone fractures; severe cytopenia and malabsorption. A subvariant of ASM is Lymphadenopathic Systemic Mastocytosis with Eosinophilia, definited by a increased of number of eosinophil (>1500/mm3 ). In this particular form the FIP1-like-1 (FIPL1)/platelet-derivated growth factor receptor alpha (PDGFRα) fusion gene is often detected and it is responsive to the tyrosine kinase inhibitor, imatinib mesylate [6]. In patients with ASM the serum tryptase level is usually over 200 μg/l. The prognosis of ASM is poor.
- Mast cell Leukemia. Mast cell leukemia (MCL) is characterized by leukemic infiltration of organs by immature or atypical neoplastic mast cells. Typically, mast cells are more than 10% of the circulating blood cells. MCL is rare and has a poor prognosis. A subvariant of MCL, aleukemic MCL, has less than 10% immature of atypical mast cells within circulating blood cells. Over the course of the disease, patients may progress from aleukemic MCL to MCL [30].
VI. MEDIATOR-RELATED SYMPTOMS IN MASTOCYTOSIS
In both CM and SM the clinical picture is often dominated by local and systemic symptoms generated by the acute or chronic release of mast cell-derived mediators. These clinical signs and symptoms are highly variable inter- and intra-individually. These symptoms depend on the release of mediators, such as histamine, heparin, tryptase, leukotrienes and cytokines. Related clinical findings are often unspecific symptoms: headache attacks, fatigue, pruritus, flushing, often in combination with hypotensive crises, syncope and tachycardia, gastrointestinal complaints (nausea, abdominal pain, diarrhea, vomiting, gastritis, peptic ulcers), bone pain and rarely even neurologic-psychiatric symptoms. Such symptoms can occur in all patients with mastocytosis, but in patients with SM these symptoms are often recurrent, severe and require continuous medical treatment. Almost all patients report variable worsening of symptoms after intensive physical activity, consumption of alcohol, infections, nonsteroidal anti-inflammatory drugs but also during emotional stress.
In cutaneous mastocytosis flushing, pruritus, erythema and swelling are frequent signs and symptoms and they may occur spontaneously or be induced by specific triggers. The intensity of skin involvement can be variable. In majority of patients the typical maculae and plaques develop primarily on the trunk and limbs; the head, palms and soles are usually spared. In children, in contrast, typically the head and lateral face are involved.
Gastrointestinal symptoms can be prominent in patients with mastocytosis and these symptoms can be caused by mast cell proinflammatory mediators and/or mast cell organ infiltration. The mediator related symptoms are diarrhea, abdominal pain, peptic ulcer disease and gastrointestinal bleeding; while in ASM patients mast cell tissue infiltration caused severe malabsorption, chronic diarrhea, weight loss, organomegaly (spleen, liver) with dysfunction, ascites due to periportal fibrosis up to hepatic failure.
Patients can also develop osteopenia or osteoporosis with bone pain and possible pathologic fractures. Pathologic fractures are considered a marker of advanced mastocytosis specially when they are associated with high levels of serum tryptase; while osteoporosis usually depend on the intensity of mast cell mediator release. Indeed, heparin, tryptase and IL-6 can be activated in vitro RANK-L (receptor activator of nuclear factor-kB ligand) [31].
Patients with mastocytosis have an increased risk of anaphylaxis. In adults with mastocytosis, the cumulative prevalence of anaphylaxis has been reported to be 22% to 50%, and in children 6% to 10% [32]. Anaphylactic reactions have been reported in all forms of mastocytosis, but patients with systemic disease have an increased risk of anaphylaxis as compared with patients with only cutaneous disease. The most frequently reported elicitor of anaphylaxis is hymenoptera venom (wasp stings) [33].
Other factors that caused anaphylaxis are some drugs (opiates, including morphine and codeine), food, radiological contrast media containing ionic iodine and substances administered during general anesthesia (e. g. muscle relaxants). These factors can be activate mast cells directly by non immunologic mechanisms.
VII. DIAGNOSIS OF MASTOCYTOSIS
The diagnosis of cutaneous mastocytosis can usually be made based on history and clinical criteria with careful skin inspection. The diagnosis is based on the demonstration of characteristic skin lesions (major criterion) and on the presence of one minor or two minor criteria: a) demonstrate on the skin biopsy of (multifocal or diffuse) aggregates of mast cells in the papillary dermis extending into the reticular dermis; b) presence of D816V mutations. The Darier sign with urticarial swelling after mechanical rubbing of macules or papules is almost always positive. Inspection of the skin is supplemental by palpation of peripheral lymph node stations and abdominal organs. In children bone marrow study is recommended only if there is suspicion of progression of disease: in case of organomegaly or significant lymphadenopathy, abnormalities on peripheral blood counts, elevated serum tryptase levels, severe recurrent systemic mast cell mediator-related symptoms or persistence of skin lesions after puberty.
For the diagnosis of systemic mastocytosis should be fulfilled the WHO criteria proposed in 2001 (Table III). These consist of one major and four minor criteria. In order to satisfy the diagnosis of systemic mastocytosis, either one major criterion and one minor criterion or at least three minor criteria should be present.
TABLE III.
WORLD HEALTH ORGANIZATION DIAGNOSTIC CRITERIA FOR SYSTEMIC MASTOCYTOSIS
| Major criterion* | Multifocal dense infiltrates of mast cells (tryptase positive) in bone marrow and/or other extracutaneous tissues (aggregates of more than 15 mast cells) |
| Minor criteria* |
|
* One major and one minor, or three minor criteria are needed for the diagnosis of systemic mastocytosis
The WHO major criterion as well as 2 of 4 WHO minor criteria are morphological criteria, so that both careful collection of samples (especially bone marrow biopsy and aspirates) as well as sufficient experience in the histo- and molecular pathological evaluation of the material are essential for making the diagnosis. The recommended method of evaluation of mast cells in bone marrow biopsy is immunohistochemical staining with tryptase. Neoplastic mast cell generally express CD25 and/or CD2, and the abnormal expression of at least one of these two antigens counts as a minor criterion toward the diagnosis of SM [34]. Recently, CD30 was detected in mast cells in aggressive (advanced) systemic mastocytosis. Specifically, it has been described that CD30 is preferentially expressed in the cytoplasm of neoplastic mast cells in ASM and MCL, but not in ISM. Notably, strong expression of CD30 in the cytoplasm of most neoplastic mast cells is in favor of the diagnosis ASM or MCL. Thus, CD30 may be a forthcoming gradingand prognostic marker in systemic mastocytosis [35, 36].
The determination of serum tryptase levels (normal value under 11.4 μg/l) is in principle a good diagnostic and differential diagnostic parameter, also in evaluation of the disease course. The level correlates with the mast cell proliferation and its activation [37-39]. Elevated serum tryptase levels are therefore not pathognomonic for SM, as elevated levels can be detected also in patients with acute (40 % of patients have elevated levels) or chronic myeloid leukemia and more rarely also in myelodysplastic disorders. In patients with elevated tryptase levels after questionable anaphylactic reactions, determining the course after 4 to 8 weeks is recommended for a differential diagnosis of SM. When SM is suspected and serum tryptase is elevated, bone marrow biopsy should always be performed. Even with only slightly elevated tryptase levels and unclear complaints, bone marrow biopsy is recommended.
SM without skin involvement is rare and is then also often difficult to recognize. In face of the often diffuse complaints of patients many diseases must be included in the differential diagnosis. Patients who present a severe anaphylactic reaction, e. g. after an insect sting – even without the detection of specific IgE – or who develop an unclear (idiopathic) anaphylactic reaction are diagnostically problematic. Here in every case further mastocytosis diagnostics should be performed. A lack of skin involvement is more often associated with aggressive forms (ASM, mast cell leukemia). Extremely rare and therefore difficult to diagnose routinely are mast cell sarcoma, mastocytosis of the spleen and benign extracutaneous mastocytoma.
A careful internal medicine work-up is always recommendable. Organ-related symptoms e. g. of the gastrointestinal tract result in specific diagnostics (e. g. endoscopy with step-by-step biopsies and mast cell-specific immunohistochemical and molecular pathological processing). Differentiation between symptoms due to mast cell infiltrates and symptoms due to massive mediator release can be problematic in the individual case. With strict observation of the major and minor criteria and a standardized step-by-step diagnostic approach mastocytosis can almost always be clearly identified and subtyped [8]. In ISM with a stable course only annual Laboratory controls (routine serology, differential blood count, serum tryptase) and more invasive studies only in case of clinical/ laboratory chemical alterations are recommended.
VIII. PROGNOSIS OF MASTOCYTOSIS
The variants and subvariants of mastocytosis, with their wide range of clinical manifestations, have relatively defined prognosis. Most forms of CM in children spontaneously remit by puberty [40]. The prognosis of patients with mastocytosis depends on the degree of organ infiltration by mast cells, the evolution of associated hematologic disorders, the occurrence of anaphylaxis and the associated osteoporosis with pathological fractures. In adults, the prognosis of indolent systemic mastocytosis is generally good and these patients usually have a normal life expectancy; while advanced systemic mastocytosis and mast cell leukemia are associated with a severe prognosis.
Several different clinical, sierological, cytomorphological and immunological variables have been described as a prognostic findings in SM. Clinical prognostic variables include absence of skin lesions, large osteolyses, weight loss, malabsorption, enlarged liver with portal hypertension and splenomegaly with hypersplenism [41]. The absence of a skin lesion is typical for an aggressive disease variant. Low levels of albumin can be caused by hepatopathy or malabsorption. Hepatomegaly with elevated liver enzymes and/or signs of portal hypertension (such as ascites) is indicative for progressive destruction of the liver by MC infiltrates. Both progressive hepathopathy and malabsorption are markers of poor prognosis in SM.
In patients with indolent systemic mastocytosis, the β2-microglobulin level is a prognostic marker predicting an unfavorable outcome [42]. SM with eosinophilia was found to be predictive for a significantly reduced overall and event-free survival compared with patients without eosinophilia [41].
Another prognostic factor in patients with mastocytosis is the percentage of mast cells in the bone marrow smear. The majority of patients with ISM present with ≤ 5% MCs in bone marrow smear. Patients with a percentage of MCs in the bone marrow smear greater than 5%: have a lower survival [43].
As previously mentioned, CD30 is preferentially expressed on neoplastic MCs in ASM and MCL, but not ISM. The strong expression of CD30 in the cytoplasm of neoplastic MCs correlates with a poor prognosis [41,44,45].
IX. TREATMENT OF MASTOCYTOSIS
Recently, a number of therapeutic approaches for the treatment of mastocytosis have been reported [46]. Management of patients within all categories of mastocytosis includes:1) a careful counseling of patients and care providers; 2) avoidance of factors triggering acute mediator release; 3) treatment of acute mast cell mediator release; 4) treatment of chronic mast cell mediator release, and if indicated; 5) an attempt to treat organ infiltration by mast cells.
Complete information about the disease, including guide-lines for avoidance of triggering factor, and risks associated with massive mast cell mediator release may be given to the patients and/or their parents in case of children. The first arm of therapy is to avoid mast cell degranulation. Triggering factors are various: physical stimuli (heat, cold, friction of skin lesions, pressure, excessive sunlight); emotional factors (stress, anxiety, sleep deprivation); infection disease with fever; drugs (aspirin and other non steroidal anti-inflammatory drugs, morphine and derivatives, polymyxin-B, amphotericin B).
Skin lesions represent a problem in many patients. A treatment for skin lesions is oral psoralen plus UV-A (PUVA). In response to PUVA, most patients show a substantial regression of skin lesions. However, due to the limited duration of responses, long-term treatment with repeated cycles of PUVA is necessary [47].
The primary objective of all clinical forms of mastocytosis (both cutaneous and systemic mastocytosis) is the treatment of mediator-related symptoms. Non sedating histamine H1 receptor antagonists (rupatadine, levocetirizine and desloratadine) have been shown to be useful in the treatment of flushing, pruritus, wheals, swelling and the sensation of burning of the skin. Additional administration of histamine H2 receptor antagonists, such as ranitidine is suggested in cases where H1-receptor antagonists monotherapy is inadequate. Corticosteroids, administered over a short period of time, may be considered in case of recurrent and severe mediator-related symptoms.
Patients with gastrointestinal complaints such as pain, vomiting, diarrhea or meteorism profit from therapy with chromolyn sodium, antacids, proton pump inhibitors and H1 receptor antagonists and depending on the degree of indirect histamine release-induced symptoms also with combinations of H1 and H2 receptor antagonists.
Adults with mastocytosis and children with extensive cutaneous involvement are at increased risk of anaphylaxis and should carry an emergency kit for self-medication that includes epinephrine and, as warranted, antihistamine and corticosteroids. Treatment with omalizumab (anti- IgE) can be successful in selected patients with otherwise uncontrolled idiopathic anaphylaxis and in those with anaphylaxis during the initiation of specific immunotherapy [48].
Due to the increased perioperative risk, patients, anesthesiologists and surgeons should discuss comprehensively the procedure to lower the risk of anaphylaxis. As premedication antihistamines and corticosteroids in a sufficiently high dosage should be selected. Only selected drugs that are proven not to be relevant histamine releasers and do not provoke mast cell activation should be administered perioperatively.
In patients with advanced systemic mastocytosis (with the presence of “C-findings”) the prime goal is to inhibit further mast cell proliferation with cell infiltration and cell activation. In cases of SM-AHNMD the hematologic disorder must be treated.
Interferon (IFN)-α is considerate the first-line cytoreductive therapy in aggressive systemic mastocytosis. Prednisone (30-60 mg/day) is commonly added at the start of treatment to improve tolerability and response. Several studies have shown IFN-α to improve symptoms of MC degranulation, decrease bone marrow MC infiltration, and reduce mastocytosis-related ascites/hepatosplenomegaly, cytopenia, skin lesions and osteoporosis. The frequency of major response (complete resolution of one or more baseline C findings) is approximately of 50%. IFN-α treatment is frequently complicated by toxicities, including flu-like symptoms, bone pain, fever, cytopenia, depression and hypothyroidism.
Cladribine or 2-chlorodeoxyadenosine (2-CdA) has activity in all SM subtypes. 2-CdA can be used in patients who are refractory or intolerant to IFN-α. A reduction of the mast cell load could be demonstrated, but controlled studies are urgently needed. Potential toxicities of 2-CdA include myelosuppression and lymphopenia with increased risk of opportunistic infections.
In recent years various tyrosine kinase (TK) inhibitors have also been employed for SM. Imatinib mesilate (IM) inhibits a series of receptor TK including kit, abl, bcr-abl, platelet-derived growth factor receptor. Due to steric interaction of the kitD816V mutation at the receptor imatinib is effective only in patients with SM lacking this mutation. Indeed it demonstrates in vitro efficacy against wild-type KIT and certain transmembrane (F522C) and juxta-membrane (V560G) KIT mutants, but not the common kinase (D816V) domain mutants. Furthermore, imatinib showed efficacy in patients with one of subvariant of ASM, lymphadenophatic systemic mastocytosis with eosinophilia: this particular subset has a FIP1-like-1(FIPL1)/platelet-derivated growth factor receptor alpha (PDGFRα) fusion gene defect.
New tyrosine kinase inhibitors under clinical investigation for blocking KIT are dasatinib, nilotinib, masatinib mesilate and PKC412 (midostaurine). However, it is currently not clear which patients with SM will benefit from such treatments and more studies are needed to clarify the advantage of tyrosine kinase inhibitors over IFN-α or cladribine, which are currently considered first line treatment in ASM [41,50]. A possible treatment option in the future is the combination of multiple cytoreductive drugs with potential synergistic effects.
REFERENCES
- [1].Nettleship E, Tay W.Rare forms of urticaria. Br Med J 1869;2:323–330 [Google Scholar]
- [2].Ellis JM. Urticaria pigmentosa: a report of a case with autopsy. AMA Arch Pathol 1949;48:426–435 [PubMed] [Google Scholar]
- [3].Valent P, Horny HP, Escribano L, Longley BJ, Li CY, Schwartz LB, Marone G, Nuñez R, Akin C, Sotlar K, Sperr WR, Wolff K, Brunning RD, Parwaresch RM, Austen KF, Lennert K, Metcalfe DD, Vardiman JW, Bennett JM. Diagnostic criteria and classification of mastocytosis: a consensus proposal. Leuk Res. 2001; 25(7):603–625 [DOI] [PubMed] [Google Scholar]
- [4].Escribano L, Akin C, Castells M, Orfao A, Metcalfe DD. Mastocytosis: current concepts in diagnosis and treatment. Ann Hematol. 2002; 81(12):677–690 [DOI] [PubMed] [Google Scholar]
- [5].Akin C, Metcalfe DD. Systemic mastocytosis. Annu Rev Med 2004;55:419–432 [DOI] [PubMed] [Google Scholar]
- [6].Valent P, Akin C, Sperr WR, Horny HP, Arock M, Lechner K, Bennett JM, Metcalfe DD. Diagnosis and treatment of systemic mastocytosis: state of the art. Br J Haematol. 2003; 122(5):695–717 [DOI] [PubMed] [Google Scholar]
- [7].Valent P, Akin C, Sperr WR, Escribano L, Arock M, Horny HP, Bennett JM, Metcalfe DD. Aggressive systemic mastocytosis and related mast cell disorders: current treatment options and proposed response criteria. Leuk Res. 2003; 27(7):635–641 [DOI] [PubMed] [Google Scholar]
- [8].Valent P, Akin C, Escribano L, Födinger M, Hartmann K, Brockow K, Castells M, Sperr WR, Kluin-Nelemans HC, Hamdy NA, Lortholary O, Robyn J, van Doormaal J, Sotlar K, Hauswirth AW, Arock M, Hermine O, Hellmann A, Triggiani M, Niedoszytko M, Schwartz LB, Orfao A, Horny HP, Metcalfe DD. Standards and standardization in mastocytosis: consensus statements on diagnostics, treatment recommendations and response criteria. Eur J Clin Invest. 2007; 37(6):435–453 [DOI] [PubMed] [Google Scholar]
- [9].Valent P, Granata F, Triggiani M, Del Vecchio L, Marone G. Recent advances in the classification, diagnosis and treatment of mastocytosis. Giorn It Allergol Immunol Clin 2002; 12: 57–72 [Google Scholar]
- [10].Amon U, Hartmann K, Horny HP, Nowak A. Mastocytosis - an update. J Dtsch Dermatol Ges. 2010; 8(9):695–711; quiz 712 [DOI] [PubMed] [Google Scholar]
- [11].Kirshenbaum AS, Goff JP, Semere T, Foster B, Scott LM, Metcalfe DD. Demonstration that human mast cells arise from a progenitor cell population that is CD34(+), c-kit(+), and expresses aminopeptidase N (CD13). Blood 1999; 94(7):2333–2342 [PubMed] [Google Scholar]
- [12].Kirshenbaum AS, Kessler SW, Goff JP, Metcalfe DD. Demonstration of the origin of human mast cells from CD34+ bone marrow progenitor cells. J Immunol 1991; 146(5):1410–1415 [PubMed] [Google Scholar]
- [13].Sperr WR, Escribano L, Jordan JH, Schernthaner GH, Kundi M, Horny HP, Valent P. Morphologic properties of neoplastic mast cells: delineation of stages of maturation and implication for cytological grading of mastocytosis. Leuk Res 2001; 25(7):529–536 [DOI] [PubMed] [Google Scholar]
- [14].Nagata H, Worobec AS, Oh CK, Chowdhury BA, Tannenbaum S, Suzuki Y, Metcalfe DD. Identification of a point mutation in the catalytic domain of the protooncogene c-kit in peripheral blood mononuclear cells of patients who have mastocytosis with an associated hematologic disorder. Proc Natl Acad Sci U S A 1995; 92(23):10560–10564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Longley BJ, Tyrrell L, Lu SZ, Ma YS, Langley K, Ding TG, Duffy T, Jacobs P, Tang LH, Modlin I. Somatic c-KIT activating mutation in urticaria pigmentosa and aggressive mastocytosis: establishment of clonality in a human mast cell neoplasm. Nat Genet 1996; 12(3):312–314 [DOI] [PubMed] [Google Scholar]
- [16].Longley BJ, Jr, Metcalfe DD, Tharp M, Wang X, Tyrrell L, Lu SZ, Heitjan D, Ma Y.Activating and dominant inactivating c-KIT catalytic domain mutations in distinct clinical forms of human mastocytosis. Proc Natl Acad Sci U S A 1999; 96(4):1609–1614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Longley BJ, Reguera MJ, Ma Y. Classes of c-KIT activating mutations: proposed mechanisms of action and implications for disease classification and therapy. Leuk Res 2001;25:571–576 [DOI] [PubMed] [Google Scholar]
- [18].Féger F, Ribadeau Dumas A, Leriche L, Valent P, Arock M.Kit and c-kit mutations in mastocytosis: a short overview with special reference to novel molecular and diagnostic concepts. Int Arch Allergy Immunol 2002; 127(2):110–114 [DOI] [PubMed] [Google Scholar]
- [19].Orfao A, Garcia-Montero AC, Sanchez L, Escribano L.REMA. Recent advances in the understanding of mastocytosis: the role of KIT mutations. Br J Haematol 2007; 138(1):12–30 [DOI] [PubMed] [Google Scholar]
- [20].Bodemer C, Hermine O, Palmérini F, Yang Y, Grandpeix-Guyodo C, Leventhal PS, Hadj-Rabia S, Nasca L, Georgin-Lavialle S, Cohen-Akenine A, Launay JM, Barete S, Feger F, Arock M, Catteau B, Sans B, Stalder JF, Skowron F, Thomas L, Lorette G, Plantin P, Bordigoni P, Lortholary O, de Prost Y, Moussy A, Sobol H, Dubreuil P. Pediatric mastocytosis is a clonal disease associated with D816V and other activating c-KIT mutations. J Invest Dermatol 2010; 130(3):804–815 [DOI] [PubMed] [Google Scholar]
- [21].Wilson TM, Maric I, Simakova O, Bai Y, Chan EC, Olivares N, Carter M, Maric D, Robyn J, Metcalfe DD. Clonal analysis of NRAS activating mutations in KIT-D816V systemic mastocytosis. Haematologica 2011; 96(3):459–463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Uzzaman A, Maric I, Noel P, Kettelhut BV, Metcalfe DD, Carter MC. Pediatric-onset mastocytosis: a long term clinical follow-up and correlation with bone marrow histopathology. Pediatr Blood Cancer 2009; 53(4):629–634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Lim KH, Tefferi A, Lasho TL, Finke C, Patnaik M, Butterfield JH, McClure RF, Li CY, Pardanani A. Systemic mastocytosis in 342 consecutive adults: survival studies and prognostic factors. Blood 2009; 113(23):5727–5736 [DOI] [PubMed] [Google Scholar]
- [24].Akoglu G, Erkin G, Cakir B, Boztepe G, Sahin S, Karaduman A, Atakan N, Akan T, Kolemen F. Cutaneous mastocytosis: demographic aspects and clinical features of 55 patients. J Eur Acad Dermatol Venereol 2006; 20(8):969–973 [DOI] [PubMed] [Google Scholar]
- [25].Hartmann K, Henz BM. Mastocytosis: recent advances in defining the disease. British Journal of Dermatology 2001;144:682–695 [DOI] [PubMed] [Google Scholar]
- [26].Hannaford RM, Rogers M.Presentation of cutaneous mastocytosis in 173 children. Aust J dermatol. 2001;42:15–21 [DOI] [PubMed] [Google Scholar]
- [27].Bachmeyer C, Guillemette J, Blum L, Turc Y, Dhôte R, Fermand JP, Aractingi S. Telangiectasia macularis eruptiva perstans and multiple myeloma. J Am Acad Dermatol 2000;43(5 Pt 2):972–974 [DOI] [PubMed] [Google Scholar]
- [28].Hartmann K, Metcalfe DD. Pediatric mastocytosis. Hematol Oncol Clin North Am 2000; 14(3):625–640 [DOI] [PubMed] [Google Scholar]
- [29].Horny HP, Sotlar K, Sperr WR, Valent P. Systemic mastocytosis with associated clonal haematological non-mast cell lineage diseases: a histopathological challenge. J Clin Pathol 2004; 57(6):604–608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Worobec AS. Treatment of systemic mast cell disorders. Hematol Oncol Clin North Am 2000; 14(3):659–687vii. [DOI] [PubMed] [Google Scholar]
- [31].Theoharides TC, Boucher W, Spear K. Serum interleukin-6 reflects disease severity and osteoporosis in mastocytosis patients. Int Arch Allergy Immunol 2002;128:344–50 [DOI] [PubMed] [Google Scholar]
- [32].Brockow K, Jofer C, Behrendt H, Ring J. Anaphylaxis in patients with mastocytosis: a study on history, clinical features and risk factors in 120 patients. Allergy 2008; 63(2):226–232 [DOI] [PubMed] [Google Scholar]
- [33].Bonadonna P, Zanotti R, Müller U.Mastocytosis and insect venom allergy. Curr Opin Allergy Clin Immunol. 2010; 10(4):347–353 [DOI] [PubMed] [Google Scholar]
- [34].George TI, Horny HP. Systemic mastocytosis. Hematol Oncol Clin N Am 2011; 25(5): 1067–1083 [DOI] [PubMed] [Google Scholar]
- [35].Valent P, Sotlar K, Horny HP. Aberrant expression of CD30 in aggressive systemic mastocytosis and mast cell leukemia: a differential diagnosis to consider in aggressive hematopoietic CD30-positive neoplasms. Leuk Lymphoma 2011; 52(5):740–744 [DOI] [PubMed] [Google Scholar]
- [36].Sotlar K, Cerny-Reiterer S, Petat-Dutter K, Hessel H, Berezowska S, Müllauer L, Valent P, Horny HP. Aberrant expression of CD30 in neoplastic mast cells in high-grade mastocytosis. Mod Pathol 2011; 24(4):585–595 [DOI] [PubMed] [Google Scholar]
- [37].Sperr WR, Jordan JH, Fiegl M, Escribano L, Bellas C, Dirnhofer S, Semper H, Simonitsch-Klupp I, Horny HP, Valent P. Serum tryptase levels in patients with mastocytosis: correlation with mast cell burden and implication for defining the category of disease. Int Arch Allergy Immunol 2002; 128(2):136–141 [DOI] [PubMed] [Google Scholar]
- [38].van Doormaal JJ, van der Veer E, van Voorst Vader PC, Kluin PM, Mulder AB, van der Heide S, Arends S, Kluin-Nelemans JC, Oude Elberink JN, de Monchy JG. Tryptase and histamine metabolites as diagnostic indicators of indolent systemic mastocytosis without skin lesions. Allergy. 2012; 67(5):683–690 [DOI] [PubMed] [Google Scholar]
- [39].Alvarez-Twose I, Vañó-Galván S, Sánchez-Muñoz L, Morgado JM, Matito A, Torrelo A, Jaén P, Schwartz LB, Orfao A, Escribano L. Increased serum baseline tryptase levels and extensive skin involvement are predictors for the severity of mast cell activation episodes in children with mastocytosis. Allergy 2012; 67(6):813–821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Kong JS, Teuber S, Hallett R, Gershwin ME. Mastocytosis: the great masquerader. Clin Rev Allergy Immunol 2006; 30(1):53–60 [DOI] [PubMed] [Google Scholar]
- [41].Sperr WR, Valent P.Diagnosis, progression patterns and prognostication in mastocytosis. Expert Rev Hematol 2012; 5(3):261–274 [DOI] [PubMed] [Google Scholar]
- [42].Escribano L, Alvarez-Twose I, Sánchez-Muñoz L, Garcia-Montero A, Núñez R, Almeida J, Jara-Acevedo M, Teodósio C, García-Cosío M, Bellas C, Orfao A. Prognosis in adult indolent systemic mastocytosis: a long-term study of the Spanish Network on Mastocytosis in a series of 145 patients. J Allergy Clin Immunol 2009; 124(3):514–521 [DOI] [PubMed] [Google Scholar]
- [43].Sperr WR, Escribano L, Jordan JH, Schernthaner GH, Kundi M, Horny HP, Valent P. Morphologic properties of neoplastic mast cells: delineation of stages of maturation and implication for cytological grading of mastocytosis. Leuk Res 2001; 25(7):529–536 [DOI] [PubMed] [Google Scholar]
- [44].Valent P, Sotlar K, Horny HP. Aberrant expression of CD30 in aggressive systemic mastocytosis and mast cell leukemia: a differential diagnosis to consider in aggressive hematopoietic CD30-positive neoplasms. Leuk Lymphoma 2011; 52(5):740–744 [DOI] [PubMed] [Google Scholar]
- [45].Sotlar K, Cerny-Reiterer S, Petat-Dutter K, Hessel H, Berezowska S, Müllauer L, Valent P, Horny HP. Aberrant expression of CD30 in neoplastic mast cells in high-grade mastocytosis. Mod Pathol 2011; 24(4):585–595 [DOI] [PubMed] [Google Scholar]
- [46].Marone G, Spadaro G, Granata F, Triggiani M. Treatment of mastocytosis: pharmacologic basis and current concepts. Leuk Res 2001;25:583–594 [DOI] [PubMed] [Google Scholar]
- [47].Escribano L, Akin C, Castells M, Schwartz LB. Current options in the treatment of MC mediatior-related symptoms in mastocytosis. Inflamm. Allergy Drug Targets 2006; 5(1):61–67 [DOI] [PubMed] [Google Scholar]
- [48].Pitt TJ, Cisneros N, Kalicinsky C, Becker AB. Successful treatment of idiopathic anaphylaxis in an adolescent. J Allergy Clin Immunol. 2010;126:415–416 [DOI] [PubMed] [Google Scholar]
- [49].Gotlib J, De Angelo DJ, George TI, Corless CL, Linder A, Langford C, Dutreix C, Gross SH, Nikolova Z, Graubert T. KIT inhibitor midostaurin exhibits a high rate of clinically meaningful and durable responses in advanced systemic mastocytosis: Report of a fully accrued phase II trial. ASH Annual Meeting Abstract. Blood 2010;116:316 [Google Scholar]
- [50].Alvarez-Twose I, González P, Morgado JM, Jara-Acevedo M, Sánchez-Muñoz L, Matito A, Mollejo M, Orfao A, Escribano L. Complete response after imatinib mesylate therapy in a patient with well-differentiated systemic mastocytosis. J Clin Oncol 2012; 30(12):e126–129 [DOI] [PubMed] [Google Scholar]