

Abstract
Coupling therapeutic proteins to targeted nanocarriers can enhance their biodistribution. This is the case for enzyme replacement therapies where intravenously injected enzymes must avoid prolonged blood exposure while reaching body organs. We have shown enhanced tissue targeting of various lysosomal enzymes by coupling to nanocarriers targeted to intercellular adhesion molecule-1 (ICAM-1). Here, we varied design parameters to modify tissue enzyme levels without affecting specific targeting and relative biodistribution. We coupled α-galactosidase (αGal; affected in Fabry disease) to model polymer nanocarriers and varied enzyme load (50 vs. 500 molecules/particle), anti-ICAM surface density (80 vs. 180 molecules/particle), and nanocarrier concentration (1.6×1013 vs. 2.4×1013 carriers/kg) to render three formulations (45, 449, 555 μg αGal/kg). Naked αGal preferentially distributed in blood vs. organs, while nanocarriers shifted biodistribution from blood to tissues. Accumulation in brain, kidneys, heart, liver, lungs, and spleen did not vary among nanocarrier formulations, with enhanced specific tissue accumulation compared to naked αGal. The highest specificity was associated with lowest antibody density and nanocarrier concentration, but highest enzyme load; possibly because of synergistic enzyme affinity toward cell-surface markers. Variation of these parameters significantly increased absolute enzyme accumulation. This strategy may help optimize delivery of lysosomal enzyme replacement and, likely, other protein delivery approaches.
Keywords: ICAM-1-targeted nanocarriers, targeting surface-density, carrier bulk-concentration, enzyme delivery, lysosomal Fabry disease
1. Introduction
An increasing number of treatments use relatively large proteins as therapeutic agents. However, the efficacy of therapeutic proteins is often impaired by poor delivery in the body, owing to clearance by the reticulo-endothelial system with associated immunogenicity, reduced targeting to diseased organs and tissues, and insufficient transport at the subcellular level, which render suboptimal effects.1 As in the case of other therapeutics, these parameters impacting treatment efficacy could be improved by using strategies to modify circulation rates, tissue targeting potential, and intracellular transport of therapeutic proteins.2 Therefore, although relatively unexplored, applying principles of drug targeting and delivery systems to protein delivery has considerable relevance and potential for many therapeutic interventions.1
This is the case for enzyme replacement therapy (ERT) for lysosomal storage disorders (LSDs), a group of genetic diseases often caused by a lack of functional lysosomal proteins.3 Such deficiencies lead to widespread lysosomal accumulation of non-degraded metabolites through the body, leading ultimately to multi-organ failure.3 Current ERT consists of intravenous (i.v.) infusion of naked, recombinant enzymes to replace the missing lysosomal activity throughout the body, and relies on the presence of mannose or mannose-6-phosphate residues on the administered enzymes to target the corresponding clathrin-associated receptors on cells.4 However, blocking antibodies whose generation is enhanced by exposure in circulation, poor targeting affinity to diseased organs, and restricted cellular uptake via these cell surface receptors result in suboptimal ERT efficacy.5–9
Coupling to targeting moieties aimed at alternative cell receptors, either directly or by using drug nanocarriers, has proven beneficial in enhancing ERT delivery to the central nervous system and/or peripheral organs, affected in most LSDs.7, 10, 11 An example of such an approach is that of targeting intercellular adhesion molecule-1 (ICAM-1), a transmembrane glycoprotein which is expressed on the vascular endothelium and most other cell types 12, 13 and over-expressed in most pathological states. ICAM-1 targeting provides transport of drug nanocarriers across cellular barriers and into lysosomes.14–20 Models for this concept have been shown in the case of type A-B Niemann-Pick disease,15, 19–21 Pompe disease,17 and Fabry disease,16, 18 where coupling of the corresponding lysosomal enzymes to ICAM-1-targeted polymer nanocarriers improved binding, clathrin-independent uptake via cell adhesion molecule- (CAM)-mediated endocytosis, and lysosomal transport. By using this approach, delivery of said enzymes was markedly enhanced throughout the body, including the brain.15, 17, 18, 21 In addition, lysosomal enzymes can be simply coupled to the surface of nanocarriers, where they are essentially pro-drugs, because activity is turned on only upon encountering the acidic pH inside lysosomes. This prototype provides effective release in the lysosomal microenvironment with enhanced degradation of the accumulated substrate over naked enzymes.18
Targeting efficacy is largely impacted by both surface density of the targeting moiety on the nanocarrier particle and the bulk-concentration of nanocarriers injected in circulation. This has been shown previously in the case of ICAM-1-targeted nanocarriers lacking a therapeutic cargo.22 Although surface display of a lysosomal enzyme cargo is expected to also affect the surface-density of the targeting moiety (e.g., anti-ICAM antibodies), the presence of targeting residues on lysosomal enzymes4, 23 may compensate for decreased ICAM-1-targeting avidity, impacting tissue targeting in a manner similar to dual targeting strategies.20, 24 As an example, we focused on Fabry disease due to deficiency of α-galactosidase A (αGal), which causes lysosomal accumulation of the lipid globotriaosylceramide (Gb3) in multiple tissues and prominently in the vascular endothelium.25, 26 Although current ERT improves the disease outcome, effects on the vasculopathy associated to life-threatening cerebrovascular, cardiac and renal complications are still limited.27 Hence, predominant endothelial expression of ICAM-1 represents an attractive target to enhance delivery of this enzyme. Our previous study showed enhanced αGal delivery throughout the body by targeting ICAM-1, and provided lysosomal transport into cells with significant reduction of the corresponding lipid storage.18 In this work, we have explored how variation of the nanocarrier design parameters described above modify the absolute dose of enzyme accumulated in different organs without negatively affecting its relative biodistribution.
2. Materials and Methods
2.1. Antibodies and reagents
The monoclonal antibody used to target mouse ICAM-1 (anti-ICAM) was YN1.28 Coffee bean αGal (MW 32.25 kDa) was from Sigma-Aldrich (St. Louis, MO). Fluorescein isothiocyanate (FITC)-labeled, 100-nm diameter polystyrene particles were from Polysciences (Warrington, PA). 125Iodine labeling was performed with Na125I (Perkin Elmer-Analytical Sciences; Wellesley, MA) using Pierce Iodination Beads from Thermo Scientific (Rockford, IL). Antibody labeling with a near-infrared (NIR) fluorophore was done using Alexa Fluor 750 and SAIVI Rapid Antibody Labeling Kit (Invitrogen; Carlsbad, CA). Unless listed otherwise, all other reagents were purchased from Sigma-Aldrich (St. Louis, MO).
2.2. Nanocarrier preparation and characterization
Model polymer nanocarriers were prepared as described 18, by absorbing (e.g., hydrophobic interactions) either αGal, control IgG, a mix of anti-ICAM and IgG (40:60 mass ratio), or a mix of anti-ICAM and αGal (95:5 or 50:50 antibody-to-enzyme mass ratio) on the surface of 100-nm diameter, FITC-labeled polystyrene particles (αGal NCs, IgG NCs, anti-ICAM NCs, anti-ICAM/αGal NCs). Non-coated counterparts were separated by removing the supernatant after centrifugation, and coated nanocarriers were resuspended at the indicated concentration in phosphate-buffered saline supplemented with 0.3% bovine serum albumin, followed by gentle sonication, to prevent aggregation.18 Where specified, the targeting antibody or enzyme cargo were labeled with 125I for radioactive tracing or near-infrared (NIR) fluorophore for fluorescence tracing. The number of antibody and/or enzyme molecules per nanocarrier were determined by using 125I-labeled counterparts, as described.18 The nanocarrier surface was estimated to become fully covered with antibody and/or enzyme, with no BSA found on the coat (≤0.5% of total coat molecules). This protocol has been demonstrated to render active αGal capable of attenuating the corresponding lipid storage in lysosomes.18 We have previously characterized the release of the enzyme from anti-ICAM NCs, showing stable coating under storage and physiological conditions, and release under lysosomal-like conditions.18 The diameter and zeta potential of coated nanocarriers were measured via dynamic and phase analysis light scattering (Zetasizer nano-ZS90; Malvern Instruments; Westborough, MA).
2.3. Visualization of surface-coated nanocarriers by optical imaging in mice
Following approved guidelines established by the IACUC at University of Maryland Baltimore, female NIH Swiss mice (Harlan; Indianapolis, IN) were anesthetized and i.v. injected with FITC-nanocarriers coated with either control IgG/NIR-IgG or targeted anti-ICAM/NIR-IgG (40:60 unlabeled-to-labeled antibody mass ratio; 4×1013 NCs/kg or 8×1013 NCs/kg of body weight). Mice were euthanized under anesthesia 30 min after injection, and the abdominal and chest cavities opened for full body imaging of FITC-nanocarriers and their NIR-surface coat using a Xenogen IVIS-200 system (Caliper Life Sciences; Hopkinton, MA). Mouse organs (brain, heart, kidneys, liver, lungs, and spleen) were then dissected and separately imaged for fluorescence quantification.
2.4. Quantification by radio-isotope tracing of the biodistribution of free vs. nanocarrier-coupled enzyme cargo in mice
C57BL/6 mice (Jackson Laboratory; Bar Harbor, ME) were anesthetized and i.v. injected with control free 125I-αGal or equivalent amounts of 125I-αGal coated on NCs, either solely (2611 enzyme molecules/NC particle) or along with anti-ICAM (52, 524, or 433 enzyme molecules/NC particle; 1.6×1013 or 2.4×1013 NCs/kg of body weight). Blood samples were collected by retro-orbital bleeds at 1, 15, and 30 min post-injection and organs (brain, heart, kidneys, liver, lungs, and spleen) were dissected after euthanasia at 30 min. Samples were weighed and their 125I content was measured to determine: percent injected dose (%ID) to assess biodistribution, percent injected dose per gram of tissue (%ID/g) for comparison amongst organs with different size, and specificity index (SI) for comparison of the tissue-to-blood ratio of the targeted vs. non-targeted enzyme in each organ (see below). These studies followed IACUC protocols with humane care guidelines, approved by University of Maryland College Park.
2.5. Statistics
Data were calculated as mean ± standard error of the mean (SEM) for n≥3. Statistical significance was determined by Student’s t-tests.
3. Results and Discussion
3.1. Visualization of targeting by anti-ICAM nanocarriers
The goal of this work was to examine whether the targeting efficacy and, hence, dose of a model therapeutic protein (a lysosomal enzyme) delivered to tissues in the body can be modulated by varying the ratio of the targeting moiety vs. the cargo, and nanoparticle bulk-concentration of ICAM-1-targeted nanocarriers. Although we previously examined the effects of these variables in the biodistribution of anti-ICAM NCs,22 the presence of a cargo on the nanocarrier surface may further impact biodistribution, particularly in the case of lysosomal enzymes because they can also have targeting residues.23
For this purpose, we first examined targeting specificity and dose-dependency of a model formulation consisting of FITC-labeled polystyrene nanoparticles coated with anti-ICAM and tracer amounts of NIR-labeled IgG, to allow co-visualization of both nanoparticle and coat components, respectively. Since we aimed to evaluate targeting solely, we selected non-degradable polystyrene counterparts as model polymer nanocarriers to avoid potentially confounding results of degradation in the body. Validating this model, this formulation has been shown to have performance similar to that of the more clinically suitable ICAM-1-targeted poly(lactic-co-glycolic acid) (PLGA) nanoparticles in terms of binding, endocytosis, intracellular trafficking, and enzyme delivery in cell culture models, as well as circulation, organ targeting, and enzyme delivery in mice.15, 29 Since our goal was to examine targeting to organs, we selected 30 min after i.v. injection to avoid confounding results of other potential events occurring concomitantly (e.g., intracellular transport, transcellular transport, degradation, etc.). Nevertheless, our previous data have showed significant residence of αGal transported by anti-ICAM NCs in organs for longer periods of time (e.g., 150-fold pulmonary accumulation over free αGal 24 h after injection),18 and of the nanocarrier counterpart as well.30
Optical (fluorescence) imaging of the nanoparticle component showed clear and predominant accumulation of anti-ICAM NCs in lungs 30 min after i.v. injection, with lower accumulation in clearance organs such as the liver and spleen, whereas the opposite outcome was observed in the case of control IgG NCs (Fig. 1). Pulmonary accumulation suggests specificity of targeting, since this organ contains a significant fraction (1/5 - 1/4) of the total vascular endothelium in the body, has relatively high ICAM-1 expression, and has first-pass exposure to i.v. injected materials.15 This is in accord with our previous studies showing pulmonary accumulation of anti-ICAM NCs ~ 14.2-fold over control IgG NCs in wild-type mice,29 but not in ICAM-1 knockout mice discussed below (11.4-fold below wild-type mice; section 3.3). In addition, anti-ICAM NCs accumulated specifically at other sites, such as the kidneys (Fig. 1). This organ, although involved in passive clearance of some molecules, cannot effectively filter and, hence, do not accumulate relatively large nanocarriers unless provided with targeting features, as observed by comparing anti-ICAM NCs vs. IgG-coated NCs (Fig. 1).
Figure 1.

Visualization of specific and dose-dependent targeting of anti-ICAM NCs in mice. (a) Optical imaging of mice 30 min after i.v. injection with FITC-labeled polymer nanocarriers coated with control IgG or anti-ICAM. (b) Optical imaging of organs isolated from mice 30 min after i.v. injection with model polymer nanocarriers (4×1013 or 8×1013 NCs/kg) labeled with FITC and coated with either control IgG or anti-ICAM, where the coat also bears a fraction of Alexa Fluor 750-labeled IgG as a near-infrared (NIR) tracer (shown in Supp. Fig. 2). (c) Semi-quantitative measurement of nanocarrier distribution, from optical imaging results shown in (b).
Also demonstrating specific targeting, accumulation of anti-ICAM NCs over the level of control IgG NCs in lungs and kidneys was dose-dependent: e.g., 11.6-fold and 26.4-fold enhancement when using 4×1013 vs. 8×1013 NC/kg, respectively, in the case of lungs (Fig. 1c). The observed linear dependence in targeting of both organs suggests that binding saturation has not been reached at the doses tested. In contrast, liver accumulation of anti-ICAM NCs was consistently lower (0.6-fold) compared to IgG NCs (Fig. 1c).
These results, obtained by visualizing the nanoparticle moiety, confirm specific and dose-dependent targeting by anti-ICAM NCs, in accord with previous studies showing a similar outcome when the targeting-antibody coat component was traced by 125I-labeling.15, 29 Indeed, optical imaging showed lung co-localization of the FITC and NIR signals, which correspond to the nanocarrier particle and the antibody coat, respectively (Fig. 2). This confirms in vivo stability of the coat on the nanocarrier surface, validating previous works that show a similar result in models in vitro.18
Figure 2.
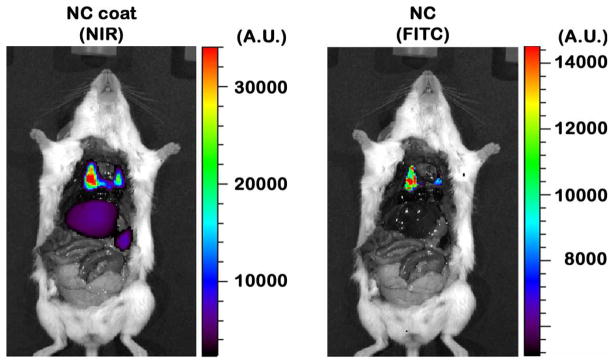
Visualization of anti-ICAM NCs and their coat in mice. Mice were injected i.v. with model FITC-labeled polymer nanocarriers coated with anti-ICAM and Alexa Fluor 750-labeled IgG as a near-infrared (NIR) tracer. Optical imaging 30 min post-injection revealed co-localization of nanocarrier particles (FITC) and their coats (NIR) in the lungs.
3.2. Comparative tissue vs. blood re-distribution of a model enzyme cargo, αGal, by anti-ICAM nanocarriers
As described, ICAM-1 targeting has shown considerable potential to improve delivery of therapeutic proteins, such as the enzymes used for lysosomal ERT,14–21 where simple coupling to the nanocarrier surface yields a pro-drug with release and effects only upon transport to the lysosomal environment.18 Also, for this type of application, targeting to both the brain and peripheral organs throughout the body is needed, to cope with multi-organ dysfunction associated with LSDs.7 As an example, we selected α-galactosidase (αGal), which is involved in catabolism of certain glycosphingolipids and is deficient in Fabry disease.25 The vascular endothelium represents a key target for therapeutic intervention of this condition.25–27 Therefore, αGal delivery is a particularly appropriate candidate for targeting via ICAM-1, as we have showed.16, 18
We prepared three different anti-ICAM NC formulations by varying (1) targeting antibody to enzyme cargo ratio, or (2) nanoparticle bulk-concentration, to examine the impact of these variations on the in vivo biodistribution of the lysosomal enzyme (Fig. 3). For two formulations, the surface-density of anti-ICAM-to-αGal was varied from ~95:5 to 50:50 mass ratio, which rendered an enzyme load of ~50 vs. 500 αGal molecules per particle. These formulations were injected at a concentration of 1.6×1013 NCs/kg, which represents final enzyme doses of ~45 (low) vs. 449 (intermediate) αGal μg/kg. In the third formulation, the coating was maintained at a 50:50 anti-ICAM-to-αGal mass ratio, yet the concentration of the injected dose was varied to 2.4×1013 NCs/kg, which represents a final enzyme dose of ~550 (high) αGal μg/kg. For all the formulations, the size, polydispersity, and zeta potential of the resulting nanocarriers was similar, ranging between 223–240 nm in diameter, 0.126 to 0.129 polydispersity index, and from −9.12 to −12.0 mV, respectively (Fig. 3).
Figure 3.

Anti-ICAM/αGal NC formulations. Model 100-nm diameter, polystyrene nanocarrier were coated with anti-ICAM as a targeting moiety and αGal as an enzyme cargo. Three formulations were prepared by varying the antibody-to-enzyme ratio on the nanocarrier surface or the final concentration of nanocarriers in solution, as indicated. These variations rendered three different enzyme doses: low, intermediate, or high. The size, polydispersity, zeta potential, surface coating, and nanocarrier bulk-concentration of these formulations are provided.
All three anti-ICAM/125I-αGal NC formulations or equivalent doses of naked, non-targeted 125I-αGal were administered separately into mice to examine their overall biodistribution after i.v. injection. In all three cases, αGal delivered by anti-ICAM NCs rapidly disappeared from the circulation (Fig. 4), while naked or free αGal remained in blood to a much greater extent. For instance (Fig. 5a), 30 min after injection, the level of αGal in circulation associated with anti-ICAM NCs ranged from ~2 to 6%ID, contrasting with ~40 to 60%ID of free αGal, a ~10-fold decrease in circulation of anti-ICAM/αGal NCs compared to the naked enzyme. A priori, short half-life in blood may seem detrimental to efficient therapy. Indeed, this is the case for delivery strategies that rely on diffusion of relatively small drug molecules from the circulation into tissues, or for drug carriers designed for delivery of cancer therapeutics.31 These strategies require passive transport into the tumor parenchyma across the leaky vasculature of tumor blood vessels, via the enhanced permeability and retention effect.31 However, rapid removal from the blood may be beneficial to avoid premature degradation and/or recognition by the immune system.5, 9 This is particularly relevant in the case of therapeutic biologics, as long as fast clearance from blood does not impair targeting of the intended tissues. On this regard, presentation of multiple copies of the enzyme on the nanocarrier surface may also pose immunological concerns, which remain to be investigated. Yet, if this was the case, encapsulating formulations can be pursued in order to ameliorate this downside of ERT strategies.
Figure 4.
Comparative circulation of naked αGal vs αGal coupled to anti-ICAM NCs. (a) Naked, 125I-αGal formulations, or (b) equivalent doses of 125I-αGal coated on anti-ICAM NC, as described in Fig. 3, were injected i.v. in mice. Blood samples were collected at 1, 15, and 30 min post-injection and measured for 125I-αGal content to determine percent of total injected dose (%ID) remaining in circulation. Intm = Intermediate dose. Data are mean ± SEM; n≥4. Comparison by Student’s t-test of each nanocarrier formulation to its naked enzyme counterpart rendered p<0.001.
Figure 5.

Anti-ICAM NCs switch biodistribution of αGal from blood to tissues. Mice were injected i.v. with each one of the anti-ICAM/125I-αGal NC formulations described in Fig. 3 or an equivalent dose of naked, non-targeted 125I-αGal. Samples of (a) blood and (b) organs (including brain, heart, kidneys, liver, lungs, and spleen) were collected 30 min after injection and measured for 125I-αGal content to calculate the percent of total injected dose (%ID) remaining in (a) circulation or (b) cumulatively found in tissues. Intm = Intermediate dose. Data are mean ± SEM; n≥4; *** p<0.001, comparing each nanocarrier formulation to naked enzyme, by Student’s t-test.
As shown in Fig. 5b, anti-ICAM/αGal NCs accumulated in tissues at a greater extent than naked αGal. Considering collectively the major organs of the body (brain, heart, kidneys, liver, lungs, and spleen), ICAM-1 targeting resulted in tissue accumulation ranging from ~60 to 70%ID, compared with only ~20%ID in the case of the free enzyme. This represents ~3.5-fold increase of global tissue distribution as a result of ICAM-1 targeting. This level of enhancement still underestimates the targeting potential of anti-ICAM NCs, because the circulating enzyme level is significantly higher for naked αGal (Fig. 5a), and this circulating fraction also contributes to the level of enzyme detected in each organ. This result is comparable to previously published work regarding other anti-ICAM/enzyme NC systems, including acid sphingomyelinase delivery for type A-B Niemann-Pick disease (67.18%ID in tissue) and α-glucosidase delivery for Pompe disease (71.62%ID), where both these formulations were prepared similarly to the low dose αGal formulation described here.15, 17
With regard to comparing the global targeting efficacy (cumulative %ID in all organs) of the different anti-ICAM/αGal NC formulations, the amount of targeting anti-ICAM on the nanocarrier surface was reduced by half (from 181 to 83 molecules per particle) when increasing the enzyme load by 10-fold (low and intermediate αGal doses). This is in line with the size difference between these molecules, e.g., ~150 kDa for anti-ICAM vs. ~32 kDa for the αGal form used. However, despite this variation of surface-density of the targeting coat, as well as the change concerning nanocarrier-bulk concentration (from 1.6×1013 to 2.4×1013 NCs/kg), no statistically significant differences were observed, and all formulations cause a similar blood-to-tissue shift of the enzyme.
3.3. Effect of varying targeting surface-density or nanocarrier bulk-concentration on αGal biodistribution
We next evaluated whether modification of any of the design parameters described above led to changes in the particular biodistribution pattern of anti-ICAM/αGal NCs. As shown in Fig. 6, very few changes were found concerning different antibody surface-densities (low vs. intermediate αGal doses; 45 vs. 449 μg αGal/kg), which occurred in the liver. For instance, a 2-fold reduction of the antibody surface-density on the nanocarrier coat resulted in an increased liver accumulation of αGal from ~12 to 24%ID/g (Fig. 6b). This seems to reflect reduced targeting, which is only noticeable in the brain, possibly due to the low magnitude of targeting to this organ as compared to other tissues. Instead, the cumulative effect of small reductions in targeting through the body was noticed in the liver, likely due to non-specific clearance. In accord with this, coating of αGal alone on the surface of nanocarriers lead to an even more enhanced liver accumulation (53.4±2.6%ID; data not shown), which is a characteristic clearance pattern for particulate objects of this size range.32
Figure 6.

Similar αGal biodistribution with varying the design parameters of anti-ICAM NCs. Mice were injected i.v. with 1.6 x1013 NCs/kg of anti-ICAM/125I-αGal NCs prepared to contain 95:5 or 50:50 antibody-to-enzyme surface-density ratios (52 vs. 524 αGal molec/NC), which rendered low (45 μg/kg) or intermediate (Intm; 449 μg/kg) αGal doses, respectively. In addition, anti-ICAM/125I-αGal NCs displaying 50:50 antibody-to-enzyme surface-density ratios was injected at 2.4×1013 NCs/kg (high αGal dose; 555 μg/kg). Organs were harvested 30 min after injection, weighed, and measured for 125I content to determine (a) percent of total injected αGal dose (%ID) in each organ and (b) relative tissue-to-blood targeting efficiency as %ID αGal per gram of organ (%ID/g) to compare among organs of different size. Data are mean ± SEM; n≥4; *p<0.05, **p<0.01, comparing to low αGal doses by Student’s t-test. Comparing intermediate and high αGal doses rendered p>0.1.
However, varying anti-ICAM surface density did not cause significant changes in the levels of enzyme accumulation in other organs, which represent Fabry targets. For example, consistent with optical imaging results using fluorescence tracing of the nanocarrier particles shown above, radioactive labeling of the enzyme coat indicated that lungs accumulated the highest dose of anti-ICAM/αGal NCs for all formulations (~35%ID). This was also the case after normalization by organ weight, with ~180%ID/g (Fig. 6b). This suggests endothelial specificity, validated by the fact that control IgG NCs or αGal NCs did not accumulate in the lungs (1.6±0.3%ID and 0.9±0.1%ID, respectively; data not shown) and this was also the case for anti-ICAM NCs injected in ICAM-1 knockout mice (2.0±0.3%ID; data not shown). Kidneys and heart accumulated ~2%ID and ~0.3 %ID αGal, equivalent to ~5%ID/g and ~2%ID/g of the enzyme, respectively, regardless of the variation in the antibody surface density.
These findings are consistent with previous results demonstrating no significant changes in the pulmonary biodistribution of cargo-free anti-ICAM NCs, along with an enhanced accumulation in the liver, when antibody surface-densities varied between 55 and 165 molecules/NC22 (surface-densities in this study are 83 vs.181 antibodies/NC). Perhaps at this range of antibody surface density, nanocarrier particles already surpass the avidity threshold that allows them to effectively bind to the target in vivo, and this makes it possible for the enzyme cargo to occupy the nanocarrier coat without impairing binding. Minimal retention in circulation described above (Fig. 4 and 5a) indeed suggests rapid targeting.
In addition, no changes where observed by varying the carrier-bulk concentration within the range tested, corresponding to intermediate vs. high αGal doses; 449 vs. 550 μg αGal/kg (note that the statistical significance associated with high dose in Fig. 6 is compared to low dose, which also encompasses different targeting surface density). The lack of biodistribution changes observed here regarding variation of the nanocarrier bulk-concentration is opposite to the outcome reported for cargo-free anti-ICAM NCs.22 However, these differences may be due to the different nanocarrier concentrations used in these two studies: 1.6×1013 and 4.8×1013 NC/kg (a 3-fold change) employed in a previous work vs. 1.6×1013 and 2.4×1013 NC/kg in this report, where the 1.5-fold concentration change may have been insufficient to cause targeting differences. This outcome may have been also affected by the allegedly lower avidity of nanocarriers used in this comparison, which displayed ~85 anti-ICAM molecules/NC vs. 209 anti-ICAM molecules/NC reported previously.22
Interestingly, the relative extent of targeting vs. clearance, exemplified by accumulation of anti-ICAM NCs in the lungs vs. liver, is significantly different between cargo-free22 and αGal-containing counterparts shown here. For instance, in a previous work, pulmonary accumulation of cargo-free anti-ICAM NCs displaying 55 vs. 165 antibody molecules/NC was 56%ID/g vs. 63%ID/g, respectively, compared to 48%ID/g vs. 46%ID/g for the liver.22 In contrast, in this study using anti-ICAM/αGal NCs displaying 83 vs.181 antibodies/NC, pulmonary accumulation was 192%ID/g vs. 175%ID/g, respectively, compared to 24%ID/g vs. 12%ID/g for the liver. It thus appears that targeting of αGal-loaded anti-ICAM NCs is enhanced. This could be due to relative differences between the anti-ICAM batch used and/or the presence of the enzyme on the nanocarrier coat, which could have conferred enhanced avidity. This is possible because most lysosomal enzymes display mannose or mannose-6-phosphate residues that can interact with their receptors and also αGal can interact with glycosphingolipid ligands on the cell surface.4, 23 In particular, as described above, pulmonary accumulation of αGal NCs was negligible (0.9±0.1%ID) and similar to that of control IgG NCs (1.6±0.3%ID), indicating a synergistic rather that additive effect. It is likely that avidity of multivalent αGal is still relatively low as to confer relevant targeting by itself, yet after initial binding to the endothelium via ICAM-1, additional αGal interactions may occur. Similar effects have been observed for strategies where one nanocarrier particle is targeted to multiple receptors;20, 24 yet in this study the cargo, not a second targeting moiety, would provide this ability.
3.4. Specificity and absolute αGal delivery of by anti-ICAM NCs
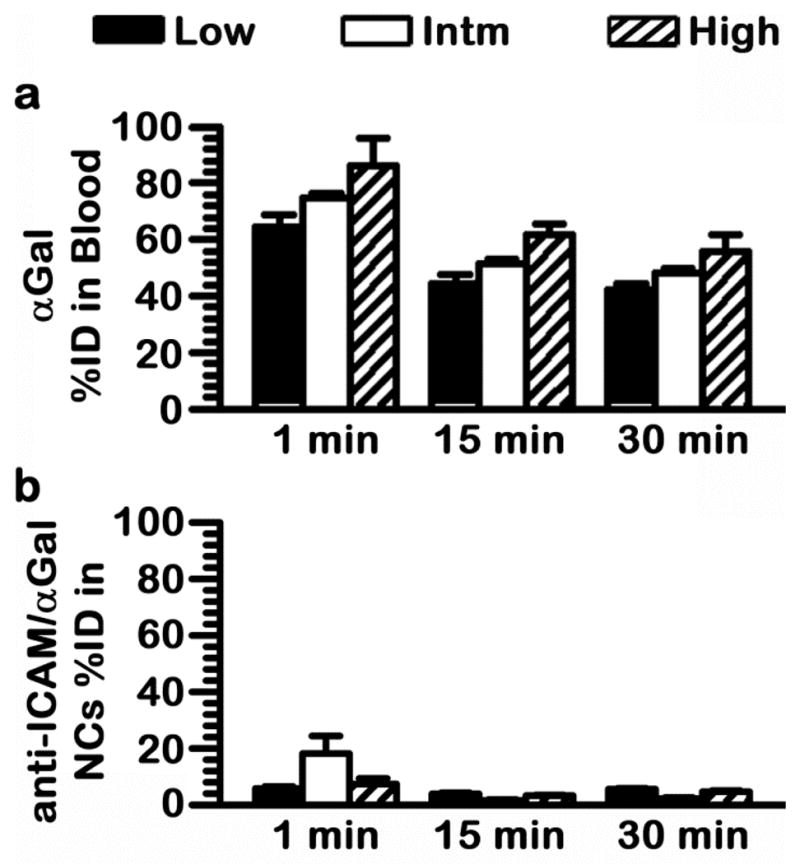
All anti-ICAM NC formulations resulted in similar overall shift αGal from the blood to tissues (Fig. 5), with a similar relative biodistribution in organs (Fig. 6). Since dose may also affect the biodistribution of naked αGal, we next examined the specificity offered by ICAM-1-targeting. This was achieved by determining the effect of varying antibody surface-density on the nanocarrier particle or the nanocarrier bulk-concentration (described above) on the specificity index (SI) of anti-ICAM/αGal NCs. SI compares the organ-to-blood targeting ratio of anti-ICAM/αGal NCs over naked αGal, hence, correcting for the different circulating fractions in these formulations (see Methods).
For all three formulations, SI was greater than 1 in all organs tested, reflecting a specific increase on enzyme targeting as a result of coupling it to anti-ICAM NCs, across variations of the injected enzyme dose from ~45 to 555 μg/kg (Fig. 7a). Counter-intuitively, the formulation displaying a greater targeting specificity in each organ was the one with reduced antibody surface-density and nanocarrier bulk-concentration (intermediate αGal dose of 449 μg/kg). For this formulation, anti-ICAM NCs enhanced specific targeting of αGal from 4-fold (in kidneys) up to 981-fold (in lungs) over the equivalent naked counterpart. This could be explained if the range of anti-ICAM surface-density used in this study already surpassed the threshold for effective targeting in vivo, and αGal presented on the nanocarrier surface could provide a synergistic targeting effect, as discussed above.
Figure 7.

Enhanced specificity and absolute αGal targeting by varying nanocarrier design parameters. Mice were injected i.v. with anti-ICAM/125I-αGal NCs displaying varying antibody-to-enzyme surface-density ratios, at varying nanocarrier bulk-concentrations, rendering low (45 μg/kg), intermediate (Intm; 449 μg/kg), or high (555 μg/kg) αGal doses, or with equivalent doses of naked, non-targeted 125I-αGal. Blood samples and organs were harvested, weighed, and measured for 125I content 30 min after injection. (a) Specificity index was determined by comparing %ID/g per gram of organ over % ID per gram of blood (tissue-to-blood localization ratio) of anti-ICAM/125I-αGal NCs to the equivalent parameter for naked, non-targeted αGal. This parameter describes the fold enhancement of αGal targeted to tissue via anti-ICAM NCs. (b) Total dose (ng) of 125I-αGal accumulated in each organ. Data are mean ± SEM; n≥4; *p<0.05, **p<0.01, comparing intermediate vs. high αGal doses by Student’s t-test. Comparing intermediate or high to low αGal doses rendered p<0.001.
A reduced enhancement in tissue targeting specificity was seen when varying nanocarrier bulk-concentration between 1.6×1013 and 2.4×1013 NC/kg. Judging by the similar biodistribution (Fig. 6) but not specificity (Fig. 7) patterns observed for these two formulations, it appears a change in the nanocarrier concentration within this range does not impact binding in the case of high-avidity multivalent anti-ICAM/αGal NCs. Instead, it is plausible that such a change in concentration enhances binding of allegedly low-avidity, monovalent naked αGal. If this was the case, the outcome would be a decreased SI for anti-ICAM/αGal NCs, as observed. Nonetheless, even in this situation ICAM-1 targeting offered a marked and significant enhancement in the specific tissue accumulation of αGal, with SI values ranging from 3.5 to 496 for high dose, observed in the kidneys and lungs, respectively.
Importantly, modulation of either antibody surface-density or nanocarrier bulk-concentration, both resulted in an increase in the absolute amount of αGal accumulated in each organ (Fig. 7b). Since variation of these nanocarrier design parameters did not negatively or significantly impact the potential for ICAM-1 targeting through the body, the increasing dose of αGal carried by these three formulations resulted in a corresponding increase in the enzyme delivered. Anti-ICAM/αGal NCs displaying ~500 αGal molecules/NC (449 μg αGal/kg; intermediate dose) enhanced αGal accumulation in organs (cumulatively) over 10-fold vs. counterparts displaying ~50 αGal molecules/NC (45 μg αGal/kg; low dose). Anti-ICAM/αGal NCs displaying ~500 αGal molecules/NC and administered at 2.4×1013 NCs/kg (550 μg αGal/kg; high dose) enhanced cumulative αGal targeting by 20% vs. administration of the same nanocarriers at 1.6×1013 NCs/kg (449 μg αGal/kg; intermediate dose). Total αGal delivered to the lungs 30 min after i.v. injection increased from ~270 ng to ~5800 ng for these three formulations. Significant enhancement was also found even for organs that are difficult to target, such as the brain, where αGal delivery increased ~20-fold when comparing high to low dose (max. brain accumulation was 17.8 ng). This was also the case for the heart and kidneys, key Fabry organs, with maximum accumulation of ~48 ng and ~435 ng, respectively.
Notably, total amount of αGal delivered to tissues increased from 0.45 μg (low dose) to 7.7 μg (intermediate dose) to 10.8 μg (high dose), which is very significant compared to the initial administered dose of ~0.73 μg, ~11.7 μg, and ~15.8 μg of αGal, respectively. This represents a consistent pattern of 65% of the injected dose accumulating in the six harvested organs. Although these data was obtained 30 min after injection, as mentioned above, previously published results indicated that a significant amount of αGal transported by anti-ICAM NCs remain in the targeted organs 24 h after administration (e.g., 150-fold pulmonary accumulation over free αGal at this time).18, 30 This result suggests that these variations of the nanocarrier formulation have a linear effect on the enzyme delivery, rendering a relatively controllable system for delivering such a cargo. From a translational perspective, current ERT for Fabry disease in the clinic typically consists of i.v. infusions of 1 mg/kg every two weeks, associated with high probability for development of resistance due to generation of anti-αGal blocking antibodies.9 The lowest dose of 45 μg of αGal/kg tested in this study is ~20-fold lower than current clinical doses, yet ICAM-1 targeting provides targeting enhancement compared to the naked enzyme.
4. Conclusion
Both the surface density of the targeting moiety on ICAM-1-targeted nanocarriers and nanocarrier bulk-concentration injected in circulation can be varied to regulate the total amount of a surface-loaded cargo (such as the case of therapeutic lysosomal enzyme pro-drugs) that can reach target organs in the body, without negatively impacting targeting specificity and biodistribution. The presence on the nanocarrier surface of a cargo that has affinity toward cell-surface markers seems to somehow compensate for the potential decay in avidity associated with the reduced surface-density of the (antibody) targeting moiety. Hence, this strategy can be used to enhance tissue delivery of lower lysosomal enzyme doses which, along with fast removal from the circulation, may help improve the efficacy of lysosomal ERT and, likely, similar therapeutic approaches.
Acknowledgments
The authors would like to thank Dr. Stuart Martin in the Department of Physiology and Greenbaum Cancer Center, University of Maryland Baltimore, for graciously allowing us to use the IVIS system. This work was funded by NIH R01-HL09816 (S.M.).
Abbreviations
- αGal
α-galactosidase
- BSA
bovine serum albumin
- ERT
enzyme replacement therapy
- FITC
fluorescein isothiocyanate
- ICAM-1
intercellular adhesion molecule-1
- IgG
immunoglobulin G
- Intm
Intermediate dose
- i.v
intravenous
- LSD
lysosomal storage disorder
- NC
nanocarrier
- %ID
percentage of injected dose
- %ID/g
percentage of injected dose per gram
- SI
specificity index
References
- 1.Solaro R, Chiellini F, Battisti A. Targeted Delivery of Protein Drugs by Nanocarriers. Materials. 2010;3:1928. [Google Scholar]
- 2.Torchilin VP. Multifunctional nanocarriers. Adv Drug Deliv Rev. 2006;58:1532. doi: 10.1016/j.addr.2006.09.009. [DOI] [PubMed] [Google Scholar]
- 3.Futerman AH, van Meer G. The cell biology of lysosomal storage disorders. Nat Rev Mol Cell Biol. 2004;5:554. doi: 10.1038/nrm1423. [DOI] [PubMed] [Google Scholar]
- 4.Neufeld EF. The uptake of enzymes into lysosomes: an overview. Birth Defects Orig Artic Ser. 1980;16:77. [PubMed] [Google Scholar]
- 5.Brooks DA. Immune response to enzyme replacement therapy in lysosomal storage disorder patients and animal models. Mol Genet Metab. 1999;68:268. doi: 10.1006/mgme.1999.2894. [DOI] [PubMed] [Google Scholar]
- 6.Dhami R, Schuchman EH. Mannose 6-phosphate receptor-mediated uptake is defective in acid sphingomyelinase-deficient macrophages: implications for Niemann-Pick disease enzyme replacement therapy. J Biol Chem. 2004;279:1526. doi: 10.1074/jbc.M309465200. [DOI] [PubMed] [Google Scholar]
- 7.Muro S. Strategies for delivery of therapeutics into the central nervous system for treatment of lysosomal storage disorders. Drug Delivery and Translational Research. 2012;2:169. doi: 10.1007/s13346-012-0072-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sun B, Li S, Bird A, Yi H, Kemper A, Thurberg BL, Koeberl DD. Antibody formation and mannose-6-phosphate receptor expression impact the efficacy of muscle-specific transgene expression in murine Pompe disease. J Gene Med. 2010;12:881. doi: 10.1002/jgm.1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wilcox WR, Linthorst GE, Germain DP, Feldt-Rasmussen U, Waldek S, Richards SM, Beitner-Johnson D, Cizmarik M, Cole JA, Kingma W, Warnock DG. Anti-alpha-galactosidase A antibody response to agalsidase beta treatment: data from the Fabry Registry. Mol Genet Metab. 2012;105:443. doi: 10.1016/j.ymgme.2011.12.006. [DOI] [PubMed] [Google Scholar]
- 10.Hsu J, Muro S. In: Nanomedicine and drug delivery strategies for treatment of genetic diseases in Human genetic diseases. Plaseska-Karanfilska D, editor. InTech; Rijeka, Croatia: 2011. p. 241. [Google Scholar]
- 11.Muro S. New biotechnological and nanomedicine strategies for treatment of lysosomal storage disorders. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2010;2:189. doi: 10.1002/wnan.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Marlin SD, Springer TA. Purified intercellular adhesion molecule-1 (ICAM-1) is a ligand for lymphocyte function-associated antigen 1 (LFA-1) Cell. 1987;51:813. doi: 10.1016/0092-8674(87)90104-8. [DOI] [PubMed] [Google Scholar]
- 13.Muro S. In: Intercellular Adhesion Molecule-1 and Vascular Cell Adhesion Molecule-1 in Endothelial biomedicine. Aird WC, editor. Aird W. C.s, Cambridge University Press; New York: 2007. p. 1058. [Google Scholar]
- 14.Bhowmick T, Hsu J, Dhami R, Burks SR, Kao JPY, Schuchman EH, Muro S. Controlled Release Society, Journal of Controlled Release. National Harbor, Maryland, U.S.A: 2011. ICAM-1-targeted nanocarriers provide efficient targeting, transport across the BBB, and uniform distribution and effects of therapeutic enzymes in the brain; p. 34. [Google Scholar]
- 15.Garnacho C, Dhami R, Simone E, Dziubla T, Leferovich J, Schuchman EH, Muzykantov V, Muro S. Delivery of acid sphingomyelinase in normal and niemann-pick disease mice using intercellular adhesion molecule-1-targeted polymer nanocarriers. J Pharmacol Exp Ther. 2008;325:400. doi: 10.1124/jpet.107.133298. [DOI] [PubMed] [Google Scholar]
- 16.Ghaffarian R, Bhowmick T, Muro S. Transport of nanocarriers across gastrointestinal epithelial cells by a new transcellular route induced by targeting ICAM-1. J Control Release. 2012;163:25. doi: 10.1016/j.jconrel.2012.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hsu J, Northrup L, Bhowmick T, Muro S. Enhanced delivery of alpha-glucosidase for Pompe disease by ICAM-1-targeted nanocarriers: comparative performance of a strategy for three distinct lysosomal storage disorders. Nanomedicine. 2012;8:731. doi: 10.1016/j.nano.2011.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hsu J, Serrano D, Bhowmick T, Kumar K, Shen Y, Kuo YC, Garnacho C, Muro S. Enhanced endothelial delivery and biochemical effects of alpha-galactosidase by ICAM-1-targeted nanocarriers for Fabry disease. J Control Release. 2011;149:323. doi: 10.1016/j.jconrel.2010.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Muro S, Schuchman EH, Muzykantov VR. Lysosomal enzyme delivery by ICAM-1-targeted nanocarriers bypassing glycosylation- and clathrin-dependent endocytosis. Mol Ther. 2006;13:135. doi: 10.1016/j.ymthe.2005.07.687. [DOI] [PubMed] [Google Scholar]
- 20.Papademetriou IT, Garnacho C, Schuchman EH, Muro S. In vivo performance of polymer nanocarriers dually-targeted to epitopes of the same or different receptors. Biomaterials. 2013;34:3459. doi: 10.1016/j.biomaterials.2013.01.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Papademetriou J, Garnacho C, Serrano D, Bhowmick T, Schuchman EH, Muro S. Comparative binding, endocytosis, and biodistribution of antibodies and antibody-coated carriers for targeted delivery of lysosomal enzymes to ICAM-1 versus transferrin receptor. J Inherit Metab Dis. 2013;36:467. doi: 10.1007/s10545-012-9534-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Calderon AJ, Bhowmick T, Leferovich J, Burman B, Pichette B, Muzykantov V, Eckmann DM, Muro S. Optimizing endothelial targeting by modulating the antibody density and particle concentration of anti-ICAM coated carriers. J Control Release. 2011;150:37. doi: 10.1016/j.jconrel.2010.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gary-Bobo M, Nirde P, Jeanjean A, Morere A, Garcia M. Mannose 6-phosphate receptor targeting and its applications in human diseases. Curr Med Chem. 2007;14:2945. doi: 10.2174/092986707782794005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Saul JM, Annapragada AV, Bellamkonda RV. A dual-ligand approach for enhancing targeting selectivity of therapeutic nanocarriers. J Control Release. 2006;114:277. doi: 10.1016/j.jconrel.2006.05.028. [DOI] [PubMed] [Google Scholar]
- 25.Desnick RJ, Ioannou YA, Eng CM. α-Galactosidase A Deficiency: Fabry Disease. In: Scriver C, Beaudet A, Sly W, Valle D, Childs B, Kinzler K, Vogelstein B, Bunz F, Scriver C, Beaudet A, Sly W, Valle D, Childs B, Kinzler K, Vogelstein B, Bunzs F, editors. The Metabolic and Molecular Bases of Inherited Disease. Vol. 3. McGraw-Hill; 2001. p. 3733. [Google Scholar]
- 26.Park JL, Whitesall SE, D’Alecy LG, Shu L, Shayman JA. Vascular dysfunction in the alpha-galactosidase A-knockout mouse is an endothelial cell-, plasma membrane-based defect. Clin Exp Pharmacol Physiol. 2008;35:1156. doi: 10.1111/j.1440-1681.2008.04984.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Desnick RJ. Enzyme replacement therapy for Fabry disease: lessons from two alpha-galactosidase A orphan products and one FDA approval. Expert Opin Biol Ther. 2004;4:1167. doi: 10.1517/14712598.4.7.1167. [DOI] [PubMed] [Google Scholar]
- 28.Jevnikar AM, Wuthrich RP, Takei F, Xu HW, Brennan DC, Glimcher LH, Rubin-Kelley VE. Differing regulation and function of ICAM-1 and class II antigens on renal tubular cells. Kidney Int. 1990;38:417. doi: 10.1038/ki.1990.221. [DOI] [PubMed] [Google Scholar]
- 29.Muro S, Dziubla T, Qiu W, Leferovich J, Cui X, Berk E, Muzykantov VR. Endothelial targeting of high-affinity multivalent polymer nanocarriers directed to intercellular adhesion molecule 1. J Pharmacol Exp Ther. 2006;317:1161. doi: 10.1124/jpet.105.098970. [DOI] [PubMed] [Google Scholar]
- 30.Rossin R, Muro S, Welch MJ, Muzykantov VR, Schuster DP. In vivo imaging of 64Cu-labeled polymer nanoparticles targeted to the lung endothelium. J Nucl Med. 2008;49:103. doi: 10.2967/jnumed.107.045302. [DOI] [PubMed] [Google Scholar]
- 31.Matsumura Y, Maeda H. A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986;46:6387. [PubMed] [Google Scholar]
- 32.Muro S, Garnacho C, Champion JA, Leferovich J, Gajewski C, Schuchman EH, Mitragotri S, Muzykantov VR. Control of endothelial targeting and intracellular delivery of therapeutic enzymes by modulating the size and shape of ICAM-1-targeted carriers. Mol Ther. 2008;16:1450. doi: 10.1038/mt.2008.127. [DOI] [PMC free article] [PubMed] [Google Scholar]